

Abstract
The DNA cytosine deaminase APOBEC3B (A3B) is a newly recognized endogenous source of mutations in a range of human tumors, including head/neck cancer. A3B inflicts C-to-T and C-to-G base substitutions in 5’-TCA/T trinucleotide motifs, contributes to accelerated rates of tumor development, and affects clinical outcomes in a variety of cancer types. High-risk human papillomavirus (HPV) infection causes A3B overexpression, and HPV-positive cervical and head/neck cancers are among tumor types with the highest degree of APOBEC signature mutations. A3B overexpression in HPV-positive tumor types is caused by the viral E6/E7 oncoproteins and may be an early off-to-on switch in tumorigenesis. In comparison, less is known about the molecular mechanisms responsible for A3B overexpression in HPV-negative head/neck cancers. Here, we utilize an immunohistochemical approach to determine whether A3B is turned from off-to-on or if it undergoes a more gradual transition to overexpression in HPV-negative head/neck cancers. As positive controls, almost all HPV-positive oral epithelial dysplasias and oropharyngeal cancers showed high levels of nuclear A3B staining regardless of diagnosis. As negative controls, A3B levels were low in phenotypically normal epithelium adjacent to cancer and oral epithelial hyperplasias. Interestingly, HPV-negative and low-grade oral epithelial dysplasias showed intermediate A3B levels, while high-grade oral dysplasias showed high A3B levels similar to oral squamous cell carcinomas. A3B levels were highest in grade 2 and grade 3 oral squamous cell carcinomas. In addition, a strong positive association was found between nuclear A3B and Ki67 scores suggesting a linkage to the cell cycle. Overall, these results support a model in which gradual activation of A3B expression occurs during HPV-negative tumor development and suggest that A3B overexpression may provide a marker for advanced grade oral dysplasia and cancer.
Keywords: APOBEC3B, DNA cytosine deamination, head and neck cancer, human papillomavirus HPV, oral premalignancies, tumor cell proliferation
Introduction
Head and neck cancers comprise the 7th most common human malignancy worldwide and represent a heterogeneous group of neoplasms in regard to their etiology, location, and molecular basis (1, 2). In 2018, head/neck cancers accounted for approximately 890,000 newly diagnosed cases and 450,000 deaths globally (3), comprising 3% of all malignancies (4). The American Cancer Society estimates that in 2020 the number of new head/neck cancer cases will be 53,260, and 10,750 deaths will occur as a result of the disease (5).
Anatomically, tumors of the head/neck involve the oral cavity and oropharynx, nasopharynx, hypopharynx, larynx, paranasal sinuses, and salivary glands. Microscopically, more than 92% of head/neck malignancies are squamous cell carcinomas (HNSCC) (6, 7). Smoking tobacco and alcohol are primary risk factors and show a synergistic effect on the development of HNSCC (8, 9). Despite advances in diagnosis and treatment, recurrent and/or metastatic disease develops in greater than 65% of patients with HNSCC (10). Moreover, 5-year survival rates of patients with HNSCC remain approximately 50–60% (1, 2).
Independently of alcohol and tobacco exposure, transcriptionally active high-risk human papillomavirus (HPV) infection, especially HPV16, has been strongly associated with oropharyngeal SCC (OPSCC) (1, 2, 11). Interestingly, HPV-related OPSCC shows improved 3-year survival (approximately 82% versus 57% for HPV-negative) and distinct biologic behavior from non-viral tumors (4, 12, 13). HPV16 is also implicated in the development of oral potentially malignant disorders (oral epithelial dysplasias) and invasive tumors of the oral cavity (14, 15). However, the percentage of oral SCC (OSCC) with presence of HPV16 is relatively low (3.7%) (16) and, in contrast to OPSCC, such low numbers have precluded determinations of prognostic utility (13, 16).
The past decade has enabled a remarkable view of human genomic DNA sequences and, therefore, also of the overall mutation landscape of head and neck cancers (17, 18). One of the most important discoveries from this work was the observation that almost all HPV-positive and many HPV-negative head/neck tumors have a large fraction of mutations attributable to members of the apolipoprotein B mRNA editing enzyme catalytic subunit-like protein 3 (APOBEC3) family of single-stranded DNA cytosine deaminases (17, 19–23). The APOBEC3 single base substitution mutation signature is defined by C-to-T and C-to-G changes in 5’-TCA and 5’-TCT trinucleotide motifs (single base substitution signatures SBS2 and SBS13, respectively) (22, 23). The APOBEC3 mutation signature also dominates other cancer types including those of cervical, bladder, breast, and lung tissues (21–26). The strongest evidence thus far that APOBEC3 enzymes drive tumor evolution is positive associations with poor clinical outcomes (27, 28) and large increases in the proportion of APOBEC3 signature mutations from primary to metastatic disease (29, 30).
The human APOBEC3 family is comprised of seven enzymes, A3A-D and A3F-H, that function to provide innate immune protection from infection by DNA-based viruses including retroviruses (e.g. HIV-1), herpesviruses (e.g. EBV), and human papillomaviruses (e.g. HPV16) (31–33). However, most successful DNA-based viruses (ie., those that are pathogenic in humans) have had to evolve mechanisms to blunt restriction by APOBEC3s such as proteosomal degradation by HIV-1 Vif and direct neutralization and re-localization by EBV BORF2 (34, 35). Although an APOBEC3 counteraction mechanism has yet to be discovered for HPV, an overwhelming body of evidence suggests ongoing viral mutagenesis by APOBEC3A (A3A) and APOBEC3B (A3B), and that the same two enzymes are the most likely sources of the overall APOBEC mutation signature in both HPV-positive and HPV-negative head/neck carcinomas. First, high-risk HPV infections induce expression of both A3A and A3B by mechanisms dependent upon the viral E6 and E7 oncoproteins (36–40). Second, APOBEC3 signature mutations are frequent in both transmitting HPV isolates as well as variants within individual patients (41, 42). Third, the APOBEC3 mutation signature in HPV genomes is strikingly similar to that in head/neck tumor genomes (17, 18, 20, 43–45). These observations support a mechanism in which HPV infection triggers A3A and A3B upregulation, perhaps to promote viral genetic diversification with collateral genomic DNA damage contributing to the overall process of tumorigenesis.
Viral infection and A3A/B upregulation in HPV-positive OPSCC suggests that the APOBEC3 mutagenesis may be an early and perhaps even an essential feature in carcinogenesis (i.e., upregulation coincident with infection). However, the role of APOBEC3 mutagenesis in HPV-negative head/neck tumors including OSCC is less clear. By analogy to HPV-positive OPSCC, perhaps APOBEC3 upregulation in HPV-negative OSCC is also an early/initiating event that drives all stages of carcinogenesis. Alternatively, APOBEC3 levels may gradually rise during the transition from low- and high-grade oral epithelial dysplasia (OED) to OSCC. To distinguish between these models, we used our newly developed rabbit anti-human A3B monoclonal antibody (mAb) (46) and applied immunohistochemistry approaches to a range of pre-neoplastic and fully neoplastic oral mucosal tissues.
