

Abstract
BACKGROUND:
Small cell-derived extracellular vesicles (EVs) can affect endothelial function. We previously found that patients with sickle cell disease (SCD) have greater numbers of circulating EVs than subjects without the disease, and the EVs differentially disrupt endothelial integrity in vitro. Because endothelial disruption is a critical component of acute chest syndrome (ACS), we hypothesized that EVs isolated during ACS would induce greater endothelial damage than those isolated at baseline.
METHODS:
Nine pediatric subjects had plasma isolated at baseline and during ACS from which EVs were isolated. Cultured microvascular endothelial cells were treated with EVs and then studied by immunofluorescence microscopy to localize VE-cadherin and F-actin.
RESULTS:
The EVs had a diameter of 95 nm. They contained CD63 and flotillin-1, which were increased in SCD patients (5–13-fold compared to control) and further increased between baseline and ACS (24–57%). The EVs contained hemoglobin, glycophorin A, and ferritin. Treatment with baseline EVs caused modest separation of endothelial cells, while ACS EVs caused substantial disruptions of the endothelial cell monolayers. EVs from subjects with ACS also caused a 50% decrease in protein levels of VE-cadherin.
CONCLUSIONS:
These results suggest that circulating EVs can modulate endothelial integrity contributing to the development of ACS in SCD patients by altering cadherin-containing intercellular junctions.
INTRODUCTION
Acute Chest Syndrome (ACS) is one of the most severe complications of sickle cell disease (SCD) and one of the leading causes of death among children with this disease (1–3). Unfortunately, there is no method for predicting which patients will develop ACS, whether evaluated when subjects are at baseline or when they are admitted to the hospital (4). Nevertheless, damage to the endothelium and abnormal cellular interactions with the vessel wall are central to the pathophysiology of ACS and many other complications of SCD (5–8).
Recent studies have implicated circulating extracellular vesicles (EVs) of a variety of sizes in the dysregulation of endothelial function in diseases of inflammation and ischemia-reperfusion injury (9–13) EVs include a variety of vesicles (defined by different sizes) that are released by cells through membrane budding or via the ESCRT (Endosomal Sorting Complex Required For Transport) (14,15). Several different groups of investigators have studied EVs of the larger category (microparticles) isolated from the blood of patients with SCD (16,17).Although the methods of isolation and analysis have differed across the studies, most agree that the total numbers of large EVs are elevated in subjects with SCD. The SCD microparticles can affect endothelial cells by acting as heme transporters and may contribute to the inflammatory milieu in SCD (18,19). In contrast, there have been few studies of small EVs in relation to the pathophysiology of SCD.
We have focused our studies on the small EVs (often termed exosomes) that facilitate specific intercellular signaling. We believe that their relevance in SCD will be critical, because they undergo regulated release and uptake, and they contain unique proteins and nucleic acids (including mRNAs and miRNAs) (20). Despite their small size of 80–120 nm, they can have significant variability in their contents based on cellular origin and the conditions of their production (15,21,22). We and others have found that patients with SCD have increased numbers of circulating small EVs as compared with control subjects (23,24). At baseline, patients with SCD have similar abundances of small EVs in their plasma regardless of their clinical history. However, their function differs: small EVs from subjects with a history of ACS cause significantly increased in vitro endothelial damage (24).
We hypothesized that if circulating small EVs contribute to the endothelial damage of SCD, then EVs isolated during an episode of ACS might cause more endothelial damage than those isolated from the same patient at baseline. Therefore, we studied the effects of EVs from SCD patients on in vitro endothelial integrity and compared the effects of EVs isolated from the same patients at baseline or during episodes of ACS.
METHODS
Subjects and event characteristics
The sickle cell disease biobank at the University of Chicago Comer and La Rabida Children’s Hospital was queried for subjects that had samples obtained both at baseline and at the beginning of an admission for ACS, prior to transfusion. Nine such patients were identified. The subjects enrolled in the biobank had been prospectively enrolled, with informed consent provided by parents or patients greater than 18 years of age and assent obtained from subjects 9–18 years of age. Subjects were excluded if less than 2 years of age. All subjects were in a steady state of disease (free of infection, new pain, or transfusion for at least 4 weeks prior to their blood draw) when their baseline samples were isolated. Control subjects (n=6) were recruited from the general pediatric clinic; they had a BMI <85th percentile, did not carry a diagnosis of asthma or other inflammatory disorder, and were having blood drawn for screening or monitoring of iron deficiency. Clinical and demographic characteristics of subjects with SCD and controls are shown in Table 1.
Table 1.
