

Abstract
MicroRNAs (miRNAs) regulate gene expression by post-transcriptional inhibition of target genes. Proangiogenic small extracellular vesicles (sEVs; popularly identified with the name “exosomes”) with a composite cargo of miRNAs are secreted by cultured stem cells and present in human biological fluids. Lipid nanoparticles (LNPs) represent an advanced platform for clinically approved delivery of RNA therapeutics. In this study, we aimed to (1) identify the miRNAs responsible for sEV-induced angiogenesis; (2) develop the prototype of bioinspired “artificial exosomes” (AEs) combining LNPs with a proangiogenic miRNA, and (3) validate the angiogenic potential of the bioinspired AEs. We previously reported that human sEVs from bone marrow (BM)-CD34+ cells and pericardial fluid (PF) are proangiogenic. Here, we have shown that sEVs secreted from saphenous vein pericytes and BM mesenchymal stem cells also promote angiogenesis. Analysis of miRNA datasets available in-house or datamined from GEO identified the let-7 family as common miRNA signature of the proangiogenic sEVs. LNPs with either hsa-let-7b-5p or cyanine 5 (Cy5)-conjugated Caenorhabditis elegans miR-39 (Cy5-cel-miR-39; control miRNA) were prepared using microfluidic micromixing. let-7b-5p-AEs did not cause toxicity and transferred functionally active let-7b-5p to recipient endothelial cells (ECs). let-7b-AEs also improved EC survival under hypoxia and angiogenesis in vitro and in vivo. Bioinspired proangiogenic AEs could be further developed into innovative nanomedicine products targeting ischemic diseases.
Keywords: nanomedicine, extracellular vesicles, microRNAs, angiogenesis, endothelial cells, let-7b-5p
Graphical abstract
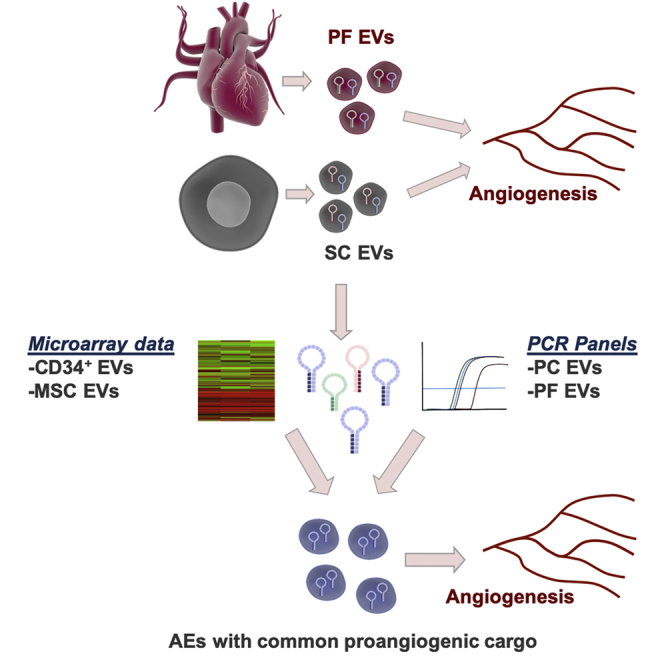
Robust therapeutic angiogenesis approaches are needed for patients with ischemic disease. Exosomes, which are the vectors of stem cell paracrine actions, command proangiogenic responses, often mediated by microRNA transfer. We have designed and functionally validated a stem cell exosome-inspired synthetic proangiogenic exosome prototype based on clinically compatible LNPs and microRNAs.
Introduction
Ischemic disease in the heart and lower extremities is the leading cause of death around the world.1,2 Stem cells (SCs), including mesenchymal SCs (MSCs) and bone marrow (BM) CD34-positive (CD34+) mononuclear cells (MNCs), have been proposed to improve post-ischemic angiogenesis and promote the recovery of blood flow to inadequately perfused areas. However, several approaches based on autologous transplantation of proangiogenic SCs have, so far, failed to convince when trialed clinically in ischemic patients.3,4 This could be partially accountable to the intrinsic defects of the patient-derived cells, which were trialed as autologous remedies in severely compromised patients.
Exosomes are membrane-bound small extracellular vesicles (sEVs), which are produced in the endosomal compartment of most eukaryotic cells. They contribute in cell-to-cell communication mechanisms, underpinning homeostatic, pathogenetic, and reparative processes.5,6 Recently updated guidelines of the International Society for Extracellular Vesicles (ISEV) on minimal information for studies of EVs (MISEVs) recommend the term “extracellular vesicle” or “EV” as the consensus generic term for lipid bilayer-delimited particles released from the cell.7 Consequently, in the most recent literature, exosomes are often identified as sEVs. We will adhere to this nomenclature when describing the endogenous nanovesicles released by cells in this paper. Notwithstanding, in recognition of the popular use of “exosomes,” we have adopted the term of “artificial exosomes” (AEs) when referring to the bioinspired synthetic version we have developed with this study. sEVs are 30−150 nm in size and contain a cargo of biologically active microRNAs (miRNAs, miRs) but also proteins, lipids, other RNA molecules, DNA, and metabolites. Depending on the cellular sources, sEVs have been shown to modulate angiogenesis in opposite directions.8,9 Similar to their parent cells, sEVs originated from healthy MSCs and CD34+ cells have a proangiogenic potential, which is mediated, at least in part, by miRNAs.10, 11, 12, 13, 14, 15 Proangiogenic sEVs can also be found in human biological fluids, including the human pericardial fluid (PF) from cardiac surgery patients who are not suffering from ischemic disease.16 We revealed that PF-derived sEVs promote angiogenesis and improve post-ischemic blood flow recovery in mice by transferring miRNA let-7b-5p (let-7b) to endothelial cells (ECs).16 Moreover, we showed that human saphenous vein pericytes (hSVPs), which represent a source of vascular progenitor cells, increase post-ischemic angiogenesis via paracrine actions.17 As part of this new study, we have characterized the role of sEVs in mediating the angiogenic properties of hSVPs and identified the miRNAs contained in the pericyte-derived sEVs. We have also validated the proangiogenic capacity of sEVs prepared from BM-MSCs in our laboratory.
Allogenic sEVs, also identified with the name of exosomes, are currently considered to represent a more manipulable, biological alternative to SC-based therapeutics. In fact, allogenic sEVs are not supposed to be rejected by the host immune system after transplantation.18,19 Moreover, sEVs can help in mitigating the risks of both microvascular occlusion and cancerous growth, which are intrinsically linked with the transplant of therapeutic SCs.20 According to https://www.clinicaltrials.gov/ , there are currently 148 clinical trials using sEVs as therapy and/or diagnosis. Out of them, twelve studies are testing sEVs as innovative therapeutic approaches (revised in Floriano et al.21). However, the production of sEVs from SCs requires laborious cell-handling tasks. The complexity of the cargo of endogenous sEVs together with the limited amount of sEVs with a reproducible composition and function that can be prepared from cultured SCs represents major challenges, which currently hamper the translation of endogenous sEVs from basic and translational research studies to off-the-shelf “pharmaceutical products.” We reasoned that among the individual miRNAs composing the cargo of proangiogenic sEVs, some may be more relevant than others to ensure the proangiogenic response. So, bioinspired AEs incorporating prototypical proangiogenic miRNAs and devoid of anti-angiogenic and angiogenesis-neutral miRNAs could afford proangiogenic responses, while ensuring feasibility for mass production.
When designing clinical-relevant AEs, an important consideration must be given to their lipid membrane composition. Lipid nanoparticles (LNPs) containing ionizable amino lipids are the most advanced non-viral delivery platforms for negatively charged nucleic acids, such as small interfering RNAs (siRNAs) and miRNAs. LNPs can ensure in vivo delivery of nucleic acids with less or no immunogenic responses compared to viral gene delivery.22, 23, 24, 25 A recent US Food and Drug Administration (FDA)-approved RNAi-based drug Onpattro (patisiran), used for contrasting polyneuropathy induced by a genetic form of amyloidosis, is comprised of the ionizable amino lipid Dlin-MC3-DMA (MC3) as part of the LNP structure.26
In our study, we have identified the proangiogenic miRNA signature shared by proangiogenic sEVs from 4 different human sources but absent in the sEVs that do not induce angiogenesis. This new knowledge has been used to design a proangiogenic AE prototype encapsulating let-7b in MC3-based LNPs. Finally, the proangiogenic potential of the bioinspired AEs has been tested in vitro and in vivo.
