

Abstract
Cachexia syndrome develops in patients with diseases such as cancer and sepsis and is characterized by progressive muscle wasting. While iNOS is one of the main effectors of cachexia, its mechanism of action and whether it could be targeted for therapy remains unexplored. Here, we show that iNOS knockout mice and mice treated with the clinically tested iNOS inhibitor GW274150 are protected against muscle wasting in models of both septic and cancer cachexia. We demonstrate that iNOS triggers muscle wasting by disrupting mitochondrial content, morphology, and energy production processes such as the TCA cycle and acylcarnitine transport. Notably, iNOS inhibits oxidative phosphorylation through impairment of complexes II and IV of the electron transport chain and reduces ATP production, leading to energetic stress, activation of AMPK, suppression of mTOR, and, ultimately, muscle atrophy. Importantly, all these effects were reversed by GW274150. Therefore, our data establish how iNOS induces muscle wasting under cachectic conditions and provide a proof of principle for the repurposing of iNOS inhibitors, such as GW274150 for the treatment of cachexia.
Keywords: cachexia, cancer, inflammation, iNOS, metabolism
Subject Categories: Metabolism, Musculoskeletal System
Cachexia is a condition marked by severe skeletal muscle atrophy in patients affected by diseases such as cancer or sepsis. The inflammation‐induced factor inducible nitric oxide synthase (iNOS) was found to promote muscle wasting by causing mitochondrial dysfunction and energetic stress.
The paper explained.
Problem
Cachexia is a syndrome that accompanies several overlying diseases, including cancer, sepsis, and AIDS, all of which promote severe, systemic inflammation. It is debilitating to patients and marked by uncontrolled and severe muscle loss, leading to a dramatic decrease in quality of life and an increase in mortality. Cachectic patients are also significantly less responsive to their treatments, thereby increasing hospitalization duration for these patients. Unsuccessful attempts at treating cachexia have focused on individually targeting each of the many inflammatory factors that cause the condition. Therefore, identifying and targeting downstream effectors is needed to address these ineffective strategies’ setbacks.
Results
Here, we identify one such effector, named inducible nitric oxide synthase (iNOS), as a druggable molecular target to treat muscle wasting. Previous works by our group and others have highlighted the importance of iNOS as a promoter of cachexia. Here, we found that iNOS induces cachexia‐induced muscle wasting by promoting mitochondrial dysfunction and metabolic crisis in skeletal muscle. Importantly, we demonstrate that inhibition of iNOS function prevents muscle wasting in sepsis‐ and cancer‐induced models of cachexia using the clinically tested iNOS inhibitor GW274150.
Impact
This study highlights the feasibility of using iNOS inhibitors to treat cachexia‐induced muscle wasting. The repurposing of the iNOS inhibitor GW274150 to treat muscle wasting in the clinic is now a promising prospect in alleviating this previously untreatable condition.
Introduction
Cachexia is a debilitating wasting syndrome that arises in numerous chronic conditions such as cancer, chronic obstructive pulmonary disorder (COPD), chronic heart failure (CHF), HIV infection, and sepsis (Farkas et al, 2013). It is characterized by a dramatic, involuntary loss of both lean muscle and adipose tissue mass (Fearon et al, 2011; Blum et al, 2014). Development of cachexia is associated with a significant increase in morbidity and mortality in patients due to loss of skeletal muscle function (Takayama et al, 2016; Naito et al, 2017; Vigano et al, 2017; Gannavarapu et al, 2018; Zhou et al, 2018). Despite its significant impact on patient quality of life and chance for survival, there are currently no effective therapies for the treatment of cachexia. It is well accepted that cachexia is triggered by chronic inflammation and the upregulation of several pro‐inflammatory cytokines (Fearon et al, 2012; Baracos et al, 2018). However, monotherapies targeting various cytokines have not successfully treated the condition due to its multifactorial nature (Jatoi et al, 2007; Wiedenmann et al, 2008; Jatoi et al, 2010). Downstream effectors of these pro‐inflammatory cytokines within affected tissues, therefore, represent a more viable avenue for the development of effective therapies.
Inflammatory cytokines promote cachexia‐induced muscle wasting by triggering a catabolic state in muscle (Hall et al, 2011; Argilés et al, 2013; Cohen et al, 2015). Many cytokines induce the expression of muscle‐specific E3 ligases, such as MuRF1 and Atrogin‐1/MAFbx, that promote atrophy by targeting protein substrates for degradation (Fearon et al, 2012; Baracos et al, 2018). Reduced anabolic signaling and protein synthesis have also been demonstrated in murine models of cachexia and human patients (Emery et al, 1984; Smith & Tisdale, 1993; White et al, 2011; White et al, 2013; Puppa et al, 2014; Brown et al, 2018; Hall et al, 2018). Recent observations have suggested that, in addition to alterations in protein homeostasis, cachectic muscle has significant impairment in metabolic functions (Fearon et al, 2012; Argiles et al, 2015; Porporato, 2016; Hall et al, 2018) and metabolome (Der‐Torossian et al, 2013b; QuanJun et al, 2015; Tseng et al, 2015b; Cui et al, 2019; Kunzke et al, 2019; Pin et al, 2019a; Lautaoja et al, 2019b). The mitochondrial dysfunction observed under these cachectic conditions is likely due to impaired oxidative phosphorylation (OXPHOS) that is associated with reduced electron transport chain (ETC) complex activity (Julienne et al, 2012; Puppa et al, 2012; Der‐Torossian et al, 2013b; Fermoselle et al, 2013; Padrao et al, 2013; McLean et al, 2014; Brown et al, 2017; Hall et al, 2018; VanderVeen et al, 2018; Pin et al, 2019a). In addition, other mitochondrial defects, such as increased uncoupling of the proton gradient from ATP production, altered fission, and fusion, and abnormal morphology have been described (Shum et al, 2012; White et al, 2012; Fontes‐Oliveira et al, 2013; Tzika et al, 2013; Antunes et al, 2014). This loss of mitochondrial function is correlated with a reduction in the rate of ATP production, suggesting that the energy balance of muscle is negatively impacted (Constantinou et al, 2011; Tzika et al, 2013; Antunes et al, 2014; Pin et al, 2019a). Many studies investigating metabolic impairment in cachectic muscle have demonstrated that these changes are due to the activation of the NF‐κB pathway by pro‐inflammatory cytokines such as TNF‐α and IL‐6 (White et al, 2012; Der‐Torossian et al, 2013b; Fermoselle et al, 2013), and the NF‐κB‐dependent mechanisms that drive cytokine‐mediated mitochondrial dysfunction in cachexia, however, have yet to be delineated.
Inducible nitric oxide synthase (iNOS; NOS2), which is highly expressed in cachectic muscle, is a known downstream effector of the NF‐kB pathway (Buck & Chojkier, 1996; Di Marco et al, 2005; Ramamoorthy et al, 2009; Hall et al, 2011; Di Marco et al, 2012; Ma et al, 2017; Hall et al, 2018). Nitric oxide synthases (NOS) produce nitric oxide (NO) from the breakdown of L‐arginine to L‐citrulline (Strijdom et al, 2009). iNOS is one of three isoforms of NOS, together with endothelial (eNOS) and neuronal (nNOS). While eNOS and nNOS are associated with physiological NO production and cellular signaling, iNOS expression and activity are strongly induced by inflammation. Although the iNOS/NO pathway has been previously linked to the onset of cachexia‐induced muscle loss (Buck & Chojkier, 1996; Barreiro et al, 2005; Di Marco et al, 2005; Szabo et al, 2007; Hall et al, 2011; Di Marco et al, 2012; Wiseman & Thurmond, 2012; Ma et al, 2017; Hall et al, 2018), the mechanisms whereby excessive NO contributes to muscle wasting are still poorly understood.
In this study, we demonstrate that the iNOS/NO pathway is a trigger of the metabolome derangement, mitochondrial dysfunction, and energy crisis associated with the onset of inflammation‐induced muscle wasting. Specifically, iNOS mediates these effects by impairing mitochondrial metabolic processes such as the TCA cycle, the ETC, and acylcarnitine metabolism in skeletal muscle. Importantly, our data demonstrate that interfering with iNOS function pharmacologically or via a genetic depletion prevents all the metabolic defects described above as well as muscle wasting. We suggest that iNOS inhibition is a viable therapeutic strategy to prevent cachexia‐induced metabolic derangement and, ultimately, muscle loss.
Results
Genetic deletion of the iNOS gene prevents inflammation‐induced energy crisis and muscle wasting
An accumulating body of evidence points to iNOS as one of the main drivers of inflammation and cancer‐induced muscle wasting (Di Marco et al, 2005; Der‐Torossian et al, 2013b; Fermoselle et al, 2013; He et al, 2013). Since pathological production of NO by iNOS has been linked to metabolic dysfunction in inflammatory diseases (Anavi & Tirosh, 2019), we tested whether this can also be the case in mouse models of inflammation‐induced muscle loss.
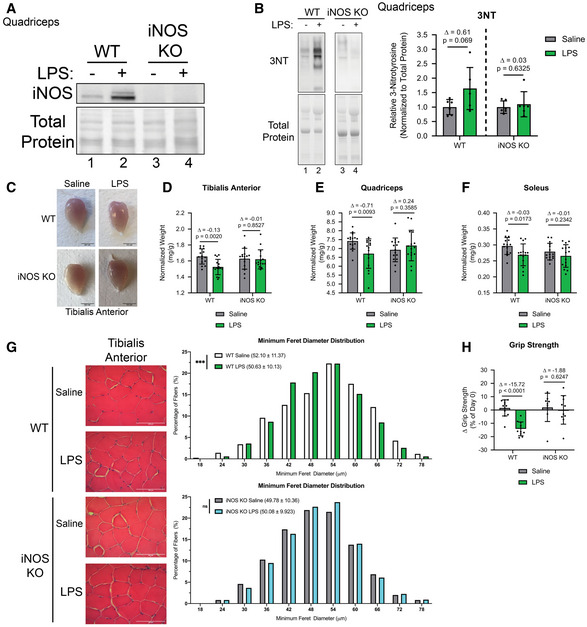
We assessed muscle loss and the associated metabolic response in wild‐type (WT) and iNOS knockout (KO) mice in a lipopolysaccharide (LPS)‐induced model of septic cachexia. Male wild‐type (WT) and iNOS KO C57BL/6 mice were injected or not with a dose of LPS previously shown to quickly induce muscle wasting over an 18‐hour period (Jin & Li, 2007; Braun et al, 2013; Hall et al, 2018). First, we assessed iNOS expression and activity in the muscles of WT and iNOS knockout mice treated with or without LPS. We confirmed, as expected, that the WT mice, but not the iNOS KO mice, expressed iNOS protein in response to LPS (Fig 1A). The activation of the iNOS/NO pathway in these muscles was determined by assessing the formation of 3‐nitrotyrosine (3NT)‐modified proteins (Chatterjee et al, 2003; Nanetti et al, 2007; Ahsan, 2013; Wei et al, 2015). We demonstrated that the levels of 3‐NT‐modified proteins were only increased in LPS‐treated WT but not iNOS KO mice (Fig 1B).
Figure 1. iNOS knockout mice are resistant to LPS‐driven muscle wasting.
-
AQuadriceps from saline or LPS‐treated, WT, or iNOS KO mice were isolated and used for Western blot analysis with anti‐iNOS (n = 4). Ponceau S was used for total protein staining.
-
B(left) Quadriceps from saline or LPS‐treated, WT, or iNOS KO mice were isolated and used for Western blot with anti‐3NT (n = 6). Total protein levels are also shown. (right) Quantification of the 3NT‐to‐total protein ratio. Ratios are expressed relative to the saline‐treated controls.
-
CRepresentative images of tibialis anterior muscles. Scale bars represent 0.5 cm.
-
D–FWeight of tibialis anterior (D), quadricep (E), and soleus (F) muscle normalized to initial body weight (n = 15).
-
G(left) Representative photomicrographs of H&E‐stained tibialis anterior muscle sections from control and iNOS KO mice injected with or without LPS. Scale bars = 100 μm. (right) Frequency histogram showing the distribution of muscle fiber minimum Feret diameter in the tibialis anterior muscles from saline‐ or LPS‐treated (top) WT control and (bottom) iNOS KO mice (n = 4). A total of 300 fibers per muscle were used for the analysis. Statistical comparisons, mean, and standard deviation of the mean are shown in the histogram legend.
-
HChange in grip strength between day of injection and endpoint of experiment. (WT saline n = 12, WT LPS n = 11, iNOS KO saline n = 9, and iNOS KO LPS n = 10).
Data information: Individual data points represent values from individual mice. Error bars represent the standard deviation (SD) of the mean. (B) Statistical comparisons were made between saline‐treated controls and LPS‐treated mice of same genotype. Plotted data were relativized to saline controls of corresponding genotype. Δ indicates the difference in mean values, and P‐values were calculated with Student’s t‐test. (D–F and H) For statistical comparisons, Δ indicates the difference in mean values and P‐values were calculated with an ANOVA followed by Fisher's LSD test. (G) P‐values were calculated with a Kolmogorov–Smirnov test (***P < 0.001). Non‐statistically significant comparisons (P > 0.05) are indicated as non‐significant (ns).
Source data are available online for this figure.
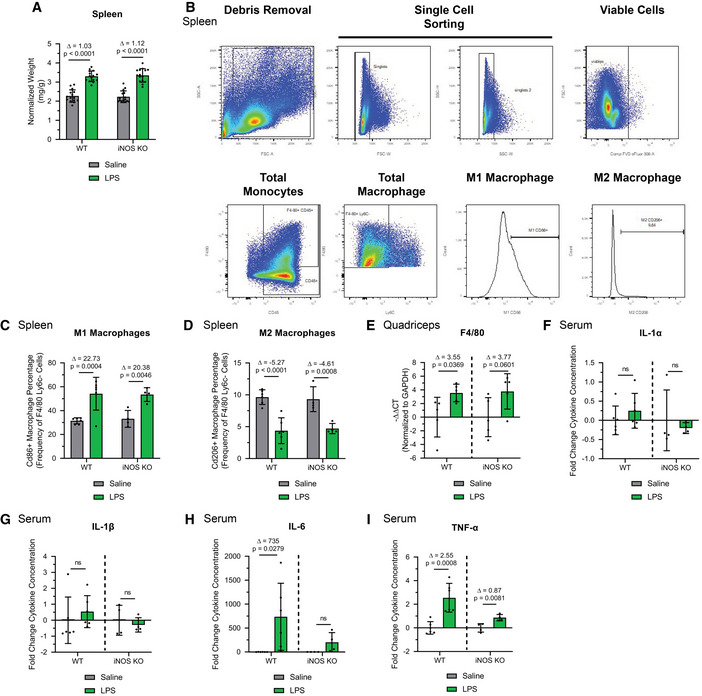
We then assessed activation of the immune response in these two mouse strains treated with or without LPS for 18 h, with particular attention to macrophage polarization and levels due to their prominent role in the LPS response (Beutler & Rietschel, 2003; Ginhoux et al, 2016). We observed that LPS had similar effects on the mass of the spleen (Fig EV1A), the population of splenic M1/M2 macrophages (Fig EV1, EV2, EV3, EV4B–D), and the accumulation of macrophages in the muscle (Fig EV1E) of WT and iNOS KO mice. Despite this, the LPS‐induced systemic levels of the pro‐cachectic cytokines IL‐6 and TNF‐α (but not IL‐1β, and IL‐1α) were significantly lower in iNOS KO mice compared with their WT counterparts (Dataset EV1 and Fig EV1, EV2, EV3, EV4F–I). Our results demonstrate that iNOS affects the immune response by mediating the secretion of systemic pro‐inflammatory cytokines such as IL‐6 and TNF‐α without affecting the polarization of macrophages.
Figure EV1. Characterization of the LPS‐induced immune response in WT and iNOS KO mice.
-
ASpleen weight normalized to initial body weight (n = 15).
-
BGating strategy for flow cytometry analysis of spleen M1 and M2 macrophages.
-
CPercentage of CD86+ M1 splenic macrophages relative to the total F4/80+Ly6C‐ macrophage population (WT saline n = 6, WT LPS n = 6, iNOS KO saline n = 4, and iNOS KO LPS n = 4).
-
DPercentage of CD206+ M2 splenic macrophages relative to the total F4/80+Ly6C‐ macrophage population (WT saline n = 6, WT LPS n = 6, iNOS KO saline n = 4, and iNOS KO LPS n = 4).
-
EQuadriceps from saline or LPS‐treated, WT or iNOS KO mice were isolated and used for RT–qPCR analysis for F4/80 mRNA expression. The – Δ Δ CT was plotted relative to the saline conditions of each background and normalized to GAPDH mRNA levels (n = 5).