Methods
Tissue procurement and HPV status assessment
Following Institutional Review Board approval, formalin-fixed paraffin-embedded (FFPE) archival tissue blocks of HPV-positive OED (n=13) and OPSCC (n=2), HPV-negative low-grade (n=43) and high-grade OED (n=45), OSCC (n=46), as well as epithelial hyperplasias (hyperkeratosis; OEH, n=13) were obtained from the Oral Pathology Laboratory, School of Dentistry, University of Minnesota and HPV-positive OPSCC (n=44), from the Department of Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta. All human tissues were derived from incisionally or excisionally biopsied intraoral or oropharyngeal lesions and were classified according to the most recent W.H.O. Classification of Head and Neck Tumors (1). Hematoxylin and eosin (H&E) stained-slides were reviewed to confirm the diagnosis and to assess the cytologic and histomorphologic features of each specimen. OEDs were subgrouped according to the binary classification system (1, 47). The cut-off between low- and high-grade OED was 4 architectural and 5 cytological changes, irrespective of the extent of the dysplastic changes within the epithelium (47). OSCCs were classified according to the level of keratinization, cellular atypia and pleomorphism, number of mitoses, and presence of perineural and/or lymphovascular invasion as grade 1 (low-grade; n=11), grade 2 (intermediate grade; n=17) and grade 3 (high-grade; n=10). The remaining 8 cases represented superficially invasive OSCCs and, thus, histograde was not reported. The epidemiologic characteristics of each patient (age and gender), smoking history (where available) and location of the lesion were retrieved and tabulated (Supplementary Tables S1 and S2).
All cases of HPV-positive OPSCC and OEDs showed strong nuclear and cytoplasmic immunohistochemical expression of the surrogate biomarker of high-risk HPV E7 oncoprotein function, p16 (p16 CIN, clone INK4α, mouse mAb) in greater >70% of the cells within the lesion (48).
A3B and Ki67 immunohistochemical (IHC) staining
IHC staining was performed following a previously described protocol (46, 49). FFPE tissues were sectioned at 4 μm, mounted on positively charged, adhesive slides and allowed to air-dry for at least 24 hrs. To deparaffinize and rehydrate the samples, slides were baked in a 65°C oven for 20 min, washed 3 times with CitrisolvTM (Decon Labs, #1601) for 5-min/each, soaked in graded alcohols (100% × 2, 95% and 80% for 3 min/each), and then rinsed in running water for at least 5 min. Epitope retrieval was performed using Reveal Decloaker (BioCare Medical, #RV1000M) in a steamer for 35 min, followed by a 20 min “cool-down” period. Then, slides were rinsed with running tap water for 5 min and transferred to TBST for 5 min. Endogenous peroxidase activity was quenched by placing the slides in 3% H2O2 in TBST for 10 min at RT, followed by a 5-min rinse under running water. To block non-specific binding of primary antibody, sections were covered with Background Sniper (BioCare Medical, #BS966MM) for 15 min at RT. After blocking, serial sections of each specimen were incubated overnight at 4°C with a custom made rabbit α-human A3A/B/G mAb (5210-87-13) (46) diluted 1:350 and rabbit α-human Ki-67 mAb (Sigma Aldrich, clone SP6) 1:400 dil. in 10% Sniper in TBST.
Following overnight incubation with primary antibody, sections were rinsed in TBST for 5 min, and completely covered with anti-rabbit poly-HRP-IgG (Leica Biosystems, Novolink Polymer, #RE7260-K) for 30 min at RT. The reaction product was developed using the Novolink DAB substrate kit (Leica Biosystems, # RE7230-K) at RT for 3–5 min, rinsed in tap water for 5 min, counterstained in Mayer’s hematoxylin solution (Electron Microscopy Sciences, # 26252–01) for up to 5 min, dehydrated in graded alcohols and CitrisolvTM, and cover-slipped using Permount mounting media.
IHC Quantification and Histoscore
A3B and Ki67 stained slides were scanned at 40x magnification and immunoreactive proteins were visualized with the Aperio ScanScope XT (Leica Biosystems) and quantified using the Aperio Nuclear Algorithm software. The Aperio Nuclear Algorithm identifies each nucleus in selected areas of the specimen and analyzes the immunostaining as negative, weak-positive, moderate-positive, or strong-positive. A3B histoscore (H-score) was calculated via the linear formula: H-score = 1x(%weak-positive cells) + 2x(%moderate-positive cells) + 3x(%strong-positive cells) as described (50, 51). For Ki67 staining the percentage of positive cells was very strongly correlated with Ki67 H-score (R2=0.95, data not shown) and was utilized for further analyses. Because data were not distributed normally, statistical differences between groups were calculated using Kruskal-Wallis one-way non-parametric tests and median and ranges were reported. Comparisons between paired patient samples were conducted using Wilcoxon signed-rank test. Correlations between A3B H-score and % of Ki67+ cells were determined by Spearman correlation test; coefficients and 95% confidence intervals were reported. P-values <0.05 were considered statistically significant.
TCGA head/neck data analysis
RNA-sequencing and clinical data for all primary head and neck tumor samples were downloaded from the Firehose GDAC resource through the Broad Institute analysis pipeline (http://gdac.broadinstitute.org/). Specifically, the FireBrowse resource was used to download scaled estimate mRNAseq data previously generated by the TCGA analysis workflow, including initial two-pass alignment with STAR (52). Transcript quantification and normalization was performed using established methods (53). All normal tissue samples and metastatic specimens were removed prior to analysis. Tumor grade was determined from the clinical files associated with primary tumors, specifically using the “grade” metric. Of the 528 total data sets available in TCGA, 499 specimens had grade information and therefore only this subset was used in our analysis of A3B mRNA levels. Expectation-maximization (RSEM) values normalized to the housekeeping gene TBP were used for A3B expression calculations. Comparisons of A3B expression levels between various tumor grades were conducted using Kruskal-Wallis tests.
Results
IHC detection of endogenous A3B protein in p16-positive (HPV-related) oral epithelial dysplasias and oropharyngeal cancers
We previously reported the creation of a rabbit mAb, 5210-87-13, that binds to the human DNA deaminase A3B in several immunoassays including IHC with patient-derived tissues (46, 49). High-risk HPV infection has been shown to cause upregulation of A3B mRNA levels in model cellular systems (36, 38, 54) and HPV-positive head/neck tumors show remarkably high A3B mRNA levels and APOBEC mutation signature loads (22, 24, 25, 55). Therefore, we first sought further validation by using this mAb to compare endogenous A3B protein levels in HPV-positive OPSCCs and OEDs, as well as HPV-negative non-neoplastic oral tissues. Multiple cases of oral hyperplastic epithelium (frictional hyperkeratosis, OEH) lacking evidence of HPV infection were mostly negative for A3B (weak immunostaining of the nuclei of some cells of the basal and suprabasal epithelial layers; n=13; H-score range = 1.4 – 45.4, median = 18.4; Figure 1A and D).