Patient characteristics, demographics and hematologic values
| SCD (n=9) | P-value% | Controls (n=6) | P-value^ | ||
|---|---|---|---|---|---|
| Baseline (n=9) | ACS (n=9) | ||||
| Demographic Data | |||||
| Age, median (range) | 6 (3,19) | 7 (4,18) | 11 (4,16) | NS | |
| Sex, n (%) | NS | ||||
| Male | 6 (66.7%) | 2 (33.3%) | |||
| Female | 3 (33.3%) | 4 (66.7%) | |||
| Clinical Characteristics | |||||
| Hematologic Values | |||||
| Median (25th, 75th) | Median (25th, 75th) | Median (25th, 75th) | |||
| White blood cell count (x10*3/uL) | 14.8 (11.8, 17.3) | 22.6 (12.4, 24) | NS | 5.3 (4.2, 6.5) | 0.001 |
| Hemoglobin (g/dL) | 8.5 (7.5, 9.7) | 7.5 (6.5, 8.1) | 0.001 | 10.9 (10.1, 10.9) | 0.002 |
| MCV (fL) | 93 (89, 96) | 85 (83, 89) | 0.008 | 70 (65, 78) | 0.008 |
| Absolute retic (K/uL) | 365 (259, 432) | 469 (344, 597) | NS | ND | |
| Platelet (x10*3/uL) | 434 (408, 481) | 415 (259, 449) | NS | 264 (220, 294) | <0.0001 |
| Hemoglobin Genotype | |||||
| SS | 9 | ||||
| Other | 0 | ||||
| Hydroxyurea (%) | 9 (100%) | ||||
| Asthma, # (%) | 6 (66.7%) | ||||
| OSA (%) | 2 (22.2%) | ||||
| Splenectomy (%) | 0 | ||||
| Cholecystectomy (%) | 0 | ||||
| Rate of ACS (# events/age) | 0.6 (0.25, 1.1) | 0.5 (0.27, 1.3) | NS | ||
| Absolute # ACS | 3 (2, 6) | 3 (2.5, 7) | NS | ||
| Rate of Pain (# events/age ) | 0.2 (0.12, 0.37) | 0.27 (0.03, 0.38) | NS | ||
| Absolute # Pain | 2 (1, 2.5) | 2 (0.5, 3) | NS | ||
Student t-test performed comparing values between Baseline and ACS values.
Controls were compared only to Baseline SCD values
ACS was defined as a new infiltrate on chest x-ray accompanied by fever, supplemental oxygen requirement, tachypnea, wheezing, cough, or chest pain. Admissions for vaso-occlusive crisis (VOC) were defined by use of parenteral opioids to treat pain without the presence of another etiology. The rate of VOC or ACS was calculated by dividing the total number of events by the number of patient-years at the time of the blood draw. Asthma was defined by either electronic medical record documentation of an ICD code for asthma or prescription of a long-acting controller asthma medication. Splenectomy and cholecystectomy were identified by procedure codes. Obstructive sleep apnea was diagnosed by sleep study.
All protocols were approved by the IRB (protocol # 14–0466 and 15–0263) and were conducted in accordance with the guidelines set by the Declaration of Helsinki.
Blood collection and EV Isolation
Baseline SCD and control subject blood samples were obtained at the time of a clinic visit; time of day and fasting/non-fasting status were not determined. ACS samples were obtained from fasting subjects at an early morning blood draw during the first 24 hours of a hospital admission. Otherwise all blood collection and EV preparation were identical. Blood (~ 6ml) was collected into EDTA-containing (lavender top) tubes by professional phlebotomists. Blood samples were centrifuged at 2,000g at 4oC for 20 min, and platelet-free plasma was removed (without disturbing the buffy coat) and frozen (in aliquots) at −80°C until further use. After thawing, samples were again centrifuged at 3,000g for 20 min at room temperature prior to isolation of EVs.
For most experiments, EVs were isolated from platelet-free plasma using the Total Exosome Isolation kit (Thermo Fisher Scientific Inc., Waltham, MA) according to the manufacturer’s guidelines, as we have done previously (23,24). Each isolation started with 100 μl of plasma; EV pellets were resuspended in a final volume of 50 μl of phosphate buffered saline (PBS).
For some experiments, EVs were isolated by size exclusion, using qEV-35nm single columns (Izon Science, Medford, MA) according to the vendor’s recommended protocol, starting with 200 μl of plasma. Twenty-two 200 μl fractions were collected. Fractions 6–8 were identified as containing the greatest abundance of exosomal markers (CD63 and flotillin-1) by immunoblotting. These fractions were combined and concentrated to 50 μL using Microcon 30K centrifugal filter devices (SIGMA-Aldrich, Saint Louis, MO) for use in functional experiments.
Nanoparticle Tracking Analysis
Nanoparticle tracking analysis was performed using the Nanosight NS300 (Malvern Panalytical Inc., Westborough, MA). Isolated EVs were diluted with PBS (1:100–1:150) and injected in the 488 nm laser chamber with a constant output controlled by a syringe pump. Three recordings were performed for each sample. Nanoparticle tracking analysis software was used to measure nanoparticle size.
Heme assay
The level of heme in plasma or EVs was measured using QuantiChrom Heme Assay Kit (DIHM-250; Bioassay Systems, Hayward, CA).
Primary endothelial cell culture
Human dermal microvascular endothelial cells (CC-2543); HMVEC-D (purchased from Lonza Walkersville, Inc.; Allendale, NJ) were maintained according to the manufacturer’s instructions in endothelial growth medium (EGM-2MV Bullet Kit; Lonza) and incubated at 37°C, 5% CO2 in cell culture incubator. All experiments were performed at passage 10.
