

SUMMARY
Nuclear envelope assembly during late mitosis includes rapid formation of several thousand complete nuclear pore complexes (NPCs). This efficient use of NPC components (nucleoporins or “NUPs”) is essential for ensuring immediate nucleocytoplasmic communication in each daughter cell. We show that octameric subassemblies of outer and inner nuclear pore rings remain intact in the mitotic endoplasmic reticulum (ER) after NPC disassembly during prophase. These “inherited” subassemblies then incorporate into NPCs during post-mitotic pore formation. We further show that the stable subassemblies persist through multiple rounds of cell division and the accompanying rounds of NPC mitotic disassembly and post-mitotic assembly. De novo formation of NPCs from newly synthesized NUPs during interphase will then have a distinct initiation mechanism. We postulate that a yet-to-be identified modification marks and “immortalizes” one or more components of the specific octameric outer- and inner-ring subcomplexes that then template post-mitotic NPC assembly during subsequent cell cycles.
Keywords: nuclear pore complex, nuclear envelope, cell division, inheritance, live-cell imaging, spinning disk confocal microscopy, lattice light sheet microscopy, FIB-SEM
eTOC blurb
Chou et al. shows that cells inherit inner and outer ring nuclear pore subassemblies from their great-great-great-great-great-great-great-great grandmothers. These “immortal” structures, retained during mitosis in highly fenestrated ER sheets, template post-mitotic nuclear pore complex (NPC) assembly as ER sheets recoat the chromatin masses in each of the two daughter cells.
INTRODUCTION
Nuclear pores are essential pathways of communication between the genome and the cytoplasm (for recent review, see (Ungricht and Kutay, 2017). The nuclear pore complex (NPC)–the multi-component assembly that lines the double-membrane aperture – carries all molecular traffic in and out of the nucleus. In a cell undergoing open mitosis, the nuclear envelope retracts into the endoplasmic reticulum (ER), and the nuclear pore complexes dissociate into subassemblies of their constituent nucleoporins (NUPs) (Beaudouin et al., 2002; Daigle et al., 2001; Dultz et al., 2008; Ellenberg et al., 1997; Yang et al., 1997). Protein synthesis ceases during mitosis (Wheatley and Gray, 1988). Efficient post mitotic nuclear import and export, essential for synthesis of many new proteins including new pore constituents, thus requires prompt reincorporation of the existing NUPs into functional pores. New microscopies allow us to probe the mechanism of this critical postmitotic reassembly.
At the onset of mitosis, the ER reorganizes as it incorporates the dissolving nuclear envelope (Anderson and Hetzer, 2007; Kumar et al., 2019; Lu etal.,2009; 2011; Poteryaev et al., 2005; Puhka et al., 2012; 2007; Wang et al., 2013); the extent of remodeling varies with cell type, but fenestrated sheets appear to be a common feature (Puhka et al., 2012). As cells exit mitosis, ER sheets and to a lesser extent tubules attach to segregating chromosomes, and within a few minutes a complete nuclear envelope has reformed around the decondensing chromatin (Dultz et al., 2008; Lu et al., 2011). Interactions with chromatin and inner nuclear membrane proteins direct this process(Anderson et al., 2009; Ulbert et al., 2006), which originates at peripheral, fenestration and pore-component rich, “non-core regions” of the assembling nuclear membrane (off-axis regions free of residual mitotic spindle) and spreads towards the initially pore-free “core regions” along the axis between the spindle pole and the central spindle (Bilir et al., 2019; Dultz et al., 2008; Lu et al., 2011; Otsuka et al., 2018). These regional differences disappear during cell cycle progression.
Alternative descriptions of how elements of the mitotic ER regenerate nuclear membranes. One set of observations has led to the suggestion that membrane tubules extending from the ER network first contact chromatin, flatten, and expand to form the nuclear envelope sheet and to enclose nuclear pore complexes assembling directly on the chromatin surface (Anderson and Hetzer, 2007; LaJoie and Ullman, 2017; Wandke and Kutay, 2013). Other, more recent, observations suggest that ER sheets attach to decondensing chromatin and that nuclear pores appear within these attached sheets (Bilir et al., 2019; Lu et al., 2011). The attaching sheets include highly fenestrated parts of the mitotic ER; the diameter of the openings shrinks to about 40 nm as nuclear envelope assembly proceeds (Otsuka et al., 2018).The reduced fenestrations are probably sites of post-mitotic NPC assembly, which therefore will not require the membrane fusion events essential for de novo pore formation during interphase (Otsuka et al., 2016). When the spindle disassembles late in mitosis, ESCRT-III complexes mediate nuclear envelope closure at sites in the core region where spindle microtubules had created gaps (Appen et al., 2020; Vietri et al., 2015).
The NPC is an eightfold symmetric structure, with striking nuclear and cytoplasmic specializations attached to a scaffold with essentially identical, oppositely directed halves (for recent reviews, see (Hampoelz et al., 2019; Lin and Hoelz, 2019)). An “inner ring” occupies the center of pore aperture; nuclear and cytoplasmic “outer rings” decorate its outer rims. Fitting reconstructions from sub-tomogram averaged electron cryo-tomography with high-resolution structures derived from x-ray crystallography has provided a first-approximation molecular picture of the entire scaffold (Allegretti et al., 2020; Appen et al., 2015; Bui et al., 2013; Kim et al., 2018; Kosinski et al., 2016; Lin et al., 2016; Stuwe et al., 2015), and a significantly improved view obtained from single particle analysis of cryo-EM images of X. laevis at 0.6-0.8 nm resolution has recently been published (Huang et al., 2020; Zhang et al., 2020). The stable, 6-10 component assembly, known from its shape as the Y-complex, is the outer ring building block (Kelley et al., 2015). In vertebrates, each outer ring contains sixteen Y-complexes organized in two radially concentric annuli of eight Y-complexes each (Appen et al., 2015; Huang et al., 2020); thus, in vertebrates, a nuclear pore contains 32 Y-complexes. Each side of the inner ring likewise comprises sixteen subcomplexes, with the two sides in tight contact around the waist of the NPC (Appen et al., 2015; Zhang et al., 2020). An inner ring component, Nup155, associates with an outer ring component, Nup160, to form a set of eight bridges between each outer ring and the corresponding edge of the inner ring (Appen et al., 2015), and a second inner-ring component, Nup205 (and presumably its yeast homolog Nup192), extends into the outer ring, also pairing corresponding concentric annuli (Huang et al., 2020). The entire scaffold structure thus comprises 32 copies of each subunit, 16 facing towards the nucleoplasm and 16 facing toward the cytoplasm. Some additional complexities (e.g., a pair of paralogous inner ring proteins) may create exceptions to an exact counting. A separate set of membrane-embedded proteins anchors the scaffold firmly in the nuclear envelope (Huang et al., 2020).
Post-mitotic NPC assembly begins on the nuclear side (for recent reviews, see (LaJoie and Ullman, 2017; Ungricht and Kutay, 2017; Weberruss and Antonin, 2016)). Presence of a nucleoplasmic outer ring opposite a 40 nm double-membrane aperture is the earliest event readily detectable by fixed-cell electron tomography (Otsuka and Ellenberg, 2017). The protein ELYS, an NPC assembly factor, is thought to be a critical link between this initial outer ring and the underlying chromatin (Franz et al., 2007; Rasala et al., 2006). Subsequent morphological events are inner ring completion and cytoplasmic outer ring assembly, but the precise connection between these events and the sequential pathway of assembly derived from fluorescence microscopy (Dultz et al., 2008) remains to be established.
The use of lattice light-sheet microscopy (LLSM) across the complete volume of a cell affords an opportunity to dissect the NPC assembly pathway in living cells with greater sensitivity and higher time resolution than previously achievable (Aguet et al., 2016; Cai et al., 2017; Chen et al., 2014; Fritz-Laylin et al., 2017). We show here, in two different human cell lines, that parts of the NPC remain assembled during mitosis as large subcomplexes and that these surviving subcomplexes then appear in postmitotic NPCs. In particular, we show that an octamer of Y-complex subunits, which we propose to be an annulus of essentially complete Y-complexes, associates with the fenestrated sheets of the mitotic ER and moves with these ER regions as they initiate formation of a nuclear envelope in late anaphase. Most post-mitotic NPCs then contain one such octamer. We further show, by HaloTag pulse labeling, that this octameric subcomplex remains intact through subsequent rounds of cell division. An inner ring subassembly has similar properties. We suggest that block incorporation of these outer and inner ring subcomplexes, stabilized by an appropriate modification or other specialization and associated during mitosis with fenestrated ER, ensure the rapid assembly of post-mitotic NPCs.
RESULTS
Genome-edited cells expressing fluorescently tagged NUPs
Previous work has shown incorporation into nuclear pores of functional fluorescent chimeras of outer ring Y-complex Nup107 and Nup133 (Dultz et al., 2008; Lu et al., 2011; Otsuka et al., 2014) and inner ring Nup205 components (Otsuka et al., 2018). For the quantitative imaging studies reported here, we used CRISPR/CAS to homozygously replaced the NUP alleles of Nup107, Nup133 or Nup205 with NUP fused to eGFP or with the self-labeling HaloTag protein (Los et al., 2008), aiming to obtain physiological expression levels of the chimeras and to eliminate untagged copies of the target protein. The HaloTag chimeras were fluorescently tagged by incubation of Halo-NUP expressing cells with JF-HaloTag ligands (Grimm et al., 2015; 2017). We genome edited SVG-A human fetal glial cells to express eGFP-Nup133 and SUM159 human breast-cancer cells to express eGFP-Nup133, Halo-Nup107, Halo-Nup133 or Halo-Nup205, and eGFP-Nup133 together with either Halo-Nup107 or Halo-Nup205; examples of representative images can be found in Figs. 1, 2, 3 and Figs. S1–3). The nuclear pores in the genome-edited cells appeared to be fully functional and equivalent to those in parental cells, as judged by the extent of nuclear import of NLS-RFP bearing a nuclear localization signal (Fig. S3C) and by the similar time required to achieve post-mitotic nuclear separation (Fig. S3D,E). Absence of detectable nuclear deformation and normal cell growth kinetics were also used as proxies for the presence of functional nuclear pores, and we confirmed earlier reports of centromeric association of Nup107 and Nup133 (Figs. 1A, 2A, 3A, 3G, Figs. S2B, yellow arrows) but not of Nup205 (Fig. 3H, S2B) (Belgareh et al., 2001; Dultz et al., 2008; Loïodice et al., 2004; Lu et al., 2011; Zuccolo et al., 2007).
Figure 1. Cellular distribution of Nup133 during cell division. See also Figures S1, S3, S4 and Video S1.