Fig. 1. Endogenous A3B protein expression in HPV-related oral epithelial dysplasias and oropharyngeal cancers.
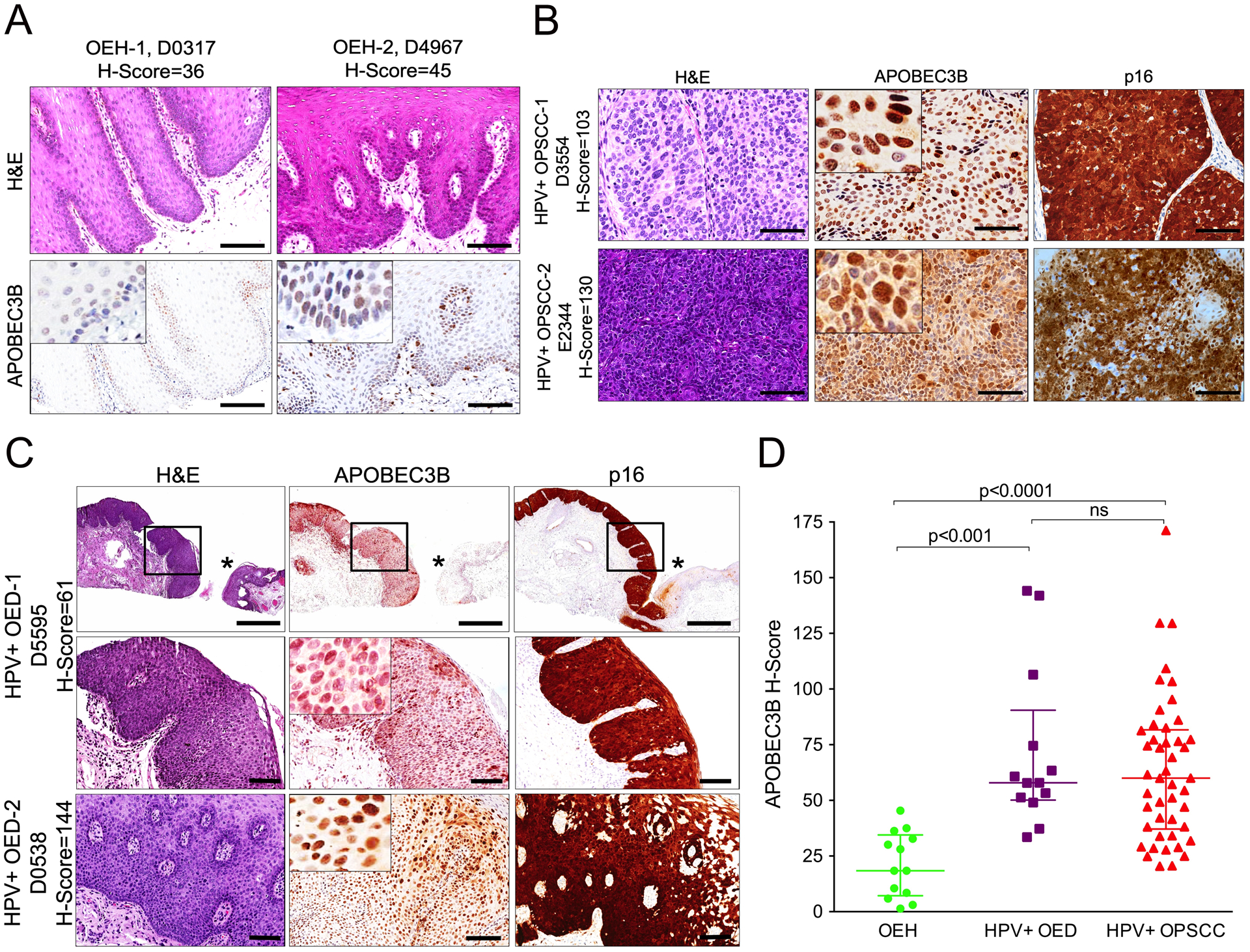
(A) A3B staining and corresponding H&E-stained photomicrographs of representative oral epithelial hyperplasias (OEHs). Scale bars are 100 μm and inset images are magnified 8-fold.
(B) A3B immunophenotype and corresponding H&E-stained photomicrographs of representative oropharyngeal squamous cell carcinomas (OPSCCs). Strong and diffuse, nuclear and cytoplasmic staining for the surrogate marker p16 is confirmatory of transcriptionally active high-risk HPV infection. Scale bars are 60 μm and inset images are magnified 4-fold.
(C) Histopathologic and A3B immunohistochemical characteristics of HPV-related epithelial dysplasias of the oral mucosa (OED). For the first row of low-power photomicrographs scale bars are 600 μm. Images on the second row represent a high-power magnification of the region marked in the black box in the upper low-power image. The asterisks point to uninfected, normal epithelium adjacent to HPV-driven (p16+) dysplasia. In contrast to the p16+ dysplastic tissue which exhibits strong A3B nuclear expression throughout the thickness of the lesion, normal mucosa is overtly negative for A3B. Scale bars for the second and third row are 100 μm and inset images are magnified 8-fold.
(D) Collective presentation of quantified A3B IHC score (H-score) of OEH (n=13, control), HPV-positive OED (n=13) and HPV-positive OPSCC (n=46). The H-score median and interquartile range for each group are shown. Statistical significance for key comparisons is indicated (Kruskal-Wallis test).
In contrast, A3B protein expression was markedly increased in p16-positive (HPV-related) OPSCCs (Figure 1B). OPSCCs generally showed strong and focal-to-diffuse, nuclear only, immunoreactivity in the majority of neoplastic cells (n=46 tumors; H-score range = 20.5 – 171, median= 60; Figure 1D). Similarly, HPV-driven OEDs exhibited elevated levels of A3B immunostaining throughout the thickness of the dysplastic epithelium (H-score range = 33.5 – 144, median = 57.9; Figure 1C and D). Remarkably, in both HPV-related OEDs and OPSCCs, the A3B immunophenotype mirrored p16 immunostaining (Figure 1B and C). This concordance is particularly clear in Figure 1C where a p16/A3B-positive dysplastic lesion occurs next to normal epithelial tissue that is negative for both of these proteins. Collectively, A3B protein levels were significantly higher in p16-positive/HPV-related OEDs and OPSCCs when compared to uninfected OEHs (p<0.001 and p<0.0001, respectively, by Kruskal-Wallis test; Figure 1D). In addition, A3B upregulation appears to be closely linked to HPV status rather than diagnosis, as p16-positive/HPV-related epithelial dysplasias and invasive tumors demonstrated similarly high ranges of A3B levels (p=0.6 by Kruskal-Wallis test; Figure 1D). Clinical information for these specimens is summarized in Table 1 and Supplementary Table S1.
Table 1.
