

Abstract
Abnormal cholesterol/lipid homeostasis is linked to neurodegenerative conditions such as age-related macular degeneration (AMD), which is a leading cause of blindness in the elderly. The most prevalent form, termed “dry” AMD, is characterized by pathological cholesterol accumulation beneath the retinal pigment epithelial (RPE) cell layer and inflammation-linked degeneration in the retina. We show here that the cholesterol-regulating microRNA miR-33 was elevated in the RPE of aging mice. Expression of the miR-33 target ATP-binding cassette transporter (ABCA1), a cholesterol efflux pump genetically linked to AMD, declined reciprocally in the RPE with age. In accord, miR-33 modulated ABCA1 expression and cholesterol efflux in human RPE cells. Subcutaneous delivery of miR-33 antisense oligonucleotides (ASO) to aging mice and non-human primates fed a Western-type high fat/cholesterol diet resulted in increased ABCA1 expression, decreased cholesterol accumulation, and reduced immune cell infiltration in the RPE cell layer, accompanied by decreased pathological changes to RPE morphology. These findings suggest that miR-33 targeting may decrease cholesterol deposition and ameliorate AMD initiation and progression.
Keywords: microRNA, ABCA1, cholesterol, age-related macular degeneration, inflammation, retinal pigment epithelial cells, geographic atrophy
Graphical abstract
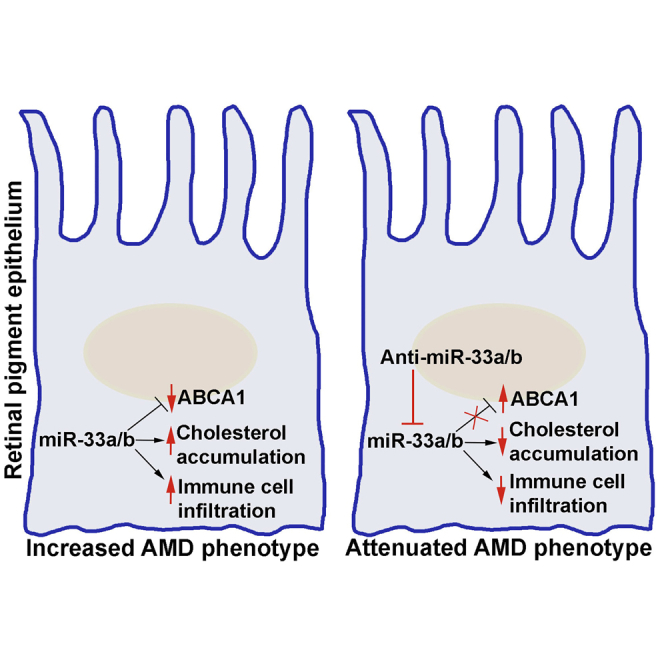
Age-related macular degeneration (AMD) is an irreversible blinding condition. Currently, no defined therapies are particularly available to treat the dry form of AMD. Here, Gnanaguru and colleagues demonstrate that anti-sense oligonucleotide targeting of microRNA-33a/b isoforms reduces cholesterol accumulation and immune cell infiltration and ameliorates pathologies associated with dry AMD in clinically relevant non-human primates.
Introduction
Defective cholesterol/lipid homeostasis is linked to neurodegenerative conditions, including age-related macular degeneration (AMD).1 In particular, deposition of cholesterol and cholesterol-containing drusen in the retinal pigment epithelial (RPE) and sub-RPE layers is strongly associated with the development of AMD.2, 3, 4 Moreover, genome-wide association studies (GWAS) of genetic risk factors linked to AMD have identified single-nucleotide polymorphisms near genes involved in cholesterol and lipid regulation, such as ABCA1, APOE, CETP, and LIPC.5, 6, 7, 8 A recent report indicated that a Western-type dietary pattern in humans also increases the risk of developing late-stage AMD by three-fold.9 Even though normal mice do not develop signature human AMD pathology, studies with high-fat/cholesterol-enriched diet-fed aging C57BL/6J or ApoE4 knock-in mice and ApoB100/Ldlr−/− mice also demonstrate that cholesterol accumulation induces RPE atrophy and basal laminar deposit formation,10, 11, 12, 13, 14, 15 providing further support for the hypothesis that abnormal cholesterol accumulation in the retina represents a prominent pathological feature.
The pathological progression of early-stage AMD can lead to advanced-stage wet AMD or dry AMD (also known as geographic atrophy).16 At present, some therapeutic options are available for the less-frequent wet AMD.17 While there is currently no treatment for geographic atrophy (which accounts for 85%–90% of all AMD cases), targeting mechanisms linked to cholesterol accumulation may be a viable therapeutic strategy in dry AMD.18
RPE cells are key regulators of cholesterol homeostasis in the retina,19 and ATP-binding cassette transporter (ABCA1) expressed by RPE cells facilitates the efflux of cholesterol.20,21 Mice that lack Abca1 along with Abcg1 in the RPE layer develop AMD-like pathology that includes cholesterol accumulation, RPE and photoreceptor degeneration, and inflammation.20 Additionally, a previous study revealed that expression of Abca1 is downregulated during aging in macrophages and linked the corresponding decreased cholesterol efflux capacity in aging macrophages to elevated retinal inflammation in a laser-induced model for choroidal neovascularization (CNV).22
MicroRNAs (miRNAs) are short (∼18–24 nucleotides) regulatory non-coding RNAs with diverse functions in development, metabolism, and disease.23 Aberrant expression or function of miRNAs has been linked to a number of diseases, and inhibition of several disease-associated miRNAs with anti-miR antisense oligonucleotides (ASOs) has recently been explored as a therapeutic intervention.24,25
We have for a number of years investigated conserved mechanisms by which the sterol regulatory element-binding protein (SREBP) family of transcription factors governs cholesterol/lipid and metabolic homeostasis.26, 27, 28, 29, 30 We and others discovered that the human SREBP genes surprisingly harbor intronic miRNAs, miR-33a/b.31, 32, 33, 34, 35, 36, 37, 38 Intriguingly, our studies and those of others have revealed that miR-33a/b act to modulate several interconnected metabolic circuits, while also cooperating with the SREBP transcription factors to promote elevated intracellular cholesterol/fatty acids and other lipids. Importantly, injection of ASOs directed against miR-33 results in significantly increased hepatic and macrophage ABCA1 expression and elevated circulating high-density-lipoprotein-cholesterol (HDL-C) in mice and non-human primates (NHPs) fed a Western-type diet (WTD) and decrease atherosclerosis in Ldlr−/− and Apoe−/− mice.31, 32, 33, 34,38, 39, 40
We have also carried out a systematic analysis of GWAS involving >188,000 individuals, associating common SNPs with abnormal plasma lipids, resulting in the identification of 69 miRNAs. Two miRNAs, miR-128-1 and miR-148a, emerged from these analyses as the top microRNAs with predicted and verified targets involved in cholesterol/lipid and metabolic homeostasis, including ABCA1.41 As with miR-33, we found that these miRNAs are also critical regulators of ABCA1-dependent cholesterol efflux from macrophages.41 Moreover, we recently showed that miR-128-1 is more broadly regulating lipid and metabolic circuits, linked to human positive selection.42 These studies together provide evidence that miRNAs may serve as key regulators of cholesterol/lipid homeostasis, with important implications for cholesterol/lipid disorders.
Interestingly, a previous study found that miR-33a expression is elevated in aging mouse macrophages and linked this to increased inflammation and CNV in the mouse eye, hallmarks of the wet form of AMD.22 However, the potential roles of cholesterol/lipid-regulating miRNAs in animal models recapitulating aspects of the predominant dry form of AMD have not been explored. Taken together with the apparent cholesterol accumulation in AMD and strong genetic connections with cholesterol regulators, this then provided the impetus to explore more deeply the potential broader functional association of miRNAs with dry AMD. Here, we describe our evaluation of miRNA regulation of ABCA1 and high-fat/cholesterol diet-induced cholesterol accumulation and AMD-like pathologies in the eye of clinically relevant NHPs and in mice.