-
F–ISerum from saline or LPS‐treated, WT or iNOS KO mice was collected. Fold change in IL‐1α (F), IL‐1β (G), IL‐6 (H), and TNF‐α (I) levels in LPS‐treated mice was plotted relative to saline condition (WT saline n = 6, WT LPS n = 6, iNOS KO saline n = 4, and iNOS KO LPS n = 4).
Data information: Individual data points represent values from individual mice. Error bars represent the standard deviation (SD) of the mean. (A, C, D) For statistical comparisons, Δ indicates the difference in mean values and P‐values were calculated with an ANOVA followed by Fisher's LSD test. (E‐I) Statistical comparisons were made between saline‐treated controls and LPS‐treated mice of same genotype. Plotted concentration data were relativized to saline controls of corresponding genotype. Δ indicates the difference in mean values, and P‐values were calculated with Student’s t‐test.
Figure EV2. Characterization of the LPS‐induced effects on muscle integrity/composition in WT and iNOS KO mice.
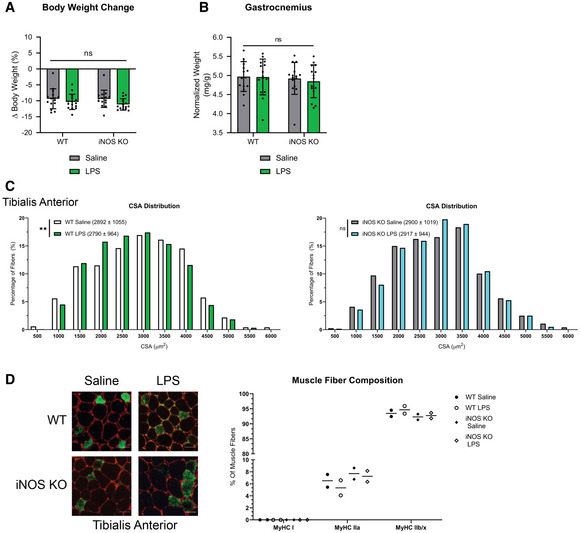
- Percent body weight change from time of injection to endpoint of experiment. (n = 15).
- Gastrocnemius weight normalized to initial body weight (n = 15).
- Frequency histogram showing the distribution of muscle fiber CSA in the tibialis anterior muscles from (left) WT control and (right) iNOS KO mice (n = 4). A total of 300 fibers per muscle were used for the CSA analysis. Statistical comparisons, mean, and standard deviation of the mean are shown in the histogram legend.
- (left) Photomicrographs of immunofluorescence images of tibialis anterior muscle sections from WT control and iNOS KO mice injected with or without LPS. Sections were stained against MyHC I (Blue), MyHC IIa (Green), and laminin (Red). Unstained fibers were classified as MyHC IIb/x. Scale bars = 50 μm. (right) Percentage of type I, type IIa, and type IIb/IIx fibers in the whole tibialis anterior muscles from control and iNOS KO mice (n = 2).
Data information: Individual data points represent values from individual mice. Error bars represent the standard deviation (SD) of the mean. (C) P‐values were calculated with a Kolmogorov–Smirnov test (**P < 0.01). Non‐statistically significant comparisons (P > 0.05) are indicated as non‐significant (ns).
Figure EV3. Effect of GW on the immune response as well as muscle integrity/composition in C26‐tumor‐bearing mice.
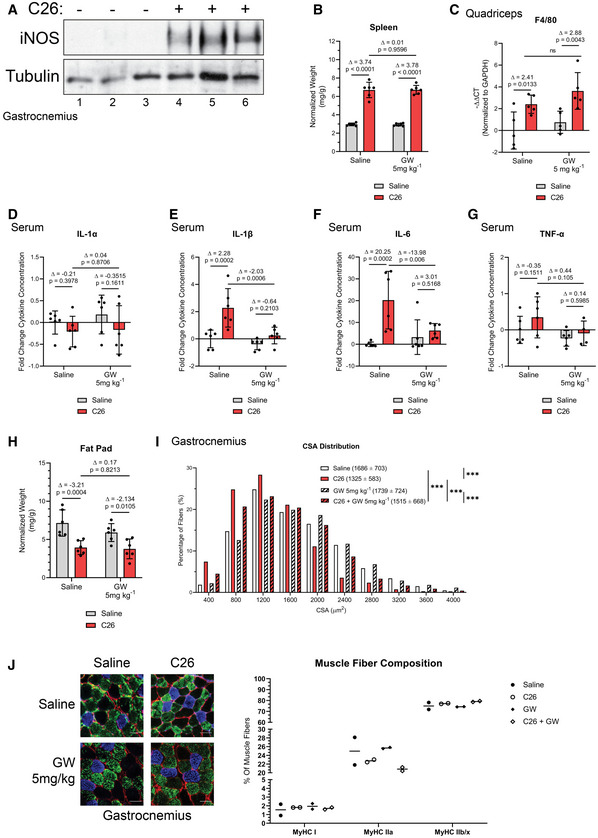
-
AiNOS and tubulin (loading control) protein levels in the gastrocnemius muscle were assessed by Western blot (n = 3).
-
BSpleen weight normalized to initial body weight.
-
CQuadriceps from control and C26 mice treated with or without GW were isolated and used for RT–qPCR analysis for F4/80 mRNA expression. The – Δ Δ CT is plotted relative to the saline condition and normalized to GAPDH mRNA levels (n = 5).
-
D–GSerum from saline and C26‐tumor‐bearing mice treated with or without GW was collected. Fold change in IL‐1α (D), IL‐1β (E), IL‐6 (F), and TNF‐α (G, n = 4 for C26 + GW) levels was plotted relative to saline condition.
-
EInguinal fat pad weight normalized to initial body weight.
-
FFrequency histogram showing the distribution of muscle fiber CSA in the gastrocnemius muscles from control and C26 mice treated with or without GW (saline n = 4, C26 n = 4, GW 5 mg/kg n = 3, and C26 GW 5 mg/kg n = 4). A total of 600–700 fibers per muscle were used for the CSA analysis. Statistical comparisons, mean, and standard deviation of the mean are shown in the histogram legend.
-
G(left) Photomicrographs of immunofluorescence images of gastrocnemius muscle sections from control and C26 mice treated with or without GW. Sections were stained against MyHC I (Blue), MyHC IIa (Green), and laminin (Red). Unstained fibers were classified as MyHC IIb/x. Scale bars = 50 μm. (right) Percentage of type I, type IIa, and type IIb/IIx fibers in the whole gastrocnemius muscles from control and C26 mice treated with or without GW (n = 2).
Data information: Individual data points represent values from individual mice, with a total of six mice per cohort (n = 6) unless stated otherwise. Error bars represent the standard deviation (SD) of the mean. (B–H) For statistical comparisons, Δ indicates the difference in mean values and P‐values were calculated with an ANOVA followed by Fisher's LSD test. (I) P‐values were calculated with a Kolmogorov–Smirnov test (***P < 0.001).
Source data are available online for this figure.
Figure EV4. Cytokine‐mediated loss of complex II and IV integrity is reversed with aminoguanidine.
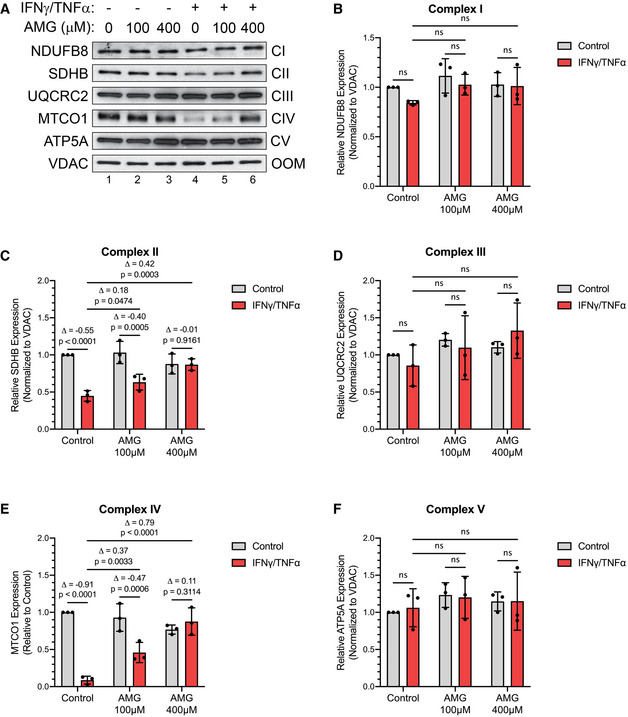
-
AWestern blot analysis for ETC protein complex subunits.
-
B–FQuantification of complex subunits normalized to VDAC (outer mitochondrial membrane; OMM) and relative to untreated control. (B) NDUFB8 (complex I; CI), (C) SDHB (complex II; CII), (D) UQCRC2 (complex III; CIII), (E) MTCO1 (complex IV; CIV), and (F) ATP5A (complex V; CV).
Data information: Individual data points are from three independent experimental replicates (n = 3). Error bars represent the standard deviation (SD) of the mean. For statistical comparisons, Δ indicates the difference in mean values and P‐values were calculated with an ANOVA followed by Fisher's LSD test. Non‐statistically significant comparisons (P > 0.05) are indicated as non‐significant (ns).
Source data are available online for this figure.
Next, we evaluated whether the genetic ablation of iNOS affects LPS‐induced changes in body weight and muscle mass. As previously described, the mass of skeletal muscle and tissues was normalized to the initial body weight (Michaelis et al, 2017; Parajuli et al, 2018; Pin et al, 2019a; Pin et al, 2019b). LPS, as previously shown, is known to affect food consumption rates in mice (Braun et al, 2013; Hall et al, 2018). Therefore, the saline‐treated WT as well as the saline and LPS‐treated iNOS KO mice were pair‐fed to match the consumption rate of the LPS‐treated WT mice in order to account for variations which may occur due to differences in food consumption. We observed that although pair feeding caused a 10–12% decrease in body weight in both saline‐treated WT and iNOS KO mice, treatment with LPS did not further exacerbate this loss of mass (Fig EV2A). The genetic ablation of iNOS, however, did prevent the LPS‐induced wasting of the tibialis anterior (TA), soleus, and quadriceps muscles but not the gastrocnemius muscle (Fig 1C–F and EV2B). The iNOS‐induced loss of muscle mass was further confirmed by assessing the minimum Feret diameter (Fig 1G) and cross‐sectional area (CSA) (Fig EV2C) of the muscle fibers. It is well‐established that inflammation‐induced muscle wasting is often associated with a functional deficit in muscle strength (Murphy et al, 2012). By measuring grip strength, we found that the iNOS KO mice, but not their WT counterparts, were completely protected from LPS‐mediated loss of muscle strength (Fig 1H). These observations clearly show that genetic disruption of iNOS protects mice from sepsis‐induced muscle wasting.
To determine whether the impact of iNOS on muscle wasting is mediated by changes in muscle metabolism, we next performed LC‐MS/MS to determine the levels of 119 key metabolites including biogenic amines, amino acids, acylcarnitines, glycerophospholipids, and organic acids (Dataset EV2) in tibialis anterior (TA) muscles collected from the WT and iNOS KO mice treated or not with LPS. The raw metabolite concentration data obtained were analyzed globally using the MetaboAnalyst 4.0 software and were mean‐centered and range‐scaled to allow for comparison of metabolites within their biological range (Chong et al, 2018; Chong et al, 2019a; Chong & Xia, 2018; Chong et al, 2019b; van den Berg et al, 2006; Xia et al, 2012; Xia et al, 2009; Xia et al, 2015; Xia & Wishart, 2011a,b, 2016). We observed, through the multivariate approach of partial least squares discriminant analysis (PLS‐DA), that the metabolomic profile of the TAs from WT mice is distinct from their iNOS KO counterparts and that LPS causes metabolomic shifts in both mouse backgrounds (Fig 2A). Analysis of heatmaps comparing the WT saline cohort to the iNOS KO saline cohort and the WT LPS cohort to the iNOS KO LPS cohort further confirmed the presence of the distinct metabolic profiles (Appendix Fig S1A–C). To assess whether differences in the observed distinct metabolic profiles found in our four treatment groups were correlated with differences in muscle fiber type composition, we assessed the distribution of type I, type IIA, and type IIB/IIX fibers in the TAs of these mice. We observed no appreciable differences between the four groups, indicating that other factors were responsible for the metabolic differences between them (Fig EV2D). Pathway analysis using the KEGG library in Mus musculus identified several pathways, including arginine metabolism (as expected), glycolysis, and tricarboxylic acid (TCA) cycle, that were altered in WT mice. These pathways, however, were not affected in the iNOS KO mice (Fig 2B). The impairment of several pathways involving energy production suggests that iNOS triggers an energetic crisis in wasting muscles. An increase in the AMP/ATP ratio, due to energetic stress, is known to activate AMPK (5’‐activated protein kinase), an important regulator of energy homeostasis, through phosphorylation at Thr172 (pAMPK; Stein et al, 2000; Hardie et al, 2012). Western blot analysis of the quadriceps muscle showed that LPS activates AMPK in WT but not iNOS KO mice (Fig 2C). Collectively, these data strongly suggest that LPS‐induced iNOS expression, in skeletal muscle, disrupts normal energy metabolism prompting energetic stress.
Figure 2. Genetic ablation of iNOS prevents LPS‐driven deregulation of the TCA cycle and energy production.
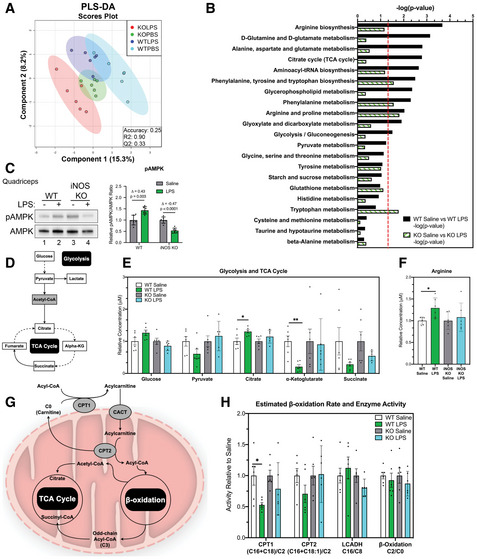
- Scores scatter plot of partial least square discriminant analysis (PLS‐DA) model of metabolites from WT and iNOS KO mice treated with or without LPS. Metabolomic data were range‐scaled and mean‐centered.
- Pathway analysis using MetaboAnalyst 4.0 Software comparing significantly altered pathways from WT saline to LPS as well as iNOS KO saline to LPS. Pathways are ranked by their significance and filtered based on a Pathway Impact Score > 0.1. Metabolomic data were range‐scaled and mean‐centered. P‐values were obtained using GlobalTest, and the −log(P‐value) corresponding to a P‐value of 0.05 is indicated by the red dashed line.
- (left) Western blot analysis of pThr172‐AMPK (pAMPK) and total AMPK (AMPK) in quadriceps muscle. (right) Quantification of the pAMPK‐to‐AMPK ratio. Ratios are expressed relative to the saline‐treated controls.
- Schematic of glycolysis and the TCA cycle. White boxes denote detected metabolites. Gray boxes denote undetected metabolites. Dashed arrows indicate the presence of multiple reactions between metabolites.
- Relative concentrations of metabolites involved in glycolysis and TCA cycle.
- Relative concentration of arginine.
- Schematic of acylcarnitine metabolism. Gray circles denote key enzymes involved in metabolic processes highlighted.
- Relative estimated activity of CPT1, CPT2, LCADH, and β‐oxidation.
Data information: Individual data points represent values from individual mice, with a total of six mice per cohort (n = 6). (B) P‐values were obtained using GlobalTest. (C–H) Error bars represent the standard deviation (SD) of the mean. Statistical comparisons were made between saline‐treated controls and LPS‐treated mice of same genotype. Plotted concentration data were relativized to saline controls of corresponding genotype. P‐values were calculated with Student’s t‐test (*P < 0.05; **P < 0.01). (C) Δ indicates the difference in mean values.
Source data are available online for this figure.