Antibodies
VE-cadherin was detected using mouse monoclonal antibodies (sc-9989, Santa Cruz, Biotechnology, Inc.) at 1:2,000 dilution for immunoblotting and at 1:100 dilution for immunofluorescence. Other antibodies for immunoblotting included: anti-flotillin-1 (sc-133153, Santa Cruz, Biotechnology, Inc.) mouse monoclonal antibodies used at 1:200 dilution; anti-CD63 (sc-5275, Santa Cruz, Biotechnology, Inc.) at 1:500 dilution; anti-ApoA-I (sc-376818, Santa Cruz, Biotechnology, Inc.) mouse monoclonal antibodies at 1:200 dilution; anti-hemoglobin mouse monoclonal antibodies (ab77125 Abcam, Cambridge, MA) at 1:2,000 dilution; anti-glycophorin A antibodies (ab134111, Abcam) at 1:5,000 dilution; anti-ferritin heavy chain mouse monoclonal antibodies (MAB9354, Novus Biologicals, Littleton, CO) at dilution 1:1,000. AlexaFluor 488 goat anti-mouse IgG and horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse IgG antibodies were obtained from Jackson ImmunoResearch (West Grove, PA) and used according to manufacturer’s instructions.
Immunoblotting of EVs and endothelial cell lysates
Cell homogenates were prepared from 60 mm dishes, as described earlier (25). Either 5 μg of protein from cell homogenates or equal volume aliquots (2–5 μL) of EVs were separated by SDS-PAGE (8% acrylamide for VE-cadherin, 10% for flotillin-1, CD63, and glycophorin A, or 14% for hemoglobin and ferritin). Polyacrylamide gels were run under reducing conditions except for CD63 and VE-cadherin, which required nonreducing conditions (26). The protein concentrations of EVs and cell homogenates were determined using the Bradford method (27). Proteins were blotted onto Immobilon-P membranes (Millipore, Bedford, MA). ProSieve Protein Colored Markers standards (Lonza) were used to calibrate the gels. MEM Code Reversible Protein Stain Kit for PVDF membranes (Thermo Fisher Scientific Inc., Waltham, MA) was used for staining proteins after transfer to the membranes when equal loading had to be confirmed. Immunoblots were developed with ECL Prime chemiluminiscence reagents (GE Healthcare Biosciences, Pittsburgh, PA) except for glycophorin A blots, which were reacted with SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific Inc.). Subsequently, blots were exposed to X-ray film. Blots were performed for two or three independent experiments for each sample and were quantified by densitometry.
Immunohistochemistry of Endothelial Cells
HMVEC-D cells were grown to confluence on glass coverslips that had been treated for 5 minutes with 5 μg/mL fibronectin in 0.02% gelatin (SIGMA-Aldrich, St. Louis, MO) in a 12-well dish. Once confluent, cells were treated for 48 h with full growth media or EVs from controls or subjects with SCD diluted in this medium. Slides were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature (RT) and then washed with PBS. Cell membranes were permeabilized with 1% Triton X-100 in PBS for 10 min at RT. After washing with PBS, samples were blocked twice for 30 min each at RT with 1% Triton X-100 and 10% normal goat serum. Cells were incubated with mouse monoclonal anti-VE-cadherin antibodies overnight at 4°C. After three 5 min washes with PBS, cells were incubated with Alexa 488 anti-rabbit secondary antibodies for 1 h. In order to stain filamentous actin (F-actin), cells were incubated with Phalloidin–Tetramethylrhodamine B isothiocyanate (SIGMA-Aldrich) (1:500) for 40 min, followed by washes with PBS. For nuclear counter staining, cover slips were incubated for 15 min with 500 μg/mL DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) (Thermo Fisher Scientific Inc.) and again washed extensively with PBS. Coverslips were mounted on glass slides using Prolong Gold anti-fade reagent (Thermo Fisher Scientific Inc.). Slides were sealed and stored in darkness at 4°C. Cells were imaged using the 10X or 40X Plan Apochromat objective in an Axioplan 2 microscope (Carl Zeiss Meditec, Munich, Germany). Images were captured with a Zeiss Axiocam digital camera using Zeiss AxioVision software. To avoid bias, microphotography and image analysis were performed by a team member who was not aware of the source of EVs used for the treatment of cells.
Image J software (http://rsb.info.nih.gov/ij/) was used to analyze images, and monolayer disruption was quantified by determining the percentage of each image occupied by intercellular space as detailed and illustrated in Supplemental Fig. S1.
Isolation of RNA and quantification of VE-cadherin mRNA levels
RNA was isolated from HMVEC-D cells (grown in 6-well tissue culture plates) using the miRNeasy Mini Kit (Qiagen, Germantown, MD). The quality of RNA was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA). All samples had RNA Integrity Number (RIN) values in the excellent quality range (7.7 – 10.0). Levels of VE-cadherin mRNA were quantified using real-time quantitative PCR (RT-qPCR) as previously described (26). cDNA was prepared from total RNA (1 μg) using oligo-dT and random primers with QuantiTect Reverse Transcription Kit (Qiagen). RT-qPCR analysis was performed using SYBR green (Thermo Fisher Scientific Inc.) in 96-well plates in a 7500 FAST Applied Biosystems instrument. All reactions were run in triplicate: amplified at 95°C for 20 seconds, followed by 40 cycles of 95°C for 3 seconds and 60°C for 30 seconds. The primers for amplification of VE-cadherin, QuantiTect Primer Assay, Cat no. QT00013244, were purchased from Qiagen. After testing the human housekeeping gene primer set (HHK-1; Real Time Primers, LLC, Elkins Park, PA), human ribosomal protein L13a, RPL13A was chosen as a housekeeping gene. Each experiment included negative control samples lacking template or reverse transcriptase. The relative expression of mRNA was calculated using the ΔΔCT method and normalized to the expression of RPL13A as previously described (26).