Results
miR-33a/b regulates RPE ABCA1 expression and cholesterol efflux
As human SREBP genes harbor intronic miR-33a/b,31,38 we first investigated the expression of miR-33a/b in primary human RPE cells and then further characterized the role of miR-33a/b in RPE cholesterol efflux using the human RPE-derived cell line ARPE-19. Analysis of primary human RPE cells show that they express miR-33a and miR-33b isoforms (Figure 1A). Furthermore, introduction of miR-33a and/or miR-33b precursors resulted in decreased ABCA1 levels in primary human RPE (Figure 1B), whereas inhibition of endogenous miR-33a or miR-33b using anti-miR ASOs in primary human RPE increased ABCA1 levels (Figure 1C).
Figure 1.

miR-33 modulated ABCA1 expression and cholesterol efflux in RPE cells
(A) The expression of miR-33a and miR-33b was analyzed by quantitative RT-PCR in primary human RPE cells (n = 3). (B) Western blot showing the expression of ABCA1 in primary human RPE cells 72 h after transfection with precursor miR control, miR-33a, or miR-33b (n = 3). (C) Western blotting demonstrating ABCA1 in primary human RPE cells 72 h post-transfection with control, anti-miR-33a, or anti-miR-33b (n = 3). (D) Western blotting demonstrating ABCA1 in ARPE-19 cells 72 h post-transfection with control, anti-miR-33a, anti-miR-33b, or anti-miR33a/b (n = 3). (E) TopFluor cholesterol efflux was measured in ARPE-19 cells transfected with precursor control miR, miR-33a, or miR-33b. (F) TopFluor cholesterol efflux was assessed in ARPE-19 cells ∼60 h after transfection with scrambled control, anti-miR-33a, anti-miR-33b, or-miR-33a/b ASO. pC, precursor scrambled control miR; aC, anti-miR control. All error bars represent ±SEM. (B–D) Expression levels were normalized to α-tubulin loading control, and statistical significance between groups was calculated by unpaired t test. (E and F) Each experiment was performed in quadruplicate and repeated ≥3 times, and statistical significance between groups was calculated by unpaired t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We then determined if the elevated expression of miR-33a and miR-33b would have a functional effect on cholesterol handling by the RPE. Transfection of miR-33a or miR-33b precursors significantly reduced cholesterol efflux to carrier ApoA1 lipoprotein as compared with scrambled precursor miR control (Figure 1E). The inhibition of endogenous miR-33a and miR-33b with anti-miR-33a/b ASOs produced an additive positive effect on ABCA1 expression (Figure 1D) and significantly improved cholesterol efflux (p = 0.001) in ARPE-19 cells as compared to anti-miR-33a (p = 0.04) or anti-miR-33b (p = 0.02) alone, or control ASO (Figure 1F).
In addition to ABCA1, miR-33 has been shown to target the lipid regulator SIRT6 in human cells.43 Transfection of ARPE-19 cells with miR-33a or miR-33b precursors significantly reduced SIRT6 levels compared to scrambled miR transfected cells (Figure S1A). Conversely, transfection of anti-miR-33a, anti-miR-33b, or anti-miR-33a/b ASOs significantly increased SIRT6 levels, compared to control ASO-transfected ARPE-19 cells (Figure S1B).
miR-33 inhibition suppresses cholesterol accumulation and inflammation in the RPE layer of retina in mice fed a WTD
In contrast with rodents, primates carry a high-visual-acuity macular region,44 suggesting that NHPs represent an appropriate model for the development of therapies for AMD.44,45 Moreover, while humans and NHPs have both miR-33a and miR-33b isoforms, mice only harbor miR-33a,31,38 further limiting translational relevance of mouse models. Nevertheless, before studies in an NHP AMD model with anti-miR-33 treatment, we wished to gain proof-of-concept data employing a previously reported dry AMD model with WTD-fed aging mice to investigate the effect of miR-33a inhibition on RPE phenotypes in vivo.12,13,46 We first determined the expression of Abca1 and miRNAs targeting Abca1 in freshly isolated murine RPE cells from C57BL/6J mice at different ages. Quantitative real-time PCR analysis revealed that the level of Abca1 mRNA was markedly decreased in RPE cells as mice age (Figure S1C). Intriguingly, of the Abca1-targeting miRNAs evaluated (miR-33a, miR-128-1, miR-148a, miR-130b, and miR-301b), only miR-33a expression was increased in RPE cells from aging mice (Figures S1D–S1G).
We then tested if feeding 12-month-old C57BL/6J male mice a high-fat/cholesterol WTD for 8 weeks would result in cholesterol accumulation in the RPE layer and whether inhibition of miR-33a by subcutaneous delivery of an anti-miR-33a locked nucleic acid (LNA) ASO31,34 for 4 weeks (while on the WTD) would reduce cholesterol deposition. Consistent with previous reports,31,34 anti-miR-33a LNA ASO treatment significantly increased total serum cholesterol (predominantly HDL-C) levels as compared to mice that received scrambled control LNA ASO (Figure S2A), without significantly affecting circulating liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST),43 and with very moderate effects on bilirubin and creatinine (Figure S2B), suggesting the LNA ASO treatment was well tolerated, as previously observed.31,34 We collected RNA from RPE cells obtained from the dissected retinas of animals that were treated with scrambled control or anti-miR-33a LNA ASO to examine the effect on miR-33 target gene expression levels, Abca1 protein localization, and cholesterol deposition in the RPE cell layer. In agreement with on-target effects of anti-miR-33a, expression of several miR-33 target genes (e.g., Cpt1a, Abca1, Prkaa1, Sirt6, and Sik134,43) were modestly increased in the RPE of anti-miR-33a LNA ASO-treated mice as compared to scrambled control LNA ASO treatment (Figure S3A).
We next analyzed Abca1 expression in cryosections of eyecups by immunofluorescence, and, consistent with the effects on Abca1 mRNA levels, we found that there was stronger Abca1 staining in the RPE cell layer, as well as in choroid blood vessels, of eyecups from animals treated with anti-miR-33 LNA ASO as compared to control LNA ASO treatment (Figure S3B).
To study the effect of LNA ASO treatment on cholesterol deposition in the RPE layer, retinal sections were stained with the cholesterol-trophic dye filipin III. Analysis of the eyecups from animals treated with scrambled control LNA ASO showed strong filipin III staining in the RPE layer closer to the optic nerve head (ONH), center, and periphery (Figure S3C), whereas eyecups from animals treated with anti-miR-33a LNA ASO exhibited significant reduction of filipin III staining in the RPE layer closer to the optic nerve head region and the central region, but not in the periphery (Figure S3C). We speculate that the lesser effect of the LNA ASO treatment on cholesterol accumulation in the retina periphery might be due to the LNA ASO not effectively reaching the peripheral RPE layer as compared to the RPE cell layer closer to the optic nerve head region and the central region.
Cholesterol accumulation causes structural changes in RPE and induces sub-RPE deposit formation.12,13 Electron microscopic imaging revealed that the integrity of the RPE basal structure is severely affected and the presence of large vacuole-like structures are observed in control LNA ASO-treated mice (Figure S3D) in comparison to mice that received anti-miR-33a LNA ASO (Figure S3D). As AMD is associated with immune cell infiltration in the retina,47,48 we then examined whether cholesterol accumulation in the RPE layer is linked to inflammatory cell recruitment, as judged by staining with an antibody directed against Iba1 (macrophage/microglia cell marker). The results show that the average number of Iba1-positive cells in the RPE cell layer of anti-miR-33a LNA ASO-treated mice was markedly and significantly lower (8 ± 3 cells/retinal section) as compared with scrambled control LNA ASO-treated mice (22 ± 2 cells/retinal section) (Figure S4C). In addition, marked infiltration of Iba1-positive cells into the photoreceptor nuclear layer was observed in the control LNA ASO-treated mice (Figure S4A), but not in anti-miR-33a LNA ASO-treated mice (Figure S4A), consistent with a potent anti-inflammatory effect of anti-miR-33a LNA ASO treatment in the eye of aging WTD-fed mice. These results led us to further investigate the therapeutic value of simultaneous ASO targeting of miR-33a and miR-33b in reducing cholesterol accumulation and inflammation in WTD-fed male NHPs (cynomolgus monkeys).