Collective presentation of the clinicopathologic, epidemiologic and A3B immunohistochemical characteristics of the studied cases.
| Lesions | Gender | Age (years) | Location | A3B H-Score |
|---|---|---|---|---|
| OEH (n=13) |
5M: 8F | Mean=57.3 Range=30–96 |
Tongue (n=7) Buccal mucosa (n=3) Mandibular Vestibule (n=2) Palate (n=1) |
Median=18.4 Range=1.4–45.4 |
| Low-grade OED (n=43) |
25M:18F | Mean=64.1 Range=28–92 |
Tongue (n=32) Buccal mucosa (n=3) Mandibular vestibule (n=3) Alveolar mucosa/gingiva (n=2) Palate (n=2) Floor of mouth (n=1) |
Median=32.4 Range=4.5–94.5 |
| High-grade OED (n=45) |
22M:23F | Mean=63.1 Range=35–88 |
Tongue (n=36) Floor of mouth (n=4) Buccal mucosa (n=2) Mandibular vestibule (n=2) Palate (n=1) |
Median=63.1 Range=11.4–108 |
| OSCC (n=46) |
22M:24F | Mean=66.2 Range=34–93 |
Tongue (n=23) Alveolar mucosa/gingiva (n=8) Floor of mouth (n=5) Mandibular vestibule (n=5) Palate (n=4) Lower lip (n=1) |
Median=49 Range=10.1–117 |
| HPV+ (p16+) OED (n=13) |
12M:1F | Mean=62.8 Range=51–87 |
Floor of mouth (n=4) Tongue (n=4) Buccal mucosa (n=2) Alveolar mucosa/gingiva (n=1) Base of tongue (n=1) Palate (n=1) |
Median=57.9 Range=33.5–144 |
| HPV+ (p16+) OPSCC (n=46) |
44M:2F | Mean=59 Range=43–82 |
Base of tongue (n=21) Tonsils (n=17) Glossotonsillar sulcus (n=8) |
Median=60 Range=20.5–171 |
A3B, APOBEC3B; OEH, oral epithelial hyperplasia; OED, oral epithelial dysplasia; OSCC, oral squamous cell carcinoma; OPSCC, oropharyngeal squamous cell carcinoma
Assessment of A3B protein levels in HPV-negative OEDs and invasive cancers
As described above, high A3B protein levels were anticipated in HPV-positive lesions because the oncoproteins of this DNA tumor virus stimulate A3B transcription (36, 38, 54). To address whether a similar off-to-on mechanism occurs in virus-negative lesions, A3B protein levels were investigated in non-HPV-related, keratinizing tumors of the oral cavity that lack histopathologic or phenotypic variations suggestive of HPV infection (Figure 2A). As above for OEH, A3B levels were absent or weak and confined to the basal epithelial layer in non-neoplastic epithelial tissues adjacent to cancer (EAC; H-score range = 5.2 – 48; median = 21.7; Figure 2A and B). In contrast, A3B levels were significantly elevated in patient-matched OSCCs (H-score range = 28.2 – 110, median = 88.1; p<0.01 by Wilcoxon signed-rank test; Figure 2A and B). In addition, marked intratumoral heterogeneity was observed for A3B immunostaining in OSCCs. For instance, carcinoma cells within the same lesion exhibited a range of focal or diffuse, low (weak) to moderate and/or high (strong) A3B nuclear expression levels (inset images in Figure 2A).
Fig. 2. A3B levels increase during progression of HPV-negative oral carcinogenesis.
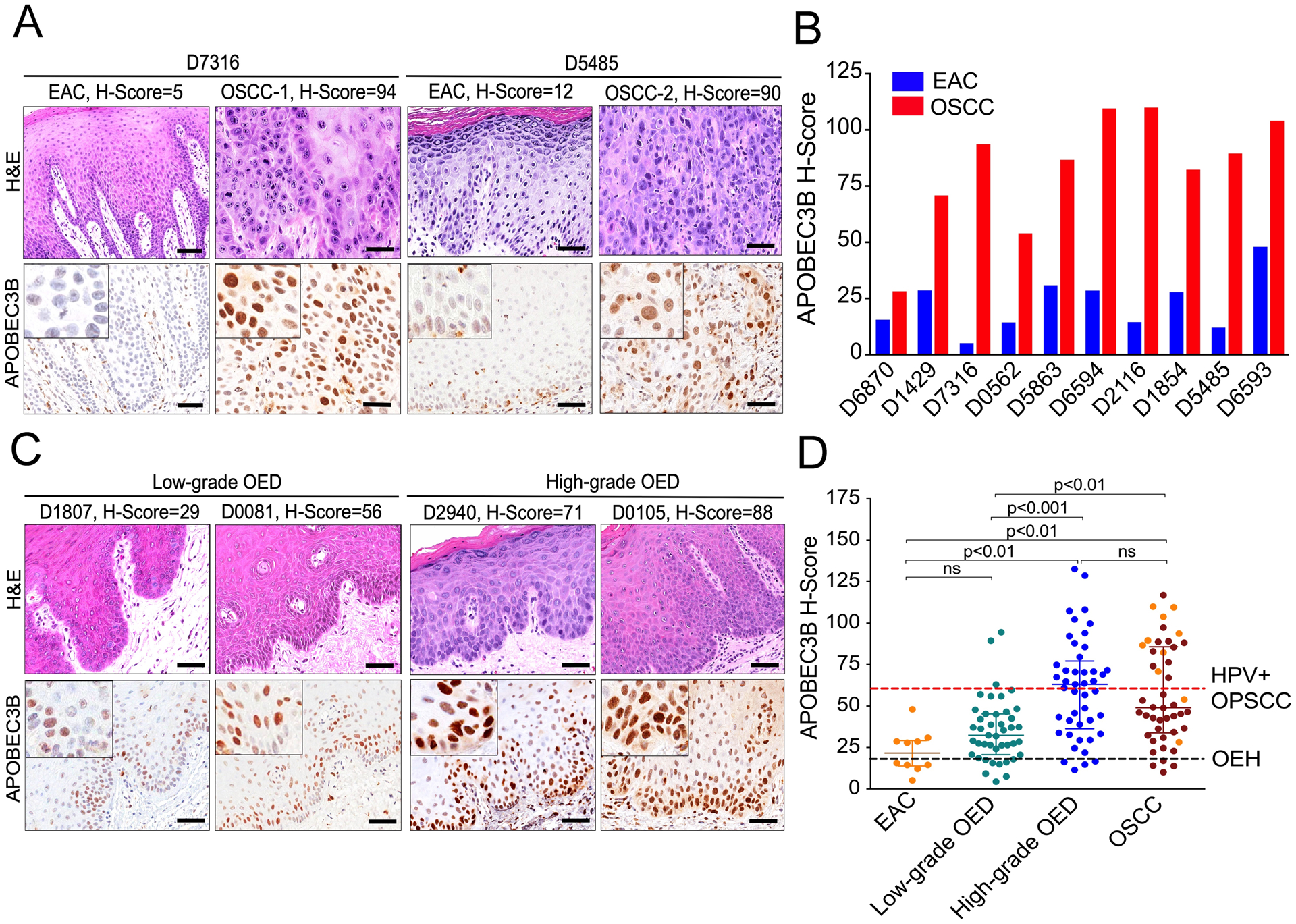
(A) A3B staining and corresponding H&E-stained photomicrographs of representative oral squamous cell carcinomas (OSCC) and paired, non-neoplastic, epithelial tissues adjacent to cancer (EAC). Scale bars are 60 μm and inset images are magnified 4-fold.