RESULTS
Subject Characteristics
We queried the SCD biobank for subjects who had samples drawn both at baseline and at the beginning of a hospitalization for ACS; Nine such subjects were identified. Six age-matched controls (including both males and females) were selected for comparison. Table 1 shows the demographic and clinical characteristics of all subjects. Subjects did not differ significantly in white blood cell, reticulocyte, or platelet counts, but subjects were more anemic when admitted for ACS (with smaller mean corpuscular volumes (MCV)).
Properties of Extracellular Vesicles
We examined the size distributions of the EVs isolated from the plasma of several control and SCD subjects by Nanoparticle Tracking Analysis. The EVs had similar profiles between subject groups, distributing in a single peak (see example shown in Fig. 1a) with a mode diameter of 95 ± 2 nm.
Fig. 1.

Properties of isolated EVs. (a) Representative Nanoparticle Tracking Analysis profile of EVs isolated from platelet-free plasma. The mode peak size of the EVs is 95 nm. Equal volumes of EVs were subjected to SDS-PAGE. Flotillin-1 (b) and CD63 (c) levels were assessed as indicated in controls, and SCD subjects at baseline and during ACS. Representative examples are shown on the left. Bands were analyzed by densitometry; values are normalized to the abundance of protein in the control sample. (# = paired t-test compared to control, p < 0.05; * = paired t-test within SCD subjects, p < 0.05)
In order to characterize the EVs, we performed immunoblots for the presence of proteins found in exosomes, or small EVs. Both flotillin-1 (a calveolar protein that is typically cytosolic and recovered in EVs) and CD63 (a tetraspanin) were present in all samples (Fig. 1b,c). Quantification of blots by densitometry revealed that flotillin-1 was increased 13-fold and CD63 was increased 5-fold in SCD subjects at baseline as compared with controls (significance determined by ANOVA across the group). In subjects with SCD, flotillin-1 was further increased by 57% and CD63 was increased by 24% between baseline and ACS (Fig. 1b,c). Independent of the subject source, the EVs appeared free of endoplasmic reticulum contamination, since they did not contain Grp94 or calnexin which are resident proteins within this compartment. We also analyzed some samples for contamination with lipoproteins by reacting immunoblots of EV samples with antiApoA-1 anti-bodies; there was trace contamination of the EVs, but the apoprotein was ~100-fold more abundant in plasma than in the EVs.
Since patients with SCD produce increased EVs derived from erythrocytes or their precursors or fragments, we assayed heme, hemoglobin, and glycophorin A in our samples (17,18,28,29). Although free heme was present in the plasma (~90 μM in plasma of subjects with SCD and ~30 μM in plasma of controls based on a colorimetric assay) (Sup Fig. S2), it was undetectable (< 5 μM) in the isolated EVs. Immunoblots showed similar hemoglobin contents of the EVs isolated from control subjects and from subjects with SCD at baseline (Fig. 2a). However, there was 3-times as much hemoglobin in the EVs isolated from subjects during ACS as compared to baseline (p < 0.05). Immunoblots also showed that the abundance of glycophorin A was 66% (ACS) to 90% (baseline) higher in subjects with SCD than in controls (Fig. 2b). Immunoblots showed that all EVs contained a small amount of ferritin (that required high sensitivity immunoblotting for detection) that did not differ significantly between samples (Fig. 2c).
Fig. 2.

Hemoglobin content of EVs increases during ACS, but glycophorin A and ferritin do not. Equal volumes of EVs were analyzed by immunoblotting for hemoglobin (a), glycophorin A (b), and ferritin (c). A representative example of each blot is shown as an insert on the left for each protein. Blots were analyzed by densitometry; values are normalized to the abundance of protein in the control sample. Values represent the mean ± SEM. (* = paired t-test, p < 0.05)
EVs isolated during ACS disrupt endothelial monolayers in vitro
We have previously studied the effects of plasma EVs from subjects with SCD (at baseline) on the integrity of endothelial cell monolayers by electric cell-substrate impedance sensing (ECIS) (23,24). In the current study, we sought to visualize and quantify this damage by microscopy and to see if there was increased damage caused by EVs from plasma obtained during an ACS episode. We initially determined the dose- and time-dependent effects of plasma EVs on the integrity of endothelial monolayers. We treated confluent HMVEC-D cells for 24 hours or 48 hours with increasing amounts of isolated vesicles (diluted 1:100, 1:67, 1:50, or 1:25). We observed no detectable effects of any samples at 24 h. At 48 h, 1:100 and 1:66 dilutions of all samples had minimal effects; 1:50 dilutions showed damage caused by some EV samples; and, 1:25 dilutions of all ACS samples produced dramatic effects. Based on these experiments, we chose to perform the remaining experiments at 48 hours using a 1:50 dilution.