Next, we sought to determine the mechanism by which iNOS impairs cellular energy production in skeletal muscles undergoing wasting. We examined the concentrations of specific metabolites in key pathways involved in cellular energy production, including glycolysis, the TCA cycle, and acylcarnitine metabolism (Fig 2D and G). Analysis of heatmaps revealed that most of the detected metabolites involved in glycolysis and the TCA cycle are altered by LPS treatment of WT but not the iNOS KO mice (Appendix Fig S2A and B). Within glycolysis, we observed that iNOS induced a trend toward an increase in glucose levels as well as a trend toward a decrease in pyruvate levels, suggesting a potential reduction in glucose utilization in WT LPS‐treated mice. We additionally observed a dramatic iNOS‐dependent decrease in the TCA cycle intermediate α‐ketoglutarate (α‐KG). We also detected a trend toward a decrease in succinate levels, as well as an increase in citrate (Fig 2D–E). Taken together with our pathway analysis, these iNOS‐dependent alterations to TCA cycle intermediates also suggest an involvement of iNOS in glutamine metabolism during LPS‐induced muscle wasting (Fig 2B and D–E). The levels of arginine, which is known to regulate the TCA cycle and oxidative metabolism (El‐Gayar et al, 2003; Lee et al, 2003; Chaturvedi et al, 2007; Xu et al, 2016; Kunzke et al, 2019), were also significantly increased in response to LPS in an iNOS‐dependent manner (Fig 2F). We then investigated β‐oxidation, which is used to produce energy by several tissues, including muscle (Henique et al, 2015). Carnitine palmitoyltransferase 1 (CPT1) and CPT2 catalyze the rate‐limiting steps of β‐oxidation. Acyl‐CoA molecules are converted to acylcarnitine species by CPT1 to allow for their transport into the mitochondria, where acylcarnitines are converted back to their CoA equivalents by CPT2 (Zhou et al, 2012; Wu et al, 2017; Li et al, 2019). Once converted back to their CoA analog, fatty acids enter β‐oxidation through long‐chain Acyl‐CoAs dehydrogenase (LCADH; Zhou et al, 2012; Wu et al, 2017; Li et al, 2019) (Fig 2G). We observed a global decrease in long, even‐chained acylcarnitine species in LPS‐treated WT mice, but not in iNOS KO mice (Appendix Fig S3). We next estimated the activities of CPT1, CPT2, and LCADH as well as β‐oxidation by calculating the ratios of various acylcarnitine combinations (Zhou et al, 2012; Wu et al, 2017; Li et al, 2019). We observed that in response to LPS treatment, the global activity of CPT1 was decreased in an iNOS‐dependent manner (Fig 2H). CPT2 activity also tended to decrease (Fig 2H), suggesting that during cachexia, iNOS impairs the activities of CPT1 and CPT2.
Together, our data suggest that LPS treatment interferes with cellular energy production and mitochondrial function in an iNOS‐dependent manner. iNOS mediates these effects by impairing glycolysis and disrupting the entry of both pyruvate (via its decrease) and fatty acids (via inhibition of CPT1 and CPT2) into the TCA cycle.
The iNOS inhibitor GW274150 prevents cancer‐induced muscle wasting
The data outlined above raise the possibility that targeting iNOS could represent a viable option to prevent inflammation‐induced muscle wasting. To provide a proof of principle for this possibility, we looked for an iNOS inhibitor that is efficient in an in vivo setting without having major side effects. GW274150 (GW) has been established as a highly specific inhibitor for iNOS (over eNOS and nNOS) and has been tested in phase II clinical trials to treat migraines, rheumatoid arthritis, and asthma. Although GW was found to be safe, its effectiveness was not better than pre‐existing drugs against these conditions (Høivik et al, 2010; Seymour et al, 2012; Vitecek et al, 2012).
Therefore, we decided to investigate the possibility of repurposing GW to treat muscle wasting. We first used our sepsis mouse model to test whether GW could mimic the effects of iNOS depletion in preventing LPS‐induced muscle wasting. GW was administered at a dose of 5 mg kg−1, which was previously shown to reduce collagen‐induced arthritis symptoms in mice to an extent similar to that in iNOS knockout mice (Cuzzocrea et al, 2002). When compared to the results obtained in Figs 1D and H, Fig EV1A, and Fig EV2A on WT and iNOS KO mice treated with or without LPS, we demonstrated that this dose of GW, similar to that was observed in iNOS KO, protected mice against LPS‐induced muscle wasting (Appendix Fig S4A–D).
Next, we assessed the efficacy of GW‐mediated inhibition of iNOS in the C26 adenocarcinoma mouse model of cancer cachexia (Di Marco et al, 2012; Murphy et al, 2012; Der‐Torossian et al, 2013b; Hall et al, 2018; Lautaoja et al, 2019a; Pin et al, 2019a; Pin et al, 2019b). For these experiments, we injected male BALB/C mice with C26 colon cancer cells, a treatment known to induce muscle wasting over the course of 14–19 days (Di Marco et al, 2012; Bonetto et al, 2016; Hall et al, 2018). First, we confirmed that iNOS expression was induced in the muscle of C26‐bearing mice (Fig EV3A), as we have previously shown (Di Marco et al, 2012). Since iNOS inhibition has been reported to impair tumor growth (Kostourou et al, 2011; Granados‐Principal et al, 2015; Garrido et al, 2017), mice were treated with GW 5 days post‐injection of the C26. We demonstrated that GW, under these conditions, did not have any effect on tumor burden (Fig 3A). To assess the effectiveness of GW in impairing the iNOS/NO pathway in skeletal muscle, we next measured levels of 3NT‐modified protein as described above (Chatterjee et al, 2003; Nanetti et al, 2007; Ahsan, 2013; Wei et al, 2015). We observed a C26‐mediated increase in 3NT‐modified protein levels that were significantly decreased by GW treatment, demonstrating that GW impaired iNOS activity in muscle (Fig 3B). We then characterized the immune response of C26 tumor‐bearing mice treated with or without GW by assessing spleen mass, muscle macrophage content (assessed by verifying F4/80 expression), and serum cytokine levels (Dataset EV3). We observed that although spleen mass and muscle macrophage content increased in C26‐tumor‐bearing mice when compared to their saline‐control counterparts, these changes were not affected by treatment with GW (Fig EV3B and C). The C26 model of cancer‐induced muscle wasting is known to be mediated by elevated levels of pro‐inflammatory cytokines such as IL‐6 (Narsale & Carson, 2014). Interestingly, we observed that although the serum levels of several cytokines including IL‐6, TNF‐α, and IL‐1β (but not IL‐1α) increased in C26‐tumor‐bearing mice, these effects were prevented by treatment with GW (Fig EV3, EV4D–G). Similar to our observations in the septic model of cachexia, the above data suggest that GW affects the C26‐mediated immune response by preventing the secretion of these factors by inflammatory cells.
Figure 3. GW274150 treatment reduces muscle wasting in the C26 model.
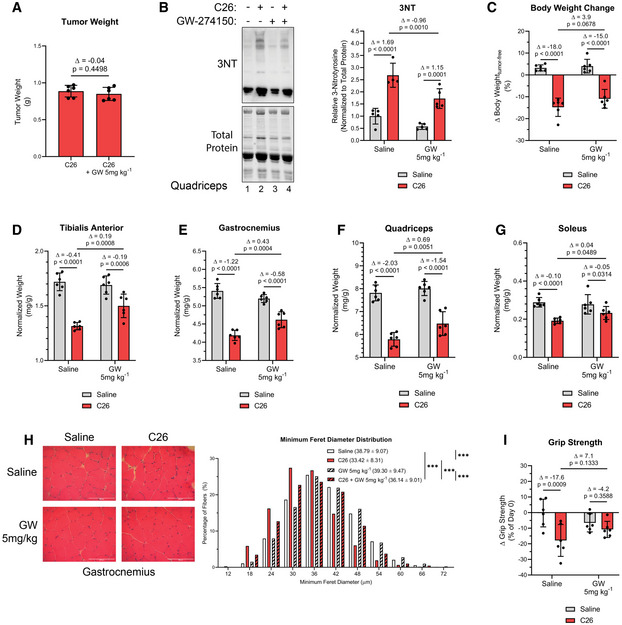
-
AEffect of GW on tumor weight.
-
B(left) Representative image of Western blot analysis of 3NT staining and stain free imaged total protein levels from quadriceps muscle. (right) Quantification of the 3NT‐to‐total protein ratio (saline n = 5, C26 n = 4, GW 5 mg/kg n = 5, and C26 GW 5 mg/kg n = 5).
-
CPercent body weight change from day 0 to day 16.
-
D–GWeight of tibialis anterior (D), gastrocnemius (E), quadricep (F), and soleus (G) muscle normalized to initial body weight.
-
H(left) Representative photomicrographs of gastrocnemius muscle sections from control and C26 mice treated with or without GW taken after H&E staining. Scale bars = 100 μm. (right) Frequency histogram showing the distribution of muscle fiber minimum Feret diameter in the gastrocnemius muscles from control and C26 mice treated with or without GW (saline n = 4, C26 n = 4, GW 5 mg/kg n = 3, and C26 GW 5 mg/kg n = 4). A total of 600‐700 fibers per muscle were used for the analysis. Statistical comparisons, mean, and standard deviation of the mean are shown in the histogram legend.
-
IChange in grip strength from before tumor cell injection (day 0) and before endpoint collection (day 16).
Data information: Individual data points represent values from individual mice, with a total of six mice per cohort (n = 6) unless stated otherwise. Error bars represent the standard deviation (SD) of the mean. (A) Δ indicates the difference in mean values, and P‐values were calculated with Student’s t‐test. (B–G, I) For statistical comparisons, Δ indicates the difference in mean values and P‐values were calculated with an ANOVA followed by Fisher's LSD test. (H) P‐values were calculated with a Kolmogorov–Smirnov test (***P < 0.001).
Source data are available online for this figure.
Next, we evaluated the effect of GW on C26‐induced cachexia. In both cohorts (with and without GW), we observed significant body weight loss, although less severely in the GW‐treated mice (Fig 3C). GW treatment did not affect adipose tissue wasting, which likely contributed to the observed overall body weight change (Fig EV3H). However, GW treatment partially protected against the C26‐mediated atrophy of the TA, soleus, quadriceps, and gastrocnemius muscles (Fig 3D–G). These data were corroborated through analyses of the minimum ferret diameter and CSA of gastrocnemius muscle fibers, which demonstrated a protective effect of GW against C26‐induced atrophy (Figs 3H and EV3I). We observed a significant decrease in grip strength in C26‐treated mice (Fig 3I). Although GW treatment alone tended to decrease grip strength, it did partially rescue (albeit non‐significantly) the loss of strength observed in C26‐treated mice (Fig 3I). Therefore, these observations suggest that GW‐mediated inhibition of iNOS protects mice from cancer‐induced muscle atrophy and loss of muscle function.
The iNOS inhibitor GW274150 prevents deleterious effects on energy production observed during cancer‐induced muscle wasting
Given the protective effect of GW against C26‐induced muscle wasting and the reported metabolomic derangement of skeletal muscle of C26‐treated mice (Murphy et al, 2012; Der‐Torossian et al, 2013b; Lautaoja et al, 2019a; Pin et al, 2019a; Pin et al, 2019b), we next assessed the impact of GW and its inhibitory effect on iNOS function on the skeletal muscle metabolome in our model. LC‐MS/MS metabolomic analysis on TA muscles (Dataset EV4) showed that GW alone had minimal effects on the skeletal muscle metabolome, only affecting arginine biosynthesis and D‐glutamine/D‐glutamate metabolism (Appendix Fig S5A–C). We next used the same metabolomic approach to compare metabolite profiles on TA samples collected from control or C26‐treated mice treated with or without GW (Dataset EV5). PLS‐DA analysis showed global cancer‐induced shifts in the overall metabolome (Fig 4A). Pathway analysis showed that, in skeletal muscle undergoing wasting, GW targeted a few key pathways that were affected by C26 tumors (Fig 4B). Similar to our observations in the LPS model, these pathways include those associated with energy production (glycolysis and pyruvate metabolism) and amino acid metabolism (Fig 4B and Appendix Fig S6A and B). Immunofluorescence analysis of the expression levels of myosin heavy chains I and IIa as well as laminin indicated that these effects on the metabolic profiles were not due to a shift in the distribution of muscle (type I, type IIA, and type IIB/IIX) fiber types in the gastrocnemius muscles of the treated mice (Fig EV3J). Next, we evaluated the impact of iNOS on the global energetic status of these mice by measuring AMPK activation. In line with our observations in the sepsis model, although AMPK was activated in muscle of C26‐tumor‐bearing mice, this effect was partially prevented by GW (Fig 4C). GW treatment alone, however, did not have any effect on AMPK activation (Fig 4C). Therefore, our data suggest that iNOS activity disrupts normal energy metabolism in skeletal muscle both in a sepsis and in a cancer‐induced cachexia model.
Figure 4. Pharmacological inhibition of iNOS reduces C26‐induced derangement of amino acids and impairment of energy production.
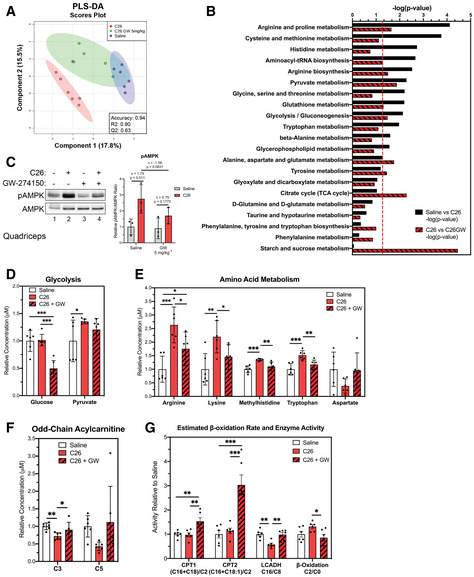
- Scores scatter plot of partial least square discriminant analysis (PLS‐DA) model of metabolites from saline, C26, and C26 + GW‐treated mice. Metabolomic data were range‐scaled and mean‐centered.
- Pathway analysis using MetaboAnalyst 4.0 Software comparing significantly altered pathways from saline to C26 as well as C26 to C26 + GW. Pathways are ranked by their significance and filtered based on a Pathway Impact Score > 0.1. Metabolomic data were range‐scaled and mean‐centered. P‐values were obtained using GlobalTest, and the −log(P‐value) corresponding to a P‐value of 0.05 is indicated by the red dashed line.
- (left) Western blot analysis of pThr172‐AMPK (pAMPK) and total AMPK (AMPK) in quadriceps muscle (n = 3). (right) Quantification of the pAMPK‐to‐AMPK ratio relative to the saline‐treated control (n = 3).
- Relative concentrations of metabolites involved in glycolysis.
- Relative concentrations of amino acids.
- Relative concentrations of odd‐chain acylcarnitines.
- Relative estimated activity of CPT1, CPT2, LCADH, and β‐oxidation.
Data information: Individual data points represent values from individual mice, with a total of six mice per cohort (n = 6) unless otherwise stated. (B) P‐values were obtained using GlobalTest. (C–G) Error bars represent the standard deviation (SD) of the mean. P‐values were calculated with an ANOVA followed by Fisher's LSD test (*P < 0.05; **P < 0.01; ***P < 0.001). (C) Δ indicates the difference in mean values.
Source data are available online for this figure.
Next, we determined how iNOS promotes the energy crisis in cancer cachexia. Although the C26 tumor did not affect the relative concentration of glucose, GW significantly decreased glucose levels in C26‐tumor muscles (Fig 4D). In addition, GW partially prevented an increase in arginine levels and other iNOS‐dependent amino acid derangements in C26‐tumor mice (Fig 4E). In particular, lysine, methylhistidine, and tryptophan levels were increased, and aspartate trended toward a decrease in C26‐tumor mice, effects which were recovered due to GW treatment. These changes in amino acid levels in muscle are consistent with a number of recent studies assessing the metabolome of cancer cachectic muscle (Der‐Torossian et al, 2013a; QuanJun et al, 2015; Tseng et al, 2015a; Kunzke et al, 2019; Lautaoja et al, 2019b). It has been suggested that elevation of methylhistidine in cachectic conditions is associated with muscle protein breakdown (QuanJun et al, 2015). Therefore, the recovery of normal methylhistidine levels in C26‐tumor‐bearing mice treated with GW could indicate a protective effect of iNOS inhibition against protein catabolism. Interestingly, increased lysine and arginine levels in cachectic muscle have been linked to defects in energy production and energetic stress (Kunzke et al, 2019). The recovery of these amino acid levels in GW‐treated mice indicates, therefore, a possible protection against energetic crisis in our model.