Results
Pericyte-sEVs are proangiogenic
We previously showed that hSVPs improve cardiac function in the mouse myocardial infarction model through a paracrine mechanism involving the secretion of proangiogenic miR-132.17 To investigate the role of sEVs in this paracrine action, sEVs were isolated from the conditioned medium (CM) of hSVPs. Nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM), and dynamic light scattering (DLS) analyses confirmed the presence of sEVs in the preparations (Figures 1A and S1). By in vitro Matrigel assay, we demonstrated that hSVP-sEVs, especially at the concentration of 2.5 μg/mL, promote tubule formation in hypoxic (1% O2) human umbilical vein ECs (HUVECs) (Figures 1B and 1C). hSVP-sEVs also reduced caspase activity and induced proliferation of hypoxic HUVECs compared with hSVP EV-free CM and untreated cell groups (Figure 1D). Both EV-free CM and hSVP-EVs improved EC metabolism (adenosine triphosphate [ATP] production) and viability compared with the untreated cell group (Figure 1D).
Figure 1.

Characterization and activity of hSVP EVs
(A) NTA results show that hSVP EVs can be isolated from cell culture media using Exo-Spin columns (n = 3). (B) Images of Matrigel assay and evaluation of different parameters in Matrigel assay using ImageJ angiogenesis analyzer tool. hSVP EVs increase HUVEC tubule-like formation on Matrigel under hypoxic conditions (1% O2). Scale bars, 200 μm. hSVP EV concentration: 2.5 μg/mL. (C) Evaluation of different parameters in Matrigel assay using ImageJ angiogenesis analyzer tool. (D) hSVP EVs increase cell proliferation and decrease caspase activity compared with other groups at 2.5 μg/mL concentration. In all graphs, values are given as average ± SEM (n = 5-10). ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001. Comparisons between EV and EV-free groups were performed with Mann-Whitney U test.
Bioinformatic analyses reveal let-7a-5p (let-7a) and let-7b as common proangiogenic miRNA cargos
In order to understand whether there is a common miRNA signature for the proangiogenic sEVs, we performed computational analysis integrating miRNA datasets obtained in-house (BM-CD34+ MNCs and BM-MNC;14 hSVP-sEVs; PF-sEVs) (Tables S1 and S2) or available through GEO (MSCs). As non-angiogenic controls, we used sEV miRNA datasets obtained from unsorted BM-MNCs and the PF of ischemic patients who were operated on for coronary artery bypass graft (CABG). Recent evidence shows that PF-sEVs from CABG patients do not promote angiogenesis (unpublished data). Our analysis revealed the enrichment of 21 common miRNAs in the proangiogenic EVs (Figure 2A). Due to the importance of the let-7 family in angiogenesis and because we detected a high expression of these miRNAs in all of the proangiogenic sEVs analyzed in this study, we focused on the miRNAs let-7a and let-7b in the following sections. Proangiogenic potential of sEVs prepared from MSCs (passage 4 [P4]) was also validated in our lab (Figure S2). Additionally, we showed that the function of MSC-sEVs was highly dependent on the passage number of the mother cells (Figure S3). Finally, we confirmed the presence of let-7a and let-7b in MSC-sEVs obtained from MSCs at P4 (Figure S4).
Figure 2.

Preparation and internalization of AEs
(A) let-7a and let-7b are among the top common miRNAs in all angiogenic EVs tested. (B) miRNA-loaded AEs with the size of ∼50 nm can be prepared by microfluidic micromixing. Scale bar is 200 nm in TEM picture. (C) Flow cytometry analysis of Cy5-cel-miR-39-AEs internalized by HUVECs under hypoxia (1% O2). (D and E) AEs can pass their cargo (cel-miR-39 in the graphs) to HUVECs under both hypoxia (1% O2) and normoxia (20% O2). Relative expression was given as 2−ΔΔCt (normalization to untreated cell group). In all graphs, values are given as average ± SEM (n = 2−4).
Members of the let-7 family have proangiogenic properties
We next validated the angiogenic potential of let-7a and let-7b on hypoxic ECs (HUVECs cultured in 1% O2). For this purpose, we transfected HUVECs using pre-miR molecules or scrambled controls. Our results confirmed that scrambled controls do not affect cell behavior in a negative way (Figure S5). To understand whether we can improve the therapeutic capacity of these miRNAs by using them together, we also transfected the cells both with let-7a and let-7b simultaneously (co-transfection). The transfection of cells and downregulation of gene targets were confirmed by qPCR (Figure S6). When the cells were transfected with either let-7a or let-7b, cell viability was improved compared with the scramble control group (Figure S7A). let-7a also increased cell proliferation in a dose-dependent manner (Figure S7A). Contrarily, the co-transfection caused a decrease in cell viability and metabolism (Figure S7B).
In vitro Matrigel assay showed that let-7a and let-7b, given either together or via co-transfection, increase sprout formation of hypoxic HUVECs (1% O2) on Matrigel (Figures S8–S10). However, the percentage of increase for different parameters was lower after co-transfection of the 2 miRNAs when compared with individual transfections. Therefore, we decided to focus on let-7b alone for the next part of the study, including the formulation of AEs and proof-of-concept studies.
AEs can be prepared by microfluidic micromixing
sEVs might have different lipidic structures depending on their parent cells and the environmental conditions of these cells. However, it is known that the sEV membranes have more cholesterol and less phosphatidylcholine in comparison with their mother cells.27,28 Similarly, our AE formulation has more cholesterol and less phosphatidylcholine compared with the widely used liposome formulations in the literature. AEs encapsulating either let-7b or fluorescence (cyanine 5 [Cy5])-conjugated cel-miR-39-3p (cel-miR-39) (a miRNA present in C. elegans but not in vertebrates) were prepared using a microfluidic mixing system as previously described (Figure 2B).29, 30, 31, 32 The size of AEs was around 48.41 ± 0.45 for let-7b-AEs and 56.95 ± 3.37 for Cy5-cel-miR-39-AEs with a narrow size distribution (polydispersity index [PDI] ∼ 0.2−0.3) as measured by DLS (Figure 2B). ζ-potential measurements showed a slight negative surface charge, as expected, at physiological pH.33 TEM analysis of the LNPs indicated a globular shape and size distribution in accordance with the DLS measurements (Figure 2B).
AEs can be internalized by and transfer miRNA to ECs
Flow cytometry analyses showed that AEs are internalized by HUVECs cultured under hypoxic conditions (1% O2) (Figure 2C). In order to measure the percentage of cells internalizing AEs, cells were treated with Cy5-cel-miR-39-AEs. After different time points, cells were washed to remove AEs that were not internalized or bound and finally, analyzed by flow cytometry. Even after 4 h, almost 100% of the cells had internalized the miRNA content of the AEs (Figure 2C). After 12 h, all cells were labeled with AE-derived, Cy5-positive cel-miR-39, and this percentage did not change up to 24 h. The capacity of the cells to internalize the AE-delivered miRs was also validated by PCR for cel-miR-39-AEs (Figure 2D for normoxia and Figure 2E for hypoxia).
AEs do not induce toxicity in vascular cells
After confirming the internalization of the AEs by HUVECs, we checked whether they cause any cytotoxicity in the potential recipient cells. Metabolically active cells produce ATP that reflects their viability. Hence, we used an ATP production assay to show any possible cytotoxic effects of AEs on the cells. HUVECs were cultured with different concentrations of AEs (both Cy5-cel-miR-39-AEs and let-7b-AEs) up to 48 h. After 24 h, a slight decrease in the ATP production of the cells treated with especially higher concentrations of Cy5-cel-miR-39-AEs was observed (Figure 3A). However, after 48 h with AEs, the cells recovered and showed even higher levels of ATP production, indicating higher cell numbers (next sections give more detailed analysis about the behavior of the cells treated with the AEs) (Figure 3A). Similar to the results obtained from HUVECs, cardiac human microvascular ECs (cHMVECs) showed a slight decrease in the ATP production at the higher concentrations of the AEs after 24 h but recovered and increased cell number after 48 h with AEs (Figure S11A). In hSVPs, higher concentrations of AEs slightly decreased ATP production after 48 h with AEs (Figure S11B).
Figure 3.