We examined the changes in confluent monolayers of endothelial cells treated with EVs by fluorescence microscopy, including localization of VE-cadherin (by immunofluorescence), F-actin (by staining with fluorescent phalloidin), and nuclei (by staining with DAPI). Representative examples are shown in Fig. 3. We compared cells treated with no EVs with cells treated with EVs from controls or subjects with SCD at baseline or during ACS. Cells treated with no EVs or with those from control subjects looked similar (Fig. 3a–f). VE-cadherin was abundant in a continuous distribution along cell membranes at points of contact between cells. There were no apparent spaces between cells. Phalloidin staining illustrated that F-actin was primarily located along the cell membranes and in some stress fibers within the cells. Endothelial cells treated with EVs from subjects with SCD at baseline showed very rare disruptions in the monolayers; VE-cadherin was continuous around the cells, but the width of the staining at many cell-cell contacts was thickened and more convoluted; F-actin localization was generally similar to that in controls, but there was an impression of increased intracellular stress fibers (Fig. 3b, e, h, k). Cells treated with EVs isolated from subjects during an episode of ACS looked markedly different. There were frequent spaces between cells; VE-cadherin localization was thick, convoluted, and had frequent discontinuities (Fig. 3j); phalloidin staining was increased and labeled many stress fibers within cells (Fig. 3k). DAPI staining of nuclei appeared similar in all cells regardless of treatments (Fig. 3c, f, i, l). We counted DAPI-stained nuclei in low magnification micrographs to quantify cell numbers, and we found no difference in numbers of nuclei between treatments (Sup Fig. S3), suggesting that the EVs did not differentially affect cell growth or death.
Fig. 3.
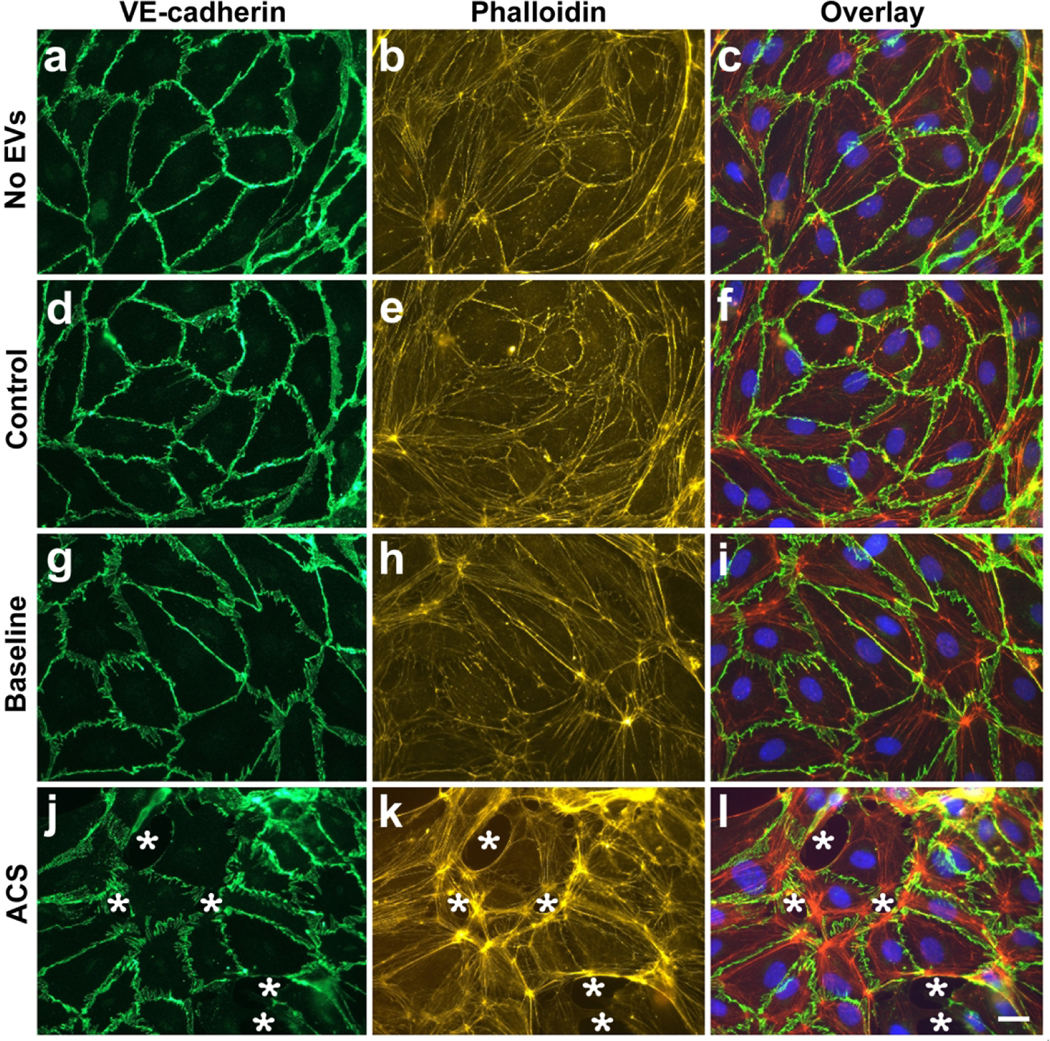
Localization of VE-cadherin, F-actin, and nuclei in endothelial cells treated with EVs. Representative photomicrographs are shown for HMVEC-D cells 48 h following treatment with no EVs (a-c), EVs from a control subject (d-f), EVs from a subject with SCD at baseline (g-i), and EVs from the same subject at the beginning of an episode of ACS (j-l). VE-cadherin was detected by immunofluorescence (green), F-actin by staining with TRITC-phalloidin (red), and nuclei by staining with DAPI (blue). White stars indicate spaces between cells. Scale bar is 20 μm.