As acylcarnitine metabolism was significantly affected by iNOS in our LPS model, we assessed whether this is also the case in our C26 model. We observed a decrease in saturated, long, even‐chain acylcarnitines in C26 tumor mice, similar to WT mice treated with LPS (Appendix Fig S7A and B). Interestingly, GW treatment either recovered normal levels or increased the concentration of many long‐chain acylcarnitines (e.g., C16 and C18) in C26 tumor‐bearing mice (Appendix Fig S7A and B). Mice treated with GW alone also exhibited increases in many long‐chain acylcarnitines, indicating that the GW‐specific effect occurs independently of the tumor environment (Appendix Fig S8A). Short, odd‐chain acylcarnitines were also decreased in cancer cachectic mice and recovered with GW treatment (Fig 4F). It is well‐established that these acylcarnitine chains act as anapleurotic metabolites, which feed into the TCA cycle through conversion into succinyl‐CoA (Brunengraber & Roe, 2006; Pfeuffer & Jaudszus, 2016). As GW widely affected the acylcarnitine profile of C26 tumor mice, we determined the activity levels of CPT1, CPT2, LCADH, and β‐oxidation. Although activity of CPT1 and CPT2 was not significantly affected between control and C26 tumor mice, we observed an increase in their activity in the C26 + GW cohort (Fig 4G). In addition, LCADH and β‐oxidation appeared to have iNOS‐dependent alterations to their activity, as C26 tumors decreased LCADH activity and increased β‐oxidation, both of which were prevented by GW (Fig 4G). Of note, treatment of mice with GW alone affected CPT1, CPT2, and LCADH activity and β‐oxidation similarly to what was observed when used to treat C26 tumor‐bearing mice (Appendix Fig S8B). Our data therefore indicate that iNOS, in the C26‐adenocarcinoma model, affects acylcarnitine‐mediated energy production in the mitochondria by interfering with the relationship between acylcarnitine metabolism and the TCA cycle as well as deregulating critical enzymes in β‐oxidation.
iNOS activity affects the wasting of myotubes treated with pro‐cachectic cytokines
The above metabolomic analysis demonstrated that iNOS drives muscle wasting by impairing cellular metabolism that leads to an energetic crisis. It is well‐established that the induction of energy crisis is directly related to ATP levels, which are controlled by rates of glycolysis and oxidative phosphorylation, ultimately dictated by the enzymes carrying out these processes (Stein et al, 2000; Hardie et al, 2012; Mookerjee et al, 2017, 2018). Therefore, we wanted to understand whether and how iNOS affect ATP production rates from glycolysis and oxidative phosphorylation.
We attempted to test ATP production rates from glycolysis and oxidative phosphorylation in muscle fibers isolated from LPS‐treated and C26 tumor mice using the Seahorse Extracellular Flux Analyzer (Schuh et al, 2012). However, the muscle fibers did not survive long enough to assess their means of energy production by this technique. We then decided to assess ATP production in IFNγ/TNF‐α‐treated C2C12 myotubes that are amenable to Seahorse analysis (Mookerjee et al, 2017, 2018; Hall et al, 2018). We have routinely used this in vitro cell model of muscle wasting to mimic the effect of cytokines on muscle fibers observed, in vivo, during inflammation‐induced muscle wasting (Di Marco et al, 2005; Di Marco et al, 2012; Ma et al, 2017; Hall et al, 2018). We began by characterizing the validity and relevance of C2C12 myotubes in relation to our in vivo models by assessing the role of iNOS after cytokine treatment. As previously shown, IFNγ and TNF‐α induce the wasting of C2C12 myotubes over 48 h (Fig 5A and B) (Cramer et al, 2018; Hall et al, 2018; Mubaid et al, 2019). The wasting of these myotubes is associated with significant increases in both the expression of iNOS protein and nitrite levels in the media as well as deregulation of the TCA cycle and anapleurotic metabolites that feed into it (Dataset EV6 and Fig 5C and D). Consistent with our observations in the in vivo models, inhibition of iNOS with GW prevented cytokine‐induced atrophy of C2C12 myotubes (Fig 5A and B; Di Marco et al, 2005; Di Marco et al, 2012). Although GW does not affect iNOS protein levels in cytokine‐treated myotubes, it did inhibit, in a dose‐dependent manner, iNOS‐induced NO production (Fig 5C). These results were confirmed using another specific iNOS inhibitor, aminoguanidine (AMG), that has not underwent clinical development (Appendix Fig S9) (Thornalley, 2003). While cytokine treatment activated AMPK, this effect was prevented when the cells were co‐treated with GW or AMG (Fig 5E and F and Appendix Fig S10A). These results were furthermore confirmed by assessing the phosphorylation of ACC, a downstream marker of AMPK (Fig 5E and F). In addition, we observed a reduction in approximately 35% in cellular ATP levels in cytokine‐treated myotubes, which was prevented by GW and AMG (Fig 5G and Appendix Fig S10B). One consequence of energetic stress is the inhibition of protein synthesis via the central regulator of protein translation, mammalian target of rapamycin (mTOR; Heberle et al, 2015). AMPK activation suppresses mTOR activity and reduces protein synthesis; moreover, we have previously demonstrated that mTOR is heavily suppressed due to IFNγ/TNF‐α treatment (White et al, 2013; Hall et al, 2018). Here, our experiments showed that GW restored the phosphorylation of mTOR target ribosomal protein S6 kinase (S6K) and its target, ribosomal protein S6 (S6) (Fig 5H and I).
Figure 5. Cytokine treatment of C2C12 myotubes alters the levels of TCA cycle intermediates and activates AMPK in an iNOS‐dependent manner.
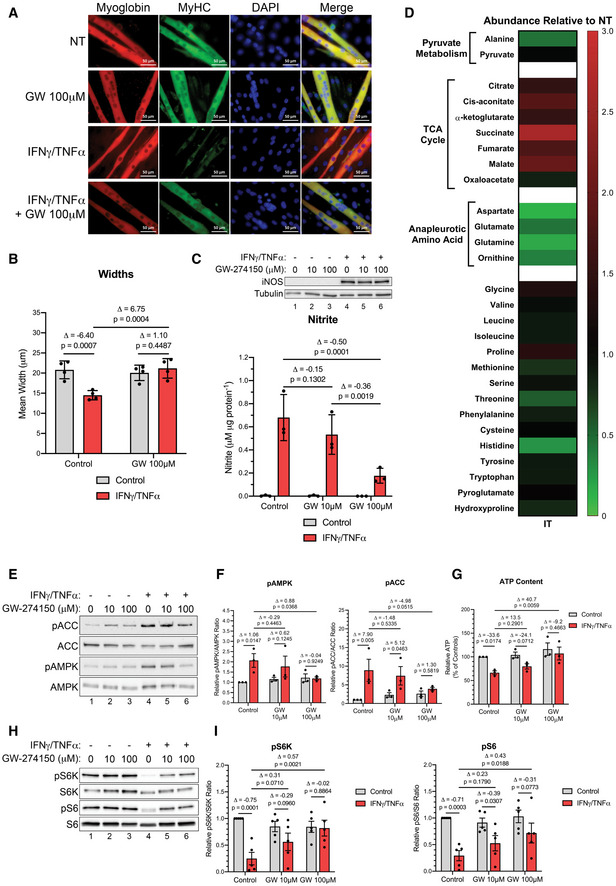
- Representative immunofluorescence imaging for myoglobin and myosin heavy chain (MyHC) in not treated (NT) controls and IFNγ/TNF‐α (IT) samples treated with or without GW. Nuclei were visualized with DAPI staining (n = 4).
- Quantification of mean fiber widths (n = 4).
- (top) Western blot analysis for iNOS and tubulin (n = 3). (bottom) Media nitrite levels (n = 3).
- Heatmap visualizing mean concentration corresponding to metabolites of IFNγ/TNF‐α (IT)‐treated samples relative to not treated (NT) controls (n = 3). Red and green indicate an increase or decrease in metabolite levels, respectively.
- Western blot analysis for pThr172‐AMPK (pAMPK), total AMPK (AMPK), pSer79‐ACC (pACC), and total ACC (ACC).
- Quantification of the pAMPK‐to‐AMPK ratio (left) and the pACC‐to‐ACC ratio (right) relative to the untreated control (n = 3).
- Cellular ATP content quantified as a percentage of the untreated control (n = 3).
- Western blot analysis for pThr389‐S6K (pS6K), total S6K (S6K), pSer235/236‐S6 (pS6), and total S6 (S6) (n = 5).
- Quantification of the (left) pS6K‐to‐S6K ratio and the (right) pS6‐to‐S6 ratio relative to the untreated control (n = 5).
Data information: Individual data points represent independent experimental replicates. Error bars represent the standard deviation (SD) of the mean. For statistical comparisons, Δ indicates the difference in mean values and P‐values were calculated with an ANOVA followed by Fisher's LSD test.
Source data are available online for this figure.
These data demonstrate that C2C12 myotubes treated with pro‐cachectic cytokines recapitulate our findings on sepsis‐ and tumor‐induced cachexia in mice. Inhibition of iNOS using GW, however, reverts these effects.
iNOS activity disrupts the mitochondrial electron transport chain
We next investigated whether the iNOS‐dependent induction of energy crisis in myotubes undergoing wasting is caused by dysfunctions in glycolysis or OXPHOS, or both. Using the Seahorse Extracellular Flux Analyzer, we assessed oxygen consumption rate (OCR) and extracellular acidification rate (ECAR), which are correlated with oxidative respiration and glycolysis, respectively (Appendix Figs S11A and B and S12A and B). We determined the theoretical basal and maximal rates of oxidative and glycolytic ATP production (J‐ATP) of C2C12 myotubes treated or not with cytokine and iNOS inhibitors (Mookerjee et al, 2017, 2018). Determination of the initial J‐ATP rates, when the cells are primarily consuming exogenous glucose, allows for the assessment of the basal energetic state (J‐ATPtotal) of the cell, as well as preferences for either glycolytic (J‐ATPglycolytic) or mitochondrial OXPHOS (J‐ATPoxidative) ATP production. The theoretical maximal J‐ATPoxidative and J‐ATPglycolytic rates, respectively, can be determined after FCCP and monensin treatment (Mookerjee et al, 2017, 2018). When plotted on perpendicular axes, the area defined by these maximal rates represents the bioenergetic profile, with the intersection of these two rates defining the maximal bioenergetic capacity of the cell. Our analysis revealed that while control C2C12 myotubes have a highly oxidative metabolism, IFNγ/TNF‐α caused a dramatic shift in both basal and maximal metabolic activity away from oxidative respiration toward glycolysis (Fig 6A and B and Appendix Fig S13A and B). This shift was prevented by treatment with GW or AMG in a dose‐dependent manner (Fig 6A and B and Appendix Fig S13A and B). Interestingly, cytokine‐treated cells had decreased basal rate of J‐ATP production, indicating their glycolytic energy production was not sufficient to compensate for the loss of oxidative capacity (Fig 6B and Appendix Fig S13B). Importantly, J‐ATP production was recovered by GW and AMG (Fig 6B and Appendix Fig S13B). These changes were reflected in the glycolytic index, a measure of how much of the total basal ATP production comes from glycolytic activity. In IFNγ/TNF‐α‐treated cells, the glycolytic index was 60%‐95%, a dramatic increase from approximately 10% observed in control untreated cells (Fig 6C and Appendix Fig S13C). Although treatment with GW or AMG had no effects on the glycolytic index in untreated cells, it did significantly reduce it in cytokine‐treated myotubes (Fig 6C and Appendix Fig S13C). Notably, the total bioenergetic capacity in cytokine‐treated myotubes was significantly reduced (Fig 6D and Appendix Fig S13D). However, treatment with GW, in addition to reverting the metabolism profile back to a more oxidative state, was able to partially recover the bioenergetic capacity, allowing for restoration of the basal ATP production rate (Fig 6B and D). Although AMG was also able to restore basal ATP production, it did not have such a robust effect on bioenergetic capacity as GW (Appendix Fig S13B and D). Overall, our data indicate that inhibition of iNOS prevents the shift toward glycolytic metabolism and loss of bioenergetic capacity induced by cytokines.
Figure 6. GW274150 prevents a cytokine‐induced shift to aerobic glycolysis in C2C12.
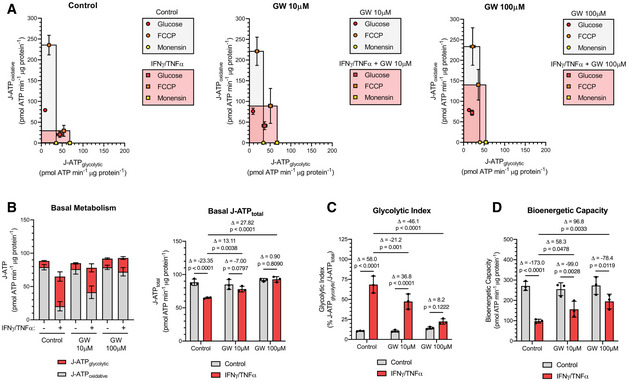
- Bioenergetic profiles. Highlighted squares are defined by the theoretical maximal J‐ATPoxidative and J‐ATPglycolytic rates.
- (left) Basal J‐ATPglycolytic and J‐ATPoxidative rates. (right) Total basal J‐ATP rate.
- Glycolytic index of basal metabolism.
- Total bioenergetic capacity.
Data information: Individual data points represent three independent experiments (n = 3). The data points for each experiment are calculated from the average of technical triplicates. Error bars represent the standard deviation (SD) of the mean. For statistical comparisons, Δ indicates the difference in mean values and P‐values were calculated with an ANOVA followed by Fisher's LSD test.
To determine how iNOS impairs OXPHOS directly in muscle fibers, we used Western blot analysis to assess the integrity of the ETC complexes by determining the expression levels of the subunits of each complex that are known to be labile when the complexes are not assembled (Fig 7A and B, Appendix Fig S14A–C, and Fig EV4A–F). Cytokine treatment significantly reduced the levels of MTCO1 and SDHB, subunits of cytochrome c oxidase (COX; complex IV) and succinate dehydrogenase (SDH; complex II), by approximately 90% and 60%, respectively. The levels of these subunits were partially re‐established by treatment with GW or AMG (Figs 7A and B and EV4A, C, E). This indicates that the integrity of complexes II and IV in the ETC is compromised during cytokine treatment in an iNOS‐dependent manner, suggesting that the activity of these two complexes is inhibited by iNOS in myotubes undergoing wasting.
Figure 7. Inflammation‐mediated loss of complex II and IV integrity are reversed with GW274150.
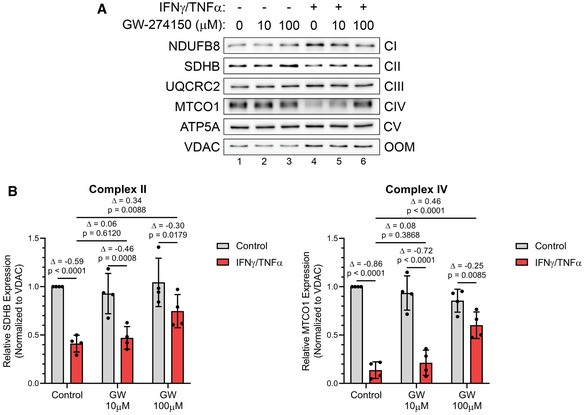
- Western blot analysis for ETC protein complex subunits.
- (left) Quantification of SDHB (complex II; CII) normalized to VDAC (outer mitochondrial membrane; OMM) and relative to untreated control. (right) Quantification of MTCO1 (complex IV; CIV) normalized to VDAC and relative to untreated control.
Data information: Individual data points are from four independent experimental replicates (n = 4). Error bars represent the standard deviation (SD) of the mean. For statistical comparisons, Δ indicates the difference in mean values and P‐values were calculated with an ANOVA followed by Fisher's LSD test.
Source data are available online for this figure.
iNOS activity disrupts the mitochondrial structure in wasting skeletal muscle
Both our in vivo metabolomic study and our in vitro analyses point to impairment of mitochondrial energy production as a common feature behind inflammation‐induced muscle wasting. As mitochondrial morphology and number are strongly linked to its function and cellular energy production (Cogliati et al, 2016; Li et al, 2020), we assessed the impact of iNOS on these parameters in our in vivo models of cachexia. Using transmission electron microscopy (TEM), we observed, in our sepsis‐induced muscle wasting model, that LPS did not appear to affect the number of mitochondria in both WT and iNOS KO mice (Fig 8A left). Interestingly, however, we observed a loss of cristae in the majority of LPS‐treated WT mice compared with their controls and that this loss of cristae was absent in the LPS‐treated iNOS KO mice (Fig 8A right). As cristae are the site of OXPHOS and significantly increase the surface area of the inner mitochondrial membrane (IMM) to house more OXPHOS complexes (Gilkerson et al, 2003; Cogliati et al, 2016; Li et al, 2020), this loss of cristae in the LPS‐treated WT mice suggests that the oxidative energy production of their muscles is significantly impaired during wasting. We next analyzed mitochondrial morphology and number in our cancer‐induced muscle wasting model where saline‐ and C26‐injected mice were treated with or without GW. We observed that, when compared to control conditions, the C26 tumor environment drastically decreased the mitochondrial content in muscle fibers (Fig 8B left), causing fragmentation and loss of cristae in the mitochondria that remained (Fig 8B right). We found that treatment with GW alone did not alter the mitochondrial morphology, but tended to increase the number of mitochondria in the muscles (Fig 8B). In the context of C26 treatment, GW partially prevented the loss of mitochondria in the muscle (Fig 8B left) and maintained normal mitochondrial structure (normal cristae) (Fig 8B right). Therefore, the C26‐mediated loss of mitochondrial content and disruption of structure indicates that oxidative energy production is impaired in these muscles in an iNOS‐dependent manner.