(B) A3B H-scores in 10 pairs of EAC and patient-matched OSCCs. Collectively, A3B expression was significantly higher in OSCCs than EAC (p<0.01 by Wilcoxon signed-rank test).
(C) Histopathologic and A3B immunophenotypic characteristics of representative examples of low- and high-grade, oral epithelial dysplasias (OED). Scale bars are 60 μm and inset images are magnified 4-fold.
(D) Collective presentation of quantified A3B H-scores of EAC (n=10), low-grade OED (n=43), high-grade OED (n=45) and OSCC (n=46). The black horizontal dotted line indicates the OEH (control) A3B median (18.4), while the red one the A3B median (60) of HPV-positive OPSCC. Use of same color highlights matched pairs of EAC and OSCC. The H-score median and interquartile range for each group are shown and statistical significance for key comparisons is indicated (Kruskal-Wallis test).
By IHC, low-grade OEDs exhibited primarily weak-to-moderate A3B staining in the basal third of the epithelial thickness similar to control tissues (H-score range = 4.5 – 94.5; median = 32.4; inset images in Figure 2C). A3B levels were modestly but not significantly higher in low-grade OEDs than OEH and EAC (Figure 2D). A3B staining was stronger in high-grade OEDs and extended higher into the spinous cell layer of the epithelium (Figure 2C); A3B protein expression was significantly increased (H-score range = 11.4 – 108, median = 63.1) when compared to low-grade OED (p<0.001 by Kruskal-Wallis test; Figure 2D). OSCCs showed significantly higher A3B H-scores (H-score range = 10.1 – 117, median = 49; p<0.01 by Kruskal-Wallis test) than low-grade OEDs. However, A3B levels were similarly elevated in high-grade OED and OSCC (Figure 2D and inset images in Figure 2A and C). Clinical information for these specimens is shown in Table 1 and Supplementary Table S2.
Collectively, these results support a model in which expression of the mutating enzyme A3B increases gradually during the continuum of non-viral oral carcinogenesis reaching a peak in histologically advanced dysplasias and tumors. Surprisingly, A3B levels in HPV-negative, high-grade OEDs and OSCCs did not significantly differ from the HPV-positive specimens (Figure 2D). These results strongly indicate the presence of a distinct molecular mechanism(s) that, independently of high-risk HPV infection, promotes A3B upregulation in head/neck cancers.
A3B upregulation is associated with histologic grading in HPV-negative oral cancers
Overall, as shown above, the DNA mutating enzyme A3B is overexpressed in HPV-negative tumors of the oral mucosa. However, considerable heterogeneity in A3B levels was observed with apparent low and high staining groups of tumors and we wondered whether this might be linked to tumor grade. OSCCs in this study were subclassified according to histopathologic grade as grade 1 (n=11), grade 2 (n=17), and grade 3 (n=10) tumors; eight superficially invasive (microinvasive) OSCCs could not be assigned grades (Methods, Supplementary Table S2). Grade 1 OSCCs showed relatively low A3B protein levels (H-score range = 10.1 – 67.1, median = 28.2; Figure 3A and B). In sharp contrast, grade 3 tumors showed high A3B protein levels (H-score range = 14 – 117, median = 89.1; p<0.01 by Kruskal-Wallis test; Figure 3A and B). Interestingly, intermediate grade 2 tumors, which displayed a combination of low- and high-grade characteristics, stained most heterogeneously for A3B with some showing weak and others exhibiting high staining intensities. However, A3B levels in grade 2 OSCCs were still significantly higher than grade 1 neoplasms (H-score range = 28.9 – 110, median = 57; p<0.05 by Kruskal-Wallis test) but not statistically different than those in grade 3 tumors (p=0.08, Figure 3B). Median A3B expression in superficially invasive OSCCs was 44.7 (H-score range = 13.8 – 85.5). Similar results were obtained upon analysis of A3B mRNA levels in 499 head/neck tumor data sets from TCGA (Kruskal-Wallis test; Figure 3C). These independent data sets combine to suggest that either A3B mRNA or A3B protein levels, as assessed by IHC, may serve as an indicator of tumor grade.
Fig. 3. A3B upregulation associates with histopathologic grading in HPV-negative oral cancers.

(A) H&E-stained and corresponding A3B immunohistochemical photomicrographs of representative grade 1, grade 2, and grade 3 OSCCs. Scale bars are 60 μm and inset images are magnified 4-fold.
(B) Collective presentation of quantified A3B H-scores of grade 1 (n=11), grade 2 (n=17), and grade 3 (n=10) OSCCs. The H-score median and interquartile range for each subgroup are shown. Statistical significance for key comparisons is indicated (Kruskal-Wallis test).
(C) Genomic analysis of A3B mRNA levels in 499 head/neck tumors classified as grade 1 (n=63), grade 2 (n=311) and grade 3 (n=125). Data sets available through TCGA. Statistical significance for key comparisons is indicated (Kruskal-Wallis test).
The proliferation marker Ki67 is associated with A3B upregulation in HPV-negative head/neck cancers
We next investigated whether A3B upregulation may somehow be linked to cellular proliferation in non-viral OED and OSCC. This was done by immunostaining the various groups of lesions described above for the proliferation marker Ki67, which is expressed in all active phases of the cell cycle (56). Interestingly, similar to A3B, levels of proliferation marker Ki67 increased from normal to dysplasia and, finally, cancer (Figure 4A, B, and C). The lowest Ki67 levels were observed in OEH (range = 9.7 – 42.9, median = 34.3) and low-grade OED (range = 19 – 67.7, median = 33.9; Figure 4B and C). The percentage of Ki67+ cells was significantly higher in high-grade OED (range = 22.4 – 80, median = 41.9; p<0.05) and OSCC (range = 29 – 86.7, median = 47.1; p<0.0001 by Kruskal-Wallis test; Figure 4C). No statistically significant difference was observed between the OSCC and HPV-positive OPSCC groups in regard to Ki67 expression (range = 30.3 – 85.6, median = 53.8; p = 0.14 by Kruskal-Wallis test; Figure 4C). In the majority of cases the distribution of Ki67 staining resembled the A3B immunostaining pattern (Figure 4A), with most Ki67+ cells also featuring A3B reactivity (inset images in Figure 4A).
Fig. 4. The proliferation marker Ki67 is associated with A3B upregulation in HPV-negative head/neck cancers.