We performed multiple experiments using the EVs prepared from all 9 subjects with SCD (isolated both at baseline and during an ACS episode) and took photomicrographs similar to those shown in Fig. 3. We quantified the extent of damage to the endothelial monolayers by calculating the area of intracellular space as a percentage of the total image area (as described and illustrated in Sup Fig. S1). Endothelial cells treated with no EVs or with EVs from control subjects showed almost no open spaces within the monolayers (Fig. 4a). Treatment with EVs collected from subjects with SCD at baseline caused a minimal amount of disruption of monolayer integrity. EVs isolated from the same subjects during episodes of ACS caused almost 15-fold more disruption of the endothelial monolayer compared to the EVs isolated at baseline (Fig. 4a). While the monolayer disruption experiments were performed with all 9 subjects with SCD, one subject could not be included in the final calculations because the damage caused by this ACS sample was very severe at 48 hours. Regardless, it still supports the rest of the data that show increased disruption of the endothelial monolayer during ACS episodes as compared to baseline.
Fig. 4.

EVs isolated during ACS disrupt endothelial monolayers in vitro. Endothelial monolayers were treated with EVs for 48 h, and then the percentage of monolayer disruption was calculated. (a) Graph shows the average percentage of disruption (± SEM). EVs isolated from subjects during an ACS episode caused significantly more disruption (# = p < 0.0005, ANOVA; * = p < 0.05, paired t-test within SCD subjects) (b) Graph shows the percent disruption in individual patients at baseline or during ACS (with results from the same patient connected by lines). With the exception of one subject, all SCD patients showed an increase in monolayer disruption from baseline to ACS.
We compared the extent of monolayer disruption caused by samples from the same subject at baseline or during ACS (Fig. 4b). EVs isolated from all subjects except for one caused increased disruption during ACS.
We also isolated EVs from some subjects by an alternative method (size exclusion). Nanoparticle tracking analysis showed that this approach also produced particles of ~100–120 nm, but the yield of particles was somewhat lower than produced by the precipitation method. Immunoblotting and protein determinations showed that several fractions contained marker proteins for small EVs, CD63 and flotillin (not shown), but very little protein or ApoA-1 (Fig. 5a). While endothelial cells were unaffected by treatment with buffer alone or control EVs (Fig. 5b,c), they responded to size exclusion isolated EVS from a SCD subject in ACS with robust monolayer disruption similar to that produced by EVs isolated by precipitation (Fig. 5d,e).
Fig. 5.

Endothelial disrupting EVs can also be purified by size exclusion chromatography from the plasma of a SCD subject obtained from a during an episode of ACS. (a) 200 μl of plasma was applied to a size exclusion column and eluted fractions were tested for CD63, ApoA-1, and total protein. Immunoblot detection of CD63 and ApoA-I is shown below the elution profile. The peak of CD63 was found in fractions 6–8, while the bulk of the protein and lipoprotein ApoA-I were present in much later fractions. (b-e) Representative photomicrographs show the localization of VE-cadherin (green in b-e) and nuclei (blue in c,e) in endothelial cells 48 h following treatment with no EVs (b, c) or EVs from a subject with SCD during ACS purified by size exclusion chromatography (d, e). In the example shown, the monolayer disruption was 10.7%. White stars indicate spaces between cells. Scale bar, 20 μm.
EVs isolated during ACS cause decrease in VE-cadherin protein levels
Because our microscopy suggested that EVs caused disruption of adherens junctions between endothelial cells, we also examined the RNA and protein levels for a major component of these junctions, VE-cadherin. We prepared homogenates from HMVEC-D cells after treatment for 48 hours with EVs from control and subjects with SCD and isolated RNA for RT-qPCR analysis or performed immunoblotting. Endothelial VE-cadherin mRNA levels did not differ between controls or subjects with SCD (Fig. 6a). Immunoblots revealed no differences in VE-cadherin levels among cells treated with no EVs or those treated with control or SCD-baseline EVs (Fig. 6b). In contrast, VE-cadherin protein levels were decreased to 50% of control levels after treating cells with EVs obtained during ACS (Fig. 6b).
Fig. 6.

EVs isolated during ACS cause a decrease in levels of endothelial VE-cadherin protein in vitro. Endothelial cells were grown to confluence and then treated for 48 h with no EVs, EVs from controls, EVs from patients with SCD at baseline, or EVs from the same patients during an ACS episode. (a) Graph shows VE-cadherin mRNA levels. These levels did not differ regardless of treatment. (b) Graph shows densitometry of immunoblots. There was a dramatic decrease in VE-cadherin protein levels in cells treated with ACS-EVs. (* = paired t-test within SCD subjects, p < 0.05) (c) Representative immunoblot and MEM Code Reversible Protein Staining of PVDF membrane to show comparability of protein loading and electrotransfer.