Figure 8. Inflammation‐mediated loss of mitochondrial integrity is reversed with genetic and pharmacological inhibition of iNOS.
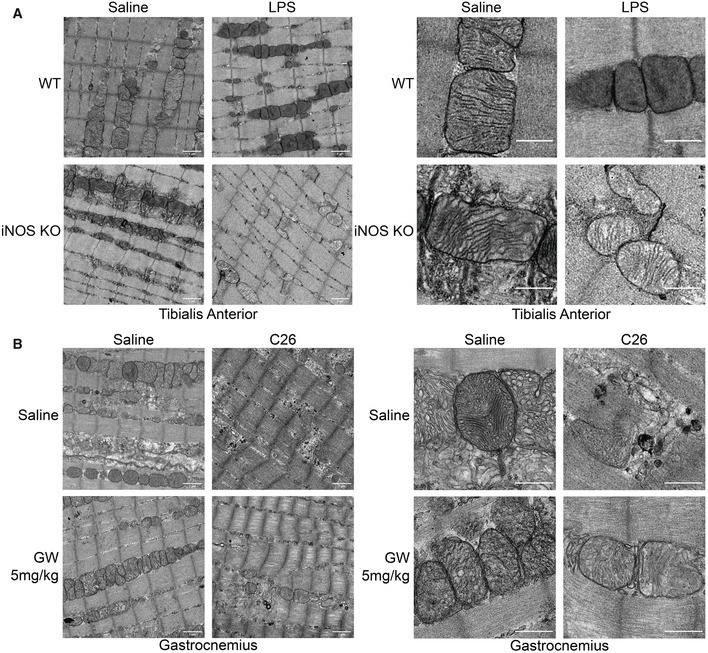
- Male C57BL/6 wild‐type (WT) and iNOS knockout (KO) mice were intraperitoneally injected with 1 mg kg−1 LPS or an equivalent volume of carrier solution. Control WT, control KO, and LPS‐treated KO cohorts were pair‐fed (PF) to the WT LPS‐treated cohorts. After 18 h, mice were euthanized, and tibialis anterior muscles were imaged by transmission electron microscopy. (left) Representative micrograph of tibialis anterior muscle (n = 2). Scale bar = 1 µm. (right) Zoomed section of representative image to highlight mitochondria. Scale bar = 0.5 µm.
- Male BALB/C mice were injected subcutaneously with C26 cells (1.25 × 106 cells) or an equivalent volume of saline. After 5 days, and everyday thereafter, saline‐ and C26‐injected mice were treated with or without GW (5 mg kg−1). After 16 days, mice were euthanized and gastrocnemius muscles were imaged by transmission electron microscopy. (left) Representative micrograph of gastrocnemius muscle (n = 2). Scale bar = 1 µm. (right) Zoomed section of representative image to highlight mitochondria. Scale bar = 0.5 µm.
Data information: Images representative of 2 individual mice.
Together, our models of muscle wasting demonstrate that genetic ablation of iNOS or inhibition of its activity enhances mitochondrial health and function by preventing both the loss of structure and the loss of capacity for oxidative phosphorylation by maintaining complex protein integrity.
Discussion
In this study, we have shown that iNOS is a central downstream effector of cachexia, both in sepsis and cancer models, and that its inhibition with the clinically tested drug GW274150 can successfully ameliorate cancer‐associated cachexia. In addition, the efficacy of GW confirms that iNOS is a central player in cachexia.
Notable attempts at treating cachexia have focused on ameliorating individual symptoms with little success (Speck et al, 2010; Murphy et al, 2011; Stene et al, 2013; Baldwin, 2015; Baracos et al, 2018). For example, nutritional supplementation and physical exercise to treat weight loss and loss of muscle mass demonstrated distinct beneficial outcomes but were unable to reverse cachexia (Speck et al, 2010; Murphy et al, 2011; Stene et al, 2013; Baldwin, 2015; Argiles et al, 2018). These treatments were also hindered by their dependence on patient compliance and the timing of intervention, which can limit their feasibility and effectiveness (Argilés et al, 2012; Baracos et al, 2018). Due to the lack of efficacy of the above approaches, therapeutic strategies were devised to target the inflammatory signals responsible for cachexia. Inhibition of these pro‐cachectic cytokines, such as IL‐6 and TNF‐α, was not effective in treating cachexia and caused unwanted side effects in these patients (Wiedenmann et al, 2008; Jatoi et al, 2010; Bayliss et al, 2011). These setbacks emphasize the multifactorial nature of cachexia, where numerous pro‐cachectic cytokines work together to promote the condition (Fearon et al, 2012; Baracos et al, 2018). Therefore, it is important to identify effectors of common upstream pathways that can constitute a better candidate to be targeted for treatment. iNOS is one such effector whose importance in muscle wasting has been suggested in previous studies (Buck & Chojkier, 1996; Di Marco et al, 2005; Di Marco et al, 2012; Ma et al, 2017; Hall et al, 2018). Despite this, the mechanism driving the pro‐cachectic effect of iNOS in muscles undergoing wasting has remained elusive (Buck & Chojkier, 1996; Di Marco et al, 2005; Di Marco et al, 2012; Hall et al, 2018), and the possibility of targeting iNOS to treat this deadly condition has never been explored.
Our data demonstrate that iNOS causes atrophy by impairing energy production and inducing energetic crisis in cachectic muscle which were reversed with GW or genetic ablation of iNOS (Fig 9). Indeed, this metabolic derangement is in line with previously observed cachexia‐mediated impairment of oxidative metabolism and mitochondrial dysfunction in models of cachexia (Julienne et al, 2012; Puppa et al, 2012; Der‐Torossian et al, 2013b; Fermoselle et al, 2013; Padrao et al, 2013; McLean et al, 2014; Brown et al, 2017; VanderVeen et al, 2018; Kunzke et al, 2019; Pin et al, 2019a; VanderVeen et al, 2019). In our cancer‐ and sepsis‐induced cachexia models, we observed distinct alterations to metabolite profiles, which could arise from the numerous differences between the conditions of our models including the severity of the cachectic stimulus, the duration of the cachexia, and the background of the mice used. Despite this, many of the metabolic perturbations in both models were shown to be both iNOS‐dependent and deleterious to the function of energy production pathways leading to energetic stress in skeletal muscle. Our data show that in muscles undergoing wasting, iNOS promoted metabolic defects, at least in part, by disrupting mitochondrial processes such as the TCA cycle by either altering metabolite levels such as α‐KG and succinate or affecting anapleurotic metabolite levels such as the odd short‐chain acylcarnitines. In addition, iNOS impairs acylcarnitine utilization, contributing to ineffective oxidative metabolism and lower ATP production. The alterations to mitochondrial metabolism and energy production in muscle appear to be a result of iNOS‐mediated reduction in mitochondrial content and loss of cristae of the inner mitochondrial membrane that could disrupt enzymes conducting these processes as seen by the impairment of CPT1, CPT2, and OXPHOS complexes. Indeed, previous studies in inflammation‐activated macrophages have shown that iNOS causes metabolic reprogramming mirroring our observations through inhibition of enzymes involved in mitochondrial processes and dysregulating mitochondrial structure (Gao et al, 2017; Bailey et al, 2019; Li et al, 2020; Palmieri et al, 2020). Therefore, we suggest that iNOS drives deleterious metabolic reprogramming in inflamed muscle to lead to energetic deficit and atrophy despite its important physiological role in activated macrophages.
Figure 9. Schematic depicting the role of the iNOS/NO pathway in promoting energy crisis during cachexia‐induced muscle wasting.
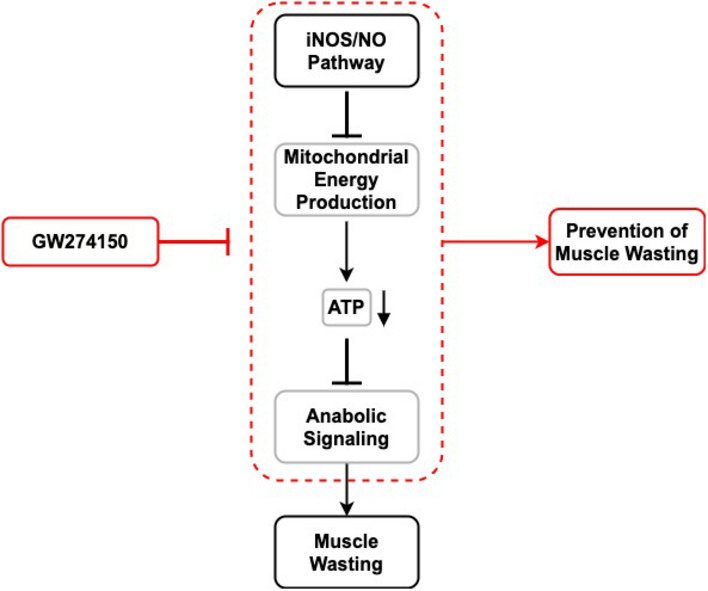
(outlined in black) Upon activation by underlying diseases such as cancer, the iNOS/NO pathway is activated. In turn, iNOS inhibits mitochondrial energy production in skeletal muscles reducing ATP production which leads to muscle wasting. As a result of these effects, anabolic signaling is reduced and muscle loss is induced. (highlighted in red) On the hand, pharmacological inhibition of iNOS activity with drugs such as GW274150 (GW) reverses these effects, preventing muscle wasting.
Through metabolomic approaches, we identify a signature of amino acids that are altered by iNOS activity in muscle afflicted by sepsis‐ and cancer‐induced cachexia. In our cancer model, we observed significant increases in arginine, lysine, tryptophan, and methylhistidine, which were recovered by iNOS inhibition with GW. Interestingly, the observed cancer‐induced shifts in amino acid levels above are consistent with a number of recent studies (Der‐Torossian et al, 2013a; QuanJun et al, 2015; Tseng et al, 2015a; Kunzke et al, 2019; Lautaoja et al, 2019b). This body of evidence raises the possibility that increases in the intramuscular concentration of these amino acids correlate with the progression of this syndrome and protein breakdown. Indeed, increases in methylhistidine and other amino acids can be markers of the muscle protein catabolism that contributes to wasting (QuanJun et al, 2015). Furthermore, in a recent study on a mouse genetic intestinal cancer model of cachexia, increases in lysine and arginine levels correlated with not only the breakdown of ETC complex proteins, but also lower overall energy levels (Kunzke et al, 2019). The detected increases in arginine levels in both the C26 and LPS models were of note as arginine is the substrate of iNOS in its production of NO and has been shown to promote iNOS translation (El‐Gayar et al, 2003; Lee et al, 2003; Chaturvedi et al, 2007). The iNOS‐dependent accumulation of arginine suggests that iNOS downregulates pathways that consume this metabolite as well as promotes its biosynthesis in order to facilitate iNOS expression and activity. Further investigations are needed to delineate how iNOS promotes amino acid accumulation in cachectic muscle and its consequences.
Due to the hyper‐inflammatory nature of cachexia, the contribution of immune cells to muscle wasting and the overall cachectic phenotype cannot be ignored. Indeed, recent studies have demonstrated roles for cancer cachexia‐induced macrophage populations in promoting muscle wasting and regulating adipose wasting (Erdem et al, 2019; Shukla et al, 2020). iNOS expression and NO secretion from inflammatory cells in the muscle micro‐environment are likely contributors to muscle wasting in vivo. Our perturbations to iNOS, whether the full‐body knockout mouse used in our septic model or our pharmacological inhibition of iNOS in our cancer model, impaired iNOS in both muscle and associated inflammatory cells. Nevertheless, we observed increases in iNOS activity visualized by 3NT levels in whole muscle lysates from cachectic mice which was associated with metabolic dysfunction in the whole tissue and mitochondrial deformation in muscle fibers. Importantly, these effects were protected against by GW and iNOS knockout. Due to the prominent role of iNOS in immunity (Bogdan, 2001), it is also likely that iNOS will affect immune cell function in our systems. Indeed, we found that cachexia‐mediated induction of IL‐6 and TNF‐α, two key pro‐cachectic factors, was dampened by iNOS inhibition, despite not affecting alterations in macrophage levels, macrophage polarization, spleen swelling, and tumor size. These data suggest that the anti‐inflammatory effects of GW and iNOS KO could, in part, contribute to the protection against cachexia and its associated metabolic defects further supporting the efficacy of iNOS impairment in treating the condition. Further studies are required to distinguish the contributions of the systemic anti‐inflammatory effects of iNOS inhibition against the localized effects focusing on mitochondrial and metabolic recovery.
Increased iNOS expression has been shown in the skeletal muscle of cancer, AIDS, chronic heart failure, and COPD cachexia patients, suggesting that iNOS may be involved in the onset of cachexia under a variety of overlying conditions (Adams et al, 2003; Agusti et al, 2004; Ramamoorthy et al, 2009). The inhibitor used in our studies, GW274150, has been developed clinically for other inflammatory conditions, such as asthma, arthritis, and migraines (Vitecek et al, 2012). While GW274150 was not more effective than pre‐existing treatments in reverting the pathology of these diseases, the drug was well tolerated and effective at inhibiting iNOS, with good pharmacokinetics and minimal adverse events (Vitecek et al, 2012). As such, drug repurposing of GW274150, or the development of other iNOS inhibitors, may be warranted for the treatment of cachexia. Specifically, the use of iNOS inhibitors may be particularly effective in cancer cachexia, given the potential pro‐tumorigenic function of iNOS. Indeed, several studies have shown that iNOS inhibition can reduce tumor growth and metastasis (Kostourou et al, 2011; Granados‐Principal et al, 2015; Garrido et al, 2017). Further clinical studies are warranted to firmly establish the therapeutic potential of iNOS inhibitors in cancer cachexia, where inhibition may play a dual role in the treatment of both cancer and muscle wasting.
Materials and Methods
Reagents and antibodies
IFNγ (485‐MI) and TNF‐α (410‐MT) were purchased from R&D systems. GW274150 (HY‐12119) was purchased from MedChemExpress. Aminoguanidine (396494), DAPI (10236276001), oligomycin (75351), Rotenone (R8875), antimycin A (A8674), and monensin (M5273) were purchased from Sigma‐Aldrich. ECL Western Blotting Detection Reagent (RPN2106) was purchased from GE Healthcare Life Sciences.
The iNOS antibody (610431) was purchased from BD Transduction Laboratories. The tubulin antibody (DSHB Hybridoma Product 6G7; deposited by Halfter, W.M.), myosin heavy chain (DSHB Hybridoma Product MF20; deposited by Fischman, D.A.), MyHC I (DSHB Hybridoma Product BA‐D5; deposited by Schiaffino, S.), and MyHC IIa (DSHB Hybridoma Product SC‐71; deposited by Schiaffino, S.) antibodies were obtained from the Developmental Studies Hybridoma Bank (DSHB), created by the NICHD of the NIF and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. The myoglobin (ab77232) and 3‐NT (ab61392) antibodies as well as the Total OXPHOS Rodent Antibody Cocktail (ab110413) (containing five antibodies, one against a subunit of each ETC complex) were purchased from Abcam. VDAC (48665), pThr172‐AMPKα (2535), total AMPKα (2603), pSer79‐ACC (3661), total ACC (3662), pThr389‐S6K (9205), total S6K (2708), pSer235/236‐S6 (2211), and S6 (2317) antibodies were purchased from Cell Signaling Technology. The F4/80 conjugated to PE/Cy7 (123114), Ly6c conjugated to APC‐fire750 (128046), CD206 conjugated to PE (141706), and CD86 conjugated to BV421 (105032) antibodies were purchased from BioLegend. The CD45 conjugated to BV786 (564225) antibody was purchased from BD. Laminin (L9393) antibody was purchased from Sigma‐Aldrich. Horseradish peroxidase (HRP)‐conjugated goat secondary antibodies against mouse (315‐035‐003) and rabbit (111‐035‐003) primary antibodies were obtained from Jackson ImmunoResearch Laboratories. Alexa FluorTM 488‐conjugated goat anti‐mouse secondary (A11029), Alex FluorTM 594‐conjugated goat anti‐rabbit secondary (A11072), Alexa FluorTM 594‐conjugated goat anti‐mouse secondary (A11032), Alexa FluorTM 647‐conjugated goat anti‐rabbit secondary (A21244), Alexa FluorTM 488‐conjugated goat anti‐mouse IgG1 secondary (A21121), and Alexa FluorTM 594‐conjugated goat anti‐mouse IgG2b secondary (A21145) antibodies were purchased from Thermo Fisher Scientific.