miR-33a/b inhibition ameliorates WTD-induced AMD-like phenotypes in NHPs
NHPs were fed a WTD for 20 months and then switched to a regular chow diet (to mimic dietary intervention in humans with cardiometabolic diseases) and concomitantly treated with anti-miR-33a/b ASO or vehicle control for 6 weeks. Plasma lipid profiling showed that total cholesterol and HDL-C levels were significantly increased in anti-miR-33a/b ASO-injected NHPs as compared to vehicle-treated NHPs (Figure 2A), as expected.32,34 There was no significant difference between treatment groups in plasma levels of triglycerides, low-density lipoprotein cholesterol (LDL-C), or very-low-density lipoprotein cholesterol (VLDL-C), nor in levels of liver enzymes ALT/AST and kidney damage markers creatinine and blood urea nitrogen (BUN), indicating that the anti-miR-33a/b ASO treatment exhibited specific effects and was well tolerated (Figure 2B; Figure S5). The eyes collected from NHPs treated with anti-miR-33a/b ASO or vehicle were then evaluated. In RPE cells of NHPs treated with anti-miR-33a/b ASO, the expression levels of miR-33a/b target genes (ABCA1, PRKAA1, CPT1A, CROT, SIRT6, and SIK1) were increased as compared to vehicle-injected NHPs (Figure 3A). As miR-33a and miR-33b suppress ABCA1 protein translation by targeting ABCA1 mRNA, examination of RPE-choroid lysates extracted from anti-miR-33a/b ASO-injected NHPs showed that the inhibition of miR-33a and miR-33b resulted in significant (more than two-fold) increase in ABCA1 level in comparison to vehicle-injected samples (Figure 3B). Complementing the western blot data, analysis of retinal cryosections demonstrated that ABCA1 protein was markedly increased in the RPE cell layer of anti-miR-33a/b ASO-treated NHPs as compared to the vehicle-treated NHPs (Figure 3C; Figure S9), further supporting a direct effect of the anti-miR-33a/b ASO in the RPE layers of NHPs.
Figure 2.

Anti-miR-33a/b ASO treatment improves plasma HDL-C and total cholesterol in NHPs
(A) Plasma HDL-C and total cholesterol were measured in NHPs fed a high-fat/cholesterol diet for 20 months and then switched to a regular chow diet and injected with anti-miR-33 ASO or vehicle for 6 weeks (n = 12/group). (B) Serum AST, ALT, bilirubin, and uric acid levels were measured to monitor liver and kidney functions in mice that were fed a high-fat/cholesterol diet and injected with scrambled control or anti-miR-33 LNA ASO (n = 12/group). All error bars represent ±SEM. Statistical differences between vehicle control and anti-miR-33a/b ASO-injected mice were calculated by unpaired t test. ∗p < 0.05 and ∗∗p < 0.01.
Figure 3.

Anti-miR-33a/b ASO treatment increased miR-33 target gene expression levels and ABCA1 protein localization in NHP RPE cell layer
(A) Expression levels of ABCA1, PRKAA1, CPT1A, CROT, SIRT6, and SIK1 were measured by quantitative RT-PCR in RPE cells isolated from NHPs injected with anti-miR-33 ASO or vehicle for 6 weeks (n ≤ 4). mRNA expression levels were normalized to PPIH or HPRT1. (B) Protein blot showing the ABCA1 protein expression in RPE/choroid lysate extracted from vehicle or anti-miR-33 injected NHP (n = 3) and the bar graph showing the relative fold change. (C) Retinal cryosections prepared from NHPs that were treated with vehicle or anti-miR-33 ASO by subcutaneous injections were immunostained for ABCA1 and DAPI nuclear stain (n = 3). Scale bar: 10 μm. (D) Free and esterified cholesterol levels were measured in the lipid extracts from RPE/choroid of NHPs that were injected with anti-miR-33 ASO or vehicle for 6 weeks (n = 3). (E) NHP retinal sections showing four highlighted regions (R1–4) from the fovea to the periphery were chosen to quantify filipin III staining in the RPE cell layer of vehicle- or anti-miR-33 ASO-treated NHP retinal sections. Arrow points to fovea; scale bar: 1 mm. (F) Retinal sections of NHPs that were injected with anti-miR-33a/b ASO or vehicle for 6 weeks were stained with filipin III to label unesterified cholesterol. Four regions (R1–4) from the fovea to the periphery shown in (E) chosen to quantify filipin III staining in the RPE cell layer of vehicle- or anti-miR-33 ASO-treated NHP retinal sections (n = 9). Scale bar: 50 μm. (G) Retinal sections of NHPs that were injected with anti-miR-33 ASO or vehicle for 6 weeks were pretreated with cholesterol esterase and then stained with filipin III to label esterified cholesterol. Four regions (R1–4) from the fovea to the periphery shown in (E) chosen to quantify filipin III staining in the RPE cell layer of vehicle- or anti-miR-33 ASO-treated NHP retinal sections (n = 4). Scale bar: 50 μm. All error bars represent ± SEM. Statistical differences between vehicle- and anti-miR-33 ASO-injected NHPs were calculated by unpaired t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Since unesterified and esterified cholesterol accumulation in the RPE and sub-RPE layers has been postulated to contribute to AMD pathogenesis,49 we evaluated whether miR-33a/b inhibition reduced cholesterol levels in the RPE-choroid of NHPs. Total lipids were extracted from the RPE-choroid of vehicle- and anti-miR-33a/b-treated NHPs, followed by measurement of free and esterified cholesterol levels. Results showed that free cholesterol levels in the RPE-choroid of anti-miR-33a/b-treated NHPs were significantly reduced (p < 0.03) as compared with vehicle-treated NHPs (Figure 3D). Similarly, cholesterol ester levels were also reduced in the RPE-choroid of anti-miR33a/b-injected NHPs as compared to vehicle controls (Figure 3D).
To further evaluate the effect of cholesterol accumulation in the RPE and sub-RPE layers, we systematically examined the cholesterol deposition from the fovea up to the peripheral retina of NHPs treated with vehicle or anti-miR-33a/b ASO (Figure 3E). NHP retinal sections of vehicle- or anti-miR-33 ASO-injected groups were prepared with and without cholesterol esterase treatment followed by filipin III staining to visualize esterified and unesterified cholesterol, respectively. The data revealed that anti-miR-33a/b ASO-treated NHPs exhibited significantly reduced unesterified cholesterol in the fovea (p = 0.01), parafovea (p = 0.01), perifovea (p = 0.003), and periphery (p = 0.001), as compared with vehicle-treated NHPs (Figure 3F; Figure S7A). With respect to esterified cholesterol, the filipin III staining of esterified cholesterol was mostly detected in the sub-RPE layer from fovea to the central retina (R1–R3) of vehicle- or anti-miR-33a/b ASO-treated NHPs (Figure S8), while the staining of esterified cholesterol was very weak in the periphery (Figure S8). In the fovea to the central retinal RPE layer, unesterified cholesterol staining was moderately decreased in the anti-miR-33a/b ASO-treated NHPs compared to the vehicle-treated group (Figure 3G; Figure S8). Taken together, these results show that retinal cholesterol levels and key cholesterol trafficking proteins are beneficially impacted upon therapeutic targeting of miR-33a/b in WTD-fed NHPs, in agreement with a pathological role for miR-33a/b in contributing to cholesterol-related AMD-like phenotypes in mammals.
In AMD, RPE cells have been shown to enlarge and undergo morphological changes preceding cell death and geographic atrophy.50 To assess whether miR-33a/b might contribute to high-fat/cholesterol-diet-induced RPE morphological changes, RPE flatmounts were prepared from vehicle- or anti-miR-33a/b ASO-treated NHPs and stained for phalloidin to visualize the actin cytoskeleton and quantify the area of each RPE cell in the regions closer to the optic nerve head, center, and periphery in the retina. In the eyes of vehicle-treated NHPs, a number of RPE cells displayed enlarged morphology, and the normal hexagonal RPE shape was severely altered (Figure 4). Quantitative analysis of cell size revealed that the number of enlarged RPE cells was significantly increased in the RPE in vehicle-treated NHPs, as compared to RPE of anti-miR-33a/b ASO-treated NHPs (Figure 4). Furthermore, RPE cells in the anti-miR-33a/b ASO-treated NHPs showed well-preserved hexagonal morphology in comparison to vehicle controls (Figure 4). These results suggest that miR-33a/b contribute to WTD-induced AMD-like RPE abnormalities in NHPs.