The effects of let-7b-AEs on HUVEC activity
(A) AEs are not toxic for HUVECs. For toxicity studies, cells were cultured under normoxic conditions (20% O2) (n = 5−10). (B) let-7b-AEs decrease caspase activity and increase proliferation in hypoxic (1% O2) HUVECs (n = 5). (C) let-7b-AEs increase sprout formation on Matrigel (n = 5). Scale bars, 200 μm. (D) let-7b-AEs pass active let-7b to the cells and (E−G) decrease let-7b gene target expression. (E) Results are given for 60 nM let-7b-AEs. Relative expression was given as 2−ΔΔCt (normalization to untreated group in D and to cel-miR-39-AE group in E−G). (D−G) n = 4. In all graphs, values are given as average ± SEM, and “n” indicates completely independent biological replicates. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001. Comparison across subgroups was performed with Kruskal-Wallis test and between groups with Mann-Whitney U test.
let-7b-loaded AEs improve the survival and tubule-like formation capacities of ECs
let-7b-AEs reduced caspase activity and increased proliferation in hypoxic (1% O2) HUVECs compared with Cy5-cel-miR-39-AEs after 48 h (Figure 3B). They slightly increased caspase-3 activity and cell proliferation in hSVPs at 60 nM concentration and decreased cell metabolism in cHMVECs, both cultured under 1% O2 (Figure S12).
Next, we checked whether the transfer of let-7b can increase the angiogenic response of the cells under hypoxia (1% O2). Our results showed that let-7b-AEs could increase sprouting of HUVECs both in 2.5D Matrigel and 3D fibrin gel bead assays (Figures 3C, 4, S13, and S14) and of cHMVECs in Matrigel assay (Figure 5). Although higher concentrations of let-7b-AEs had a higher impact on the tubule-like formation in HUVECs, lower let-7b-AE concentrations favored sprout formation in cHMVECs (Figures 3C and 5).
Figure 4.

Fibrin gel bead assay to evaluate the angiogenic potential of let-7b-AEs in HUVECs under hypoxic conditions (1% O2)
(A and B) Representative images (A) and quantification of sprout length and number of sprouts (n = 4) (B) after 30 h. 30 nM let-7b-AEs were used in the experiments. White arrows indicate the sprouts. In all graphs, values are given as average ± SEM (n = 5−10). ∗∗p ≤ 0.01. Scale bars, 300 μm. Comparison across subgroups was performed with Kruskal-Wallis test.
Figure 5.

Matrigel analysis of cHMVECs treated with cel-miR-39-AEs or let-7b-AEs under hypoxic conditions (1% O2)
(A) Representative Matrigel photos. Scale bars, 200 μm. (B and C) Analysis of different parameters by ImageJ angiogenesis analyzer. In all graphs, values are given as average ± SEM (n = 5). ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001. Comparisons between cel-miR-39-AE and let-7b-AE groups were performed with Mann-Whitney U test.
AEs pass let-7b to ECs and decrease the expression of its gene targets
Internalization studies clearly showed that AEs are taken up by ECs (Figures 2C−2E). However, as important as being internalized by the target cells, AEs also must be able to pass their miRNA payload in a functionally active form to the recipient cells (Figure 3D). Our PCR results indicated that AEs can increase the let-7b levels in recipient ECs in a time-dependent manner (Figure 3D), while subsequently decreasing the expression of a pool of let-7b target genes (Figures 3E−3G). Interestingly, let-7b-AEs could downregulate CASP3 more efficiently under hypoxic conditions than normoxic conditions after 4 h (Figure 3E). This can explain the decrease in caspase activity in HUVECs (Figure 3B). At 15 nM concentration, let-7b-AEs could downregulate gene targets in HUVECs as effectively as let-7a or let-7b Lipofectamine complexes (Figures 3F and S6).
let-7b-AEs induce formation of microvessels in vivo
To test the therapeutic effect of let-7b-AEs in vivo, we used an in vivo Matrigel plug assay.34 Matrigel xenografts supplemented with let-7b-AEs-transfected HUVECs increased the density of vessel-like structures in comparison to cel-miR-39-AEs and untreated cell control groups. The plug vascularization in the let-7b-AE group was comparable to the vascular endothelial growth factor (VEGF)-positive control (Figures 6A, 6B, and S15). Although statistical significance (defined as p < 0.05) was barely missed (p = 0.0571), we could detect a clear trend suggesting the capacity of let-7b-AEs to increase the number of CD31+ ECs (in comparison with negative controls) (Figure 6C).
Figure 6.

The let-7b-AE group has a tendency to increase HUVEC survival in the Matrigel plug assay in vivo
(A and B) Representative images (A) and quantification of number of vessel-like structures per image (B). One-way ANOVA with Bonferroni post hoc test was used for statistical analysis. ∗∗p ≤ 0.01. (C) Quantification of number of nuclei/CD31 area. Comparison between untreated and let-7b-AE groups was performed with Mann-Whitney U test (p = 0.0571). In CD31 images, pseudocolor was used (red channel shown in green) to make cells easier to see. All data are expressed as mean ± SEM, n = 4. Scale bars, 50 μm.
Discussion
This study has developed a prototype of clinically compatible proangiogenic, synthetic sEVs. Our AEs are formed by LNPs containing ionizable amino lipids, which are the most advanced non-viral delivery platforms available for negatively charged nucleic acids, such as miRNAs. Ionizable amino lipid is a critical component of LNP functionality. Notably, the amino lipid MC3 used to build our AEs also composes the LNP structure of a recently FDA-approved, RNAi-based drug, Onpattro (patisiran).26 In addition, the lipidic composition of our LNP-based AEs is different from widely used liposomes and has more cholesterol and less phosphatidylcholine as the natural sEV exosomes.27,28 The microfluidic mixing technology based on the NanoAssemblr device used by us is compatible with scaling up in the good manufacturing practice (GMP) of clinical-grade nanoparticle-based therapeutics.
As part of the current study, we have tested the in vivo angiogenic capacity of our AE prototype using the Matrigel plug model, which does not propose biodistribution and off-targeting issues. In fact, ECs were exposed to AEs in vitro before being mixed with Matrigel and injected in immunocompromised mice. Future studies will employ animal models of ischemia to test the biodistribution and therapeutic angiogenesis potential of AEs. In details, we will trial the capacity of the let-7b-AEs to promote the formation of functional vessels, improving post-ischemic blood flow recovery. Such clinically relevant investigations are deemed essential to progress our translational pathway, and we aim at developing them with a refined AE formulation. Especially, we will functionalize the bioinspired proangiogenic AEs to improve their targeting to ECs within ischemic tissues. This should be feasible considering we have recently showed that it is possible to develop ionizable lipids for effective in vivo siRNA delivery and gene silencing in hard-to-transfect cells, such as T-leukocyte subsets.25 The structure of ionizable lipids is divided into three major parts: hydrophilic amine head group, hydrophobic lipid chain, and a linker region, which connect these two parts. Our recent leukocyte-targeting study has identified the possibility to improve siRNA-based gene silencing in a leukocyte subset by synthesizing ionizable lipids based on the linkers ethanolamine, hydrazine, and hydroxylamine.25 A similar approach can be developed to improve in vivo targeting of LNPs carrying either siRNAs or miRNAs to ECs.
Scientists working at sEV characterization have so far mostly focused on the miRNA and protein content of sEVs. Although we have not performed lipidomic analyses of the proangiogenic and antiangiogenic sEV used in the paper, this is part of our future plans. We regard the characterization of the lipidome of endogenous sEV to further advance our understanding of endogenous sEV and to improve the therapeutic use of sEVs and exosome mimetics.
The miRNA payload of the proangiogenic AEs developed here consists of let-7b, which has emerged from unbiased analyses of the miRNAs shared across proangiogenic endogenous sEVs. However, the miRNA cargo can be easily modified in line with a personalized medicine approach, for example, to supply tailored pro- and anti-angiogenic therapies in a different group of patients. Moreover, the AEs could also be modified to deliver anti-miRNAs and siRNAs to ECs.
Liposomes have been already employed to deliver miRNAs in vivo.35,36 However, the use of miRNA-carrying liposomes to modulate angiogenesis in vivo is still very limited, with the absence of major reports in the recent literature. In vivo biodistribution of liposomes has been shown to be significantly influenced by changes in their surface potentials.37 By contrast, our formulation offers the advantage of maintaining the distribution profile based on the design of the headgroup moieties and despite differences in surface potentials. This is a significant advantage for RNA-based therapeutics because RNA molecules are negatively charged, and addition of different RNA sequences to AEs can influence the nanoparticle ζ-potential. However, in our formulation, ζ-potential is mostly affected by the composition of LNP structural lipids and less from the encapsulated RNA.