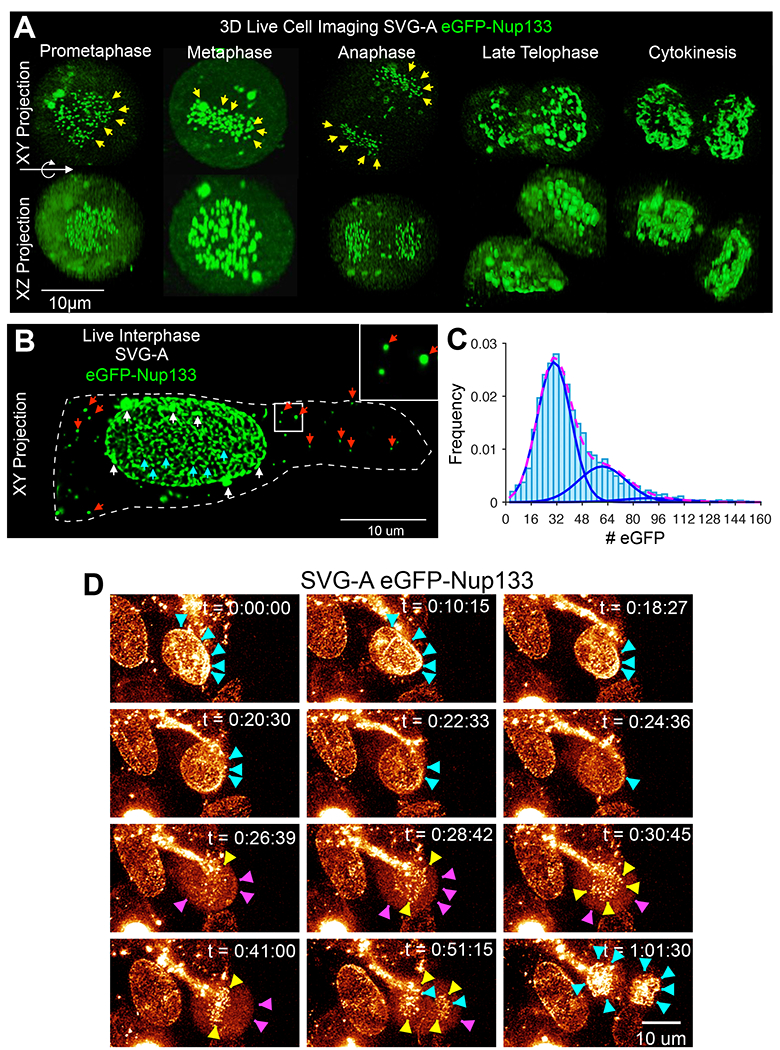
(A-C) Examples of live 3D LLSM images acquired during division of genome-edited SVG-A cells expressing eGFP-Nup133. (A) Examples corresponding to xy- and xz-projections along the optical axis during mitosis; eGFP-Nup133 at kinetochores (yellow) is highlighted. Scale bar represents 10 um. (B) Example of xy-projection obtained during interphase showing diffraction-limited spots of eGFP-Nup133 on the nuclear envelope (cyan) or in the cytosolic volume (red) and larger clusters located on the nuclear envelope (white). The spots in the cytosolic volume disappear during mitosis and reappear in telophase. Dotted line corresponds to the outline of the cell membrane where it contacts the glass coverslip. Enlarged view is from boxed region at left. Scale bar, 10 um.(C) Distribution of the number of eGPF-Nup133 molecules associated with the diffraction-limited spots on the nuclear envelope as in the example in (C). Data from 3691 diffraction-limited spots in three interphase cells obtained using calibrated LLSM. The fits from a simple Gaussian model correspond to contributions of 30 ± 11 and 61 ± 15 molecules per spot (mean ± SD).
(D) Relocalization of Nup133 during cell division. Related to Video S1. Snapshots from a time series, shown as xy-projections along the optical axis obtained by live cell 3D LLSM of a genome-edited SVG-A cell expressing eGFP-Nup133 undergoing cell division. Arrowheads indicate fluorescent Nup133 spots on the nuclear envelope of the cell during interphase (cyan), at kinetochores (yellow), and in the cytosolic volume excluded by the chromosome mass (magenta) during mitosis. Time stamps for each snapshot are indicated. Scale bar, 10 um.
Figure 2. Subassemblies containing Nup107, Nup133 or Nup205 during mitosis and interphase. See also Figures S2, S7, S8 and Video S2.
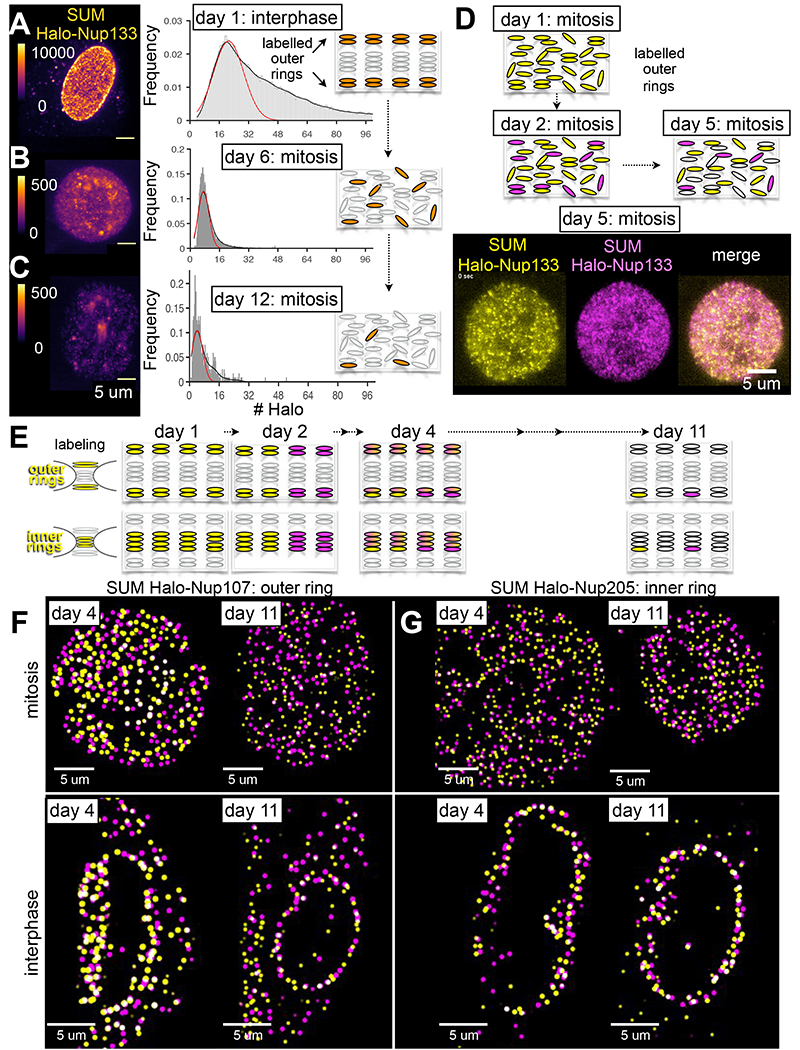
(A-C) Representative images and fluorescence intensity of spots in genome-edited SUM159 cells expressing Halo-Nup133 visualized at different days after brief incubation with Janelia Fluor ® 549 dye using 3D LLSM with single-molecule sensitivity. The images correspond to xy-projections derived from a single z-scan of a cell during interphase imaged immediately after dye labeling on day one (A) or during mitosis on days six (B) or 12 (C). The color-coding represents relative fluorescence intensity. Scale bar, 5 um. The histograms represent distribution of the number of Janelia Fluor ® 549 molecules associated with diffraction limited spots in the nuclear envelope of interphase cells (A) or in the cytosolic volume, excluding the chromosome mass, of metaphase cells (B,C). The fits correspond to the most prominent normal distributions centered at 21 ± 8 (51931 spots, 12 cells), 7 ± 3 (4642 spots, one cell) and 4 ± 3 molecules (466 spots, one cell). The cartoons highlight labeling with Janelia Fluor ® 549 of all outer ring NUPs during the first day (black), and detection in subsequent days of labeled octameric subassemblies (black) in the volume excluded from chromatin in cells undergoing mitosis.
(D) Snapshot from a time series taken by single-plane imaging of a metaphase genome-edited SUM159 cell expressing Halo-Nup133 that was subjected to brief incubation on the first day with Janelia Fluor ® 549 (yellow), with Janelia Fluor ® 635 (magenta) on the second day, and visualized on day five by LLSM. Scale bar, 5 um. The schematic representations highlight outcomes for different days obtained from the optical pulse-chase experiment.
(E) Schematic representation of pulse-chase experiments in which asynchronous populations of genome-edited SUM159 cells expressing Halo-Nup107 or Halo-Nup205 were subjected to brief incubation on the first day with Janelia Fluor ® X549 (yellow), with Janelia Fluor ® X646 (magenta) on the second day, and visualized volumetrically on days 4 and 11 by LLSM after chemical fixation; the models reflect the observed inheritance of inner and outer ring subassemblies in NPCs imaged during interphase.
(F, G) The images correspond to approximately equatorial single-plane 2D views showing 3D localized spots from z-scans acquired with LLSM of chemically fixed cells visualized during mitosis or interphase on the indicated days after the first labeling following the steps described in (E). Scale bars, 5 um.
Figure 3. Octameric subassemblies of Nup107, Nup133, and Nup205 in mitotic cells. See also Figure S3.
(A) Representative example of orthogonal views of an xz-projected image obtained using 3D live cell LLSM imaging during metaphase of a genome-edited SVG-A cell expressing eGFP-Nup133 and ectopically expressing the ER marker mCherry-Sec61β (magenta). The weak fluorescent spots (magenta) correspond to diffraction limited, octameric, eGFP-Nup133 objects; the bright green spots (yellow) correspond to eGFP-Nup133 associated with kinetochores. Enlarged view is from boxed region at upper left of the cell. Scale bar, 10 um.
(B) Histogram representing the distribution of number of eGFP molecules determined by LLSM calibrated for fluorescence intensity. Data from two cells are from 939 diffraction-limited spots in the volume excluded from them nuclear region. The fits correspond to a linear combination of normal distributions; the more prominent is centered at 9 ± 2 molecules; the less prominent, at 19 ± 3 molecules.
(C, D) Representative, approximately equatorial, single-plane 2D views after 3D deconvolution from z-scans acquired with LLSM obtained from chemically fixed genome-edited mitotic SUM159 cells expressing (C) Halo-Nup107 or (D) Halo-Nup205 (yellow) together with ectopic expression of ss-YFP-KDEL (magenta) used to mark the ER. The cells were imaged the same day after Janelia Fluor ® 549 labeling. Scale bar, 10 um.
(E, F) The LLSM was focused on a single plane and the samples were subjected to continuous imaging until complete beaching has been achieved. Each histogram corresponding to data from the two cells in panels C and D represent the frequency and the cumulative frequency distribution of bleaching steps for each of the diffraction-limited spots detected in the images. The number of bleaching steps was calculated as the ratio between the starting fluorescence amplitude (before bleaching) and the bleaching step size. n, number of diffraction limited spots analyzed.