Figure 4.

Anti-miR-33a/b ASO treatment reduced abnormal RPE cytoskeletal organization in the RPE cell layer of NHPs fed a high-fat/cholesterol diet
RPE flatmounts prepared from NHPs that received subcutaneous injections of vehicle or anti-miR-33 ASO for 6 weeks were stained with phalloidin and examined for RPE cytoskeletal organization, and then RPE cell size was quantified and segmented in the area closer to the optic nerve head (ONH), center, and periphery. Arrows in the top panel indicate enlarged RPE cells (n ≤ 7). Scale bar: 100 μm. RPE cell size was quantified in field of view (FOV) and represented in the bar graphs. All error bars represent ±SEM. Statistical differences between vehicle- and anti-miR-33 ASO-injected NHPs were calculated by unpaired t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Finally, we assessed retinal inflammation by IBA1 staining (macrophage/microglia marker). Similar to our observations in WTD-fed aging mice, immune cell infiltration into the RPE-photoreceptor layer and sub-RPE layer was markedly elevated in vehicle-treated NHP retinal sections in the mid and peripheral regions (Figures 5A and 5B), as compared to anti-miR-33a/b ASO-treated NHP retinal sections. This is consistent with a potent pro-inflammatory effect of miR-33a/b in the retina, in the context of WTD feeding of NHPs, akin to what is observed in human AMD.48,51, 52, 53
Figure 5.

Anti-miR-33a/b ASO treatment reduced immune cell infiltration in RPE-photoreceptor and RPE layers
(A) IBA1 staining (magenta) and superimposed brightfield (BF) revealing IBA1 positive cells in the sub-RPE layers in vehicle-treated NHP retinal sections, while low IBA1-positive staining is seen in the sub-RPE-choroid layer of anti-miR-33 ASO-injected NHP retinal sections. Scale bar: 20 μm. POS, photoreceptor outer segment. (B) Representative IBA1 (magenta) and superimposed DAPI (blue) staining revealing IBA1-positive cell in the RPE-photoreceptor layer in vehicle-treated NHP retinal sections (white arrow), while no IBA1-positive staining is seen in the RPE-photoreceptor layer of anti-miR-33 ASO-injected NHP retinal sections. Scale bar: 20 μm. ONL, outer nuclear layer of photoreceptor cells.
Discussion
In addition to genetic susceptibility, normal aging and cholesterol deposition with age are postulated to predispose patients to develop AMD.54,55 Our studies demonstrate that feeding aging mice and NHPs a high-fat/cholesterol WTD led to cholesterol deposition in the RPE layer, induced RPE morphological and cytoskeletal changes, and elicited inflammatory cell recruitment, which are the key clinical features of dry AMD.48,50,56,57 Subcutaneous delivery of anti-miR-33 ASOs reduced cholesterol deposition in the RPE layer, decreased RPE phenotypic changes, and suppressed WTD-induced retinal inflammation in aging mice and NHPs. Macrophages are also thought to be involved in the development and progression of AMD,57 and an experimental murine CNV model suggested a possible role for miR-33a in aging macrophages in wet AMD.22 Although we cannot definitively conclude whether RPE cells and/or macrophages were the direct targets of anti-miR-33 ASO treatment, we did find that miR-33 canonical targets (e.g., ABCA1) were de-repressed and cholesterol accumulation and inflammation were significantly reduced in the RPE cell layer in response to anti-miR-33 ASO treatment, supporting a role for miR-33 action in the RPE in contributing to the dry AMD-like phenotypes in WTD-fed aging mice and NHPs.
Even though genetic variants in or near lipid genes and cholesterol/lipid accumulation in the RPE layer are associated with AMD pathogenesis,5,8,54 the role for plasma-derived cholesterol/lipids in AMD remains controversial.6,58,59 However, accumulating evidence indicates that locally RPE-derived cholesterol and lipoproteins might contribute to cholesterol-rich drusen formation.60, 61, 62 Moreover, inflammatory cues elicited by the RPE could promote immune cell infiltration.63,64 Our results together suggest that miR-33 acts locally in the retina to suppress beneficial RPE cholesterol clearance and stimulate RPE-mediated immune response. The miR-33-dependent cholesterol accumulation and inflammation in the RPE cell layer may thus play a key role in the development of dry AMD-like pathology, and therapeutic targeting of miR-33a/b could facilitate the clearance of cholesterol in the RPE cell layer, decrease inflammation, and attenuate pathologic changes leading to geographic atrophy, a hallmark of dry AMD.
Materials and methods
Reagents
Precursor miRNAs, including miR-33a, miR-33b, miR-128-1, and miR-148a, and anti-miRNAs, including miR-33a, anti-miR-33b, anti-miR-128, and anti-miR-148a, were purchased from Ambion/Thermo Fisher Scientific. Antibodies included: ABCA1 (ab18180, Abcam), SIRT6 (D8D12), GAPDH (2118), vinculin (4650) (Cell Signaling), alpha-tubulin (Calbiochem/EMD Millipore), APOE (NB110-60531), ABCA1 (NB400-105), and Iba1 (NB100-1028) (Novus Biologicals). Other reagents used were: filipin III (Cayman Chemicals), cholesterol esterase (Sigma-Aldrich), phalloidin-670 (Cytoskeleton), cell lysis reagent (Cell Signaling), protein blot blocking buffer (Li-COR Biosciences), TopFluor cholesterol (Avanti Polar Lipids), APOA1 (Alfa Aesar), and lipoprotein-deficient serum (EMD Millipore). LNA anti-miR and scrambled control oligonucleotides for in vitro and mouse studies were purchased from Exiqon (Vedbaek, Denmark; now QIAGEN).
Cell culture and transfection
Primary human RPE cells and human RPE cell line (ARPE-19 cells, ATCC) were cultured as described previously.63 Cells were transfected with precursor miRNA (33 nM final concentration), anti-miR (33 nM final concentration), or LNA antisense oligonucleotides (50 nM final concentration) using Lipofectamine RNAiMAX reagent (Life Technologies/Thermo Fisher Scientific). Cell lysates were collected 72 h post-transfection, and 25–30 μg of protein was loaded and separated on SDS-PAGE gel. Transferred protein blots were blocked, incubated with indicated primary and secondary antibodies, and examined by Odyssey Imaging System (LiCOR Biosciences). Alpha tubulin or vinculin was used as a loading control for normalization.
Cholesterol efflux assay
ARPE-19 cells were plated at a density of 5 × 105 per well in a 24-well plate. After attaching for 24 h, cells were transfected with precursor miRNA or LNA anti-miR, then washed with serum-free DMEM/F-12 (Gibco/Thermo Fisher Scientific) media and incubated with 10 or 25 μM TopFluor cholesterol for 24 h. Cells were washed in serum-free media and then incubated with phenol-red free DMEM/F-12 containing 5% lipoprotein deficient serum and 10 μM APOA1 lipoprotein. Supernatant and cell lysates were collected 4 h post treatment, and fluorescence levels were measured using microplate reader (BioTek Instruments) to calculate the percentage of efflux.
Study of miRNA and gene expression in aging mice
C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and maintained at Schepens Eye Research Institute (SERI). Eyes were enucleated at 6, 12, 15, and 18 months. Retinas were dissected out to separate RPE cells, as described previously.65 RPE cells were isolated from six to eight mice per age group. RNA from the RPE pellet was extracted using RNA-Bee (AMS Biotechnology), according to the manufacturer’s protocol. Total RNA was then reverse transcribed using iScript cDNA synthesis kit (Bio-Rad Laboratories). RT reactions were performed using SYBR Green (Roche) and quantified by real-time PCR (Lightcycler, Roche).