Cell culture
C2C12 myoblast cells (ATCC, Manassas, VA, USA) were cultured as previously described (Hall et al, 2018). Cells were grown on 0.1% gelatin‐coated culture dishes (purchased from Corning) in DMEM (Thermo Fisher 11995‐065) supplemented with 20% fetal bovine serum (Sigma‐Aldrich F1051) and 1% penicillin/streptomycin (Sigma‐Aldrich P0781). Cells were differentiated upon reaching 90‐100% confluence by switching to DMEM containing 2% horse serum (Invitrogen 16050122) and 1% penicillin/streptomycin. Myotubes were treated when visible which typically occurred three to four days after induction of differentiation. Cells were monitored for mycoplasma infection by DAPI staining.
GC‐MS metabolomic analysis
Cells were collected and lysed at the indicated time period in dry‐ice chilled 80% methanol with subsequent sonication. Both the soluble and insoluble fractions were collected. Total genomic DNA (gDNA) was extracted from the insoluble fraction using phenol:chloroform:isoamyl alcohol extraction and used later to relativize metabolite levels to cellular yield. The soluble fraction was collected, and 750 ng/sample of D‐myristic acid was added as an internal standard. Samples were dried down and then dissolved in 30 μl of methoxyamine hydrochloride (10 mg/ml) in pyridine and derivatized using 70 μl of N‐(tert‐butyldimethylsilyl)‐N‐methyltrifluoroacetamide (MTBSTFA) to generate tert‐butyldimethylsilyl (TBDMS) esters. Metabolite levels were analyzed using an Agilent 5975C GC/MS equipped with a DB‐5MS+DG (30 m × 250 μm × 0.25 μm) capillary column (Agilent J&W, Santa Clara, CA, USA). Electron impact was set at 70 eV, and a total of 1 μl of derivatized sample was injected in splitless mode with the inlet temperature set to 280°C. Helium was used as a carrier gas with a flow rate of 1.5512 ml/min (myristic acid internal standard elutes at 17.94 min). Quadrupole was set at 150°C and the interface at 285°C. The oven program was started at 60°C for 1 min, then increased at a rate of 10°C/min until 320°C, which was held for 10 min. Data were acquired with both in scan (1–600 m/z) and selected ion (SIM) modes (McGuirk et al, 2013). Mass isotopomer distribution and metabolite abundance were determined using a custom algorithm developed at McGill University (McGuirk et al, 2013). Metabolite abundance was normalized to D‐myristic acid levels and relativized to total gDNA content from the insoluble fraction.
Media nitrite levels
Media nitrite levels were measured using GRIESS reagent as previously described (Di Marco et al, 2005). GRIESS reagent 1 (1% sulphanilamide, 5% phosphoric acid) and GRIESS reagent 2 (0.1% N‐(1‐naphthyl)‐ethylenediamine dihydrochloride) were mixed in a 1:1 ratio to make GRIESS assay reagent. A standard curve was generated using a serial dilution of sodium nitrite solution. Standards and unknown media samples were mixed in a 1:1 ratio with GRIESS assay reagent and left at room temperature for 5 min. Absorbance was subsequently measured at a wavelength of 543 nm, and the nitrite concentration of unknown samples was determined by comparison with the standard curve.
Immunoblotting
Protein content was extracted from cells using a detergent lysis buffer (50 mM HEPES pH 7.0, 150 mM NaCl, 10% glycerol, 1% Triton® X‐100, 10 mM sodium pyrophosphate, 100 mM NaF, 1 mM EGTA, 1.5 mM MgCl2). Protein content from muscle was extracted by homogenizing muscle in ice‐cold muscle protein extraction buffer (PBS supplemented with 1% NP‐40, 0.5% sodium deoxycholate, 50 mM NaF, 5 mM Na4P2O7, and 0.1% SDS). The soluble protein fraction was clarified by centrifugation at 12,000 g for 5 min at 4°C and diluted with Laemmli loading dye. Western blot analysis was performed with the Bio‐Rad system. Proteins were resolved on acrylamide gels and transferred to nitrocellulose membranes using the Trans‐Blot® Turbo™ system according to the manufacturer’s instructions. Successful transfer and gel loading were confirmed by reversible Ponceau S staining or Stain‐Free imaging. Membranes were blocked in 10% skim milk and washed three times in Tris‐buffered saline containing 0.1% Tween (TBS‐T). Membranes were incubated in primary antibodies diluted in either 5% skim milk or 5% BSA containing TBS‐T overnight. Total OXPHOS Rodent antibody cocktail was diluted 1:1,000. VDAC was diluted 1:5,000. 3NT was diluted 1:2,000. All other antibodies were diluted as previously reported (Hall et al, 2018). Primary antibodies were removed by washing three times in TBS‐T. Membranes were then incubated in the appropriate HRP‐conjugated secondary antibodies diluted 1:5,000–1:10,000 in 5% skim milk, TBS‐T for 1 h at room temperature. Membranes were washed three times in TBS‐T and exposed using ECL reagent. Chemiluminescent signal was detected using either photosensitive film or a Bio‐Rad ChemiDocTM imaging system. Densitometry quantification was performed using either ImageJ software (Schneider et al, 2012) or Bio‐Rad Image LabTM software.
RT–qPCR
RT–qPCR was performed as previously described (Hall et al, 2018). Total RNA was extracted using TRIzol® (Thermo Fisher 15596018) according to the manufacturer’s instructions. RNA quality and quantity were determined using a Thermo Fisher NanoDropTM reader (ND‐1000) and agarose gel electrophoresis. Following reverse transcription, cDNA was analyzed by qPCR with SsoFastTM Evagreen® Supermix (Bio‐Rad 1725200) using primers for F4/80 (F: 5’‐GCA TCA TGG CAT ACC TGT TC‐3′ and 5′‐GAG CTA AGG TCA GTC TTC CT‐3′) and Gapdh (F: 5’‐AAG GTC ATC CCA GAG CTG AA‐3’ R: 5’‐AGG AGA CAA CCT GGT CCT CA‐3’). All levels were normalized to GAPDH and subsequently relativized to the non‐treated controls.
Extracellular flux and bioenergetics analysis
OCR and ECAR were determined using an Agilent Seahorse XFe24 Analyzer. Cells were grown and differentiated in XFe24 culture plates (1007777‐004) as previously described (Hall et al, 2018). One hour before assessment, cells were carefully switched to XF Base Medium (103575‐100) supplemented with 10 mM D‐glucose (Sigma G7528) and cultured in a CO2‐free incubator at 37°C, as described by the manufacturer. A XFe24 sensor cartridge (102340‐100), calibrated overnight in a CO2‐free 37°C incubator according to the manufacturer’s instructions, was loaded with Oligomycin (final concentration 1 µM), FCCP (final concentration 1.5 µM), Rotenone (final concentration 1 µM), Antimycin A (final concentration 1 µM), and monensin (final concentration 20 µM). After calibration, basal extracellular flux was measured in three cycles. Drugs were subsequently injected in the indicated order, with two measurement cycles between each injection. Measurement cycles consisted of a 3‐minute mix, a 2‐min wait, and a 3‐min measurement. Media buffering capacity was determined at the time of the experiment using a sequential injection of HCl in two wells and ranged from 0.061 to 0.065 mpH pmol H+ −1. Following completion of the run, individual well protein content was determined as previously described (Mookerjee et al, 2017). Cells were carefully washed three times in room temperature albumin‐free Krebs‐Ringer phosphate HEPES (KRPH) medium (2 mM HEPES, 136 mM NaCl, 2 mM NaH2PO4, 3.7 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, pH 7.4). Cells were lysed by adding 25 µl of RIPA lysis buffer (150 mM NaCl, 50 mM Tris, 1 mM EGTA, 1 mM EDTA, 1% Triton® X‐100, 0.5% sodium deoxycholate, 0.1% SDS, pH 7.4) to each well. Plates were incubated on ice for 30 min and then agitated on a plate shaker for 5 min. Plates were placed back on ice, and well protein content was determined using a BCA assay (Thermo Fisher 23225) according to the manufacturer’s instructions. Extracellular flux rates were normalized to protein content. Bioenergetic profiling was determined as detailed by Mookerjee et al (2017, 2018).
ATP content
Cellular ATP content was determined using the Invitrogen ATP Determination Kit (A22066) as described by the manufacturer. To prepare cell lysates, media was removed from a few wells at a time by manual pipetting to avoid drying. Wells were quickly washed once with room temperature PBS, and then, cells were lysed in ATP Assay Lysis Buffer (25 mM Tris–HCl, 2 mM DTT, 2 mM EDTA, 10% glycerol, 1% Triton® X‐100). After lysis buffer was added to all wells, the plate was agitated on a plate shaker for 5 min at room temperature. Plate was then placed on ice, and ATP content was determined.
Animal models
Animal experiments were carried out with approval from the McGill University Faculty of Medicine Animal Care Committee and are in accordance with the guidelines set by the Canadian Council of Animal Care. Mice were housed in a controlled environment in sterile cages with corn‐cob bedding set on a 12‐h light–12‐h dark cycle. Mice were provided commercial laboratory food (Harlan #2018; 18% protein rodent diet; Madison, WI) and had free access to water.
The C26 model of cancer cachexia was performed as previously described (Hall et al, 2018). Male BALB/C age 6–8 weeks were obtained from Jackson Laboratory. Mice were randomly assigned by cage to each treatment group. C26 adenocarcinoma cells (kindly provided by Dr. Denis Guttridge) were thawed approximately 5 days before the planned injection day. Cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were kept below 70% confluency and passed a minimal number of times before injection (typically 1–2 times). C26 was prepared in chilled, sterile PBS at a concentration of 1.25 × 107 cells ml−1. Cells were briefly warmed before injection, and 100 µl was injected subcutaneously into the right flank (1.25 × 106 cells per mouse). For saline controls, an equivalent volume of saline was injected. Tumor growth was monitored by manual measurement with calipers. GW274150 was prepared ahead of time to a final concentration of 0.5 mg ml−1 in PBS under sterile conditions and stored in single‐use aliquots at −20°C. Five days after C26 injection and every day thereafter, mice were intraperitoneally injected with 10 ml kg−1 of either GW274150 (final dose: 5 mg kg−1) or PBS. On day 14–19 post‐C26 injection, mice were anesthetized with isoflurane gas and exsanguinated by cardiac puncture. Following cervical dislocation, tissues were rapidly dissected, weighed, and snap‐frozen in liquid nitrogen.
The LPS model of septic cachexia was performed as previously described (Hall et al, 2018). Male C57BL/6 wild‐type and whole‐body iNOS knockout mice on a C57BL/6 background age 8–12 weeks were obtained from Jackson Laboratories. GW‐injected mice were intraperitoneally injected with 5 mg/kg dose of GW 24 h before LPS injection and then simultaneously as LPS injection. LPS was prepared fresh in a solution of 0.5% BSA dissolved in sterile PBS to a concentration of 0.1 mg ml−1. Mice were then intraperitoneally injected with 10 ml kg−1 to a final concentration of 1 mg kg−1 at the beginning of the dark cycle (18:30 h to 19:30 h). Before injection, mice were separated into individual cages to allow for pair‐feeding. Wild‐type, LPS‐injected mice were fed ad libitum, and all other cohorts were fed the average amount consumed by LPS‐injected mice from previous studies. Mice were euthanized approximately 18 h after injection, and tissue was collected as described above.
Grip strength
Forelimb grip strength was measured using a using a DFE II Series Digital Force Gauge (Ametek DFE II 2‐LBF 10‐N). Mice were allowed to acclimate to the machine the morning of C26 or LPS injection. Forelimb grip force was then measured by suspending the mouse by the tip of the tail and allowing them to establish a grip. The mouse was then gently pulled parallel to the meter until their grip was broken. Peak force was recorded, and the mouse was returned to their home cage for at least one minute. Four measurements were taken, and the maximum grip strength was determined. This process was repeated the morning of euthanasia, and the percent change in maximal grip strength was determined.
Fluorescence‐activated cell sorting (FACS)
Spleens from saline or LPS‐treated WT and iNOS KO mice were removed and passed through a strainer to get single cells that were kept on ice. Residual red blood cells were lysed using ACK buffer for 3 min at room temperature. Viability was assessed using fixable viability dye eFluorYM506 (65‐0866‐14) (Thermo Fisher/ eBioscience). Viable cells were then analyzed using the CD45, F4/80, Ly6c, CD206, and CD86 antibodies. BD Cytofix/CytopermTM (BD Bioscience, # 554722) was used to fix the myeloid compartment according to the manufacturer’s protocol. The CD86+ cells or CD206+ cells were gated on viable cells Cd45+, F4/80, and Ly6c−. Flow cytometry was done in the BD LSR Fortessa 4 lasers (405/488/561/633 nm) flow cytometer, and the results were analyzed using Flowjo_v 10.7.1_CL (Treestar).
Muscle freezing and sectioning
Gastrocnemius and tibialis anterior muscles were mounted on 7% tragacanth gum and snap‐frozen in liquid nitrogen‐cooled isopentane. Samples were stored at −80°C before cryosectioning. Sections (10 μm).
Histological analysis of muscle cross‐sectional area
Sections of muscles were stained using the hematoxylin and eosin (H&E) staining method. Stained samples were subsequently imaged by light microscopy. Muscle minimum Feret’s diameter and cross‐sectional areas were determined using ImageJ software to manually trace the circumference of individual fibers (Schneider et al, 2012).
Immunofluorescence
Immunofluorescence on C2C12 myotubes was performed as previously described (Hall et al, 2018). In brief, cells were fixed in 3% paraformaldehyde for 30 min, permeabilized, and blocked in 0.1% Triton® X‐100, 1% goat serum (Thermo Fisher 16210‐064), incubated in primary antibodies against myosin heavy chain (MF‐20; 1:1,000) and myoglobin (1:500) for 1 h and, after washing, Alexa Fluor® secondary antibodies (1:500) for one hour, stained with DAPI, and mounted on coverslips with VECTASHIELD® Antifade Mounting Medium (Vector Laboratories H‐1000). Samples were then blinded to the experimenter, and cells were imaged with a Zeiss Observer Z1 microscope and AxioCam MRm digital camera. Myotube diameters were measured at three points along each cell using the Carl Zeiss Zen2 (blue) software.
Immunofluorescence on sections of muscles to determine fiber type composition was performed as follows: Frozen sections were equilibrated to room temperature for 20 min. Following this, the sections were fixed in 3% paraformaldehyde for 30 min, blocked in 5% goat serum for 1 h, incubated in primary antibodies against MyHC I (1:50), MyHCIIa (1:500), and laminin (1:250) overnight, and, after washing, incubated with Alexa Fluor® secondary antibodies (1:500) for one hour. The sections were then mounted on coverslips with VECTASHIELD® HardSet Antifade Mounting (Vector Laboratories H‐1400). The entirety of the sections was then imaged with a Zeiss Observer Z1 microscope and AxioCam MRm digital camera. The images were subsequently assembled together using ImageJ software, and each fiber was then classified as type I or type IIa positive. Unstained fibers with no signal were classified as being type IIb/x.
Transmission electron microscopy
Tibialis anterior or gastrocnemius muscles were imaged at the Facility for Electron Microscopy Research at McGill University (Montreal, Quebec, Canada). The tissue samples were fixed overnight in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer. The tissues were then postfixed with 1% OsO4 + 1.5% potassium ferrocyanide in 0.1 M sodium cacodylate buffer for 2 h at 4°C and washed three times with Milli‐Q water. The tissues were dehydrated in a graded series of acetone‐dH2O up to 100%. Following dehydration, the tissues were infiltrated with a graded series of Epon‐acetone (1:1, 2:1, 3:1), embedded in 100% Epon, and polymerized at 65°C for 48 h. Ultrathin serial sections (90–100 nm thick) were prepared from the embedded tissue blocks with a Diatome diamond knife using a Leica Microsystems EM UC6 ultramicrotome, transferred onto 200‐mesh copper TEM grids, and poststained with 4% uranyl acetate for six minutes and Reynold's lead citrate for five minutes. The ultrathin sections were imaged by an FEI Tecnai G2 Spirit 120 kV TEM equipped with a Gatan UltraScan 4000 CCD Camera Model 895. The proprietary Gatan Digital Micrograph 16‐bit images (DM3) were subsequently converted to unsigned 8‐bit TIFF images.