Endogenous AEs, which are produced from different types of SCs, share the therapeutic angiogenic potential of their parent cells, thus considered as “SCs without the cells.”8 Here, we have expanded the number of SC and progenitor types able to secrete proangiogenic sEVs by demonstrating that pericytes produce highly proangiogenic sEVs in culture. Moreover, we have confirmed the angiogenic potential of MSC-sEVs.38,39 In addition to their proangiogenic capacity, MSC-sEVs can reduce infarct size,40,41 and they have also been shown to increase ATP levels, decrease oxidative stress, and activate prosurvival pathways in a rodent model of myocardial infarct and ischemia/reperfusion injury.42 Furthermore, MSC-sEVs have an attractive immunomodulatory capacity, inducing immunoregulatory, anti-inflammatory monocyte phenotypes, as evidenced in studies on different bronchopulmonary disease models.43,44 Notwithstanding, a restriction on endogenous sEV utilization for therapeutic application is their limited secretion from SCs. This bottleneck is especially relevant for MSCs, which have a limited expansion capacity and become senescent after only a few passages in culture.45 It was already known that sEVs derived from senescent SCs have impaired regenerative capacity compared to young cells,45 and we have directly validated this in our study, where we have detected dramatic differences in the EC survival and proliferative responses induced by sEVs prepared from MSCs at different passages (P4 versus P7−8). This issue, however, does not prevent deriving information on the miRNA cargo of the highly functional sEVs produced by young MSCs and then using such knowledge for designing AE counterparts.
To decide on the miRNA payload to be used to design our prototype of angiogenic AEs, we have bioinformatically integrated sEV-miRNA datasets from both cultured human cells, including MSCs, pericytes, BM-CD34+ MNCs, and total BM-MNCs14,15 and a human biological fluid in direct contact with the heart, the PF. We already showed that the PF of non-ischemic patients contains sEVs with high proangiogenic potential in vitro and in a murine limb ischemia model.16 Moreover, recent evidence shows that PF-sEVs from patients with ischemic heart disease are unable to stimulate angiogenesis responses in recipient ECs (unpublished data). The addition of miRNA datasets from PF-sEVs to this study is justified by our interest in understanding the proangiogenic miRNA cargo of sEVs, which are already in the position to interact with ECs of the heart, an organ that could particularly benefit from AE-mediated proangiogenic therapies. Our computational analyses have also included the miRNA cargo of PF-sEVs from patients with severe ischemic heart disease requiring CABG surgery. The sEV-miRNAs extracted from the PF of CABG patients have been assumed here not to be significant contributors to in vivo autonomous reparative angiogenic responses and treated as “non-proangiogenic” controls in our miRNA analyses. let-7b has emerged from unbiased bioinformatic analyses developed using predefined filter criteria for miRNA selection. It is, however, noteworthy that the role of let-7b in mediating the in vitro angiogenic potential of PF-sEVs was already demonstrated by us.16 Moreover, the capacity of other members of the let-7 family, but not let-7b, to regulate ocular angiogenesis has been recently established using a transgenic mouse model, overexpressing let-7a, -7d, and -7f.46
We have analyzed the mRNA expressional changes induced by the AE-let-7b in ECs to characterize the functional capacity of AE-derived miRNA to inhibit the miRNA target genes in recipient cells. That being said, it is possible that the deregulation of some of these genes targeted by let-7b contributed to the survival and angiogenic responses observed in ECs treated with AE-let-7b. In details, the apoptotic protease-activating factor-1 (APAF1) is a key molecule in the mitochondrial pathway of apoptosis. Its inhibition by let-7b-5b could therefore contribute to the prosurvival effect of this miR.47 The loss of STARD-13 (StAR-related lipid transfer [START] domain containing 13), also known as (aka) DLC2 (deleted in liver cancer 2), has been associated with angiogenic responses.48 Finally, CTNNB1 (catenin beta 1, aka β-catenin), as part of the Wnt/β-catenin pathway, can either promote or inhibit angiogenesis.49 The Wnt receptor Frizzled-4 (FZD4) has also a dual impact on angiogenesis regulation.50,51
In conclusion, we have provided the evidence that the let-7 family of miRNAs is highly expressed in 4 different types of proangiogenic sEVs derived from different sources and that the incorporation of let-7b into LNP-based AEs allows the efficient delivery of this miRNA to ECs regulating functional responses, which lead to angiogenesis. Our results suggest the potential of AEs as a platform in therapeutic angiogenesis.
Materials and methods
Clinical sample collection and processing
The collection and use of clinical leftover samples from cardiac surgery patients complied with the ethical principles stated in the Declaration of Helsinki and the Human Tissue Act and were covered by ethical approvals from the UK National Research Ethics Service (NRES; Research Ethics Committee [REC] references 13/OL/1687 and 10/HO107/63). The collection of long saphenous vein leftovers from CABG surgery complied with the principles stated in the Declaration of Helsinki and was approved by the Bath REC (reference 06/Q2001/197). Patients gave written, informed consent for inclusion in the study.
Preparation of PF-sEVs
PF samples from patients undergoing aortic valve replacement (AVR) and CABG were collected immediately after opening of the pericardium using a 20-mL syringe and transported to the laboratory in a sterile 50-mL container. Information for the patients used in this study is given in Table S3. PF was centrifuged at 13,500 × g, room temperature (RT), for 5 min to deplete miRNA-rich platelets. The platelet-free fluid was centrifuged at 13,500 × g, RT, for 5 min to remove any remainder of cells. Processed samples were stored at −80°C until use. EVs were isolated from the PF of the patients undergoing AVR and CABG surgeries (n = 4 in each group) using Exo-Spin columns from Cell Guidance Systems (Cambridge, UK). Briefly, 200 μL PF was filtered through a sterile 0.22-μm filter (Merck Millipore, Burlington, MA, USA) into a fresh tube. Then, the filtered PF was applied onto the Exo-Spin columns, and EV separation was performed as described by the manufacturer. EVs isolated from 200 μL PF and concentrated in 100 μL PBS were used in RNA isolation without any further modification.
Preparation of sEVs from human BM-CD34+ cells
BM-MSCs were purchased from Rooster Bio (Frederick, MD, USA). Please see Major Resources (Table S4) for more information about BM-MSCs. Cells at P4 to P8 were seeded in T225 flasks (Cellbind Flasks, Corning, NY, USA) containing 45 mL of α-minimum essential medium (MEM; Sigma-Aldrich, St. Louis, MO, USA) with 10% fetal bovine serum (FBS; seeding density of 2.9−3.7 × 103 cells/cm2). Upon reaching 98%–99% confluency, cells were washed three times with PBS and incubated for 38 h in 35 mL of α-MEM serum-free medium. EVs were isolated from a starting volume of 280 mL MSC CM. Briefly, medium was centrifuged (at 300 × g for 10 min at RT; 2,000 × g for 20 min at +4°C, and 13,000 × g for 30 min at +4°C), filtered through a sterile 0.22-μm filter (Millipore Express polyethersulfone [PES] membrane; Millipore, Burlington, MA, USA), and concentrated to a final volume of 500 μL with Centricon Plus-70 Centrifugal Filter Units 100 molecular weight cut-off (MWCO; Millipore, Burlington, MA, USA), according to the manufacturer’s protocol. sEVs were subsequently isolated by size-exclusion chromatography with the EVoriginal/35 nm Izon column (Izon Science, Christchurch, New Zealand). sEV-containing fractions (7−9) were pulled together and concentrated to a final volume of 200 μL with the Amicon Ultra-4 Centrifugal Filter Units 10 MWCO (Millipore, Burlington, MA, USA). The output of sEV preparation was quantified with the NanoSight NS300 system at 1:100 concentration. A final concentration of 1.9 × 1010 particles/mL was detected. Total protein concentration was determined using Micro bicinchoninic acid (BCA) protein assay (BosterBio, Pleasanton, CA, USA) according to the manufacturer’s protocol.