Mouse LNA ASO treatment studies
Twelve-month-old C57BL/6J mice were purchased from Jackson Laboratory and fed a WTD supplemented with 40% kcal from milkfat (Research Diets, D12079B) for 4 weeks prior to and during treatment. Mice were treated weekly during the 4 weeks with 10 mg/kg 16-mer LNA anti-miR-33a (5′-ATGCAACTACAATGCA-3′) or scrambled control LNA. LNA ASOs were dissolved in PBS (total volume of 200 μL) then administered subcutaneously. Mice were sacrificed 72 h after the last injection. Upon sacrifice, ∼1 mL of blood was obtained from mice by right ventricular puncture. Blood was centrifuged at 8,000 rpm for 5 min to obtain serum, which was frozen at −80°C. Eyes were enucleated for RNA extraction from RPE cells, cryosectioning, and electron microscopy.
Blood lipid profile and blood chemistry in mice
Total serum cholesterol, triglycerides, AST,43 ALT, bilirubin, and uric acid levels were determined with a Heska Dri-Chem 4000 Chemistry Analyzer (Heska, Loveland, CO, USA) at the MGH Center for Comparative Medicine.
NHP study
Young adult male cynomolgus monkeys (Macaca fascicularis) originated from Mauritius and were an average of 5.0 years of age (range, 4.2–6.7 years) at the onset of the study. The NHPs were housed in an American Association for Accreditation of Laboratory Animal Care (AAALAC)-accredited facility under the direct care of the University of Kentucky Division of Laboratory Animal Resources. Monkeys were housed in climate-controlled conditions with a 12-h light and dark cycle. The NHPs were initially ad libitum fed a standard NHP diet (Teklad 2050). For the study, the NHPs were singly housed from ∼08:00–15:00 each day and in the morning and afternoon received weighed portions of a semi-synthetic atherogenic diet (see composition in Table S1), which provided on average 74 kcal/kg body weight/day. After 20 months on the atherogenic diet, the monkeys were switched back to standard chow diet and were treated for 6 weeks with either vehicle or miR-33a/b antagonist RG428651, a 2′-fluoro/methoxyethyl-modified, phosphorothioate (PS)-backbone-modified, antisense oligonucleotide (5′-TGCAATGCAACTACAATGCAC-3′).32 Monkeys were injected subcutaneously with vehicle (United States Pharmacopeia [USP] grade saline) or 5 mg ASO/kg body weight twice weekly during the first 2 weeks and then once weekly during the remainder of the study. During the treatment period, animals were singly housed from ∼08:00–15:00 each day and received 12 biscuits of standard diet, which provided on average 64 kcal/kg body weight/day. At the end of the treatment period, the monkeys were fasted overnight and sedated with ketamine (25 mg/kg, intramuscularly [i.m.]) and isoflurane (3%–5% induction, 1%–2% maintenance). After an adequate depth of anesthesia was established by lack of physical response, the inferior vena cava was exposed and cut for exsanguination. A 16-gauge needle was inserted into the left ventricle of the heart, and saline was perfused to flush the body of blood. The euthanasia method was deemed acceptable by the American Veterinary Medical Association. After euthanasia, eyes were enucleated for RNA extraction from the RPE cell layer and for fixation in 10% formalin for cryosectioning and RPE flatmount preparations. The handling of the NHP eyes was performed at SERI.
Lipid and lipoprotein cholesterol analysis and blood chemistry of NHPs
After an overnight fast, monkeys were sedated with ketamine (10 mg/kg, i.m.), body weights were recorded, and blood was collected from the femoral vein into EDTA-containing or serum separation vacutainers. Plasma and serum were isolated by centrifugation at 1,500 × g for 30 min at 4°C. For determination of circulating concentrations of ALT, AST, creatinine, and BUN, serum was analyzed using the Superchem blood test (ANTECH Diagnostics). Enzymatic assays were used to measure plasma total cholesterol (C7510, Pointe Scientific) and triglycerides (T2449 and F6428, Sigma). The plasma cholesterol distribution among lipoprotein classes was determined after separation by gel filtration chromatography based upon the method described previously.32 An aliquot of plasma was diluted to 0.5 μg total cholesterol/μL in 0.9% NaCl, 0.05% EDTA/NaN3 and centrifuged at 2,000 × g for 10 min to remove any particulate debris. The supernatant was transferred to a glass insert contained in a gas chromatography vial, loaded into an autosampler at 4°C (Agilent Technologies, G1329A), and 40 μL of sample was injected onto a Superose 6 10/300 or Superose 6 Increase 10/300 (GE Healthcare Life Sciences) chromatography column. Under the control of an isocratic pump (Agilent Technologies, G1310A/B), the sample was separated at a flow rate of 0.4 mL/min with eluent containing 0.9% NaCl, 0.05% EDTA/NaN3. The column effluent was mixed with total cholesterol enzymatic reagent (C7510, Pointe Scientific), running at a flow rate of 0.125 mL/min, and the mixture was passed through a knitted reaction coil (Aura Industries, EPOCOD) in a 37°C H2O jacket. The absorbance of the reaction mixture was read at 500 nm using a variable wavelength detector (Agilent Technologies, G1314F). The signal was subsequently integrated using Agilent OpenLAB Software Suite (Agilent Technologies). VLDL-C, LDL-C, and HDL-C concentrations were determined by multiplying the total plasma cholesterol (TPC) concentration by the cholesterol percentage within the elution region for each lipoprotein class.
Quantitative RT-PCR
Total RNA and miRNA were extracted from RPE cells using TriZOL (Life Technologies/Invitrogen) and the mirVana miRNA Isolation Kit (Life Technologies/Invitrogen), respectively, according to the manufacturer’s instructions. Following extraction, total RNA and miRNA were reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit and the TaqMan MicroRNA Reverse Transcription Kit (Life Technologies/Invitrogen), respectively. RT products were quantified by quantitative real-time PCR (Lightcycler, Roche) using the TaqMan Universal PCR Master Mix. The amount of the indicated mRNA or miRNA was normalized to the amount of B2M mRNA and U6 RNA or snoRNA234 (for mice) or RNU48 (for NHPs), respectively.
NHP RPE-choroid western blot
Assessment of ABCA1 protein expression by western blot was carried out as per manufacturer’s guidelines (Minute Protein Extraction Kit for Fixed and Embedded Tissues, Invent Biotechnologies). After extracting the protein lysates from thin preparations of formalin-fixed RPE-choroid, 30 μg of protein was loaded and separated on SDS-PAGE gel. Transferred protein blots were blocked, incubated with indicated primary and secondary antibodies, and examined by Odyssey Imaging System (LiCOR Biosciences). GAPDH was used as a loading control for normalization.
Cryosectioning
Following dissection of the anterior chamber from NHP eyes, the eyecup was dissected into four quadrants and the quadrant containing the fovea was cryopreserved by serial treatment with 10%, 20%, and 30% sucrose solution. Similarly, the anterior chamber was dissected from the mouse eyes that were fixed overnight in 4% paraformaldehyde, and the posterior eyecup was cryopreserved by serial sucrose solution treatment. The cryopreserved eyecups were embedded in Tissue-Tek O.C.T compound (Sakura Finetek), frozen, and stored at −80°C. Thick retinal frozen sections (12 μm) were cut using a Leica CM3050 S cryostat. For proper comparison and consistency, retinal sections containing the fovea in all the NHPs were used for staining. In mice, retinal sections from the optic nerve head regions of all the treatment groups were used for staining.
Total cholesterol measurement in retina and RPE-choroid
Lipids were extracted from NHP retinas and RPE-choroid, and total cholesterol was quantified as directed by the manufacturer (Total Cholesterol Assay Kit, Cell Biolabs). Briefly, to 10 mg of tissue, 100 μL of lipid extraction buffer (chloroform:isopropanol:NP-40 [7:11:0.1]) was added and homogenized. Samples were spun at 15,000 × g for 10 min, and the organic phase was collected in a tube, air-dried at 50°C, and then maintained under vacuum for 30 min to remove traces of organic solvents. The dried lipids were dissolved in 30 μL of assay diluent provided by the kit, and 10 μL of the sample was mixed with the reaction buffer to quantify the total cholesterol level.