LC‐MS metabolomic analysis
Tibialis anterior muscles were analyzed using the Metabolomics Innovation Centre (TMIC) Prime Metabolomics Profiling Assay. Analysis of 119 metabolites including biogenic amines, amino acids, acylcarnitines, glycerophospholipids, and organic acids in mice muscle samples was conducted by LC/DI‐MS/MS developed by TMIC at University of Alberta (Edmonton, Alberta, Canada). For global assessments, the raw metabolite concentration data were analyzed using the MetaboAnalyst 4.0. The data were mean‐centered and range scaled before further analysis, which included PLS‐DA modeling of the data and pathway analysis using MetaboAnalyst 4.0.
Cytokine protein analysis in sera
IL‐1α, IL‐1β, IL‐6, and TNF‐α levels in serum collected from mice were measured using BioPlex 200 Mouse Cytokine Array by Eve Technologies Corp. (Calgary, Alberta, Canada).
Statistics and data processing
For in vitro studies, n indicates experimental replicates. For animal studies, n indicates the number of animals per treatment cohort. For in vitro studies, experimental replicates were excluded if the negative (non‐treated) and positive (cytokine‐treated) samples did not show an appropriate inflammatory response. For in vivo studies, mice were excluded if they developed complications unrelated to the cachexia phenotype or if they developed humane‐intervention end‐point complications early in the course of the study (e.g., ulceration of the tumor mass). Samples for measuring myotube widths and muscle fiber cross‐sectional areas were blinded before acquisition of images and during quantification. Bar graphs represent the mean, with error bars showing either the standard deviation of the mean for biological replicates or the standard deviation for technical replicates. Significance P‐values between means were computed using either using one‐way ANOVA with Fisher’s LSD test for multiple treatments groups, Student’s t‐test for comparison of two groups, or Kolmogorov–Smirnov test for comparison of frequency distributions in GraphPad Prism version 7 or 8, GraphPad Software, La Jolla California USA, www.graphpad. The pathway analysis of the metabolomics data set was conducted using MetaboAnalyst 4.0, www.metaboanalyst.ca. P‐values of significantly altered pathways were determined using GlobalTest on MetaboAnalyst 4.0. P‐values for the main figures, expanded view figures, and appendix figures can be found in Appendix Tables [Link], [Link].
Author contributions
JS and DTH were responsible for experimental design and conducted all experimental investigations, with assistance from other co‐authors. JS and DTH performed the formal analysis and visualization of experimental findings and prepared the original draft of the manuscript. BC assisted with sample collection and processing for Western blot analyses for in vitro studies and assisted with the animal studies. AO conducted the RT–qPCR analyses and the immunofluorescence staining of muscle sections, as well as assisting with the animal studies and data analysis. A‐MKT assisted with animal studies performing collection and preparation of mouse samples analyzed. VSG assisted with the flow cytometry sample processing and data analysis. WM assisted with the interpretation of the flow cytometry data. SDM assisted with experimental design and data analysis. MEB assisted with data interpretation and preparation of the original manuscript. I‐EG supervised experimental design, execution, data interpretation, and helped write and revise the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
We are thankful to Shawn McGuirk for his help in understanding the intricacies of bioenergetic profiling. This work was funded by a CIHR operating grant (MOP‐142399) and a CIHR project grant (PJT‐159618) to IEG. DTH was funded by and a recipient of the James O. & Maria Meadows Studentship, the Maysie MacSporran Award, and the Charlotte and Leo Karassik Foundation Oncology PhD Fellowship. JS was funded by CIHR Studentship Award (GSD‐164154).
EMBO Mol Med (2021) 13: e13591.
Data availability
This study includes no data deposited in external repositories.
References
- Adams V, Spate U, Krankel N, Schulze PC, Linke A, Schuler G, Hambrecht R (2003) Nuclear factor‐kappa B activation in skeletal muscle of patients with chronic heart failure: correlation with the expression of inducible nitric oxide synthase. Eur J Cardiovasc Prev Rehabil 10: 273–277 [DOI] [PubMed] [Google Scholar]
- Agusti A, Morla M, Sauleda J, Saus C, Busquets X (2004) NF‐kappaB activation and iNOS upregulation in skeletal muscle of patients with COPD and low body weight. Thorax 59: 483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahsan H (2013) 3‐Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum Immunol 74: 1392–1399 [DOI] [PubMed] [Google Scholar]
- Anavi S, Tirosh O (2019) iNOS as a metabolic enzyme under stress conditions. Free Radic Biol Med 146: 16–35 [DOI] [PubMed] [Google Scholar]
- Antunes D, Padrão AI, Maciel E, Santinha D, Oliveira P, Vitorino R, Moreira‐Gonçalves D, Colaço B, Pires MJ, Nunes C et al (2014) Molecular insights into mitochondrial dysfunction in cancer‐related muscle wasting. Biochim Biophys Acta 1841: 896–905 [DOI] [PubMed] [Google Scholar]
- Argilés JM, Busquets S, López‐Soriano FJ, Costelli P, Penna F (2012) Are there any benefits of exercise training in cancer cachexia? J cachexia Sarcopenia Muscle 3: 73–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argilés J, López‐Soriano F, Busquets S (2013) Mechanisms and treatment of cancer cachexia. Nutr Metab Cardiovasc Dis 23: S19–S24 [DOI] [PubMed] [Google Scholar]
- Argiles JM, Lopez‐Soriano FJ, Busquets S (2015) Muscle wasting in cancer: the role of mitochondria. Curr Opin Clin Nutr Metab Care 18: 221–225 [DOI] [PubMed] [Google Scholar]
- Argiles JM, Stemmler B, Lopez‐Soriano FJ, Busquets S (2018) Inter‐tissue communication in cancer cachexia. Nat Rev Endocrinol 15: 9–20 [DOI] [PubMed] [Google Scholar]
- Bailey JD, Diotallevi M, Nicol T, McNeill E, Shaw A, Chuaiphichai S, Hale A, Starr A, Nandi M, Stylianou E et al (2019) Nitric oxide modulates metabolic remodeling in inflammatory macrophages through TCA cycle regulation and itaconate accumulation. Cell Rep 28: 218–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin C (2015) The effectiveness of nutritional interventions in malnutrition and cachexia. Proc Nutr Soc 74: 397–404 [DOI] [PubMed] [Google Scholar]
- Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH (2018) Cancer‐associated cachexia. Nat Rev Dis Primers 4: 17105 [DOI] [PubMed] [Google Scholar]
- Barreiro E, de la Puente B, Busquets S, Lopez‐Soriano FJ, Gea J, Argiles JM (2005) Both oxidative and nitrosative stress are associated with muscle wasting in tumour‐bearing rats. FEBS Lett 579: 1646–1652 [DOI] [PubMed] [Google Scholar]
- Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR (2011) A humanized anti‐IL‐6 antibody (ALD518) in non‐small cell lung cancer. Expert Opin Biol Ther 11: 1663–1668 [DOI] [PubMed] [Google Scholar]
- van den Berg RA, Hoefsloot HCJ, Westerhuis JA, Smilde AK, van der Werf MJ (2006) Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genom 7: 142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B, Rietschel ET (2003) Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol 3: 169–176 [DOI] [PubMed] [Google Scholar]
- Blum D, Stene GB, Solheim TS, Fayers P, Hjermstad MJ, Baracos VE, Fearon K, Strasser F, Kaasa S, Block Lieve VD et al (2014) Validation of the Consensus‐Definition for Cancer Cachexia and evaluation of a classification model–a study based on data from an international multicentre project (EPCRC‐CSA). Ann Oncol 25: 1635–1642 [DOI] [PubMed] [Google Scholar]
- Bogdan C (2001) Nitric oxide and the immune response. Nat Immunol 2: 907–916 [DOI] [PubMed] [Google Scholar]
- Bonetto A, Rupert JE, Barreto R, Zimmers TA (2016) The Colon‐26 carcinoma tumor‐bearing mouse as a model for the study of cancer cachexia. J Vis Exp e54893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun TP, Grossberg AJ, Krasnow SM, Levasseur PR, Szumowski M, Zhu XX, Maxson JE, Knoll JG, Barnes AP, Marks DL (2013) Cancer‐ and endotoxin‐induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J 27: 3572–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Lee DE, Rosa‐Caldwell ME, Brown LA, Perry RA, Haynie WS, Huseman K, Sataranatarajan K, Van Remmen H, Washington TA et al (2018) Protein imbalance in the development of skeletal muscle wasting in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 9: 987–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Rosa‐Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA, Wiggs MP et al (2017) Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 8: 926–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunengraber H, Roe CR (2006) Anaplerotic molecules: current and future. J Inherit Metab Dis 29: 327–331 [DOI] [PubMed] [Google Scholar]
- Buck M, Chojkier M (1996) Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J 15: 1753–1765 [PMC free article] [PubMed] [Google Scholar]
- Chatterjee PK, Patel NSA, Sivarajah A, Kvale EO, Dugo L, Cuzzocrea S, Brown PAJ, Stewart KN, Mota‐Filipe H, Britti D et al (2003) GW274150, a potent and highly selective inhibitor of iNOS, reduces experimental renal ischemia/reperfusion injury. Kidney Int 63: 853–865 [DOI] [PubMed] [Google Scholar]
- Chaturvedi R, Asim M, Lewis ND, Algood HMS, Cover TL, Kim PY, Wilson KT (2007) L‐arginine availability regulates inducible nitric oxide synthase‐dependent host defense against Helicobacter pylori. Infect Immun 75: 4305–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46: W486–W494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Wishart DS, Xia J (2019a) Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr Protoc Bioinformatics 68: e86 [DOI] [PubMed] [Google Scholar]
- Chong J, Xia J (2018) MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 34: 4313–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Yamamoto M, Xia J (2019b) MetaboAnalystR 2.0: from raw spectra to biological insights. Metabolites 9: 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S, Enriquez JA, Scorrano L (2016) Mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci 41: 261–273 [DOI] [PubMed] [Google Scholar]
- Cohen S, Nathan JA, Goldberg AL (2015) Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discovery 14: 58 [DOI] [PubMed] [Google Scholar]
- Constantinou C, Fontes de Oliveira CC, Mintzopoulos D, Busquets S, He J, Kesarwani M, Mindrinos M, Rahme LG, Argiles JM, Tzika AA (2011) Nuclear magnetic resonance in conjunction with functional genomics suggests mitochondrial dysfunction in a murine model of cancer cachexia. Int J Mol Med 27: 15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer Z, Sadek J, Vazquez GG, Di Marco S, Pause A, Pelletier J, Gallouzi I‐E (2018) eIF4A inhibition prevents the onset of cytokine‐induced muscle wasting by blocking the STAT3 and iNOS pathways. Sci Rep 8: 8414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui P, Shao W, Huang C, Wu CJ, Jiang B, Lin D (2019) Metabolic derangements of skeletal muscle from a murine model of glioma cachexia. Skelet Muscle 9: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzzocrea S, Chatterjee PK, Mazzon E, McDonald MC, Dugo L, Di Paola R, Serraino I, Britti D, Caputi AP, Thiemermann C (2002) Beneficial effects of GW274150, a novel, potent and selective inhibitor of iNOS activity, in a rodent model of collagen‐induced arthritis. Eur J Pharmacol 453: 119–129 [DOI] [PubMed] [Google Scholar]
- Der‐Torossian H, Wysong A, Shadfar S, Willis MS, McDunn J, Couch ME (2013a) Metabolic derangements in the gastrocnemius and the effect of Compound A therapy in a murine model of cancer cachexia. J Cachexia Sarcopenia Muscle 4: 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der‐Torossian H, Wysong A, Shadfar S, Willis MS, McDunn J, Couch ME (2013b) Metabolic derangements in the gastrocnemius and the effect of Compound A therapy in a murine model of cancer cachexia. J Cachexia Sarcopenia Muscle 4: 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco S, Cammas A, Lian XJ, Kovacs EN, Ma JF, Hall DT, Mazroui R, Richardson J, Pelletier J, Gallouzi IE (2012) The translation inhibitor pateamine A prevents cachexia‐induced muscle wasting in mice. Nat Commun 3: 896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marco S, Mazroui R, Dallaire P, Chittur S, Tenenbaum SA, Radzioch D, Marette A, Gallouzi IE (2005) NF‐(kappa)B‐mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol Cell Biol 25: 6533–6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Gayar S, Thüring‐Nahler H, Pfeilschifter J, Röllinghoff M, Bogdan C (2003) Translational control of inducible nitric oxide synthase by IL‐13 and arginine availability in inflammatory macrophages. J Immunol 171: 4561–4568 [DOI] [PubMed] [Google Scholar]
- Emery PW, Edwards RH, Rennie MJ, Souhami RL, Halliday D (1984) Protein synthesis in muscle measured in vivo in cachectic patients with cancer. Br Med J 289: 584–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdem M, Möckel D, Jumpertz S, John C, Fragoulis A, Rudolph I, Wulfmeier J, Springer J, Horn H, Koch M et al (2019) Macrophages protect against loss of adipose tissue during cancer cachexia. J Cachexia Sarcopenia Muscle 10: 1128–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas J, von Haehling S, Kalantar‐Zadeh K, Morley JE, Anker SD, Lainscak M (2013) Cachexia as a major public health problem: frequent, costly, and deadly. J Cachexia Sarcopenia Muscle 4: 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon KC, Glass DJ, Guttridge DC (2012) Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166 [DOI] [PubMed] [Google Scholar]
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G et al (2011) Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 12: 489–495 [DOI] [PubMed] [Google Scholar]
- Fermoselle C, Garcia‐Arumi E, Puig‐Vilanova E, Andreu AL, Urtreger AJ, de Kier Joffe ED, Tejedor A, Puente‐Maestu L, Barreiro E (2013) Mitochondrial dysfunction and therapeutic approaches in respiratory and limb muscles of cancer cachectic mice. Exp Physiol 98: 1349–1365 [DOI] [PubMed] [Google Scholar]
- Fontes‐Oliveira CC, Busquets S, Toledo M, Penna F, Aylwin MP, Sirisi S, Silva AP, Orpí M, García A, Sette A et al (2013) Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 1830: 2770–2778 [DOI] [PubMed] [Google Scholar]
- Gannavarapu BS, Lau SKM, Carter K, Cannon NA, Gao A, Ahn C, Meyer JJ, Sher DJ, Jatoi A, Infante R et al (2018) Prevalence and survival impact of pretreatment cancer‐associated weight loss: a tool for guiding early palliative care. J Oncol Pract 14: e238–e250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Li Y, Wang F, Huang T, Fan K, Zhang Yu, Zhong J, Cao Q, Chao T, Jia J et al (2017) Mitochondrial dynamics controls anti‐tumour innate immunity by regulating CHIP‐IRF1 axis stability. Nat Commun 8: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido P, Shalaby A, Walsh EM, Keane N, Webber M, Keane MM, Sullivan FJ, Kerin MJ, Callagy G, Ryan AE et al (2017) Impact of inducible nitric oxide synthase (iNOS) expression on triple negative breast cancer outcome and activation of EGFR and ERK signaling pathways. Oncotarget 8: 80568–80588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson RW, Selker JM, Capaldi RA (2003) The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett 546: 355–358 [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK (2016) New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 17: 34–40 [DOI] [PubMed] [Google Scholar]
- Granados‐Principal S, Liu Yi, Guevara ML, Blanco E, Choi DS, Qian W, Patel T, Rodriguez AA, Cusimano J, Weiss HL et al (2015) Inhibition of iNOS as a novel effective targeted therapy against triple‐negative breast cancer. Breast Cancer Res 17: 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DT, Griss T, Ma JF, Sanchez BJ, Sadek J, Tremblay AMK, Mubaid S, Omer A, Ford RJ, Bedard N et al (2018) The AMPK agonist 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR), but not metformin, prevents inflammation‐associated cachectic muscle wasting. EMBO Mol Med 10: e8307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DT, Ma JF, Marco SD, Gallouzi IE (2011) Inducible nitric oxide synthase (iNOS) in muscle wasting syndrome, sarcopenia, and cachexia. Aging 3: 702–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas‐Ahner J, Wang J, Bloomston M, Muscarella P, Nau P et al (2013) NF‐kappaB‐mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 123: 4821–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberle AM, Prentzell MT, van Eunen K, Bakker BM, Grellscheid SN, Thedieck K (2015) Molecular mechanisms of mTOR regulation by stress. Mol Cell Oncol 2: e970489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henique C, Mansouri A, Vavrova E, Lenoir V, Ferry A, Esnous C, Ramond E, Girard J, Bouillaud F, Prip‐Buus C et al (2015) Increasing mitochondrial muscle fatty acid oxidation induces skeletal muscle remodeling toward an oxidative phenotype. FASEB J 29: 2473–2483 [DOI] [PubMed] [Google Scholar]