Culture of hSVPs and isolation of sEVs
Intravascular hSVPs were isolated from saphenous veins that were leftovers from CABG surgery, as described.17,52 For the isolation of the sEVs, pericytes (P6) were seeded in complete EGM-2 medium, and after the attachment of the cells, the medium was replaced by EGM-2 (without SingleQuot FBS; Lonza, Basel, Switzerland) containing 2% sEV-depleted FBS from Gibco (Waltham, MA, USA). Pericyte CM was collected from confluent pericyte cultures every 3 days and centrifuged at 300 × g for 10 min to remove cells. Then, the supernatant was transferred into a new centrifuge tube and spun at 16,000 × g for 30 min to remove any remaining cell debris. The supernatant was transferred into a new Falcon and kept at −80°C until use. On the day of the isolation, supernatants were thawed on ice, and then sEV isolation was performed from 5 mL cell culture medium using Exo-Spin columns from Cell Guidance Systems (Cambridge, UK) according to the manufacturer’s instructions. Total protein concentration was determined using Micro BCA protein assay (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. The amount of EVs used in the experiments is given as “μg EV protein/mL” (“μg/mL” in graphs). At difference with the sEV derived from the PF and MSC, pericyte-derived sEVs were not previously studied. Therefore, we have characterized them by combining TEM, NTA, and DLS as described in the next sections.
sEV TEM analyses
Frozen exosome pellet was brought to RT. Equal volumes of exosomes and 3% glutaraldehyde were mixed and kept at RT for 1 h. Osmium tetraoxide was added to the exosome solution and was kept at RT for 1 h. Final exosome solution was transferred to a formvar-coated TEM grid (EMS FCF100-Cu). The grids were observed under the electron microscope at 80 kV and stored in appropriate grid storage boxes for future use.
sEV NTA analyses
The NTA apparatus NanoSight NS300 (Malvern Panalytical, Malvern, UK), with NTA software version 3.2 (Malvern Instruments), was employed to characterize the size and concentration of the sEVs isolated from the pericyte CM.53 For the analyses, sEVs were diluted 5 times in Dulbecco’s phosphate-buffered saline (DPBS). Then, each sample was filtered using 0.22 μm Millex syringe filter unit (Millipore, Burlington, MA, USA) and injected into the flow cell of the NanoSight NS300. Once loaded, the sample was passed through the system using a syringe pump at a rate of 50 arbitrary units (a.u.), decreased gradually through 500, 300, 200, and 100 a.u. Four consecutive videos were recorded for each pericyte line (each obtained from a different donor), and 3 lines were used to calculate particle concentration per milliliter.
sEV DLS analysis
For the DLS analysis, the isolated sEVs were diluted to 1 mL using PBS (filtered via 0.1 μm filter) containing 2 mM ethylenediaminetetraacetic acid (EDTA). Dynamic light-scattering measurements were performed with a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). Intensity, volume, and distribution data for each sample were collected on a continuous basis for 1 min in sets of six. At least three different measurements were performed for the analysis. Data were presented as size distribution by number (percent) enrichment.
RNA isolation and qPCR analysis
Total RNA was extracted from cells and sEVs using the miRNeasy Mini Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions.
For the determination of miRNA expression in hSVP-sEVs and PF-sEVs, a synthetic analog of non-human cel-miR-39 (QIAGEN, Hilden, Germany) was spiked (in 10 μL of a 5-fmol/μL stock) to normalize RNA extraction efficiency. cDNA synthesis and real-time qPCR were performed using a miRCURY LNA Universal RT microRNA PCR system (Exiqon, Denmark). cDNA products were transferred to the microRNA PCR Human Panels (I + II; version 4) and run using the QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). EV miRNA data are presented in Table S1 (PF EVs) and Table S2 (hSVP EVs) and have been deposited in GEO (GEO: GSE118103 and GSE118855).
2.5 μg MSC-sEV was used to determine the expression of let-7a and let-7b. For this purpose, synthetic analog of the non-human cel-miR-39 was spiked (in 10 μL of a 5-fmol/μL stock) to normalize RNA extraction efficiency. Mature let-7b, let-7a, and cel-miR-39 levels were analyzed using miRNA TaqMan Assays and TaqMan MicroRNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA, USA). TaqMan miRNA primer IDs are given in Table S5. miRNA expression was normalized with U6 small nuclear RNA (snRNA), and relative expression was calculated using the 2−ΔCT (2[−(Ctsample − Cthousekeeping)]) method.
To determine the expression of the miRNAs in the cells, reverse transcription of individual miRNAs was performed using the TaqMan miRNA Reverse Transcription Kit and miRNA-specific stem-loop primers (Table S5) (Thermo Fisher Scientific, Waltham, MA, USA). qPCR was performed using 2 × Universal PCR Master Mix with No AmpErase UNG (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed using the QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). miRNA expression was normalized with U6 snRNA, and relative expression was calculated by normalizing data using reference samples (scrambled miRNA group for cells transfected with Lipofectamine containing let-7a, let-7b, or let-7a + let-7b or cel-miR-39-AE group for cells transfected with let-7b-AEs; 2−ΔΔCt, please see below).
For mRNA analysis, cDNA obtained using the High-Capacity RNA-to-cDNA Kit (Thermo Fisher Scientific, Waltham, MA, USA) was amplified by real-time qPCR. SYBR Green primers (from Sigma, St. Louis, MO, USA) and PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) were used to analyze the gene expression. The 2−ΔΔCt method was used for the quantification of target gene expression relative to the scrambled miRNA group (for cells transfected with Lipofectamine containing let-7a, let-7b, or let-7a + let-7b) or cel-miR-39-AE group (for cells transfected with let-7b-AEs). After calculation of ΔCT using the formula ΔCT = CT (target gene) − CT (GAPDH), ΔΔCT was calculated: ΔΔCT = ΔCT (target sample) − ΔCT (a reference sample). Then, relative expression was given as 2[−(ΔCtsample − ΔCtreference)] (2−ΔΔCt). Primer sequences are given as supporting information (Table S6). Gene targets of synergistic miRNA regulation for let-7a and let-7b were determined using TriplexRNA database54,55 and given in Table S7.
Procurement and bioinformatic analyses of miRNA datasets from proangiogenic sEVs and non-proangiogenic sEVs
For analysis of miRNAs enriched in proangiogenic sEVs, we used the following datasets: (1) miRNA array data of proangiogenic sEVs from the proangiogenic BM-CD34+ fraction of human BM MNCs (CD34+ sEVs) and from control total BM-MNC (cells inert for what concerns angiogenesis);14 (2) miRNA array data of MSC-sEVs that also have proangiogenic effects (GEO: GSE71241); (3) miRNA qPCR panels of PF EVs, which we previously proved to be angiogenic16 (GEO: GSE118103); and (4) miRNA qPCR panels of hSVP EVs that were also determined as “proangiogenic” as part of this study (vide supra) (GEO: GSE118855). In this analysis, miRNAs (1) present in proangiogenic sEVs, only (absent in angiogenesis-neutral sEVs from: BM-MNC and CABG-PF) and (2) of which expression is higher than inert BM-MNC EVs or CABG EVs were taken into consideration. After determination of miRNAs, Venn diagrams were prepared using the VENNY interactive tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html).
AE production
AEs were prepared using microfluidic micromixture (Precision NanoSystems). 1 vol of a mixture of ionizable lipid (MC3),29 with cholesterol, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and dimyristoyl glycerol-polyethyleneglycol (DMG-PEG), at a molar ratio of 50:38:10:2, at 9.64 nM total lipid concentration in ethanol and 3 vol of miRNA (1:16 w/w miRNA to lipid) in an acetate buffer (pH 4.0), was injected into the micromixer in a controlled flow rate (0.5 mL/min for ethanol and 1.5 mL/min for buffer) to form uniform AEs. miRNA sequences used in the preparation of AEs are given in Table S8. The resultant mixture was dialyzed overnight against PBS (pH 7.4) to remove ethanol. AE size distribution was determined by DLS and confirmed by NTA (both from Malvern Instruments). ζ-potential was determined by Nano ZS ζ-Sizer (Malvern Instruments). For size measurements, LNPs were diluted 1:20 in PBS. For ζ-potential measurements, AEs were diluted 1:200 in double-distilled water. Size and shape of AEs were analyzed by TEM (Philips Tecnai F20 field emission TEM operated at 200 kV). For this, AEs diluted in PBS were placed on a formvar/carbon-coated copper grid, air dried, and stained with 2% (w/v) aqueous uranyl acetate.
Determination of uptake of AEs in HUVECs by flow cytometry
HUVECs were seeded onto 1% gelatin-coated cell culture plates and incubated overnight at 37°C with 5% CO2 for cell attachment and recovery. For internalization studies, HUVECs were incubated with different concentrations of Cy5-cel-miR-39-AEs (i.e., 15 nM and 30 nM miRNA concentration). Internalization studies were performed under hypoxic conditions (1% O2), and cells were transfected with AEs for 4, 8, 12, and 24 h. After transfection of the cells with different concentrations (15 and 30 nM miRNA concentration) of AEs, cells were detached using TrypLE Express (Thermo Fisher Scientific, Waltham, MA, USA), centrifuged at 300 × g for 5 min, and stained with the Zombie Yellow Fixable Viability Kit according to the manufacturer’s instructions (BioLegend, San Diego, CA, USA). Finally, the cells were fixated using 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) and suspended in PBS. Novocyte Flow Cytometer and NovoExpress software (both from ACEA Biosciences, San Diego, CA, USA) were used for the acquisition and analysis of the data.