Filipin III staining of unesterified and esterified cholesterol
Retinal sections were washed in PBS and incubated with filipin III as recommended by the manufacturer for 2 h at room temperature to stain unesterified cholesterol. After washing, slides were mounted with ProLong Gold antifade media (Invitrogen/Thermo Fisher Scientific) and imaged using a fluorescence microscope (Nikon). To stain the esterified cholesterol, retinal sections were incubated in 70% ethanol followed by incubation with cholesterol esterase (1.65 U/mL) for 2 h at 37°C. After the enzyme treatment, retinal sections were stained with filipin III and imaged as described above.
Immunofluorescence staining
Retinal cryosections were washed in 1× PBS, blocked in PBS containing 10% goat or donkey serum and 0.05% Triton X-100 for 1 h at room temperature. After blocking, sections were incubated with indicated primary and secondary antibodies prepared in PBS containing 2% goat or donkey serum and 0.01% Triton X-100. After washing, the samples were treated with TrueBlack (Biotium, autofluorescence quencher) as suggested by the manufacturer, and then the sections were mounted with ProLong Gold antifade media with DAPI (Invitrogen/Thermo Fisher Scientific) and imaged using a fluorescence microscope (Axioscope, Carl Zeiss or Leica SP8 confocal microscope).
Electron microscopy
Eyes were enucleated and the posterior eyecup was fixed with half-strength Karnovsky’s fixative (2% formaldehyde + 2.5% glutaraldehyde, in 0.1 M sodium cacodylate buffer [pH 7.4]; Electron Microscopy Sciences, Hatfield, PA, USA) overnight at 4°C. After fixation, mouse eye samples were rinsed with 0.1 M sodium cacodylate buffer, post-fixed with 2% osmium tetroxide in 0.1 M sodium cacodylate buffer for 1.5 h, en bloc stained with 2% gadolinium (III) acetate hydrate in 0.05 M sodium maleate buffer, then dehydrated with graded ethyl alcohol solutions, transitioned with propylene oxide and resin infiltrated in tEPON-812 epoxy resin (Tousimis, Rockville, MD, USA), utilizing an automated EMS Lynx 2 EM tissue processor (Electron Microscopy Sciences, Hatfield, PA, USA). The processed samples were oriented into tEPON-812 epoxy resin inside flat molds and polymerized in a 60°C oven. Semi-thin sections were cut at 1 μm thickness, then stained with 1% toluidine blue in 1% sodium tetraborate aqueous solution for assessment by light microscopy. Ultrathin sections (80 nm) were cut from each sample block using a Leica EM UC7 ultramicrotome (Leica Microsystems, Buffalo Grove, IL, USA) and a diamond knife, then collected using a loop tool onto either 2 × 1 mm, single-slot formvar-carbon coated or 200 mesh uncoated copper grids and air dried. The thin sections on grids were stained with aqueous 2.5% aqueous gadolinium (III) acetate hydrate and Sato’s lead citrate stains using a modified Hiraoka grid staining system. Grids were imaged using a FEI Tecnai G2 Spirit transmission electron microscope (FEI, Hillsboro, OR, USA) at 80 kV interfaced with an AMT XR41 digital CCD camera (Advanced Microscopy Techniques, Woburn, MA, USA) for digital TIFF file image acquisition. Transmission electron microscopy (TEM) imaging of retina samples was assessed and digital images captured at 2,000 × 2,000 pixel, 16-bit resolution.
NHP flatmount preparation and analysis
For consistency, the retina was gently detached from the quadrant opposite to the fovea, and the RPE-choroid layer was carefully separated from the sclera. The RPE-choroid layer was incubated with phalloidin-670 overnight at 4°C, as recommended by the manufacturer. The samples were then washed with PBS and mounted with ProLong Gold antifade media (Invitrogen/Thermo Fisher Scientific). The areas closer to the optic nerve head, center, and periphery were imaged (five images per region) using a fluorescence microscope (Axioscope, Carl Zeiss). The area of each RPE cells was quantified using MATLAB as described below, and the cells were segregated based on size.
MATLAB image quantification methodology
The phalloidin stained RPE cell size was measured using the MATLAB module developed by The Nikon Imaging Center, Harvard Medical School. In brief, the images were annotated using the ImageAnnotationBot module (https://www.mathworks.com/matlabcentral/fileexchange/64719-imageannotationbot). After annotation, the following parameters were set to measure the area of each cell per image: (1) BoundariesThreshold, to obtain binary images; (2) MinAreaBoundaryComps, to eliminate small components from thresholded images; (3) DistTransfThreshold, to select markers from distance transform images; (4) RemoveBoundaryCells, to remove cells in the boundary; (5) SolidityRange, to select nearly convex cells; and (6) ExtentRange, to create area of shape. The MATLAB machine learning module used for cell size quantification will be available at https://hms-idac.github.io/MatBots/.
Statistics
All in vitro experiments were repeated at least three times. A majority of the in vivo data analyses were conducted in a masked manner (except Iba1 staining in NHPs). Based on a preliminary study, we used 10 mice for the LNA ASO study per treatment condition. Four mice from each treatment group were used for histology, and the remaining six were used for gene expression studies. None of the mice were excluded from the analysis. All NHP samples received were analyzed. There were 12 vehicle controls and 12 anti-miR-33a/b ASO samples for histological studies and nine vehicle controls and six anti-miR-33a/b ASO samples for gene-expression-related studies. All statistical analyses were conducted using GraphPad Prism software, and the error bars on the histogram represent ±SEM. Statistical differences for age-related gene or miRNA expression studies in mice were analyzed by one-way analysis of variance followed by a post Dunnett’s multiple comparisons. Statistical differences for all the other studies were measured using unpaired two-sided Student’s t test. p ≤0.05 was considered as statistically significant.
Study approval
All the procedures were approved by the Massachusetts General Hospital (MGH), SERI of Mass Eye and Ear, and University of Kentucky Institutional Animal Care and Use Committees.
Acknowledgments
Supported by the NIH National Eye Institute core grant P30EY003790 (P.A.D.), the Grimshaw-Gudewicz Charitable Foundation for AMD Research (P.A.D.), NIH grant R01 HL111932 (R.E.T.), the Stein Innovation Award from the Research to Prevent Blindness Foundation (A.M.N.), and an MGH Research Scholar Award (A.M.N.). We would like to thank Sierra Schlicht and Courtney Burkett and the Temel Lab, Saha Cardiovascular Research Center, Department of Pharmacology and Nutritional Sciences, University of Kentucky, Lexington, KY, USA, for technical assistance; Marcelo Cicconet, Department of Cell Biology, Harvard Medical School, Boston, MA, USA, for assistance with RPE image analysis; and Philip Seifert, morphology core, Schepens Eye Research Institute of Mass Eye and Ear, Boston, MA, USA.
Author contributions
G.G. performed all the in vitro studies and gene expression studies, prepared mouse and NHP retinal sections for immunostaining, analyzed data, and wrote the manuscript. A.W. designed and performed mouse LNA ASO studies, measured miRNA levels, performed serum lipid profiles in mice, and assisted G.G. with experimental design. J.O. assisted A.W. with all the in vivo studies. K.L.S.-T. assisted G.G. with immunostaining and masked analysis of RPE cell size measurements. T.L. performed NHP studies. R.E.T. designed and oversaw NHP studies. M.E.K. taught enucleation of NHP eyes to T.L. and assisted with RPE sample collection for histology and gene expression studies. A.M.N. and P.A.D. supervised the overall study and assisted with data analysis and manuscript preparation.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.03.014.
Contributor Information
Anders M. Näär, Email: naar@berkeley.edu.
Patricia A. D’Amore, Email: patricia_damore@meei.harvard.edu.