- Høivik HO, Laurijssens BE, Harnisch LO, Twomey CK, Dixon RM, Kirkham AJ, Williams PM, Wentz AL, Lunnon MW (2010) Lack of efficacy of the selective iNOS inhibitor GW274150 in prophylaxis of migraine headache. Cephalalgia 30: 1458–1467 [DOI] [PubMed] [Google Scholar]
- Jatoi A, Dakhil SR, Nguyen PL, Sloan JA, Kugler JW, Rowland KM, Soori GS, Wender DB, Fitch TR, Novotny PJ et al (2007) A placebo‐controlled double blind trial of etanercept for the cancer anorexia/weight loss syndrome: results from N00C1 from the North Central Cancer Treatment Group. Cancer 110: 1396–1403 [DOI] [PubMed] [Google Scholar]
- Jatoi A, Ritter HL, Dueck A, Nguyen PL, Nikcevich DA, Luyun RF, Mattar BI, Loprinzi CL (2010) A placebo‐controlled, double‐blind trial of infliximab for cancer‐associated weight loss in elderly and/or poor performance non‐small cell lung cancer patients (N01C9). Lung Cancer 68: 234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, Li YP (2007) Curcumin prevents lipopolysaccharide‐induced atrogin‐1/MAFbx upregulation and muscle mass loss. J Cell Biochem 100: 960–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julienne CM, Dumas JF, Goupille C, Pinault M, Berri C, Collin A, Tesseraud S, Couet C, Servais S (2012) Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J Cachexia Sarcopenia Muscle 3: 265–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostourou V, Cartwright JE, Johnstone AP, Boult JK, Cullis ER, Whitley G, Robinson SP (2011) The role of tumour‐derived iNOS in tumour progression and angiogenesis. Br J Cancer 104: 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzke T, Buck A, Prade VM, Feuchtinger A, Prokopchuk O, Martignoni ME, Heisz S, Hauner H, Janssen K‐P, Walch A et al (2019) Derangements of amino acids in cachectic skeletal muscle are caused by mitochondrial dysfunction. J Cachexia Sarcopenia Muscle 11: 226–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautaoja JH, Lalowski M, Nissinen TA, Hentila J, Shi Y, Ritvos O, Cheng S, Hulmi JJ (2019a) Muscle and serum metabolomes are dysregulated in colon‐26 tumor‐bearing mice despite amelioration of cachexia with activin receptor type 2B ligand blockade. Am J Physiol Endocrinol Metab 316: E852–E865 [DOI] [PubMed] [Google Scholar]
- Lautaoja JH, Lalowski M, Nissinen TA, Hentilä J, Shi Y, Ritvos O, Cheng S, Hulmi JJ (2019b) Muscle and serum metabolomes are dysregulated in colon‐26 tumor‐bearing mice despite amelioration of cachexia with activin receptor type 2B ligand blockade. Am J Physiol Endocrinol Metab 316: E852–E865 [DOI] [PubMed] [Google Scholar]
- Lee J, Ryu H, Ferrante RJ, Morris SM, Ratan RR (2003) Translational control of inducible nitric oxide synthase expression by arginine can explain the arginine paradox. Proc Natl Acad Sci 100: 4843–4848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Gao D, Jiang Y (2019) Function, detection and alteration of acylcarnitine metabolism in hepatocellular carcinoma. Metabolites 9: 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, He Y, Miao K, Zheng Y, Deng C, Liu T‐M (2020) Imaging of macrophage mitochondria dynamics in vivo reveals cellular activation phenotype for diagnosis. Theranostics 10: 2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JF, Sanchez BJ, Hall DT, Tremblay AK, Di Marco S, Gallouzi IE (2017) STAT3 promotes IFNgamma/TNFalpha‐induced muscle wasting in an NF‐kappaB‐dependent and IL‐6‐independent manner. EMBO Mol Med 9: 622–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuirk S, Gravel SP, Deblois G, Papadopoli DJ, Faubert B, Wegner A, Hiller K, Avizonis D, Akavia UD, Jones RG et al (2013) PGC‐1alpha supports glutamine metabolism in breast cancer. Cancer Metab 1: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean JB, Moylan JS, Andrade FH (2014) Mitochondria dysfunction in lung cancer‐induced muscle wasting in C2C12 myotubes. Front Physiol 5: 503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis KA, Zhu X, Burfeind KG, Krasnow SM, Levasseur PR, Morgan TK, Marks DL (2017) Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle 8: 824–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem 292: 7189–7207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee SA, Gerencser AA, Nicholls DG, Brand MD (2018) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J Biol Chem 293: 12649–12652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mubaid S, Ma JF, Omer A, Ashour K, Lian XJ, Sanchez BJ, Robinson S, Cammas A, Dormoy‐Raclet V, Di Marco S et al (2019) HuR counteracts miR‐330 to promote STAT3 translation during inflammation‐induced muscle wasting. Proc Natl Acad Sci 116: 17261–17270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KT, Chee A, Trieu J, Naim T, Lynch GS (2012) Importance of functional and metabolic impairments in the characterization of the C‐26 murine model of cancer cachexia. Dis Model Mech. 5: 533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RA, Yeung E, Mazurak VC, Mourtzakis M (2011) Influence of eicosapentaenoic acid supplementation on lean body mass in cancer cachexia. Br J Cancer 105: 1469–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito T, Okayama T, Aoyama T, Ohashi T, Masuda Y, Kimura M, Shiozaki H, Murakami H, Kenmotsu H, Taira T et al (2017) Unfavorable impact of cancer cachexia on activity of daily living and need for inpatient care in elderly patients with advanced non‐small‐cell lung cancer in Japan: a prospective longitudinal observational study. BMC Cancer 17: 800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanetti L, Taffi R, Vignini A, Moroni C, Raffaelli F, Bacchetti T, Silvestrini M, Provinciali L, Mazzanti L (2007) Reactive oxygen species plasmatic levels in ischemic stroke. Mol Cell Biochem 303: 19–25 [DOI] [PubMed] [Google Scholar]
- Narsale AA, Carson JA (2014) Role of IL‐6 in cachexia–therapeutic implications. Curr Opin Support Palliat Care 8: 321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrão AI, Oliveira P, Vitorino R, Colaço B, Pires MJ, Márquez M, Castellanos E, Neuparth MJ, Teixeira C, Costa C et al (2013) Bladder cancer‐induced skeletal muscle wasting: disclosing the role of mitochondria plasticity. Int J Biochem Cell Biol 45: 1399–1409 [DOI] [PubMed] [Google Scholar]
- Palmieri EM, Gonzalez‐Cotto M, Baseler WA, Davies LC, Ghesquière B, Maio N, Rice CM, Rouault TA, Cassel T, Higashi RM et al (2020) Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun 11: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli P, Kumar S, Loumaye A, Singh P, Eragamreddy S, Nguyen TL, Ozkan S, Razzaque MS, Prunier C, Thissen J‐P et al (2018) Twist1 activation in muscle progenitor cells causes muscle loss akin to cancer cachexia. Dev Cell 45: 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeuffer M, Jaudszus A (2016) Pentadecanoic and heptadecanoic acids: multifaceted odd‐chain fatty acids. Adv Nutr 7: 730–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin F, Barreto R, Couch ME, Bonetto A, O'Connell TM (2019a) Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J Cachexia Sarcopenia Muscle 10: 140–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin F, Novinger LJ, Huot JR, Harris RA, Couch ME, O'Connell TM, Bonetto A (2019b) PDK4 drives metabolic alterations and muscle atrophy in cancer cachexia. FASEB J 33: 7778–7790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato PE (2016) Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 5: e200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppa MJ, Gao S, Narsale AA, Carson JA (2014) Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma‐induced cachexia. FASEB J 28: 998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppa MJ, White JP, Velazquez KT, Baltgalvis KA, Sato S, Baynes JW, Carson JA (2012) The effect of exercise on IL‐6‐induced cachexia in the Apc ( Min/+) mouse. J Cachexia Sarcopenia Muscle 3: 117–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- QuanJun Y, GenJin Y, LiLi W, Yan H, YongLong H, Jin L, Jie L, JinLu H, Cheng G (2015) Integrated analysis of serum and intact muscle metabonomics identify metabolic profiles of cancer cachexia in a dynamic mouse model. RSC Adv 5: 92438–92448 [Google Scholar]
- Ramamoorthy S, Donohue M, Buck M (2009) Decreased Jun‐D and myogenin expression in muscle wasting of human cachexia. Am J Physiol Endocrinol Metab 297: E392–E401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh RA, Jackson KC, Khairallah RJ, Ward CW, Spangenburg EE (2012) Measuring mitochondrial respiration in intact single muscle fibers. Am J Physiol Regul Integr Comp Physiol 302: R712–R719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour M, Pétavy F, Chiesa F, Perry H, Lukey P, Binks M, Donatien P, Freidin A, Eckersley R, McClinton C (2012) Ultrasonographic measures of synovitis in an early phase clinical trial: a double‐blind, randomised, placebo and comparator controlled phase IIa clinical trial of GW274150 (a selective inducible nitric oxide synthase inhibitor) in rheumatoid arthritis. Clin Exp Rheumatol Incl Suppl 30: 254 [PubMed] [Google Scholar]
- Shukla SK, Markov SD, Attri KS, Vernucci E, King RJ, Dasgupta A, Grandgenett PM, Hollingsworth MA, Singh PK, Yu F et al (2020) Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia. Cancer Lett 484: 29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum AM, Mahendradatta T, Taylor RJ, Painter AB, Moore MM, Tsoli M, Tan TC, Clarke SJ, Robertson GR, Polly P (2012) Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer‐induced skeletal muscle wasting. Aging 4: 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KL, Tisdale MJ (1993) Increased protein degradation and decreased protein synthesis in skeletal muscle during cancer cachexia. Br J Cancer 67: 680–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck RM, Courneya KS, Mâsse LC, Duval S, Schmitz KH (2010) An update of controlled physical activity trials in cancer survivors: a systematic review and meta‐analysis. J Cancer Surviv 4: 87–100 [DOI] [PubMed] [Google Scholar]
- Stein SC, Woods A, Jones NA, Davison MD, Carling D (2000) The regulation of AMP‐activated protein kinase by phosphorylation. Biochem J 345(Pt 3): 437–443 [PMC free article] [PubMed] [Google Scholar]
- Stene GB, Helbostad J, Balstad TR, Riphagen II, Kaasa S, Oldervoll LM (2013) Effect of physical exercise on muscle mass and strength in cancer patients during treatment—a systematic review. Crit Rev Oncol Hematol 88: 573–593 [DOI] [PubMed] [Google Scholar]
- Strijdom H, Chamane N, Lochner A (2009) Nitric oxide in the cardiovascular system: a simple molecule with complex actions. Cardiovasc J Afr 20: 303–310 [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Ischiropoulos H, Radi R (2007) Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6: 662–680 [DOI] [PubMed] [Google Scholar]
- Takayama K, Atagi S, Imamura F, Tanaka H, Minato K, Harada T, Katakami N, Yokoyama T, Yoshimori K, Takiguchi Y et al (2016) Quality of life and survival survey of cancer cachexia in advanced non‐small cell lung cancer patients‐Japan nutrition and QOL survey in patients with advanced non‐small cell lung cancer study. Support Care Cancer 24: 3473–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley PJ (2003) Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys 419: 31–40 [DOI] [PubMed] [Google Scholar]
- Tseng Y‐C, Kulp SK, Lai I‐L, Hsu E‐C, He WA, Frankhouser DE, Yan PS, Mo X, Bloomston M, Lesinski GB et al (2015a) Preclinical investigation of the novel histone deacetylase inhibitor AR‐42 in the treatment of cancer‐induced cachexia. J Natl Cancer Inst 107: djv274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YC, Kulp SK, Lai IL, Hsu EC, He WA, Frankhouser DE, Yan PS, Mo X, Bloomston M, Lesinski GB et al (2015b) Preclinical Investigation of the Novel Histone Deacetylase Inhibitor AR‐42 in the Treatment of Cancer‐Induced Cachexia. J Natl Cancer Inst 107: djv274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzika AA, Fontes‐oliveira CC, Shestov AA, Constantinou C, Psychogios N, Righi V, Mintzopoulos D, Busquets S, Lopez‐soriano FJ, Milot S et al (2013) Skeletal muscle mitochondrial uncoupling in a murine cancer cachexia model. Int J Oncol 43: 886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVeen BN, Fix DK, Montalvo RN, Counts BR, Smuder AJ, Murphy EA, Koh HJ, Carson JA (2019) The regulation of skeletal muscle fatigability and mitochondrial function by chronically elevated interleukin‐6. Exp Physiol 104: 385–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVeen BN, Hardee JP, Fix DK, Carson JA (2018) Skeletal muscle function during the progression of cancer cachexia in the male Apc(Min/+) mouse. J Appl Physiol 124: 684–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigano AAL, Morais JA, Ciutto L, Rosenthall L, di Tomasso J, Khan S, Olders H, Borod M, Kilgour RD (2017) Use of routinely available clinical, nutritional, and functional criteria to classify cachexia in advanced cancer patients. Clin Nutr 36: 1378–1390 [DOI] [PubMed] [Google Scholar]
- Vitecek J, Lojek A, Valacchi G, Kubala L (2012) Arginine‐based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm 2012318087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Xiong X, Zhao H (2015) Tim‐3 cell signaling and iNOS are involved in the protective effects of ischemic postconditioning against focal ischemia in rats. Metab Brain Dis 30: 483–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, Carson JA (2011) The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS One 6: e24650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, Kostek MC, Matesic LE, Carson JA (2012) IL‐6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA (2013) Muscle mTORC1 suppression by IL‐6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab 304: E1042–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenmann B, Malfertheiner P, Friess H, Ritch P, Arseneau J, Mantovani G, Caprioni F, Van Cutsem E, Richel D, DeWitte M et al (2008) A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J Support Oncol 6: 18–25 [PubMed] [Google Scholar]
- Wiseman DA, Thurmond DC (2012) The good and bad effects of cysteine S‐nitrosylation and tyrosine nitration upon insulin exocytosis: a balancing act. Curr Diabetes Rev 8: 303–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Zheng X, Yang M, Zhao A, Li M, Chen T, Panee J, Jia W, Ji G (2017) Serum lipid alterations identified in chronic hepatitis B, hepatitis B virus‐associated cirrhosis and carcinoma patients. Sci Rep 7: 42710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS (2012) MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic Acids Res 40: W127–W133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Psychogios N, Young N, Wishart DS (2009) MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res 37: W652–W660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Han B, Wishart DS (2015) MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res 43: W251–W257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Wishart DS (2011a) Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Curr Protoc Bioinformat 34: 14.10.11‐14.10.48 [DOI] [PubMed] [Google Scholar]
- Xia J, Wishart DS (2011b) Web‐based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc 6: 743–760 [DOI] [PubMed] [Google Scholar]
- Xia J, Wishart DS (2016) Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr Protoc Bioinformat 55: 14.10.11‐14.10.91 [DOI] [PubMed] [Google Scholar]
- Xu W, Ghosh S, Comhair SAA, Asosingh K, Janocha AJ, Mavrakis DA, Bennett CD, Gruca LL, Graham BB, Queisser KA et al (2016) Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J Clin Investig 126: 2465–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Wang Q, Yin P, Xing W, Wu Z, Chen S, Lu X, Zhang Y, Lin X, Xu G (2012) Serum metabolomics reveals the deregulation of fatty acids metabolism in hepatocellular carcinoma and chronic liver diseases. Anal Bioanal Chem 403: 203–213 [DOI] [PubMed] [Google Scholar]
- Zhou T, Wang B, Liu H, Yang K, Thapa S, Zhang H, Li L, Yu S (2018) Development and validation of a clinically applicable score to classify cachexia stages in advanced cancer patients. J Cachexia Sarcopenia Muscle 9: 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Source Data for Expanded View/Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Data Availability Statement
This study includes no data deposited in external repositories.