(A) H&E-stained and corresponding A3B and Ki67 immunohistochemical photomicrographs of OEH, low-grade and high-grade OED, and OSCC. Scale bars are 60 μm and inset images are magnified 4-fold.
(B) Correlation analyses of A3B H-score and Ki67 expression in OEH, low-grade and high-grade OED, and OSCC. Spearman correlation coefficients and p values for each group are depicted.
(C) Collective presentation of quantified Ki67 positivity in the various subgroups. The median and interquartile range for each subgroup is shown. Statistical significance for key comparisons is indicated (Kruskal-Wallis test).
(D) Combined correlation analysis of A3B H-score and Ki67 expression in all head/neck lesions analyzed in this cohort. Spearman correlation coefficient and p value is shown.
Importantly, the aggregate analysis of all data in this study revealed a strong positive correlation between A3B H-score and Ki67 expression (Spearman r = 0.5, 95% CI = 0.4 – 0.6; p< 0.0001; Figure 4D). Notably, at least a moderate association between A3B protein levels and cellular proliferation was observed in the different histopathologic groups (Spearman correlation test; Figure 4B). Although elevated levels of Ki67 are expected in HPV-positive cancers due to Rb and p53 inactivation, the association between A3B and Ki67 protein levels was significantly stronger in HPV-negative, high-grade OED (Spearman r = 0.4, 95% CI = 0.2 – 0.7; p< 0.01; Figure 4B) and OSCC (Spearman r = 0.4, 95% CI = 0.1 – 0.6; p< 0.01; Figure 4B) than HPV-positive OPSCC (Spearman r = 0.3, 95% CI = 0.04 – 0.6; p< 0.05; data not shown). Collectively, these data suggest that cell-cycle events may be associated with A3B upregulation in HPV-negative oral premalignancies and cancers.
Discussion
Head and neck carcinogenesis is a multistep process instigated primarily by exposure to exogenous carcinogens, ie., tobacco and alcohol metabolites and/or high-risk HPV infection (11, 57, 58). Tumor DNA sequencing studies and subsequent analyses have identified endogenous APOBEC mutagenesis as an additional etiologic source of mutation in head and neck cancer (17, 22, 24, 25, 43). The single-stranded DNA cytosine deaminase A3B is induced by high-risk HPV infection and a likely contributor to the overall APOBEC mutation signature in both HPV-positive and HPV-negative head and neck cancers (36, 38, 54). The studies here are the first to investigate A3B protein levels in head/neck cancer. In support of prior work dependent upon mRNA quantification (40), immunostaining results here demonstrate high A3B protein levels in HPV-positive OPSCC (Figure 1). This approach also provided a unique spatial perspective demonstrating strong overlap between A3B and p16 staining, suggesting that nuclear A3B levels may be a useful additional marker for high-risk HPV infection. Interestingly, HPV-positive OEDs showed similarly high A3B protein levels consistent with a model in which viral oncoproteins E6 and E7 combine to switch A3B expression rapidly from off-to-on. Thus, high-risk HPV infection not only contributes to classical oncogenic events such as p53 and Rb inactivation but also to high nuclear A3B levels and an elevated capacity to evolve.
The IHC studies here also demonstrated that A3B protein levels in HPV-negative OSCC are high and similar to those in HPV-positive OPSCC (Figure 2). This result is interesting because it demonstrates that similarly high A3B protein levels can be achieved by both viral and non-viral mechanisms. However, an analysis of HPV-negative low-grade OEDs revealed an intermediate A3B level, higher than normal adjacent-to-cancer epithelial tissue and virus-negative hyperplasia but statistically lower than high-grade OEDs and overt OSCC. This key result supported a progression model for A3B induction in HPV-negative oral tumorigenesis, where multiple genetic and/or epigenetic events combine to gradually turn-on A3B expression. Our results also indicated that OSCC tumor grade may be associated with A3B expression levels because grade 1 tumors showed significantly lower A3B protein levels than grade 2 or grade 3 tumors (Figure 3).
Our studies here have focused on A3B and do not exclude the possibility that other APOBEC3 enzymes such as A3A and A3H may also provide mutational fuel for both HPV-positive and HPV-negative cancers of the head and neck (20, 25, 39, 54, 59, 60). To our knowledge, monoclonal antibodies have yet to be developed to specifically distinguish A3A and A3B, and antibodies for A3H (though specific) are not robust enough for IHC experiments (46, 61). A3A and the catalytic domain of A3B are 92% identical at the protein level and the C-terminal epitope recognized by our rabbit mAb 5210-87-13 is shared by these two enzymes (46). However, several lines of evidence indicate that all (or nearly all) of the IHC signal in the head/neck experiments here is due to A3B. First, mRNA-based studies have demonstrated that A3B mRNA is expressed at much higher levels than A3A mRNA in multiple tumor types including cancers of the head/neck (24, 25). Second, A3B is the only APOBEC3 family member that localizes constitutively to the nuclear compartment of cells (46, 49, 62); endogenous A3A is cytoplasmic in myeloid cell types and cell-wide when overexpressed in heterologous systems (63). Although little cytoplasmic signal was detected here, only nuclear staining was used to generate quantitative A3B H-scores. Third, our recent studies on clear cell ovarian cancer were able to quantify both mRNA and protein levels in some of the same tumor tissues, and A3B (not A3A) mRNA levels correlated positively with A3B H-scores using the same mAb as used here and identical IHC protocols (49).
In addition, one of the strongest lines of evidence in favor of A3B (and not another A3 family member) in head/neck lesions is the significant positive association between expression of A3B and the proliferation marker Ki67 by IHC (Figure 4). Indeed, A3B is the only family member under cell cycle control as evidenced by direct regulation by the Rb/E2F signal transduction pathway (64–67). In further support of this mechanistic linkage, JC and BK polyomavirus T-antigen proteins trigger A3B upregulation through a mechanism most likely requiring deregulation of the RB/E2F cell cycle regulatory pathway (66, 68), and A3B was shown to interact with the G1/S regulatory protein CDK4 to disrupt CDK4-mediated nuclear import of Cyclin D1 (69). In addition, a large proportion of APOBEC signature mutations in cancer map to the lagging-strand template and thus associate broadly with DNA replication (70, 71).
Our studies raise the possibility that A3B IHC may be a marker for oral dysplasias more likely to become malignant. For instance, as high-grade OEDs are known to undergo malignant transformation more frequently than low-grade OEDs (72), high A3B protein levels may be an additional marker to help identify the subset of high-grade OEDs that are more likely to become carcinomas. Moreover, even though most moderate to high grade dysplasias are likely to be excised surgically, A3B may be a marker for lesions that ultimately continue to evolve and re-emerge as recurrent disease. The utilization of A3B protein levels as a marker for preneoplastic lesions may also be applicable to other cancer types such as breast cancer where non-invasive lesions (lobular carcinoma in situ) have lower A3B mRNA levels and less prominent APOBEC mutation signatures than more aggressive lesions (invasive lobular carcinoma) (73). More broadly, as A3B is overexpressed and APOBEC signature mutations are apparent in a large number of cancer types, the IHC approach described here may be useful for investigating and helping to identify higher-risk precancerous lesions as well as establishing associations with clinical features such as therapy responsiveness and overall survival.