Supplemental information
References
- 1.Xu Q., Cao S., Rajapakse S., Matsubara J.A. Understanding AMD by analogy: systematic review of lipid-related common pathogenic mechanisms in AMD, AD, AS and GN. Lipids Health Dis. 2018;17:3. doi: 10.1186/s12944-017-0647-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curcio C.A., Johnson M., Huang J.D., Rudolf M. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J. Lipid Res. 2010;51:451–467. doi: 10.1194/jlr.R002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curcio C.A., Johnson M., Rudolf M., Huang J.D. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 2011;95:1638–1645. doi: 10.1136/bjophthalmol-2011-300344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreira E.F., Larrayoz I.M., Lee J.W., Rodríguez I.R. 7-Ketocholesterol is present in lipid deposits in the primate retina: potential implication in the induction of VEGF and CNV formation. Invest. Ophthalmol. Vis. Sci. 2009;50:523–532. doi: 10.1167/iovs.08-2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKay G.J., Patterson C.C., Chakravarthy U., Dasari S., Klaver C.C., Vingerling J.R., Ho L., de Jong P.T., Fletcher A.E., Young I.S. Evidence of association of APOE with age-related macular degeneration: a pooled analysis of 15 studies. Hum. Mutat. 2011;32:1407–1416. doi: 10.1002/humu.21577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pennington K.L., DeAngelis M.M. Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. (Lond.) 2016;3:34. doi: 10.1186/s40662-016-0063-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Y., Bhangale T.R., Fagerness J., Ripke S., Thorleifsson G., Tan P.L., Souied E.H., Richardson A.J., Merriam J.E., Buitendijk G.H. Common variants near FRK/COL10A1 and VEGFA are associated with advanced age-related macular degeneration. Hum. Mol. Genet. 2011;20:3699–3709. doi: 10.1093/hmg/ddr270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu Y., Reynolds R., Fagerness J., Rosner B., Daly M.J., Seddon J.M. Association of variants in the LIPC and ABCA1 genes with intermediate and large drusen and advanced age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2011;52:4663–4670. doi: 10.1167/iovs.10-7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dighe S., Zhao J., Steffen L., Mares J.A., Meuer S.M., Klein B.E.K. Diet patterns and the incidence of age-related macular degeneration in the Atherosclerosis Risk in Communities (ARIC) study. Br. J. Ophthalmol. 2020;104:1070–1076. doi: 10.1136/bjophthalmol-2019-314813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bretillon L., Acar N., Seeliger M.W., Santos M., Maire M.A., Juanéda P., Martine L., Grégoire S., Joffre C., Bron A.M., Creuzot-Garcher C. ApoB100,LDLR-/- mice exhibit reduced electroretinographic response and cholesteryl esters deposits in the retina. Invest. Ophthalmol. Vis. Sci. 2008;49:1307–1314. doi: 10.1167/iovs.07-0808. [DOI] [PubMed] [Google Scholar]
- 11.Malek G., Johnson L.V., Mace B.E., Saloupis P., Schmechel D.E., Rickman D.W., Toth C.A., Sullivan P.M., Bowes Rickman C. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc. Natl. Acad. Sci. USA. 2005;102:11900–11905. doi: 10.1073/pnas.0503015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miceli M.V., Newsome D.A., Tate D.J., Jr., Sarphie T.G. Pathologic changes in the retinal pigment epithelium and Bruch’s membrane of fat-fed atherogenic mice. Curr. Eye Res. 2000;20:8–16. [PubMed] [Google Scholar]
- 13.Roddy G.W., Rosa R.H., Viker K.B., Holman B.H., Hann C.R., Krishnan A., Gores G.J., Bakri S.J., Fautsch M.P. Diet Mimicking “Fast Food” Causes Structural Changes to the Retina Relevant to Age-Related Macular Degeneration. Curr. Eye Res. 2020;45:726–732. doi: 10.1080/02713683.2019.1694156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landowski M., Kelly U., Klingeborn M., Groelle M., Ding J.D., Grigsby D., Bowes Rickman C. Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc. Natl. Acad. Sci. USA. 2019;116:3703–3711. doi: 10.1073/pnas.1814014116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toomey C.B., Johnson L.V., Bowes Rickman C. Complement factor H in AMD: Bridging genetic associations and pathobiology. Prog. Retin. Eye Res. 2018;62:38–57. doi: 10.1016/j.preteyeres.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ambati J., Fowler B.J. Mechanisms of age-related macular degeneration. Neuron. 2012;75:26–39. doi: 10.1016/j.neuron.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holekamp N.M. Review of neovascular age-related macular degeneration treatment options. Am. J. Manag. Care. 2019;25(10, Suppl):S172–S181. [PubMed] [Google Scholar]
- 18.Vavvas D.G., Daniels A.B., Kapsala Z.G., Goldfarb J.W., Ganotakis E., Loewenstein J.I., Young L.H., Gragoudas E.S., Eliott D., Kim I.K. Regression of Some High-risk Features of Age-related Macular Degeneration (AMD) in Patients Receiving Intensive Statin Treatment. EBioMedicine. 2016;5:198–203. doi: 10.1016/j.ebiom.2016.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishida B.Y., Duncan K.G., Bailey K.R., Kane J.P., Schwartz D.M. High density lipoprotein mediated lipid efflux from retinal pigment epithelial cells in culture. Br. J. Ophthalmol. 2006;90:616–620. doi: 10.1136/bjo.2005.085076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storti F., Klee K., Todorova V., Steiner R., Othman A., van der Velde-Visser S., Samardzija M., Meneau I., Barben M., Karademir D. Impaired ABCA1/ABCG1-mediated lipid efflux in the mouse retinal pigment epithelium (RPE) leads to retinal degeneration. eLife. 2019;8:e45100. doi: 10.7554/eLife.45100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Storti F., Raphael G., Griesser V., Klee K., Drawnel F., Willburger C., Scholz R., Langmann T., von Eckardstein A., Fingerle J. Regulated efflux of photoreceptor outer segment-derived cholesterol by human RPE cells. Exp. Eye Res. 2017;165:65–77. doi: 10.1016/j.exer.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Sene A., Khan A.A., Cox D., Nakamura R.E., Santeford A., Kim B.M., Sidhu R., Onken M.D., Harbour J.W., Hagbi-Levi S. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;17:549–561. doi: 10.1016/j.cmet.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 24.Mendell J.T., Olson E.N. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rottiers V., Näär A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012;13:239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Näär A.M., Beaurang P.A., Robinson K.M., Oliner J.D., Avizonis D., Scheek S., Zwicker J., Kadonaga J.T., Tjian R. Chromatin, TAFs, and a novel multiprotein coactivator are required for synergistic activation by Sp1 and SREBP-1a in vitro. Genes Dev. 1998;12:3020–3031. doi: 10.1101/gad.12.19.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Näär A.M., Beaurang P.A., Zhou S., Abraham S., Solomon W., Tjian R. Composite co-activator ARC mediates chromatin-directed transcriptional activation. Nature. 1999;398:828–832. doi: 10.1038/19789. [DOI] [PubMed] [Google Scholar]
- 28.Walker A.K., Jacobs R.L., Watts J.L., Rottiers V., Jiang K., Finnegan D.M., Shioda T., Hansen M., Yang F., Niebergall L.J. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell. 2011;147:840–852. doi: 10.1016/j.cell.2011.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker A.K., Yang F., Jiang K., Ji J.Y., Watts J.L., Purushotham A., Boss O., Hirsch M.L., Ribich S., Smith J.J. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010;24:1403–1417. doi: 10.1101/gad.1901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang F., Vought B.W., Satterlee J.S., Walker A.K., Jim Sun Z.Y., Watts J.L., DeBeaumont R., Saito R.M., Hyberts S.G., Yang S. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature. 2006;442:700–704. doi: 10.1038/nature04942. [DOI] [PubMed] [Google Scholar]