(G,H) Representative, approximately equatorial, single-plane 2D views after 3D deconvolution from z-scans acquired with LLSM from a chemically fixed genome-edited mitotic SUM159 cells expressing (G) Halo-Nup107 (yellow) or (H) Halo-Nup205 (yellow) together with ectopic expression of ss-YFP-KDEL (magenta) used to mark the ER. The cells were imaged six days after Janelia Fluor ® X549 labeling. Scale bars, 10 um. Enlarged regions, scale bars, 3 um.
(I,J) The LLSM was focused on a single plane and the samples were subjected to continuous imaging until complete beaching has been achieved. Each histogram corresponding to data from three cells including those in panels G and H represent the frequency and the cumulative frequency distribution of number of molecules for each of the diffraction-limited spots detected in the images. The number of molecules was calculated as the ratio between the starting fluorescence amplitude (before bleaching) and the bleaching step size. n, number of diffraction limited spots analyzed.
Full incorporation into nuclear pores of fluorescently tagged Nup107, Nup133, and Nup205
Most of the detectable eGFP-Nup133 signal in genome-edited SVG-A cells was in diffraction-limited spots surrounding the nucleus (Fig. 1B, cyan arrows). As expected for the known stoichiometry of outer ring structures, the intensities of these spots peaked at ~30 molecules, with a second mode at ~61 molecules (Fig. 1C). As quantitative detection of 30 eGFP molecules was well within the experimental precision of our optical measurements (Fig. S4), we concluded that the two populations represented, respectively, optically resolved individual pores and two unresolved pores located within ~300 nm of each other. Less abundant and brighter, non-diffraction limited objects likely represented clusters of three or more pores (Fig. 1B, white arrows).
Fluorescent tag incorporation of Halo-Nup133 into interphase nuclear pores labeled with the Janelia Fluor ® 549 HaloTag ligand in SUM159 cells visualized immediately after with live-cell LLSM had a bimodal distribution, with a dominant first mode at ~21 molecules skewed towards higher values (Fig. 2A). Incorporation of less than ~32 labels is consistent with the incomplete covalent attachment of the Janelia Fluor ® 549 tag to genetically encoded Halo in living cells previously described for other Halo-tagged proteins (Lepore et al., 2019; Thevathasan et al., 2019; Urh and Rosenberg, 2012). Using live-cell LLSM we also compared the fluorescence signal of Halo-Nup107 with the inner ring Halo-Nup205 component during interphase in SUM159 cells labeled with the Janelia Fluor ® 549 fluorophore and found that they were similar (Fig. 4). LLSM visualization of chemically fixed samples of these cells, used as a way to facilitate the optical imaging, as fixation does not affect the fluorescence of Janelia Fluor ® 549, also showed bimodal distributions with first dominant modes at ~ 21 molecules. Concordance of the various molecular counting measurements with the values expected from the known nuclear-pore structure (32 copies of Nup107, Nup133 and Nup205) (for recent review, see (Schwartz, 2016)) further validates the use of these cells to follow nuclear pore breakdown and reassembly during mitosis.
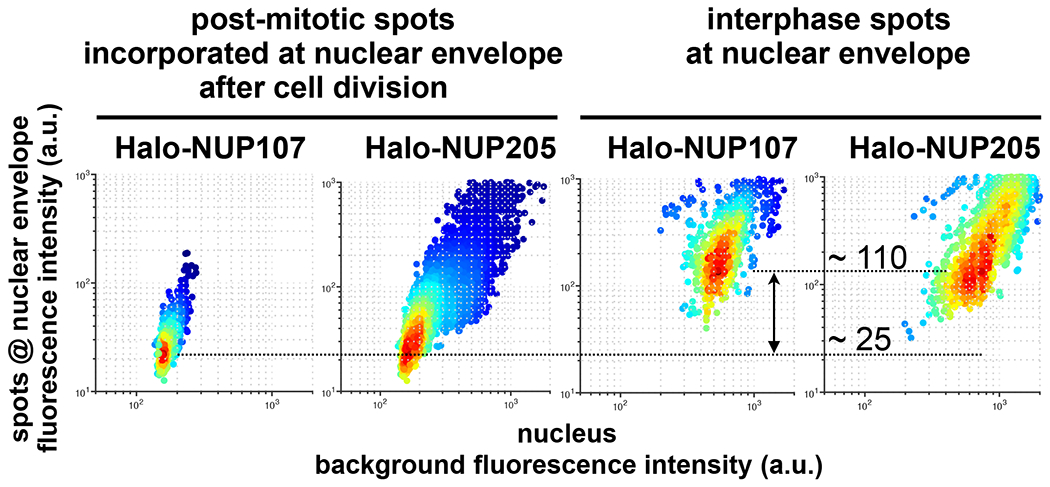
Figure 4. Nup107 and Nup205 at the nuclear margin of postmitotic and interphase cells.
Genome-edited cells expressing either Halo-Nup107 or Halo-Nup205 were briefly incubated with Janelia Fluor ® 549. Cells undergoing mitosis identified by single-plane live LLSM imaging were subjected to single-plane bleaching by 1000 consecutive exposures in the same single plane. Cells were allowed to continue through telophase and imaged by a single volumetric LLSM scan with planes spaced 1 um apart. Data from an adjacent interphase cell included in the same imaging volume are shown. The plot shows the maximum amplitude fluorescence intensity of the spots at the nuclear margin determined from the corresponding Gaussian fits and the local background. The spots are color coded for number of spots.
Mitotic disassembly of nuclear pores yields long-lasting outer and inner ring subassemblies
We followed the cellular distribution of eGFP-Nup133 during mitosis by live-cell LLSM. Volumetric projection from a time series of mitotic cells confirmed loss of the characteristic punctate fluorescence of eGFP-Nup133 at the nuclear envelope (Fig. 1D, cyan arrows and associated Video S1) and its replacement, as mitosis proceeded, by increased diffuse cytosolic fluorescence in the volume surrounding the condensed chromatin (Fig 1D, purple arrows). As expected during the postmitotic formation of the nuclear envelope (Dultz et al., 2008; Lu et al., 2011), shortly after the onset of anaphase eGFP-Nup133 reappeared as discrete patches at the non-core region of the dividing chromosomes (Fig. 1D, cyan arrows).
To probe the oligomeric state of Nup133 in the cytosol of mitotic cells, we isolated cytosol from mitotic, genome-edited SUM159 cells expressing eGFP-Nup133 and examined it by quantitative total internal reflection fluorescence (TIRF) microscopy (Fig. S4B, C) calibrated for single-molecule eGFP counting (Fig. S4D) (Cocucci et al., 2012; He et al., 2017). TIRF microscopy imaging of eGFP-Nup133 in the isolated cytosol showed that it contained diffraction-limited spots, mostly corresponding to monomers and dimers (Fig. S4B). Aiming to stabilize potentially larger oligomeric complexes of eGFP-Nup133 that might be present in the cytosol, the cells were treated with the membrane-permeable crosslinker DDS before cytosol extraction. TIRF imaging still showed spots corresponding to monomers and dimers (Fig. S4C). These observations are consistent with absence of large oligomeric complexes of Nup133 (and by extension, of other outer ring NUPs) in the cytosol of mitotic cells.
Since live-cell LLSM imaging is sensitive enough to detect moving fluorescent objects that would blur into diffuse signals when imaged by the slower regimes of conventional confocal microscopy, we used this approach to examine the oligomeric state of Nup133 in the cytosol of intact mitotic cells. We found, as expected, a large number of relatively bright, mobile, diffraction-limited, fluorescent eGFP-Nup133 spots in the volume of mitotic SVG-A cells excluded by chromatin (Fig. 3A, white arrows) but rather than representing monomers or dimers, the spot intensities had a principal mode corresponding to 8-10 eGFP-Nup133 molecules (Fig. 3B). Most of these spots colocalized with the mitotic ER, identified by ectopic expression of mCherry-Sec61β (Fig. 3A).
We confirmed that these spots were outer-ring octameric subcomplexes by labeling an asynchronous population of SUM159 cells expressing Halo-Nup107 with Janelia Fluor ® 549 HaloTag ligand and visualizing them after chemical fixation in order to facilitate the optical imaging. We searched for round mitotic cells and imaged them by multiplane LLSM. We detected diffraction-limited fluorescent spots in the volume excluded by chromatin and confirmed their colocalization with the mitotic ER, as identified by expression of ss-YFP-KDEL (Fig. 3C). We then configured the LLSM was to perform extensive single-plane acquisitions in order to bleach that plane completely during an extended series of exposures. A bleaching event represents the most probable step size from bleaching a single molecule; we can therefore calculate the total number of molecules within a spot from the ratio between the starting fluorescence intensity and the total number of bleaching steps (Coffman and Wu, 2014). The Halo-Nup107 spots typically bleached fully in 5-8 steps, indicating the presence of 8 copies of Nup107 (Fig. 3E). Less than this number of bleaching steps associated with a given spot can result from a combination of factors: the stochasticity of the fluorescence tagging of the Nup107 population, with an estimated 70-80% HaloTag labeling efficiency, and the reduced contribution from spots with weaker fluorescence signal from out of focus objects. Events with more than 8 bleaching steps might reflect the contribution of larger oligomers or optically unresolved objects.
We infer from these results that during mitosis, Nup107 and Nup133 remain together in Y-complex assemblies, which contain tightly associated single copies of Nup107 and Nup133 (Kelley et al., 2015; Schwartz, 2016). The most plausible structural interpretation of the eightfold multiplicity, further discussed below, is that the mitotic oligomers located in the chromatin-excluded volume correspond to one of the two octameric Y-complex annuli from a complete outer ring.
We also investigated the extent of mitotic disassembly of the inner ring, by visualizing Halo-Nup205 in the volume excluded by chromatin of chemically fixed mitotic, genome-edited SUM159 cells (Fig. 3D). As just described for Halo-Nup107, most diffraction-limited Halo-Nup205 spots also colocalized with the mitotic ER identified by expression of ss-YFP-KDEL (Fig. 3D) and on average they bleached in 6-8 steps (Fig. 3F), corroborating the presence of 8 copies of this component, leading us also to propose the presence of inner-ring mitotic subassemblies in the chromatin-excluded volume.