Supplementary Material
Acknowledgements
We thank Brian Dunnette for sharing expertise regarding the use of the Aperio ScanScope XT and Ashley Gunderson and Alexandra Blixt, University of Minnesota Oral Pathology Laboratory, for assistance with specimen procurement and clinical information collection. These studies were supported by P01-CA234228 (to RSH). Salary support for PEW was provided by the UMSOD Summer Research Fellowship program. Salary support for MCJ was provided in part by T32-CA009138 and subsequently F31-CA243306. RSH is the Margaret Harvey Schering Land Grant Chair for Cancer Research, a Distinguished McKnight University Professor, and an Investigator of the Howard Hughes Medical Institute.
Footnotes
Declarations of Interest
R.S.H is a co-founder, shareholder, and consultant of ApoGen Biotechnologies Inc. The other authors have no conflicts to declare.
Supplementary information is available at Modern Pathology’s website.
References
- 1.El-Naggar AK, Chan JKC, Takata T, Grandis JR, Slootweg PJ. Tumours of the oral cavity and mobile tongue. In: World Health Organization (WHO) Classification of Head and Neck Tumours. International Agency for Research on Cancer (IARC). 4th Edition. Lyon: 2017; pp105–115. [Google Scholar]
- 2.Cohen N, Fedewa S, Chen AY. Epidemiology and demographics of the head and neck cancer population. Oral Maxillofac Surg Clin North Am 2018;30:381–95. [DOI] [PubMed] [Google Scholar]
- 3.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 4.Chow LQM. Head and Neck Cancer. N Engl J Med 2020;382:60–72. [DOI] [PubMed] [Google Scholar]
- 5.American Cancer Society. Key Statistics for Oral Cavity and Oropharyngeal Cancers. Available from: https://www.cancer.org/cancer/oral-cavity-and-oropharyngeal-cancer/about/key-statistics.html [Accessed April 22, 2020].
- 6.Sanderson RJ, Ironside JA. Squamous cell carcinomas of the head and neck. BMJ 2002;325:822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neville B, Damm D, Allen C, Chi A. Oral and Maxillofacial Pathology. 4th Edition ed. Elsevier, Inc: St. Louis, Missouri: 2016; pp355–93. [Google Scholar]
- 8.Mehanna H, Paleri V, West CM, Nutting C. Head and neck cancer--Part 1: Epidemiology, presentation, and prevention. BMJ 2010;341:c4684. [DOI] [PubMed] [Google Scholar]
- 9.Khariwala SS, Hatsukami D, Hecht SS. Tobacco carcinogen metabolites and DNA adducts as biomarkers in head and neck cancer: potential screening tools and prognostic indicators. Head Neck 2012;34:441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet 2008;371:1695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dok R, Nuyts S. HPV Positive Head and Neck Cancers: Molecular pathogenesis and evolving treatment strategies. Cancers (Basel) 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thibaudeau E, Fortin B, Coutlée F, Nguyen-Tan P, Weng X, Audet ML, et al. HPV prevalence and prognostic value in a prospective cohort of 255 patients with locally advanced HNSCC: A single-centre experience. Int J Otolaryngol 2013;2013:437815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pytynia KB, Dahlstrom KR, Sturgis EM. Epidemiology of HPV-associated oropharyngeal cancer. Oral Oncol 2014;50:380–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woo SB, Cashman EC, Lerman MA. Human papillomavirus-associated oral intraepithelial neoplasia. Mod Pathol 2013;26:1288–97. [DOI] [PubMed] [Google Scholar]
- 15.Lerman MA, Almazrooa S, Lindeman N, Hall D, Villa A, Woo SB. HPV-16 in a distinct subset of oral epithelial dysplasia. Mod Pathol 2017;30:1646–54. [DOI] [PubMed] [Google Scholar]
- 16.Lingen MW, Xiao W, Schmitt A, Jiang B, Pickard R, Kreinbrink P, et al. Low etiologic fraction for high-risk human papillomavirus in oral cavity squamous cell carcinomas. Oral Oncol 2013;49:1–8. [DOI] [PubMed] [Google Scholar]
- 17.The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillison ML, Akagi K, Xiao W, Jiang B, Pickard RKL, Li J, et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genome Res 2019;29:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cannataro VL, Gaffney SG, Sasaki T, Issaeva N, Grewal NKS, Grandis JR, et al. APOBEC-induced mutations and their cancer effect size in head and neck squamous cell carcinoma. Oncogene 2019;38:3475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faden DL, Thomas S, Cantalupo PG, Agrawal N, Myers J, DeRisi J. Multi-modality analysis supports APOBEC as a major source of mutations in head and neck squamous cell carcinoma. Oral Oncol 2017;74:8–14. [DOI] [PubMed] [Google Scholar]
- 21.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013;494:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet 2013;45:977–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 2013;45:970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012;149:979–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Law EK, Sieuwerts AM, LaPara K, Leonard B, Starrett GJ, Molan AM, et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci Adv 2016;2:e1601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meng F, Zhang L, Ren Y, Ma Q. The genomic alterations of lung adenocarcinoma and lung squamous cell carcinoma can explain the differences of their overall survival rates. J Cell Physiol 2019;234:10918–25. [DOI] [PubMed] [Google Scholar]
- 29.Bertucci F, Ng CKY, Patsouris A, Droin N, Piscuoglio S, Carbuccia N, et al. Genomic characterization of metastatic breast cancers. Nature 2019;569:560–4. [DOI] [PubMed] [Google Scholar]
- 30.Angus L, Smid M, Wilting SM, van Riet J, Hoeck A, Nguyen L, et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet 2019;51:1450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol 2004;4:868–77. [DOI] [PubMed] [Google Scholar]
- 32.Harris RS. Enhancing immunity to HIV through APOBEC. Nat Biotechnol 2008;26:1089–90. [DOI] [PubMed] [Google Scholar]
- 33.Harris RS, Dudley JP. APOBECs and virus restriction. Virology 2015;479–480:131–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002;418:646–50. [DOI] [PubMed] [Google Scholar]
- 35.Cheng AZ, Yockteng-Melgar J, Jarvis MC, Malik-Soni N, Borozan I, Carpenter MA, et al. Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat Microbiol 2019;4:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vieira VC, Leonard B, White EA, Starrett GJ, Temiz NA, Lorenz LD, et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio 2014;5:e02234–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mori S, Takeuchi T, Ishii Y, Kukimoto I. Identification of APOBEC3B promoter elements responsible for activation by human papillomavirus type 16 E6. Biochem Biophys Res Commun 2015;460:555–60. [DOI] [PubMed] [Google Scholar]