DISCUSSION
In this paper, we have presented data confirming that subjects with SCD produce small EVs that cause damage to endothelial cells in vitro. Most importantly, the damage is more severe when the EVs are isolated from SCD subjects during an episode of ACS. We performed substantial characterization of the EVs isolated from the plasma of subjects with SCD. These EVs have a diameter of 95 nm (mode) as measured by NTA, and they contain flotillin-1 and CD63. Comparable effects are elicited by EVs isolated using two different procedures. While similar EVs have been called exosomes in other studies, the vesicles that we isolate are most appropriately termed “small EVs” based on the MISEV2018 recommendations regarding vesicular nomenclature (30)Immunoblots showed that hemoglobin and glycophorin A were readily detectable in the EVs of both controls and subjects with SCD, implying that many of the EVs originated from red blood cells or their precursors. Our blots showing that glycophorin A was more abundant in SCD patients than in controls suggest that erythroid-derived small EVs may be more abundant in the subjects with SCD than in controls, consistent with results found for larger extracellular vesicles. A variety of previous studies have shown that SCD patients have increased circulating vesicles of various sizes and cell origin (including small EVs/exosomes and medium/large EVs/microparticles) (16,18).
Our data and previous studies give some indications of the abundance of small EVs in control subjects and those with SCD. While we (like others) found nanoparticle tracking to be unreliable for determining precise numbers/concentrations of EVs, the EV samples prepared from subjects with SCD contained much more flotillin-1 and CD63 than samples prepared from equal volumes of the plasma of control patients (Fig. 1). The simplest interpretation of this result is that the subjects with SCD had much greater numbers of circulating EVs. This conclusion is consistent with our previous studies (23,24). The abundances of flotillin-1 and CD63 were only moderately increased between subjects with SCD at baseline vs. during ACS, suggesting that there is only a moderate further increase in EV numbers during ACS. This implies that the functional differences that we observed in induction of endothelial monolayer damage must primarily reflect qualitative differences in the EVs (i.e., their contents). The significant difference in hemoglobin content between baseline and ACS (Fig. 2) provides evidence for differences in the protein contents of EVs under the two clinical conditions.
Several studies have implicated free heme and free hemoglobin in the vascular dysfunction or disruptions associated with hemolytic anemias like SCD (28,31). As expected, the plasma of our subjects with SCD did contain increased plasma-free heme. However, we do not believe that functional impacts of our EV preparations were due to these components, since free heme was undetectable in them, and the hemoglobin levels were very low. Indeed, when we treated endothelial monolayers with similar or higher concentrations of hemoglobin to that present in the EVs, we saw no effects
Our immunofluorescence studies showed that EVs from subjects with SCD caused damage to endothelial cell monolayers. The EVs from subjects with SCD at baseline caused a small amount of monolayer disruption, consistent with our previous work (24). These cellular changes represent a microscopic visualization of the changes of endothelial monolayer impedance that we previously detected by ECIS in response to baseline SCD EVs. Even this relatively small amount of intercellular spaces would allow substantial trans-endothelial passage of small molecules, ions, and even larger plasma components. Excitingly, there was one subject that we thought was at baseline based on clinical presentation, but whose EVs caused more disruption of the endothelial monolayer at baseline than expected. This subject went on to develop ACS 10 days after their clinic visit, suggesting there may be a component of the EVs that can be utilized to predict complications before they become severe.
The EVs from the subjects with SCD that were obtained during ACS episodes caused more severe effects on the endothelium. The abundant intercellular spaces that opened up would allow easy passage (extravasation) of ions, small molecules, plasma proteins, and cells. This phenomenon could certainly contribute to the vascular pathophysiology of ACS. It will be critical to determine contents of the ACS EVs that are responsible for this damage, whether they produce similar effects in vivo, and whether we can develop treatments to prevent or antagonize their effects. We believe that miRNA contents could contribute to this phenomenon and are working on sequencing them currently, however there are most likely additional membrane and protein changes in the small EVs.
The EVs isolated during an ACS episode produce damage to the endothelial cells through a complex cellular process. The effects require intact vesicles, since mechanical disruption of the EVs by sonication abolished monolayer disruption. The effects cannot represent a rapid, pharmacological effect of the EVs, since effects were not detectable by microscopy until two days following EV application. Detection of changes in trans-monolayer impedance also required many hours (23,24). Our photomicrographs of phalloidin staining suggest that the EVs caused retraction and shape changes of the cells involving the actin cytoskeleton. It likely increased tension on the adherens junctions (into which actin is anchored), as evidenced by the broadening and distortion of the VE-cadherin staining. The cells have pulled apart at many sites. The disruption of cell contacts is likely exacerbated by degradation of VE-cadherin (as implied by its reduced protein levels). In contrast, expression of VE-cadherin mRNA was not affected.
Our work is one of the first to implicate regulation of VE-cadherin and adherens junctions in the vasculopathy associated with SCD. VE-cadherin and adherens junctions are important in many other vasculopathies. VE-cadherin is the primary transmembrane protein of the endothelial adherens junction, it is upregulated during angiogenesis, and its abundance/distribution regulates vascular permeability (32). Interestingly, other studies have shown that VEGF, which is increased in SCD, causes increased phosphorylation of VE-cadherin resulting in alterations in endothelial permeability and changes to the cytoskeleton (33,34).
Our new data increase the body of evidence linking small EVs to the pathophysiology of SCD complications, particularly ACS. Detailed mechanisms will require additional studies of the contents of these EVs and of the cell biological mechanisms of disruption of endothelial monolayer integrity. It will also be critical to determine whether EVs might also contribute to the vasculopathy associated with other complications of SCD like vaso-occlusive crises.