In vitro functional studies
Please see Major Resources (Table S4) in the Supplemental information for information regarding HUVECs and cHMVECs. Cells were transfected with EVs or individual pre-miRs or scramble miRNAs or AEs. In detail, cells were seeded onto 1% gelatin-coated cell culture plates and incubated overnight at 37°C with 5% CO2 for cell attachment and recovery. For Matrigel assay and RNA isolation, HUVECs were seeded as 4 × 104 cells/well in 24-well plates or as 8 × 104 cells/well in 12-well plates. For cell viability, metabolism, and proliferation experiments, cells were seeded as 1 × 104 cells/well in 96-well plates. After overnight incubation at 37°C with 5% CO2, cells were transferred to a hypoxia incubator (1% O2) to mimic ischemic conditions. Following 1 day’s incubation under hypoxic conditions, cells were transfected using different test substances (EVs, pre-miRs, or AEs). Lipofectamine RNAiMax (Invitrogen, Carlsbad, CA, USA) was used according to the manufacturer’s protocols for the transfection with pre-miRs or scramble miRNAs. Overexpression of individual let-7 family members was achieved by transfection of cells with 7.5−30 nM pre-miRs (Ambion, Carlsbad, CA, USA). 7.5−30 nM pre-miR scramble control from Ambion was used as control in all transfection experiments. For AE studies, AEs with 15−60 nM miRNA were used unless otherwise stated. After an additional 2 days of incubation under hypoxic conditions (1% O2), analyses of the cells were performed as described in the sections below. Only the toxicity studies of AEs were performed under normoxic (20% O2) conditions.
Cell metabolism, caspase activity, and cell viability measurements
Cell metabolism was determined by the CellTiter-Glo ATP production assay and apoptosis by Caspase-Glo 3/7 assay (all from Promega, Madison, WI, USA) according to the manufacturer’s instructions with modifications. For CellTiter-Glo and Caspase-Glo 3/7 assays, 50 μL of PBS and 50 μL CellTiter-Glo or Caspase-Glo 3/7 reagents were mixed in each well and incubated at RT for 5 min on an orbital shaker to induce cell lysis. PBS without cells, with CellTiter-Glo or Caspase-Glo 3/7 reagents, was used as a blank control.
Cellular viability was determined by the RealTime-Glo MT Cell Viability Assay as described by the manufacturer without any modifications. Briefly, 2× RealTime-Glo reagent was prepared by diluting the MT cell viability substrate and NanoLuc enzyme in 37°C cell culture medium to a 2× concentration for each reagent. To prepare 1 mL of 2× RealTime-Glo reagent, 2 μL of MT cell viability substrate (1,000×) and 2 μL of NanoLuc enzyme (1,000×) were added to 996 μL of cell culture medium. 2× RealTime-Glo reagent was added onto the cells and incubated for 1 h at 37°C, 5% CO2. Cell culture medium without cells, with 2× RealTime-Glo reagent, was used as blank control. For all experiments, the measurements were done using a GloMax-96 microplate luminometer (Promega, Madison, WI, USA).
Cell proliferation measurements
The cell proliferation ELISA, bromodeoxyuridine (BrdU; colorimetric) (Roche, Mannheim, Germany), was used according to the manufacturer’s instructions for the determination of cell proliferation. Briefly,10 μL/well BrdU labeling solution (final concentration:10 μM BrdU) was added, and cells were re-incubated for an additional 4 h at 37°C. After removal of the labeling medium and drying of the labeled cells, plates were stored for up to 1 week at +4°C. 200 μL/well FixDenat solution was added to the cells and incubated for 30 min at RT. FixDenat solution was removed, and 100 μL/well anti-BrdU-POD working solution was added into the plates. Following incubation for 90 min at RT, antibody conjugate was removed by flicking off, and wells were washed three times with 200 μL/well washing solution (PBS, 1×). Washing solution was removed, 100 μL/well substrate solution was added, and plates were incubated at RT until color development is sufficient for photometric detection (5−30 min). Finally, 25 μL 1 M H2SO4 (R&D Systems, Minneapolis, MN, USA) was added to each well, and plates were incubated for approximately 1 min on the shaker at 300 rpm. Absorbance of the samples was measured at 450 nm using a Dynex Opsys MR Microplate Photometer (Aspect Scientific, Cheshire, UK).
2.5D-Matrigel in vitro assay
Angiogenesis μ-slides (IBIDI, Munich, Germany) were coated with Matrigel (10 μL per well; without dilution; BD Biosciences, San Diego, CA, USA) and incubated for 30 min at 37°C. Transfected (for 2 days) and non-transfected HUVECs or cHMVECs were detached using TrypLE Express (Gibco, Waltham, MA, USA), suspended in EGM-2 medium, and seeded on top of polymerized Matrigel at a concentration of 1 × 105 cells/mL in 50 μL medium. Cells were incubated under hypoxia conditions (1% O2) with 5% CO2. Pre-chilled pipette tips and slides were used in these experiments, and Matrigel was kept on ice to prevent polymerization. Sprout formation was evaluated by phase contrast microscopy (Zeiss Axiovert 200; Carl Zeiss, Oberkochen, Germany) 6 h after the cell seeding.
3D-fibrin gel bead in vitro assay
3D-fibrin gel bead assay was performed as described previously.56 Briefly, 1 × 106 HUVECs were mixed with 2,500 Cytodex-3 beads (GE Healthcare, Uppsala, Sweden) in EGM BulletKit for 4 h at 37°C. Then, beads were transferred to T25 flask in 5 mL of EGM BulletKit and left overnight. The following day, the coated beads were re-suspended in fibrinogen solution (2.0 mg/mL fibrinogen, 0.15 U/mL of aprotinin; Sigma-Aldrich, St. Louis, MO, USA) at a concentration of ∼200 beads/mL. 0.625 U/mL of thrombin was added per well of a 24-well plate followed by 0.5 mL of the fibrinogen/bead suspension per well (1 mg fibrinogen per well). The plate was then placed at 37°C for 10−15 min. After clot formation, 1 mL of EGM BulletKit was added to each well. After 30 h in hypoxia (1% O2), angiogenic sprouting was quantified by measuring cumulative sprout length and branches using NIH ImageJ software. For this analysis, pictures were taken using a Nikon Eclipse TE200 inverted microscope at 4× magnification. For each experiment, at least 10 spheroids per triplicate well were analyzed after 30 h.
In vivo Matrigel plug assay
Please see Major Resources (Table S4) in the Supplemental information for information about animals and antibodies used in Matrigel plug assay. Matrigel was thawed at 4°C overnight before implantation. 1 × 105 HUVECs were resuspended in 100 μL of medium without FBS and mixed with 100 μL of Matrigel on ice (2 times dilution of Matrigel). The Matrigel mix was then implanted subcutaneously into nude mice (Crl:CD1-Foxn1nu; Charles River, UK). After 7 days, mice were sacrificed, and Matrigel plugs were harvested for immunohistochemistry. Matrigel plugs were embedded in optimal cutting temperature (OCT) prior to staining and fixated using 4% paraformaldehyde. 20 μm cryosections were stained with anti- CD31 antibody (Abcam; cat #] ab28364; dilution factor: 1:50). Alexa Fluor 647 AffiniPure donkey anti-rabbit immunoglobulin G (IgG) (heavy + light chains [H+L]) antibody from Jackson ImmunoResearch was used as a secondary antibody (cat #711-605-152; dilution factor: 1:200). The nuclei of the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Slides were observed under a fluorescence microscope (Zeiss LSM-780 confocal microscope). 4 high-power fields/mouse (20× magnification) were captured, and the number of nuclei/CD31 area was calculated using ImageJ software. For the analysis of images, background correction was done using ImageJ. Briefly, HiLo LUT option was chosen to display zero values as blue and white values (pixel value 255) as red. Background was removed with the “Brightness/Contrast” command by slowly raising the “Minimum” value until most of the background is displayed blue. In the figures, CD31 images have pseudocolor (red channel was shown in green) to make cells easier to see.
Statistical analysis
Comparisons between two different conditions were assessed using an unpaired t test. If the normality test failed, the Mann-Whitney test was performed. Experiments with three or more experimental groups were compared by one-way analysis of variance (ANOVA). GraphPad Prism software (San Diego, CA, USA; http://www.graphpad.com) was used for the statistical analysis. Results were considered significant when p <0.05. Data are shown as mean ± SEM.