- 31.Najafi-Shoushtari S.H., Kristo F., Li Y., Shioda T., Cohen D.E., Gerszten R.E., Näär A.M. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rayner K.J., Esau C.C., Hussain F.N., McDaniel A.L., Marshall S.M., van Gils J.M., Ray T.D., Sheedy F.J., Goedeke L., Liu X. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature. 2011;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rayner K.J., Sheedy F.J., Esau C.C., Hussain F.N., Temel R.E., Parathath S., van Gils J.M., Rayner A.J., Chang A.N., Suarez Y. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rottiers V., Obad S., Petri A., McGarrah R., Lindholm M.W., Black J.C., Sinha S., Goody R.J., Lawrence M.S., deLemos A.S. Pharmacological inhibition of a microRNA family in nonhuman primates by a seed-targeting 8-mer antimiR. Sci. Transl. Med. 2013;5:212ra162. doi: 10.1126/scitranslmed.3006840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerin I., Clerbaux L.A., Haumont O., Lanthier N., Das A.K., Burant C.F., Leclercq I.A., MacDougald O.A., Bommer G.T. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J. Biol. Chem. 2010;285:33652–33661. doi: 10.1074/jbc.M110.152090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horie T., Ono K., Horiguchi M., Nishi H., Nakamura T., Nagao K., Kinoshita M., Kuwabara Y., Marusawa H., Iwanaga Y. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc. Natl. Acad. Sci. USA. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marquart T.J., Allen R.M., Ory D.S., Baldán A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rayner K.J., Suárez Y., Dávalos A., Parathath S., Fitzgerald M.L., Tamehiro N., Fisher E.A., Moore K.J., Fernández-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rotllan N., Ramírez C.M., Aryal B., Esau C.C., Fernández-Hernando C. Therapeutic silencing of microRNA-33 inhibits the progression of atherosclerosis in Ldlr-/- mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2013;33:1973–1977. doi: 10.1161/ATVBAHA.113.301732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horie T., Baba O., Kuwabara Y., Chujo Y., Watanabe S., Kinoshita M., Horiguchi M., Nakamura T., Chonabayashi K., Hishizawa M. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE-/- mice. J. Am. Heart Assoc. 2012;1:e003376. doi: 10.1161/JAHA.112.003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagschal A., Najafi-Shoushtari S.H., Wang L., Goedeke L., Sinha S., deLemos A.S., Black J.C., Ramírez C.M., Li Y., Tewhey R. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 2015;21:1290–1297. doi: 10.1038/nm.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L., Sinnott-Armstrong N., Wagschal A., Wark A.R., Camporez J.P., Perry R.J., Ji F., Sohn Y., Oh J., Wu S. A MicroRNA Linking Human Positive Selection and Metabolic Disorders. Cell. 2020;183:684–701.e14. doi: 10.1016/j.cell.2020.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dávalos A., Goedeke L., Smibert P., Ramírez C.M., Warrier N.P., Andreo U., Cirera-Salinas D., Rayner K., Suresh U., Pastor-Pareja J.C. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA. 2011;108:9232–9237. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Picaud S., Dalkara D., Marazova K., Goureau O., Roska B., Sahel J.A. The primate model for understanding and restoring vision. Proc. Natl. Acad. Sci. USA. 2019;116:26280–26287. doi: 10.1073/pnas.1902292116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yiu G., Tieu E., Munevar C., Wong B., Cunefare D., Farsiu S., Garzel L., Roberts J., Thomasy S.M. In Vivo Multimodal Imaging of Drusenoid Lesions in Rhesus Macaques. Sci. Rep. 2017;7:15013. doi: 10.1038/s41598-017-14715-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asare-Bediako B., Noothi S.K., Li Calzi S., Athmanathan B., Vieira C.P., Adu-Agyeiwaah Y., Dupont M., Jones B.A., Wang X.X., Chakraborty D. Characterizing the Retinal Phenotype in the High-Fat Diet and Western Diet Mouse Models of Prediabetes. Cells. 2020;9:464. doi: 10.3390/cells9020464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ambati J., Atkinson J.P., Gelfand B.D. Immunology of age-related macular degeneration. Nat. Rev. Immunol. 2013;13:438–451. doi: 10.1038/nri3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao X., Shen D., Patel M.M., Tuo J., Johnson T.M., Olsen T.W., Chan C.C. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol. Int. 2011;61:528–535. doi: 10.1111/j.1440-1827.2011.02695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curcio C.A., Johnson M., Huang J.D., Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog. Retin. Eye Res. 2009;28:393–422. doi: 10.1016/j.preteyeres.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ach T., Tolstik E., Messinger J.D., Zarubina A.V., Heintzmann R., Curcio C.A. Lipofuscin redistribution and loss accompanied by cytoskeletal stress in retinal pigment epithelium of eyes with age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2015;56:3242–3252. doi: 10.1167/iovs.14-16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan C.C., Ardeljan D. Molecular pathology of macrophages and interleukin-17 in age-related macular degeneration. Adv. Exp. Med. Biol. 2014;801:193–198. doi: 10.1007/978-1-4614-3209-8_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devarajan G., Niven J., Forrester J.V., Crane I.J. Retinal Pigment Epithelial Cell Apoptosis is Influenced by a Combination of Macrophages and Soluble Mediators Present in Age-Related Macular Degeneration. Curr. Eye Res. 2016;41:1235–1244. doi: 10.3109/02713683.2015.1109129. [DOI] [PubMed] [Google Scholar]
- 53.Knickelbein J.E., Chan C.C., Sen H.N., Ferris F.L., Nussenblatt R.B. Inflammatory Mechanisms of Age-related Macular Degeneration. Int. Ophthalmol. Clin. 2015;55:63–78. doi: 10.1097/IIO.0000000000000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ebrahimi K.B., Handa J.T. Lipids, lipoproteins, and age-related macular degeneration. J. Lipids. 2011;2011:802059. doi: 10.1155/2011/802059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ehrlich R., Harris A., Kheradiya N.S., Winston D.M., Ciulla T.A., Wirostko B. Age-related macular degeneration and the aging eye. Clin. Interv. Aging. 2008;3:473–482. doi: 10.2147/cia.s2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levy O., Calippe B., Lavalette S., Hu S.J., Raoul W., Dominguez E., Housset M., Paques M., Sahel J.A., Bemelmans A.P. Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol. Med. 2015;7:211–226. doi: 10.15252/emmm.201404524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cherepanoff S., McMenamin P., Gillies M.C., Kettle E., Sarks S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010;94:918–925. doi: 10.1136/bjo.2009.165563. [DOI] [PubMed] [Google Scholar]
- 58.Burgess S., Davey Smith G. Mendelian Randomization Implicates High-Density Lipoprotein Cholesterol-Associated Mechanisms in Etiology of Age-Related Macular Degeneration. Ophthalmology. 2017;124:1165–1174. doi: 10.1016/j.ophtha.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reynolds R., Rosner B., Seddon J.M. Serum lipid biomarkers and hepatic lipase gene associations with age-related macular degeneration. Ophthalmology. 2010;117:1989–1995. doi: 10.1016/j.ophtha.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson L.V., Forest D.L., Banna C.D., Radeke C.M., Maloney M.A., Hu J., Spencer C.N., Walker A.M., Tsie M.S., Bok D. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. USA. 2011;108:18277–18282. doi: 10.1073/pnas.1109703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lyssenko N.N., Haider N., Picataggi A., Cipollari E., Jiao W., Phillips M.C., Rader D.J., Chavali V.R.M. Directional ABCA1-mediated cholesterol efflux and apoB-lipoprotein secretion in the retinal pigment epithelium. J. Lipid Res. 2018;59:1927–1939. doi: 10.1194/jlr.M087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang L., Li C.M., Rudolf M., Belyaeva O.V., Chung B.H., Messinger J.D., Kedishvili N.Y., Curcio C.A. Lipoprotein particles of intraocular origin in human Bruch membrane: an unusual lipid profile. Invest. Ophthalmol. Vis. Sci. 2009;50:870–877. doi: 10.1167/iovs.08-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gnanaguru G., Choi A.R., Amarnani D., D’Amore P.A. Oxidized Lipoprotein Uptake Through the CD36 Receptor Activates the NLRP3 Inflammasome in Human Retinal Pigment Epithelial Cells. Invest. Ophthalmol. Vis. Sci. 2016;57:4704–4712. doi: 10.1167/iovs.15-18663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu J., Copland D.A., Theodoropoulou S., Chiu H.A., Barba M.D., Mak K.W., Mack M., Nicholson L.B., Dick A.D. Impairing autophagy in retinal pigment epithelium leads to inflammasome activation and enhanced macrophage-mediated angiogenesis. Sci. Rep. 2016;6:20639. doi: 10.1038/srep20639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xin-Zhao Wang C., Zhang K., Aredo B., Lu H., Ufret-Vincenty R.L. Novel method for the rapid isolation of RPE cells specifically for RNA extraction and analysis. Exp. Eye Res. 2012;102:1–9. doi: 10.1016/j.exer.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.