Supplementary Material
Category of study: basic science.
Impact
Sickle cell disease patients have circulating extracellular vesicles (EVs) that modulate endothelial integrity by altering cadherin-containing intercellular junctions.
Disruption is more severe by EVs obtained during acute chest syndrome (ACS).
These results expand our knowledge of the pathophysiology of acute chest syndrome and the vasculopathies of sickle cell disease.
ACKNOWLEDGEMENTS
The authors would like to acknowledge the support of Shelby Gruntorad and the rest of the CSCDRG in helping to enroll patients and organize collecting samples. Rebecca Sturey participated in Nanoparticle tracking analysis and is gratefully acknowledged.
Funding Support: NIH UL1 TR000430, Comer Hospital RBC Race Funds, Ted Mullin Fund.
The subjects had been prospectively enrolled, with informed consent provided by parents or patients greater than 18 years of age and assent obtained from subjects 9–18 years of age.
Footnotes
There are no financial disclosures for any of the authors.
REFERENCES
- 1.Gladwin MT & Vichinsky E. Pulmonary complications of sickle cell disease. NEJM 359, 2254–2265 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Miller ST How I treat acute chest syndrome in children with sickle cell disease. Blood 117, 5297–5305 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Howard J, et al. Guideline on the management of acute chest syndrome in sickle cell disease. Br. J. Haematol 169, 492–505 (2015). [DOI] [PubMed] [Google Scholar]
- 4.DeBaun MR, et al. Factors predicting future ACS episodes in children with sickle cell anemia. Am. J. Hematol 89, E212–E217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoppe CC Inflammatory mediators of endothelial injury in sickle cell disease. Hem/Onc. Clin. No. Amer 28, 265–286 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Sharan K, et al. Association of T-786C eNOS gene polymorphism with increased susceptibility to acute chest syndrome in females with sickle cell disease. Br. J. Haematol 124, 240–243 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Hammerman SI, et al. Endothelin-1 production during the acute chest syndrome in sickle cell disease. Am. Jo. Resp. Cr. Care Med 156, 280–285 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Hebbel RP Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hem/Onc. Clin. No. Amer 28, 181–198 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Hulsmans M. & Holvoet P. MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc. Res 100, 7–18 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Wahlund CJ, Eklund A, Grunewald J. & Gabrielsson S. Pulmonary extracellular vesicles as mediators of local and systemic inflammation. Front. Cell Dev. Bio 5, 1–8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai RC, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 4, 214–222 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Vicencio JM, et al. Plasma exosomes protect the myocardium from ischemia-reperfusion injury. J. Am. Col. Cardiol 65, 1525–1536 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Ridger VC, et al. Microvesicles in vascular homeostasis and diseases. Thromb. Haem 117, 1296–1316 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Lötvall J, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 3(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathieu M, Martin-Jaular L, Lavieu G. & Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Bio 21, 9 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Hebbel RP & Key NS Microparticles in sickle cell anaemia: promise and pitfalls. Br. J. Haematol 174, 16–36 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Shet AS, et al. Sickle blood contains tissue factor–positive microparticles derived from endothelial cells and monocytes. Blood 102, 2678–2683 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Camus SM, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 125, 3805–3814 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Awojoodu AO, et al. Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood 124, 1941–1950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valadi H, et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Bio 9, 654–659 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Salomon C, et al. Exosomal signaling during hypoxia mediates microvascular endothelial cell migration and vasculogenesis. PLoS ONE 8, e68451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pant S, Hilton H. & Burczynski ME The multifaceted exosome: Biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem. Pharm 83, 1484–1494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khalyfa A, et al. Extracellular microvesicle microRNAs in children with sickle cell anaemia with divergent clinical phenotypes. Br. J. Haematol 174, 786–798 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Lapping-Carr G, et al. Exosomes contribute to endothelial integrity and acute chest syndrome risk: Preliminary findings. Pediatr. Pulmonol 52, 1478–1485 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong X-Q, Shao Q, Lounsbury CS, Bai D. & Laird DW Functional characterization of a GJA1 frameshift mutation causing oculodentodigital dysplasia and palmoplantar keratoderma. J. Bio. Chem 281, 31801–31811 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Lin X, et al. Connexin40 and connexin43 determine gating properties of atrial gap junction channels. J. Mol. Cell. Cardiol 48, 238–245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradford MM A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 72, 248–254 (1976). [DOI] [PubMed] [Google Scholar]
- 28.Balla J, et al. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antiox. Redox Signal 9, 2119–2138 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Johnstone R, Mathew A, Mason A. & Teng K. Exosome formation during maturation of mammalian and avian reticulocytes: evidence that exosome release is a major route for externalization of obsolete membrane proteins. J. Cellul. Phys 147, 27–36 (1991). [DOI] [PubMed] [Google Scholar]
- 30.Théry C, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nouraie M, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematol. 98, 464–472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallez Y. & Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 1778, 794–809 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Lopes FC, et al. Key endothelial cell angiogenic mechanisms are stimulated by the circulating milieu in sickle cell disease and attenuated by hydroxyurea. Haematol. 100, 730–739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lampugnani MG & Dejana E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res 120, S1–S6 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.