Acknowledgments
We acknowledge the help of Dr. Andrew Herman from the Flow Cytometry Facility of University of Bristol, Valeria Alvino (University of Bristol) in the collection and preparation of human saphenous vein pericytes, and Parul Dixit (Imperial College London) for part of the Matrigel plug analyses. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health and Social Care. For this study, C.E. was supported by the British Heart Foundation (BHF) Centre of Vascular Regeneration-2, a BHF Chair award (CH/15/1/31199), the Leducq Foundation Transatlantic Network of Excellence in Vascular microRNAs, and a BHF project (PG/11/67/29067). This study was also funded/supported by the NIHR Biomedical Research Centre at University Hospitals Bristol NHS Foundation Trust and the University of Bristol. D.P. thanks the Lewis Trust Family for its generous support and the Dotan Hematological Malignancies Fund at Tel Aviv University. A.V.B. is supported by a BHF project grant (PG/18/31/33759) and the Royal Society (RGS\R1\191221). S.A. acknowledges the scholarship from BHF (SS/CH/15/1/31199).
Author contributions
S.A. designed and performed experiments, analyzed data, and wrote the manuscript. I.H.-H., A.C.-J., M.A., M.G., N.B.-L., N.D., W.S., F.C., and S.O.-T. performed experiments and analyzed data. G.D.A. and P.M. collected clinical samples under ethical approval, revised the manuscript, and obtained funds for the research. S.S. and A.V.B. analyzed data and revised the manuscript. D.P. and C.E. designed the study and the experiments, wrote the manuscript, and obtained funds for the research. All authors approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.03.015.
Contributor Information
Dan Peer, Email: peer@tauex.tau.ac.il.
Costanza Emanueli, Email: c.emanueli@imperial.ac.uk.
Supplemental information
References
- 1.Olivetti G., Quaini F., Sala R., Lagrasta C., Corradi D., Bonacina E., Gambert S.R., Cigola E., Anversa P. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J. Mol. Cell. Cardiol. 1996;28:2005–2016. doi: 10.1006/jmcc.1996.0193. [DOI] [PubMed] [Google Scholar]
- 2.Anversa P., Cheng W., Liu Y., Leri A., Redaelli G., Kajstura J. Apoptosis and myocardial infarction. Basic Res. Cardiol. 1998;93(Suppl 3):8–12. doi: 10.1007/s003950050195. [DOI] [PubMed] [Google Scholar]
- 3.Losordo D.W., Dimmeler S. Therapeutic angiogenesis and vasculogenesis for ischemic disease: part II: cell-based therapies. Circulation. 2004;109:2692–2697. doi: 10.1161/01.CIR.0000128596.49339.05. [DOI] [PubMed] [Google Scholar]
- 4.Losordo D.W., Dimmeler S. Therapeutic angiogenesis and vasculogenesis for ischemic disease. Part I: angiogenic cytokines. Circulation. 2004;109:2487–2491. doi: 10.1161/01.CIR.0000128595.79378.FA. [DOI] [PubMed] [Google Scholar]
- 5.Conde-Vancells J., Rodriguez-Suarez E., Embade N., Gil D., Matthiesen R., Valle M., Elortza F., Lu S.C., Mato J.M., Falcon-Perez J.M. Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J. Proteome Res. 2008;7:5157–5166. doi: 10.1021/pr8004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denzer K., Kleijmeer M.J., Heijnen H.F., Stoorvogel W., Geuze H.J. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000;113:3365–3374. doi: 10.1242/jcs.113.19.3365. [DOI] [PubMed] [Google Scholar]
- 7.Witwer K.W., Théry C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell. Vesicles. 2019;8:1648167. doi: 10.1080/20013078.2019.1648167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emanueli C., Shearn A.I., Angelini G.D., Sahoo S. Exosomes and exosomal miRNAs in cardiovascular protection and repair. Vascul. Pharmacol. 2015;71:24–30. doi: 10.1016/j.vph.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boulanger C.M., Loyer X., Rautou P.E., Amabile N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017;14:259–272. doi: 10.1038/nrcardio.2017.7. [DOI] [PubMed] [Google Scholar]
- 10.Chen T.S., Lai R.C., Lee M.M., Choo A.B., Lee C.N., Lim S.K. Mesenchymal stem cell secretes microparticles enriched in pre-microRNAs. Nucleic Acids Res. 2010;38:215–224. doi: 10.1093/nar/gkp857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang B., Wu X., Zhang X., Sun Y., Yan Y., Shi H., Zhu Y., Wu L., Pan Z., Zhu W. Human umbilical cord mesenchymal stem cell exosomes enhance angiogenesis through the Wnt4/β-catenin pathway. Stem Cells Transl. Med. 2015;4:513–522. doi: 10.5966/sctm.2014-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong M., Yu B., Wang J., Wang Y., Liu M., Paul C., Millard R.W., Xiao D.S., Ashraf M., Xu M. Mesenchymal stem cells release exosomes that transfer miRNAs to endothelial cells and promote angiogenesis. Oncotarget. 2017;8:45200–45212. doi: 10.18632/oncotarget.16778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang X., Zhang L., Wang S., Han Q., Zhao R.C. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J. Cell Sci. 2016;129:2182–2189. doi: 10.1242/jcs.170373. [DOI] [PubMed] [Google Scholar]
- 14.Mathiyalagan P., Liang Y., Kim D., Misener S., Thorne T., Kamide C.E., Klyachko E., Losordo D.W., Hajjar R.J., Sahoo S. Angiogenic Mechanisms of Human CD34+ Stem Cell Exosomes in the Repair of Ischemic Hindlimb. Circ. Res. 2017;120:1466–1476. doi: 10.1161/CIRCRESAHA.116.310557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sahoo S., Klychko E., Thorne T., Misener S., Schultz K.M., Millay M., Ito A., Liu T., Kamide C., Agrawal H. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ. Res. 2011;109:724–728. doi: 10.1161/CIRCRESAHA.111.253286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beltrami C., Besnier M., Shantikumar S., Shearn A.I., Rajakaruna C., Laftah A., Sessa F., Spinetti G., Petretto E., Angelini G.D., Emanueli C. Human Pericardial Fluid Contains Exosomes Enriched with Cardiovascular-Expressed MicroRNAs and Promotes Therapeutic Angiogenesis. Mol. Ther. 2017;25:679–693. doi: 10.1016/j.ymthe.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katare R., Riu F., Mitchell K., Gubernator M., Campagnolo P., Cui Y., Fortunato O., Avolio E., Cesselli D., Beltrami A.P. Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro-RNA-132. Circ. Res. 2011;109:894–906. doi: 10.1161/CIRCRESAHA.111.251546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ong S.G., Wu J.C. Exosomes as potential alternatives to stem cell therapy in mediating cardiac regeneration. Circ. Res. 2015;117:7–9. doi: 10.1161/CIRCRESAHA.115.306593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phinney D.G., Pittenger M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells. 2017;35:851–858. doi: 10.1002/stem.2575. [DOI] [PubMed] [Google Scholar]
- 20.Tsao C.R., Liao M.F., Wang M.H., Cheng C.M., Chen C.H. Mesenchymal Stem Cell Derived Exosomes: A New Hope for the Treatment of Cardiovascular Disease? Zhonghua Minguo Xinzangxue Hui Zazhi. 2014;30:395–400. [PMC free article] [PubMed] [Google Scholar]
- 21.Floriano J.F., Willis G., Catapano F., Lima P.R., Reis F.V.D.S., Barbosa A.M.P., Rudge M.V.C., Emanueli C. Exosomes Could Offer New Options to Combat the Long-Term Complications Inflicted by Gestational Diabetes Mellitus. Cells. 2020;9:675. doi: 10.3390/cells9030675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rietwyk S., Peer D. Next-Generation Lipids in RNA Interference Therapeutics. ACS Nano. 2017;11:7572–7586. doi: 10.1021/acsnano.7b04734. [DOI] [PubMed] [Google Scholar]
- 23.Semple S.C., Akinc A., Chen J., Sandhu A.P., Mui B.L., Cho C.K., Sah D.W., Stebbing D., Crosley E.J., Yaworski E. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 24.Jayaraman M., Ansell S.M., Mui B.L., Tam Y.K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J.K. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramishetti S., Hazan-Halevy I., Palakuri R., Chatterjee S., Naidu Gonna S., Dammes N., Freilich I., Kolik Shmuel L., Danino D., Peer D. A Combinatorial Library of Lipid Nanoparticles for RNA Delivery to Leukocytes. Adv. Mater. 2020;32:e1906128. doi: 10.1002/adma.201906128. [DOI] [PubMed] [Google Scholar]