- 38.Mori S, Takeuchi T, Ishii Y, Yugawa T, Kiyono T, Nishina H, et al. Human Papillomavirus 16 E6 upregulates APOBEC3B via the TEAD transcription factor. J Virol 2017;91:e02413–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westrich JA, Warren CJ, Klausner MJ, Guo K, Chang-Wei L, Santiago ML, et al. Human papillomavirus 16 E7 stabilizes APOBEC3A protein by inhibiting cullin 2-dependent protein degradation. J Virol 2018;92:e01318–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warren CJ, Xu T, Guo K, Griffin LM, Westrich JA, Lee D, et al. APOBEC3A functions as a restriction factor of human papillomavirus. J Virol 2015;89:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mirabello L, Clarke MA, Nelson CW, Dean M, Wentzensen N, Yeager M, et al. The Intersection of HPV epidemiology, genomics and mechanistic studies of HPV-mediated carcinogenesis. Viruses 2018;10:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clifford GM, Tenet V, Georges D, Alemany L, Pavón MA, Chen Z, et al. Human papillomavirus 16 sub-lineage dispersal and cervical cancer risk worldwide: Whole viral genome sequences from 7116 HPV16-positive women. Papillomavirus Res 2019;7:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zapatka M, Borozan I, Brewer DS, Iskar M, Grundhoff A, Alawi M, et al. The landscape of viral associations in human cancers. Nat Genet 2020;52:320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith NJ, Fenton TR. The APOBEC3 genes and their role in cancer: insights from human papillomavirus. J Mol Endocrinol 2019;62:R269–R87. [DOI] [PubMed] [Google Scholar]
- 45.Qin T, Zhang Y, Zarins KR, Jones TR, Virani S, Peterson LA, et al. Expressed HNSCC variants by HPV-status in a well-characterized Michigan cohort. Sci Rep 2018;8:11458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown WL, Law EK, Argyris PP, Carpenter MA, Levin-Klein R, Ranum AN, et al. A rabbit monoclonal antibody against the antiviral and cancer genomic DNA mutating enzyme APOBEC3B. Antibodies (Basel) 2019;8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kujan O, Oliver RJ, Khattab A, Roberts SA, Thakker N, Sloan P. Evaluation of a new binary system of grading oral epithelial dysplasia for prediction of malignant transformation. Oral Oncol 2006;42:987–93. [DOI] [PubMed] [Google Scholar]
- 48.Lewis JS, Beadle B, Bishop JA, Chernock RD, Colasacco C, Lacchetti C, et al. Human papillomavirus testing in head and neck carcinomas: Guideline from the College of American Pathologists. Arch Pathol Lab Med 2018;142:559–97. [DOI] [PubMed] [Google Scholar]
- 49.Serebrenik AA, Argyris PP, Jarvis MC, Brown WL, Bazzaro M, Vogel RI, et al. The DNA cytosine deaminase APOBEC3B is a molecular determinant of platinum responsiveness in clear cell ovarian cancer. Clin Cancer Res 2020; February 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bu LL, Deng WW, Huang CF, Liu B, Zhang WF, Sun ZJ. Inhibition of STAT3 reduces proliferation and invasion in salivary gland adenoid cystic carcinoma. Am J Cancer Res 2015;5:1751–61. [PMC free article] [PubMed] [Google Scholar]
- 51.Wang YF, Ma SR, Wang WM, Huang CF, Zhao ZL, Liu B, et al. Inhibition of survivin reduces HIF-1alpha, TGF-beta1 and TFE3 in salivary adenoid cystic carcinoma. PloS one 2014;9:e114051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warren CJ, Westrich JA, Doorslaer KV, Pyeon D. Roles of APOBEC3A and APOBEC3B in human papillomavirus infection and disease progression. Viruses 2017;9:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep 2014;7:1833–41. [DOI] [PubMed] [Google Scholar]
- 56.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol 2000;182:311–22. [DOI] [PubMed] [Google Scholar]
- 57.Begum S, Westra WH. Basaloid squamous cell carcinoma of the head and neck is a mixed variant that can be further resolved by HPV status. Am J Surg Pathol 2008;32:1044–50. [DOI] [PubMed] [Google Scholar]
- 58.Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J Clin Oncol 2015;33:3235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Starrett GJ, Luengas EM, McCann JL, Ebrahimi D, Temiz NA, Love RP, et al. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat Commun 2016;7:12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen TW, Lee CC, Liu H, Wu CS, Pickering CR, Huang PJ, et al. APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat Commun 2017;8:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ebrahimi D, Richards CM, Carpenter MA, Wang J, Ikeda T, Becker JT, et al. Genetic and mechanistic basis for APOBEC3H alternative splicing, retrovirus restriction, and counteraction by HIV-1 protease. Nat Commun 2018;9:4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lackey L, Demorest ZL, Land AM, Hultquist JF, Brown WL, Harris RS. APOBEC3B and AID have similar nuclear import mechanisms. J Mol Biol 2012;419:301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Land AM, Law EK, Carpenter MA, Lackey L, Brown WL, Harris RS. Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J Biol Chem 2013;288:17253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cescon DW, Haibe-Kains B, Mak TW. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc Natl Acad Sci U S A 2015;112:2841–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ng JCF, Quist J, Grigoriadis A, Malim MH, Fraternali F. Pan-cancer transcriptomic analysis dissects immune and proliferative functions of APOBEC3 cytidine deaminases. Nucleic Acids Res 2019;47:1178–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Starrett GJ, Serebrenik AA, Roelofs PA, McCann JL, Verhalen B, Jarvis MC, et al. Polyomavirus T antigen induces APOBEC3B expression using an LXCXE-dependent and TP53-independent mechanism. MBio 2009;10:e02690–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Periyasamy M, Singh AK, Gemma C, Kranjec C, Farzan R, Leach DA, et al. p53 controls expression of the DNA deaminase APOBEC3B to limit its potential mutagenic activity in cancer cells. Nucleic Acids Res 2017;45:11056–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roelofs PA, Goh CY, Chua BH, Stewart TA, Jarvis MC, McCann JL, et al. The RB/E2F pathway controls expression of the cancer genomic DNA deaminase APOBEC3B. 2020. In process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCann JL, Klein MM, Leland EM, Law EK, Brown WL, Salamango DJ, et al. The DNA deaminase APOBEC3B interacts with the cell-cycle protein CDK4 and disrupts CDK4-mediated nuclear import of Cyclin D1. J Biol Chem 2019;294:12099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA. APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep 2016;14:1273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res 2016;26:174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iocca O, Sollecito TP, Alawi F, Weinstein GS, Newman JG, De Virgilio A, et al. Potentially malignant disorders of the oral cavity and oral dysplasia: A systematic review and meta-analysis of malignant transformation rate by subtype. Head Neck 2020;42:539–55. [DOI] [PubMed] [Google Scholar]
- 73.Lee JY, Schizas M, Geyer FC, Selenica P, Piscuoglio S, Sakr RA, et al. Lobular carcinomas in situ display intralesion genetic heterogeneity and clonal evolution in the progression to invasive lobular carcinoma. Clin Cancer Res 2019;25:674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.