Octameric Nup133-containing subassemblies associate with fenestrated mitotic ER
The ER of interphase cells is a combination of membrane-bound sheets and tubes (Anderson and Hetzer, 2007; Kumar et al., 2019; Lu et al., 2009; 2011; Poteryaev et al., 2005; Puhka et al., 2007; 2012); during mitosis it becomes a structure enriched in layers of double-membrane sheets (Kumar et al., 2019; Lu et al., 2009; Puhka et al., 2012). The sheets comprise highly fenestrated regions contiguous with non-fenestrated zones (Puhka et al., 2012). Volumetric visualization by 3D block-face focused ion beam scanning electron microscopy (FIB-SEM) of mitotic parental SUM159 (Fig. 5A), parental SVG-A (Fig. S5A) or genome-edited SUM159 and SVG-A cells expressing eGFP-Nup133 (Figs. 6B,C) confirmed these structural properties. The double-membrane holes, best recognized in en face views (Fig. 5A–C and Figs. S5A, S6A), were of variable size and shape. The smaller ones, which ranged from 50 to 120 nm in diameter, appeared elliptical during prophase and metaphase (Fig. S6A) and became more circular by telophase (Figs. S5B, S6B). As previously shown (Puhka et al., 2007; 2012), interphase ER contains very few holes or fenestrae (Fig. S6C).
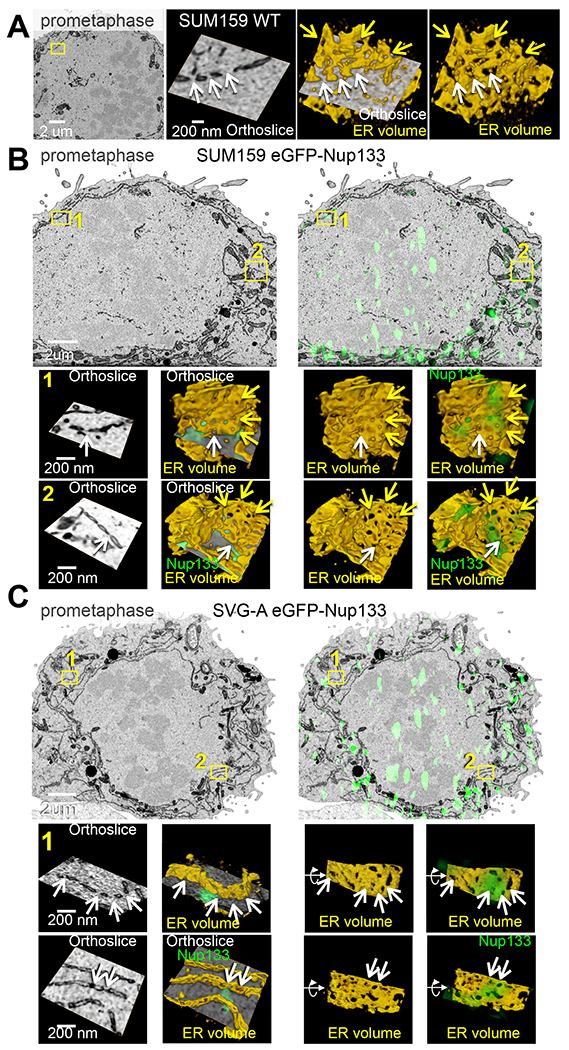
Figure 5. Ultrastructure of the ER during prometaphase imaged with 3D correlative fluorescence and electron microscopy. See also Figures S5 and S6.
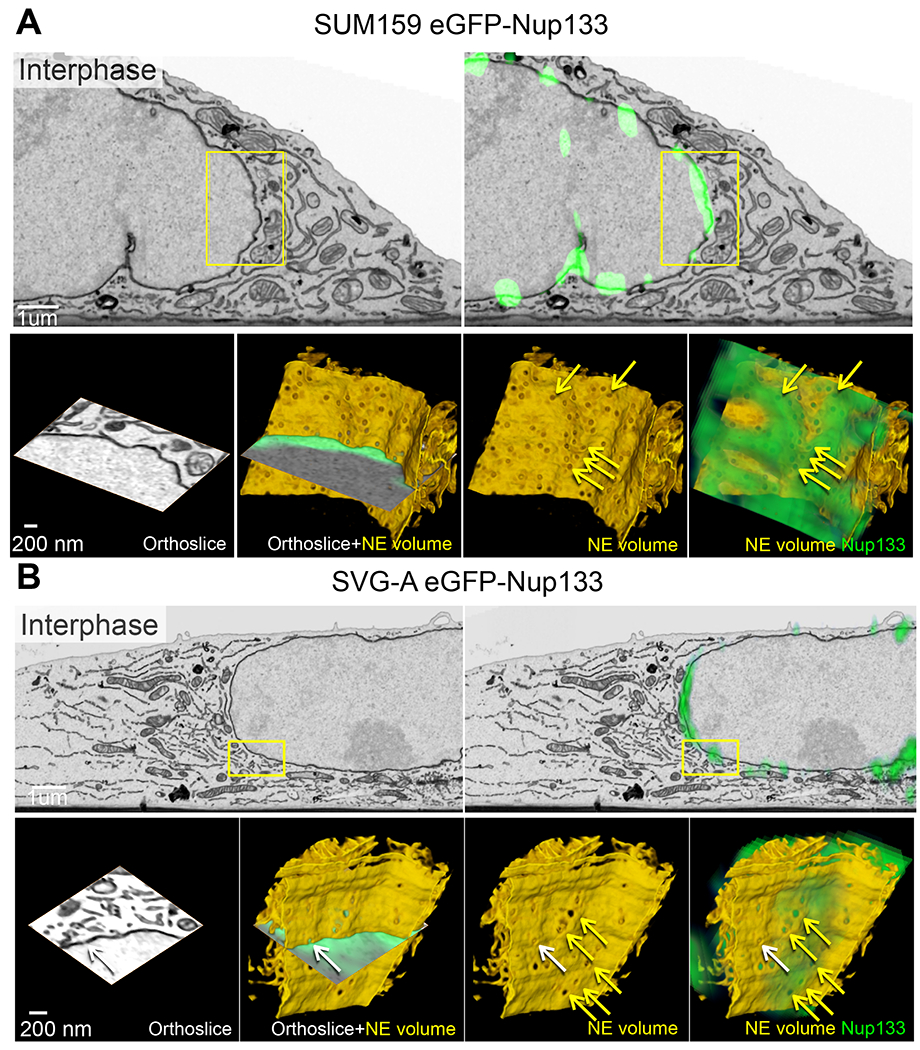
(A) 3D FIB-SEM of parental SUM159 cells during prometaphase illustrates fenestrations in ER sheets. Enlarged views are from boxed region. The yellow arrows point to fenestrations within the segmented ER sheet; the white arrows, to fenestrations at the intersection of the orthoslice and the segmented membrane. Scale bars, 1 um and 200 nm.
(B, C) Correlative 3D spinning disc confocal fluorescence imaging with FIB-SEM-CLEM of genome-edited SUM159 (B) and SVG-A (C) expressing eGFP-Nup133 during prometaphase shows colocalization of fluorescence (green) from eGFP-Nup133 with fenestrations in ER sheets. Enlarged views in (B,C) are from boxed regions; enlarged boxed regions in (C) are rotations from the en-face views. The yellow arrows point to fenestrations within the segmented ER sheet; the white arrows, to fenestrations at the intersection of the orthoslice and the segmented membrane. Larger green spots within the nuclear region are projected signals from adjacent kinetochores. Scale bars, 1 um and 200 nm for (B) and 2 um and 200 nm for (C), respectively.
Figure 6. Ultrastructure of the nuclear envelope during interphase imaged with 3D correlative light fluorescence and electron microscopy. See also Figures S5 and S6.
(A,B) Correlative 3D spinning disc confocal fluorescence imaging with FIB-SE-CLEM shows colocalization of fluorescence (green) from eGFP-Nup133 with the pore-containing nuclear envelope during interphase in genome-edited SUM159 (A) or SVG-A cells (B) expressing eGFP-Nup133. Enlarged views are from boxed regions. The yellow arrows point to nuclear pores within the segmented nuclear envelope; the white arrow, to a nuclear pore at the intersection of the orthoslice and the segmented membrane. Annulate lamellae are absent in these cells. Scale bars, 1 um and 200 nm.
We used 3D correlative light and electron microscopy (CLEM) to relate the location of the fluorescent, octameric, eGFP-Nup133 spots to ultrastructure visualized by FIB-SEM. We aligned images taken with spinning-disk confocal microscopy with FIB-SEM images as described in Methods and validated correct alignment by showing that the eGFP-Nup133 fluorescence at the nuclear margin mapped to the nuclear envelope seen in the FIB-SEM images of interphase SUM159 and SVG-A cells (Fig. 6A,B). Similarly registered fluorescence microscopy and FIB-SEM volumes of mitotic SUM159 or SVG-A cells showed positive correlation of eGFP-Nup133 fluorescent spots with fenestrated regions of the ER (Fig. 5B,C). About 80% of the fluorescent spots mapped to ER, and of those ~ 80% mapped to fenestrated sections enriched with smaller openings (Fig. S6D,E). The openings appeared smooth, but did not have features associate with fully formed nuclear pores on the nuclear envelope of interphase cells (Fig. 6B, Fig. S5C,D). Optical imprecision of the spinning-disk point-spread function (ca. 300 x 300 x 800 nm) and potential deformations introduced during the resin embedding procedure for FIB-SEM precluded direct correlation of the octameric Nup133 spots with the ER holes.
Cells progressively lost ER-associated, octameric Nup107 and Nup133 spots during late anaphase and telophase and instead acquired large, non-diffraction limited florescent patches in the non-core region of the assembling nuclear envelope, adjacent to condensed chromatin at sites distinct from centromeres, as described previously for these and other NUPs (Belgareh et al., 2001; Dultz et al., 2008; Loïodice et al., 2004; Lu et al., 2011; Zuccolo et al., 2007) (see representative example in Fig. 1D and Video S1). The patches colocalized with mCherry-Sec61β, which marked the ER and nuclear envelope (Fig. S3A). The regions with relatively weak mCherry-Sec61β fluorescence colocalizing with eGFP-Nup133 were always contiguous with regions of stronger mCherry-Sec61β signal devoid of eGFP-Nup133. Consistent with these live-cell imaging data were 3D CLEM images of cells in telophase showing that the eGFP-Nup133 patches were at non-core regions containing double membranes with densely arrayed, circular, double-membrane fenestrations (Fig. S6B), 50-100 nm in diameter (Fig. S6B), contiguous with ER and nuclear envelope devoid of nuclear pores (Fig. S6G–H). By late telophase, the Nup133 fluorescent patches had broken into smaller spots, and diffraction limited Nup133 spots appeared distributed around the chromatin (Fig. S3), in agreement with previous observations (Dultz et al., 2008; Lu et al., 2011). We infer from these observations that the octameric Y-complex subassemblies cluster into patches of newly assembled nuclear pores (see below) in late anaphase and ultimately disperse into daughter-cell nuclear pores.