- 26.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 27.Llorente A., Skotland T., Sylvänne T., Kauhanen D., Róg T., Orłowski A., Vattulainen I., Ekroos K., Sandvig K. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta. 2013;1831:1302–1309. doi: 10.1016/j.bbalip.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Rappa G., Mercapide J., Anzanello F., Pope R.M., Lorico A. Biochemical and biological characterization of exosomes containing prominin-1/CD133. Mol. Cancer. 2013;12:62. doi: 10.1186/1476-4598-12-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinstein S., Toker I.A., Emmanuel R., Ramishetti S., Hazan-Halevy I., Rosenblum D., Goldsmith M., Abraham A., Benjamini O., Bairey O. Harnessing RNAi-based nanomedicines for therapeutic gene silencing in B-cell malignancies. Proc. Natl. Acad. Sci. USA. 2016;113:E16–E22. doi: 10.1073/pnas.1519273113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belliveau N.M., Huft J., Lin P.J., Chen S., Leung A.K., Leaver T.J., Wild A.W., Lee J.B., Taylor R.J., Tam Y.K. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids. 2012;1:e37. doi: 10.1038/mtna.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen Z.R., Ramishetti S., Peshes-Yaloz N., Goldsmith M., Wohl A., Zibly Z., Peer D. Localized RNAi therapeutics of chemoresistant grade IV glioma using hyaluronan-grafted lipid-based nanoparticles. ACS Nano. 2015;9:1581–1591. doi: 10.1021/nn506248s. [DOI] [PubMed] [Google Scholar]
- 32.Ramishetti S., Kedmi R., Goldsmith M., Leonard F., Sprague A.G., Godin B., Gozin M., Cullis P.R., Dykxhoorn D.M., Peer D. Systemic Gene Silencing in Primary T Lymphocytes Using Targeted Lipid Nanoparticles. ACS Nano. 2015;9:6706–6716. doi: 10.1021/acsnano.5b02796. [DOI] [PubMed] [Google Scholar]
- 33.Leung A.K., Tam Y.Y., Cullis P.R. Lipid nanoparticles for short interfering RNA delivery. Adv. Genet. 2014;88:71–110. doi: 10.1016/B978-0-12-800148-6.00004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alajati A., Laib A.M., Weber H., Boos A.M., Bartol A., Ikenberg K., Korff T., Zentgraf H., Obodozie C., Graeser R. Spheroid-based engineering of a human vasculature in mice. Nat. Methods. 2008;5:439–445. doi: 10.1038/nmeth.1198. [DOI] [PubMed] [Google Scholar]
- 35.Scheideler M., Vidakovic I., Prassl R. Lipid nanocarriers for microRNA delivery. Chem. Phys. Lipids. 2020;226:104837. doi: 10.1016/j.chemphyslip.2019.104837. [DOI] [PubMed] [Google Scholar]
- 36.Bernstein D.L., Gajghate S., Reichenbach N.L., Winfield M., Persidsky Y., Heldt N.A., Rom S. let-7g counteracts endothelial dysfunction and ameliorating neurological functions in mouse ischemia/reperfusion stroke model. Brain Behav. Immun. 2020;87:543–555. doi: 10.1016/j.bbi.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kranz L.M., Diken M., Haas H., Kreiter S., Loquai C., Reuter K.C., Meng M., Fritz D., Vascotto F., Hefesha H. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 38.Ma T., Chen Y., Chen Y., Meng Q., Sun J., Shao L., Yu Y., Huang H., Hu Y., Yang Z. MicroRNA-132, Delivered by Mesenchymal Stem Cell-Derived Exosomes, Promote Angiogenesis in Myocardial Infarction. Stem Cells Int. 2018;2018:3290372. doi: 10.1155/2018/3290372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferguson S.W., Wang J., Lee C.J., Liu M., Neelamegham S., Canty J.M., Nguyen J. The microRNA regulatory landscape of MSC-derived exosomes: a systems view. Sci. Rep. 2018;8:1419. doi: 10.1038/s41598-018-19581-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lai R.C., Arslan F., Lee M.M., Sze N.S., Choo A., Chen T.S., Salto-Tellez M., Timmers L., Lee C.N., El Oakley R.M. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. (Amst.) 2010;4:214–222. doi: 10.1016/j.scr.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Timmers L., Lim S.K., Arslan F., Armstrong J.S., Hoefer I.E., Doevendans P.A., Piek J.J., El Oakley R.M., Choo A., Lee C.N. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem Cell Res. (Amst.) 2007;1:129–137. doi: 10.1016/j.scr.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 42.Arslan F., Lai R.C., Smeets M.B., Akeroyd L., Choo A., Aguor E.N., Timmers L., van Rijen H.V., Doevendans P.A., Pasterkamp G. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. (Amst.) 2013;10:301–312. doi: 10.1016/j.scr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Willis G.R., Fernandez-Gonzalez A., Anastas J., Vitali S.H., Liu X., Ericsson M., Kwong A., Mitsialis S.A., Kourembanas S. Mesenchymal Stromal Cell Exosomes Ameliorate Experimental Bronchopulmonary Dysplasia and Restore Lung Function through Macrophage Immunomodulation. Am. J. Respir. Crit. Care Med. 2018;197:104–116. doi: 10.1164/rccm.201705-0925OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansouri N., Willis G.R., Fernandez-Gonzalez A., Reis M., Nassiri S., Mitsialis S.A., Kourembanas S. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight. 2019;4:e128060. doi: 10.1172/jci.insight.128060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J., Bonacquisti E.E., Brown A.D., Nguyen J. Boosting the Biogenesis and Secretion of Mesenchymal Stem Cell-Derived Exosomes. Cells. 2020;9:660. doi: 10.3390/cells9030660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Q., Frost R.J.A., Anderson C., Zhao F., Ma J., Yu B., Wang S. let-7 Contributes to Diabetic Retinopathy but Represses Pathological Ocular Angiogenesis. Mol. Cell. Biol. 2017;37:e00001-17. doi: 10.1128/MCB.00001-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shakeri R., Kheirollahi A., Davoodi J. Apaf-1: Regulation and function in cell death. Biochimie. 2017;135:111–125. doi: 10.1016/j.biochi.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Lin Y., Chen N.T., Shih Y.P., Liao Y.C., Xue L., Lo S.H. DLC2 modulates angiogenic responses in vascular endothelial cells by regulating cell attachment and migration. Oncogene. 2010;29:3010–3016. doi: 10.1038/onc.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reis M., Czupalla C.J., Ziegler N., Devraj K., Zinke J., Seidel S., Heck R., Thom S., Macas J., Bockamp E. Endothelial Wnt/β-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J. Exp. Med. 2012;209:1611–1627. doi: 10.1084/jem.20111580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye X., Wang Y., Cahill H., Yu M., Badea T.C., Smallwood P.M., Peachey N.S., Nathans J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139:285–298. doi: 10.1016/j.cell.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dejana E. The role of wnt signaling in physiological and pathological angiogenesis. Circ. Res. 2010;107:943–952. doi: 10.1161/CIRCRESAHA.110.223750. [DOI] [PubMed] [Google Scholar]
- 52.Campagnolo P., Cesselli D., Al Haj Zen A., Beltrami A.P., Kränkel N., Katare R., Angelini G., Emanueli C., Madeddu P. Human adult vena saphena contains perivascular progenitor cells endowed with clonogenic and proangiogenic potential. Circulation. 2010;121:1735–1745. doi: 10.1161/CIRCULATIONAHA.109.899252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shearn A.I.U., Aday S., Ben-Aicha S., Carnell-Morris P., Siupa A., Angelini G.D., Clayton A., Boulanger C., Punjabi P., Emanueli C., Biglino G. Analysis of Neat Biofluids Obtained During Cardiac Surgery Using Nanoparticle Tracking Analysis: Methodological Considerations. Front. Cell Dev. Biol. 2020;8:367. doi: 10.3389/fcell.2020.00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai X., Schmitz U., Gupta S.K., Bhattacharya A., Kunz M., Wolkenhauer O., Vera J. Computational analysis of target hub gene repression regulated by multiple and cooperative miRNAs. Nucleic Acids Res. 2012;40:8818–8834. doi: 10.1093/nar/gks657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmitz U., Lai X., Winter F., Wolkenhauer O., Vera J., Gupta S.K. Cooperative gene regulation by microRNA pairs and their identification using a computational workflow. Nucleic Acids Res. 2014;42:7539–7552. doi: 10.1093/nar/gku465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakatsu M.N., Davis J., Hughes C.C. Optimized fibrin gel bead assay for the study of angiogenesis. J. Vis. Exp. 2007:186. doi: 10.3791/186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.