Inner and outer ring subassemblies incorporate into newly formed post mitotic nuclear pores
We used a pulse-chase labeling strategy to determine that single, stable, outer- and inner-ring octamers incorporated into individual newly formed, post-mitotic nuclear pores. We pulse-labeled with Janelia Fluor ® 549 cells expressing Halo-Nup107 and then searched for dividing cells. Immediately after a single cell division, both the octamer and monomer pools are fully labeled. Before volumetric, live-cell LLSM imaging, we carried out single-plane photobleaching by extensive consecutive imaging of the same plane of a cell during prometaphase. We then imaged in 3D the same cell during telophase, using sparse one micron z sampling. The single-plane photobleaching did not interfere with cell division, but it gave substantially enhanced signal-to-noise by effectively eliminating fluorescence from mobile dye and from the rapidly diffusing “monomeric” Y-complexes, most of which passed through the chosen plane during the bleaching period. The less mobile, ER-associated, octameric pore subassemblies were largely unaffected. The principal modes of the fluorescence intensity distribution of the spots of Halo-Nup107 appearing post-mitotically in the nuclear envelope were about 25% of the intensity of the diffraction limited spots located in the nuclear envelopes of interphase cells expressing Halo-Nup107 imaged in the same field of view (Fig. 4). This is the expected incorporation of fluorescent NUPs into NPCs if only octamers but not monomers are visible. We conclude that most daughter-cell NPCs that form post-mitotically from pre-existing NUPs during nuclear membrane reassembly include a single, stable, octameric Y-complex subassembly transmitted from the mother cell.
Using genome-edited SUM159 cells expressing Halo-Nup205 (Fig. S2) to verify that inner ring octameric subassemblies likewise incorporate post mitotically into newly formed NPCs, we carried out pulse-labeling experiments with photobleaching similar to those described above for Halo-Nup107. We found diffraction limited, fluorescent Nup205 spots in the nuclear margin of post-mitotic cells of similar intensity as Halo-Nup107 imaged after the bleach and labeling pulse (Fig. 4). Comparison of their relative fluorescence intensities with those from bona-fide NPCs in interphase cells showed a fractional intensity distribution (~ 25%), similar to that obtained for Nup107. We conclude that like the stable, octameric, outer ring subassembly, an octameric inner ring subassembly also survived breakdown and reformation of the nuclear membrane during mitosis, to then incorporate into daughter-cell nuclear pores.
Inner and outer ring subassemblies survive several cell division cycles
The observations described in the previous sections suggest that post-mitotic daughter-cell NPCs contain nuclear porins derived from two pools -- the cytosolic pool of individual subunits and the ER-associated pool of octameric inner and outer ring subassemblies. We probed the long-term stability of the outer ring octameric Y-complex subassemblies with an optical pulse-chase experiment based on transient incubation with Janelia Fluor ® dyes and live cell LLSM imaging at various times after the pulse labeling. When we briefly incubated a population of unsynchronized, primarily interphase, genome-edited SUM159 cells expressing Halo-Nup133 with Janelia Fluor ® 549 and imaged immediately by LLSM, we found in the nuclear envelope the fluorescence intensity distribution with a principal mode at ~21 molecules described above (Fig. 2A). We then allowed the cells to grow and divide for up to eleven days, during which they underwent approximately one cell cycle per day. At selected consecutive days during this period, we imaged mitotic cells, focusing our analysis on the volume excluded from chromatin.
Rapid multi-plane imagining of Halo-Nup133 expressing, mitotic SUM159 cells on days 6 and 12 post labeling with Janelia Fluor ® 549 showed fluorescent spots dispersed in the chromatin-excluded volume whose principal modes of intensity were centered on ~7 molecules (Fig. 2B) and ~ 4 molecules (Fig. 2C), respectively. This imaging could not be done immediately post labeling, because at this time point the high background fluorescence due to freely diffusing monomeric fluorescently tagged Halo-Nup133 impaired the background correction required for individual spot fluorescence quantification. The smaller number of Halo-Nup133 molecules detected 12 days post-labeling as an oligomeric complex might be due to a low level of proteolytic cleavage of the Halo domain and release of the fluorescent tag, as well as potential slow exchange within the octameric complex of tagged Nup133 with newly synthesized untagged molecules. This result suggests that the fluorescently tagged Nup133 within the octameric subassemblies did not exchange (or exchanged only very slowly) with unlabeled copies (presumably in the form of Y-complexes) synthesized during the interphase periods throughout the 11-day imaging interval and that subassemblies themselves outlive consecutive cell cycles.
Confirmation of the long-term survival of the stable subset of outer ring octameric subassemblies came from another experiment in which we pulse labeled cells expressing Halo-Nup133 with Janelia Fluor ® 549 on the first day of an imaging series, followed by pulse labeling with Janelia Fluor ® 635 on the second day (Fig. 2D). Live-cell LLSM imaging of mitotic cells on day three after second labeling showed abundant, non-overlapping Halo-Nup133 spots uniquely labeled with one or the other of the fluorophores, distributed throughout the chromatin-excluded cell volume (Fig. 2D and Video S2). This result implies that Nup133 incorporated into an octameric structure on day one did not exchange with Nup133 from substructures assembled in subsequent cell cycles. These rapidly moving spots also appeared and disappeared during the recording interval as the time series was acquired from a single optical plane; these temporal fluctuations presumably reflected the combined remodeling and movement of the ER (Guo et al., 2018) together with diffusion of the octameric complexes along the surface of the ER membranes.
We further corroborated of the stability of the outer and inner ring octameric subassemblies and their association with mitotic ER by visualizing Halo-Nup107 or Halo-Nup205 in chemically fixed mitotic, genome-edited SUM159 cells expressing ss-YFP-KDEL six days after pulse labeling with Janelia Fluor ® X549. As just described for cells imaged immediately after labeling, most diffraction-limited Halo-Nup107 and Halo-Nup205 spots colocalized with the mitotic ER identified by expression of ss-YFP-KDEL (Fig. 3G,H); they contained an average of 5-8 molecules (Fig. 3I,J).
To explore the long-term stability of the outer and inner ring subassemblies and their incorporation into newly formed, post-mitotic NPCs, we used volumetric fixed-cell LLSM imaging. We pulse-labeled with Janelia Fluor ® X549 on the first day, followed by pulse labeling with Janelia Fluor ® X646 on the second day, and imaged on days 4, 8, 11 or 14 after the first day labeling (Figs. 2E, S7A, S8A). Mitotic cells during this period showed diffraction limited spots distributed throughout the chromatin-excluded volume (Figs. 2F,G, S7B, S8B). As expected for stable subassemblies unable to exchange subunits, we observed post-labeling dilution in the number of diffraction-limited spots (Table S1). We also found unresolved spots with both tags whose abundance decreased with time (Figs. S7C, S8C). As described above for Halo-Nup133 (Fig. 2A–C), we found that while most Halo-Nup107 or Halo-Nup205 spots contained ~ 5-8 molecules on day 8, the number of detected Halo ligand molecules decreased on subsequent days (Figs. S7C, S8C, Table S1).
Analysis of data from interphase cells imaged in the same optical pulse chase experiments led to similar conclusions (Halo-Nup107; Fig. 2F, S7B,C and Halo-Nup205;Fig. 2G and S8B,C; Table S1). As expected, on day 4 many of the HaloTag signals overlapped because of the high density of labeled NPCs, but the extent of dye overlap decreased as the number of consecutive cell divisions increased (Fig. 2G, S7B,C and S8B,C, Table S1). The fluorescence intensities were similar for outer and inner ring subassemblies in the chromatin-excluded volume of mitotic cells and the NPCs at the nuclear envelope of interphase cells.
DISCUSSION
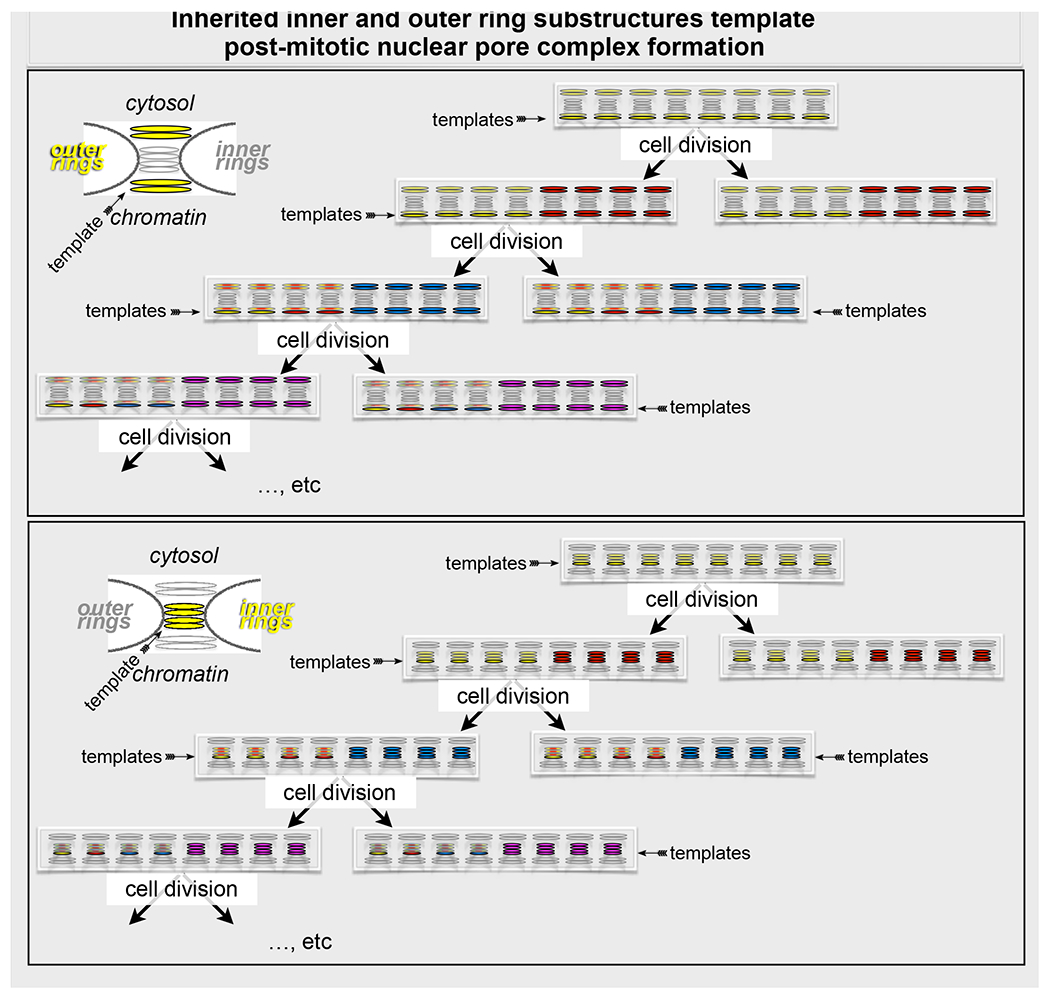
The most striking conclusion from the experiments described here is that even after ~10 cell divisions, progeny cells contain intact outer and inner ring nuclear pore subassemblies inherited from their great-great-great-great-great-great-great-great grandmothers (Fig. 7). Our work demonstrates that these structures, retained during mitosis in highly fenestrated ER sheets, participate in post-mitotic NPC assembly as ER sheets recoat the chromatin masses in the developing nuclear envelopes in each of the two daughter cells. We therefore propose that templating, by preformed, effectively “immortal” subassemblies may account for the rapidity with which freely diffusing, dissociated NUPs incorporate.
Figure 7. Inheritance mechanism for post-mitotic NPC assembly.
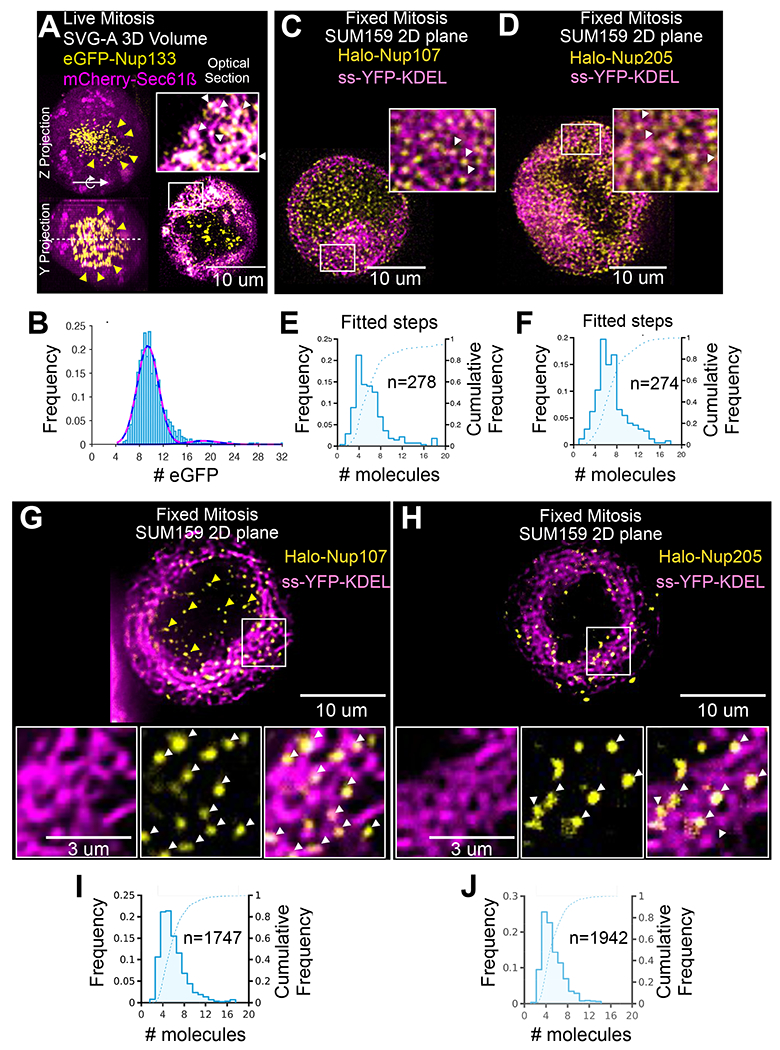
Cartoon summarizes a mechanism that explains the post mitotic formation of nuclear pore complexes. It represents the assembly of nuclear pore complexes during two sequential cycles of cell division and highlights the mechanistic differences between post-mitotic and interphase incorporation to nuclear pore complexes of inner and outer ring components. The representation does not show the location of the inner and outer-ring octameric templates that remain intact in the mitotic ER after NPC disassembly during prophase. The precise locations of the octameric outer and inner ring templates within the mature NPC remain to be determined. Even after ~10 cell divisions, progeny cells contain intact outer and inner ring nuclear pore subassemblies inherited from their great-great-great-great-great-great-great-great grandmothers. These effectively “immortal” subassemblies then template post-mitotic NPC formation, largely in regions of the nascent nuclear envelope (“non-core” regions) displaced from the residual mitotic spindle. In this inheritance mechanism, each nuclear pore in the mother cell produces the templates for one nuclear pore in one of the two daughter cells; the same template is re-used during subsequent cell divisions.
We have found two distinct pore subassemblies associated with the mitotic ER. One contains eight copies each of outer ring proteins Nup107 and Nup133, both of them components of the 10-subunit Y-complex. Because Nup133 depends on Nup107 for inclusion in the Y-complex, the two must come together. In vertebrates, the outer ring comprises two radially concentric annuli of eight Y-complexes each (Huang et al., 2020; Lin et al., 2016). We suggest that the observed subassembly corresponds to one of these annuli. The cryo-tomographic reconstruction of a human nuclear pore shows that the Nup133 subunit contacts the nuclear envelope. Association with the ER might maintain this contact. Moreover, in yeast, each outer ring has only a single annulus of eight Y-complexes; in that structure, Nup133 also contacts the surface of the nuclear envelope. The other ER-associated subassembly also contains eight copies of the inner ring protein Nup205 (Nup192 in yeast). Somewhat incomplete covalent labeling of the HaloTag limits the precision of our current measurements, but a likely inference is that this object is also an eightfold annulus. Both outer and inner ring subassemblies then appear in the nuclear pores of the post-mitotic daughter cells.
The pulse-chase experiments showed that the stable outer and inner ring subassemblies from one round of cell division survive as intact entities during subsequent rounds. In both cases, we have also shown by successive pulse labeling with two distinct fluorophores that no new subunits incorporate into the stable subassemblies at interphase. That is, during interphase synthesis and assembly of new NPCs, two octameric subassemblies, one each from inner and outer rings acquire stabilizing interactions or modifications that ensure survival during subsequent cycles of pore assembly and disassembly.
In previous work, the limited time resolution of conventional optical microscopy has led to the conclusion that mitotic nuclear pore disassembly entails full dissociation of inner and outer rings, release into the cytosol of non-membrane bound NUPs, and release into the ER of membrane bound NUPs (Dultz et al., 2008). Optical blurring, over the exposure time required, has prevented detection of the punctate, ER-associated, fluorescent objects we have now observed by use of the rapid 3D acquisition and quantitative imaging afforded by LLSM microscopy. The new observations are otherwise consistent with previous observations from optical and electron microscopy (EM).
Kinetics of post-mitotic nuclear pore assembly in the developing nuclear envelope from optical microscopy has shown that arrival of inner ring components lags that of outer ring proteins (Dultz et al., 2008; Lu et al., 2011; Otsuka et al., 2014). This sequence of events correlates with data from recently published cryo-electron subtomogram averages of pores during the first 10 mins after onset of anaphase (Otsuka et al., 2018). Presumptive structural staging of the subtomograms (all with double-membrane pores) showed that one class of images had stained density on the nucleoplasmic side, consistent with the early recruitment of outer ring Y-complexes determined by live-cell fluorescence microscopy, as well as unidentified density filling the (~40 nm diameter) aperture. A second class had additional density in the central region of the pore, next to the membrane, consistent with the presence of part or all of an inner ring. Density at this stage, which included at least some elements of a cytoplasmic-face outer ring, was sufficient to detect 8-fold symmetry (which could have been present earlier). Remaining classes showed pore dilation to a mature diameter of ~ 60 nm and density consistent with other known substructures of a functional pore (Otsuka et al., 2018).
The trans-generational stability of the class of outer and inner ring subassemblies we have found suggests both a defined location within the mature pore structure and a specific modification or composition (beyond the documented Y-complex components for the outer ring and sub complexes of the inner ring). Association of the nucleoplasmic outer ring octameric subcomplex with chromatin mediated by ELYS and (potentially) with nuclear lamins and the apparent nucleation of assembly from this position makes the nucleoplasmic outer ring seen in the early class subtomograms an attractive candidate (Otsuka et al., 2018). We have no direct information about the mode of membrane association of the 8-subunit, outer ring subassembly with the fenestrated, mitotic ER sheets. Moreover, we have no evidence to suggest that it might lie over a double-membrane pore. To reconcile any proposal that this subassembly corresponds to the initial, nucleoplasmic ring seen by tomography, the most plausible sequence of events is attachment to the chromatin surface followed by capture of a pre-existing, small aperture stabilized by the dense material seen by cryo-electron tomography, which we postulate corresponds to an inner ring octamer. The ER could slide across the captured outer ring octamer, which would then need either to open up transiently or to dissociate locally from the ER to allow positioning of the hole. Data to distinguish among these or other possibilities are not yet available. Similar questions relate to positioning of the inner ring. This order of addition is consistent with previous data from live imaging optical imaging showing that during post mitotic recruitment to the nuclear envelope, Nup133 arrives before Pom121, and Pom121 arrives before Nup93 (inner ring) (Dultz et al., 2008).
Micronuclei, often observed in cancer, are nuclear aberrations derived from lagging chromosomes embedded in the microtubule spindle during cell division (Liu and Pellman, 2020). The efficient recruitment of ‘core’ nuclear envelope proteins and inefficient recruitment of ‘non-core’ nuclear envelope proteins and minimal assembly of NPCs characterize their defective post mitotic nuclear envelope formation (Liu and Pellman, 2020). Postmitotic access of ER sheets and tubules to the core region of the nuclear envelope through the bundles of spindle microtubule is hindered (Lu et al., 2009; 2011), and the same physical barrier has been proposed for the lagging chromosomes (Liu and Pellman, 2020). We surmise that at the time when the spindle microtubules depolymerize, the octameric inner and outer ring subassemblies have been mostly used for NPC formation and are depleted from the mitotic ER. Hence, the ER mitotic remnants that can now coat the lagging chromosome are unable to provide an adequate number of templates to ensure formation of sufficient number of NPCs on the micronuclei, resulting in aberrant nuclear envelopes and subsequent DNA defects eventually leading to chromothripsis (Liu and Pellman, 2020).
Previous discussions of post-mitotic nuclear pore assembly mechanisms have focused on two alternative pictures -- either attachment of a pre-pore to chromatin, followed by lateral engulfment as the ER recoats the new nucleus, or insertion of pore components into an already deposited double membrane (Anderson and Hetzer, 2007; Bilir et al., 2019; LaJoie and Ullman, 2017; Lu et al., 2011; Wandke and Kutay, 2013). The mechanism implied by the observations presented here and by the recently published EM tomography is to some extent a hybrid of these two alternatives. The fenestrations that become mature pore apertures are likely to preexist in the ER, which also appears to bring with it stable structures that template pore formation. Once nucleated, however, the mature pore appears to complete its assembly by addition of soluble components derived from the mother cell.
Our optical pulse-chase labeling to distinguish old versus new nucleoporins is analogous to the isotope pulse-chase experiments used to distinguish parental versus newly replicated DNA (Meselson and Stahl, 1958). Inheritance of a nucleating octamer (Fig. 7) appears to ensure rapid and accurate post-mitotic NPC assembly. This mechanism requires that the postulated modifications of the surviving outer and inner ring subassemblies in NPCs assembled entirely from dissociated components (e.g., those assembled de novo in interphase) occur following their incorporation into new NPCs. These modifications are presumably different from the global mitotic phosphorylation of nuclear envelope proteins and NUPs that enable NPC disassembly and its post-mitotic reversal by phosphatases that allows subsequent nuclear envelope formation and NPC assembly (Güttinger et al., 2009; Ungricht and Kutay, 2017).
In this subassembly inheritance mechanism, each nuclear pore in the mother cell produces the template for a nuclear pore in one of the two daughter cells; the same template is re-used during subsequent cell divisions (Fig. 7). We have not found evidence to support either of two alternative mechanisms. Our observations rule out a conservative mechanism of postmitotic NPC formation analogous to centriole duplication, as it would require during mitotic disassembly that all NPC components from a single nuclear pore in the mother cell remain together to nucleate or template another nuclear pore in the daughter cell. A dispersive mechanism is equally inconsistent with our data, since it would require partial or full dissociation into individual components of all NPC assemblies during mitosis, yielding NPCs with fully randomized components, an outcome that would have been particularly apparent after many cell divisions. We note that our experiments so far do not allow us to determine whether the inherited outer- and inner-ring subassemblies stay together or reassort; our observations also do not allow us to rule out the possibility that a small fraction of post-mitotically assembled NPCs might have formed using only NUPs from the soluble pool. Centrioles and centromeric nucleosomes are related examples of such “epigenetic” inheritance of subassemblies critical for faithful transmission of genetic information from mother to daughter and reliable expression of that information once received.
Limitations of the study
First, the full composition and molecular structures of the inherited, octameric, inner- and outer-ring substructures remain to be determined.
Second, typical durations for prometaphase (nuclear membrane breakdown) and metaphase (alignment of chromosomes on the metaphase plate) are 30-60 min and 2-10 min, respectively. Practical constrains imposed by the visualization protocols (e.g., the limited number of accessible optical channels and the requirement to minimize photodamage) did not allow us to distinguish prometaphase from metaphase by monitoring directly the position and organization of chromosomes. Thus, most mitotic images were from rounded prometaphase cells (absence of nuclear envelope, misaligned metaphase plate), raising the possibility that some nuclear pores might not have dissociated completely at the time of imaging. Incomplete dissociation would alter the NUP content per spot and might affect the total number of spots. Nonetheless, it would not affect the general conclusion that intact and stable octameric outer- and inner-ring pore substructures persist for many consecutive cycles of cell division.
A third limitation comes from uncertainty in estimating the number of divisions that a given cell has undergone from the time of NUP pulse labeling until the time of imaging. As a result, we do not have an accurate count for the number of inner and outer ring subassemblies surviving as a function of the number of cell divisions. This uncertainty, together with potentially incomplete disassembly during early prometaphase, can also account for the relatively wide range in the number of spots per cell, in different cells on the same coverslip and imaged at the same time after labeling.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Tom Kirchhausen (kirchhausen@crystal.harvard.edu).
Materials Availability
All materials generated in this study are available upon request.
Data and Code Availability
This study did not generate any unique large-scale datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
The mostly diploid SUM159 human breast carcinoma cells (Forozan et al., 1999) and SVG-A human fetal astroglial cells were kindly provided by J. Brugge (Harvard Medical School) (Chou et al., 2016b) and Walter J. Atwood (Brown University), respectively. Cells were incubated at 37°C and 5% CO2 in humidified incubators and routinely verified to be mycoplasma free using a PCR-based assay.
All cells were grown at 37°C and 5% CO2. SUM159 cells were cultured in DMEM/F-12/GlutaMAX, supplemented with 5% fetal bovine serum (FBS), 100 U/ml penicillin and streptomycin, 1 μg/ml hydrocortisone, 5 μg/ml insulin, and 10 mM HEPES, pH 7.4 (SUM medium). SVG-A cells were grown in Minimum Essential Medium (MEM) supplemented with 10% heat inactivated fetal bovine serum, penicillin and streptomycin.
METHOD DETAILS
Genome editing
Double-allele genome editing of SUM159 and SVG-A cells to incorporate eGFP or Halo at the N-terminus of Nup107, Nup133 or Nup205 was done with using CRISPR/Cas9 as previously described (Aguet et al., 2016; Chou et al., 2016a; He et al., 2017; Ran et al., 2013). Briefly, parental SUM159 cells were plated in 6-well plates and subsequent to overnight growth, the cells were transfected with 800 ng each of the donor plasmid, the plasmid coding for the Streptococcus pyogenes Cas9, and the free PCR product using Lipofectamin2000 (Invitrogen) according to the manufacturer’s instructions. A subset of monoclonal cell populations was screened two weeks later for successful incorporation at the genomic locus of eGFP by PCR using GoTaq Polymerase. The following steps were used during the HaloTag genome-editing procedure. After 7-10 days of the triple transfection, cells were incubated for 15 min with the fluorescent Janelia Fluor ® 646 HaloTag ligand (Grimm et al., 2015), and fluorescent cells expressing Halo chimeras were enriched by fluorescence-activated cell sorting (FACS) (SH-800S; Sony) using 100 μm microfluidics sorting chips (Sony). The fluorescent cells were grown for 7 to 15 days, then subjected to a second round of FACS, and single fluorescent cells were sorted into 96-well plates; a subset of the monoclonal cell populations was screened for successful incorporation at the genomic locus of Halo by PCR using GoTaq Polymerase.
The donor constructs used as templates for homologous recombination to repair the Cas9-induced double-strand DNA breaks were generated by cloning into a pUC19 vector the appropriate genomic DNA fragments upstream and downstream of the CRISPR target sequences and the sequence for eGFP or Halo, assembled using the Gibson Assembly Cloning Kit (Gibson et al., 2009). As an example, PCR reactions with primers F1-R1 and F3-R3 were used to amplify ~800 bp of genomic sequences upstream and downstream of the start codon of Nup133. Primer F1 and primer R3 contain complementary sequences to the pUC19 vector and the genomic sequences; primer R1 contains complementary sequences to the 5’ end of eGFP, and primer F3 contains complementary sequences to the 3’ end of eGFP and the 3x GGS linker. The eGFP sequence containing the GGS linker was amplified using primers F2-R2 and an eGFP expression plasmid as a template. The PCR fragments (F1-R1, F2-R2 and F3-R3) and Smal linearized pUC19 vector were purified by electrophoresis in 1% agarose gel and assembled using the Gibson Assembly Cloning Kit.
The target sequences overlapping the start codon ATG (underlined) at the genomic locus recognized by the single-guide RNA (sgRNA) are 5’-AAAAGGCTTTAGCCATGGAC-3’ for Nup107 and 5’-CTCTAAGATGGCGACGCCTT-3’ for Nup205. The target sequences are located upstream of Nup133 start codon 5’-CTGTCCTAGTCGCTGCTCCT-3’. The sgRNA containing the targeting sequence was delivered as PCR amplicons containing a PCR-amplified U6-driven sgRNA expression cassette (Ran et al., 2013).
The primers sequences to generate the donor constructs were:
Nup133 up F1: gaattcgagctcggtacccCAACTGGCCAGTTTTACCAAGC
Nup133 up R1: GACTCCAAGGAGCAGCGACT
eGFP-Nup133 F2: AGTCGCTGCTCCTTGGAGTCATGGTGAGCAAGGGCGAGGAGC
eGFP-Nup133 R2: GGAACCACCAGAACCACCAG
Halo-Nup133 F2: AGTCGCTGCTCCTTGGAGTCATGGCAGAAATCGGTACTGGCTT
Halo-Nup133 R2: GGAACCACCAGAACCACCAG
Nup133 down F3: GGTTCTGGTGGTTCTGGTGGTTCCTTCCCAGCCGCCCCTTCTCC
Nup133 down R3: gtcgactctagaggatccccAACATGGTGTGTGTTGGGGGTG
Nup107 up F1: gaattcgagctcggtacccCAAAAATGTGACTGACTCCTTC
Nup107 up R1: GGCTAAAGCCTTTTCCACAC
Halo-Nup107 F2: GTGTGGAAAAGGCTTTAGCCATGGCAGAAATCGGTACTGG
Halo-Nup107 R2: GGAACCACCAGAACCACCAGAACC
Nup107 down F3: GGTTCTGGTGGTTCTGGTGGTTCCGACAGGTCAGTACTGATGGTG
Nup107 down R3: gtcgactctagaggatccccCTTTAGAACAATGGCAGTCAC
Nup205 up F1: gaattcgagctcggtacccCGTTATTGATGCTGAGTGTTTG
Nup205 up R1: CTTAGAGGCGCACTAACAGAG
Halo-Nup205 F2: CTCTGTTAGTGCGCCTCTAAGATGGCAGAAATCGGTACTGG
Halo-Nup205 R2: GGAACCACCAGAACCACCAGAACC
Nup205 down F3: GGTTCTGGTGGTTCTGGTGGTTCCGCGACGCCTTTGGCGGTAAA Nup205 down R3: gtcgactctagaggatccccCCTACTATGCGACAGCAGTG
The primers used to identify by PCR cells expressing eGFP-Nup133 or Halo-Nup133 were:
Forward: CACCTAGGCGCAGTCAAGAA
Reverse: AAGGGGCGGCTGGGAA
The primers used to identify by PCR cells expressing Halo-Nup107 were:
Forward: TGCACCTGTAGGCTGAAGT
Reverse: GCGGCTTTTTCCCCATTTGT
The primers used by PCR to identify cells expressing Halo-Nup205 were:
Forward: ACGATGAACGTCGGATCGAG
Reverse: AAGCCTCACTGCCTGGAATC
Ectopic expression
SUM 159 and SVG-A cells stably expressing mCherry-Sec61β were generated by transfection with a plasmid encoding mCherry-Sec61β (Lu et al., 2009), then grown for seven days in the presence of 1 mg/ml Geneticin (G418) and finally selected by FACS. Sorted cells were maintained in culture media supplemented with 100U/ml penicillin and streptomycin and 1 mg/ml G418.
Transduction of the retroviral vector encoding the NLS-RFP reporter protein (kind gift of Dr. Shiwei Liu and Dr. David Pellman) was used to generate cell populations of SVG-A, SVG-A-Nup133-eGFP, SUM159 or SUM159-Nup133-eGFP expressing NLS-RFP. Transduced cells were grown for eight days, followed by selection of cells expressing RFP using FACS. Sorted cells were maintained in the medium specific for SVG-A or SUM 159 cells.
Cells transiently expressing ss-YFP-KDEL were generated using ECO Transfection Reagent to transfect a plasmid encoding ss-YFP-KDEL (Valm et al., 2017).
Western Blotting
Western blot analysis for the SVG-A cells was performed as described (Aguet et al., 2016) using a rabbit monoclonal antibody specific for Nup133 at 1:2000 dilution (ab155990, Abeam) or a rabbit monoclonal antibody specific for actin at 1:2000 dilution dissolved in Tris-buffered saline (TBS, 50 mM Tris-Cl, pH 7.6; 150 mM NaCl) containing 0.05% Tween 20 (TBST) and 5% nonfat dry milk. Cells were trypsinized, washed in ice-cold PBS twice and resuspended in 4°C RIPA lysis buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with a protease inhibitor cocktail and incubated on ice for 10 minutes. The lysate was centrifuged for 15 min at 13,000 rpm at 4°C and the supernatant collected. SDS-PAGE followed by electrophoretic transfer to PVDF membranes was done using a wet transfer device (Bio-Rad) at 100V for 80 min. Afterwards, the membranes were incubated for 1 hr at room temperature with TBS including 5% non-fat dry milk and 0.05% Tween20 (TBST), followed by incubation overnight at 4°C with the antibodies. After four 15 min washes with TBST, the membranes were incubated overnight at 4°C with 1 mg/ml of Donkey anti-rabbit IgG HRP-conjugated secondary antibody. After additional, eight 15 min washes with TBST, the membranes were imaged with Amersham Imager 600 RGB system (GE Healthcare Life Sciences) using the LumiGLO Chemiluminescent Substrate (KPL).
Western blot analysis for the SUM159 cells was done as follows: cells were lysed in cell lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% (v/v) NP-40, 0.5 mM EDTA, 0.5% (v/v) sodium deoxycholate, pH 8) with Halt protease inhibitor for 30 min. The lysates were centrifuged at 14,000 × g for 15 min at 4°C, and the supernatant protein quantified with the BCA assay. Samples with the same protein concentration mixed with 1x SDS-PAGE loading buffer were denatured at 95°C for 5 min. resolved on a 10% SDS-polyacrylamide gel and transferred onto a PVDF membrane. Membranes were blocked with 5% nonfat milk in TBS containing 0.1% Tween-20 (TBST) for 30 minutes at room temperature, probed with rabbit polyclonal primary antibodies to Nup133 (1:1000 in 5% BSA/TBST; Abeam 233086) and β-actin (1:10,000 in 5% BSA/TBST; Abeam 8227) overnight at 4°C, washed thrice for 10 minutes each with TBST, incubated with goat anti-rabbit secondary antibody conjugated to horseradish peroxidase (1:10,000 in BSA/TBST) followed by washing three times for 10 minutes each with TBST. Membranes were imaged with BioRad Chemidoc MP using ECL substrate.
Validation of nuclear pore localization at the nuclear envelope
Parental and genome edited cells SVG-A or SUM 159 cells expressing eGFP-Nup133 were plated on #1.5, 25 mm diameter coverslips and incubated overnight at 37°C in the presence of 5% CO2. Cells were then fixed by incubation with 4% paraformaldehyde (PFA, Electron Microscopy Sciences) dissolved in PBS at room temperature for 10 min, followed by a brief wash with PBS and a permeabilization step using 0.1% Triton X-100 dissolved in PBS for 5 min. The cells were washed once with PBS followed by incubation in 5% BSA dissolved in PBS for 45 min, followed by incubation with a rabbit monoclonal antibody specific for Nup133 (ab155990, Abcam) diluted 1:100 in PBS containing 5% BSA for 1 hour at room temperature, followed by three consecutive washes with PBS supplemented with 5%BSA, each for 5 min. Cells were then incubated with an Alexa Fluor® 549 conjugated secondary goat-anti-rabbit antibody diluted 1:1000 in 5% BSA dissolved in PBS for 1 hour at room temperature. Finally, cells were washed 3 times with PBS, followed by incubation with Hoechst 33342 for 2 min and a final rinse with PBS. Cells were immediately imaged in 3D using the spinning disc confocal microscope (see fluorescence imaging) at 250 nm intervals in the Z direction, using filters sets for eGFP, Cy3 and Hoechst 33342. While in interphase the ab155990 antibody colocalized with eGFP-Nup133 and Halo-Nup133 on the nuclear envelope, in mitosis it failed to colocalize with the octameric sub-assemblies on the volume excluded from the chromatin.
Validation that timing of cell division is not affected
Approximately 2x104 parental or genome edited SVG-A or SUM159 cells expressing eGFP-Nup133 were plated in 8-well chambered coverglass slides per well and incubated at 37°C in the presence of 5% CO2 for 8 hours. Time-series were then acquired using a spinning disc confocal microscope built around a fully enclosed environmentally temperature-controlled chamber (see fluorescence imaging). Images were acquired using a computer-controlled piezo Z stage and a linear encoded X&Y platform (Applied Scientific Instruments), a 20x 0.5 NA PlanNeofluar objective (Carl Zeiss Microimaging, Inc.), with a 1.2x magnification lens added between the spinning disk head and the camera. Cells were imaged at 7 min interval for 24 hours and 3 fields of view were collected for each sample. The mitotic time was defined as the interval between the onset of mitosis and the end of cytokinesis (separation of two daughter cells).
Chemical crosslinking and TIRF imaging of mitotic cytosol
Genome-edited SUM159 cells expressing eGFP-Nup133 were plated into two 10 cm Petri dishes at 50% confluency with growth medium supplemented with 1mM thymidine and grown for 22 hours in order to arrest cells at G1. Thymidine was removed by three consecutive washes with pre-warmed growth media lacking thymidine, and incubated for another 20 hours in the presence of 2ug/mL Nocodazole.
Mitotic cells were detached from the Petri dishes by shaking, and the media with floating cells collected in 50mL Falcon tubes and centrifuged at room temperature (1000 rpm for 3 min); the cells were suspended in ice-cold PBS and centrifuged and resuspended three additional times. The final cell pellet was suspended in 100 uL of MES buffer (150 mM NaCl, and 25 nM 2-(N-morpholino) ethanesulfonic acid sodium salt (M5057, Sigma), pH 6.5 including 1 mM PMSF, 10 ug/ml aprotinin, 1 ug/ml leupeptin, 1 mM NaVO3, and 1 mM NaF and a protease inhibitor tablet (Roche) added to 10mL of MES buffer immediately prior to use.
Chemical crosslinking was done by adding to the resuspended cells the cell permeable crosslinker DSS (disuccinimidyl suberate) at a final concentration of 5mM and incubating at room temperature for 30 min. The crosslinking reaction was ended by addition of 1M Tris, pH 7.5 to a final concentration of 10mM and incubation at room temperature for 15 min.
Cytosol of cells subjected or not to chemical crosslinking was obtained by homogenization with a Dounce homogenizer, followed by two sequential centrifugation steps using a table-top centrifuge at 4°C at maximum speed for 10 min. The supernatant was immediately applied to the top of glass coverslips, glow discharged for 5 min and then visualized by TIRF imaging.
Lattice light sheet microscopy
Imaging of dividing cells were done by first growing the cells to confluency, followed by plating them onto 5 mm coverslips in a 35 mm culture dish at 40% confluency, 16 hour before imaging (Boucrot and Kirchhausen, 2007). The cells were imaged in phenol red free, Leibovitz’s L15 media supplemented with 20% FBS and 20 mM HEPES. SVG-A and SUM159 cells expressing fluorescently tagged Nup133 (eGFP or Halo), Nup107 (Halo), Nup205 (Halo), or mCherry-Sec61β were seeded on 5 mm round glass coverslips approximately 12 hours before imaging with the LLSM (Aguet et al., 2016). The samples were imaged as a time series in 3D using a dithered multi-Bessel lattice light sheet (details in Table 1). The inner and outer excitation NAs, number of imaging planes, sample step size, camera settings used to record fluorophore are detailed in Table 1.
Table 1. Experimental conditions used for the data acquired using lattice light sheet microscopy.
The listed data include inner and outer excitation NAs, number of imaging planes, sample step size and camera settings used to record each fluorophore.
| Figure | Camera | Mode (Lattice type) (Dithered) (Scan type) | Sample (37°C imaging temperature) | Fluorescent label | Excitation λ (nm) | Voxel volume (dx, dy, dz/ds nm3) | Image pixels (x,y,z) | (channel 1; channel 2) (Exposure time) (# time points) (imaging time + wait time /stack) |
Excitation N.A (inner, outer) |
|---|---|---|---|---|---|---|---|---|---|
| 1A 1B |
EMCCD (Andor, iXon Ultra 897) | multiBessel lattice Dithered Objective scan | SVG-A | eGFP-NUP133 mCherry-Sec61² | 488nm 560nm | 104x104x500 | 512x512x45 RAW 349x333x45 DECON | (488; 560) (47.95ms-mitosis, 97.4ms-cytokinesis; 47.95ms-mitosis) (70, 60; 70) (4.95s + 0s, 4.95s + 0s: 4.95s + 0s) | 0.505, 0.6 |
| 1C | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Objective scan | SVG-A | eGFP-NUP133 | 488nm | 104x104x500 | 512x512x45 RAW 512x410x45 DECON | 97.4ms 60 4.5s +0s | 0.505, 0.6 |
| 2 S1 Video | EMCCD (Andor, iXon Ultra 897) | multiBessel lattice Dithered Sample scan | SVG-A | eGFP-NUP133 | 488nm | 104x104x522/1000 | 512x512x100 RAW 512x1323x100 DECON | 49.62ms 71 5s + 120s | 0.35, 0.4 |
| 3A | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | SUM159 | Halo-NUP133 JF549, dye label on day1 | 560nm | 104x104x209/400 | 704x512x250 RAW 704x1329x251 DSKW | 47.4 ms 525s + 0s | 0.5, 0.55 |
| 3B | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | SUM159 | day6 after Halo-NUP133 JF549 day1 and JF635 day2 and JF635 day3 | 560nm 642nm | 104x104x209/400 | 512x1280x200 RAW 256x259x106 DSKW | 47.4 ms 172s + 20s | 0.5, 0.55 |
| 3C | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | SUM159 | day12 after Halo-NUP133JF549 day1 and JF635 day2 | 560nm 642nm | 104x104x209/400 | 704x512x250 RAW 704x1329x251 DSKW | (560nm, 642nm) (47.4 ms, 47.4ms) (5, 5) 2.5s +0s | 0.5, 0.55 |
| 3D S2 Video | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | SUM159 | day1 JF549-NUP133 and day2 JF635 NUP133 | 560nm 642nm | 104x104x150/288 | 1280x512x2 RAW | (560nm, 642nm) (47.95 ms, 47.95ms) (1000, 1000) 0.2s +0s | 0.5, 0.55 |
| 4A | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Objective scan | SVG-A | eGFP-NUP133 mCherry-Sec61β | 488nm 560nm | 104x104x500 | 512x512x45 RAW 355x365x45 DECON | (488nm, 560nm) (47.95ms ,47.95ms) (10, 10) 4.95s + 0s | 0.505, 0.6 |
| 4C | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | Fixed SUM159 | Halo-NUP107 ss-YFP-KDEL | 488nm 560nm | 104x104x209/400 | 512x488x101 RAW 227x776x102 DSKW 225x768x100 DECON | (488nm, 560nm) (47.6ms ,47.6ms) (2, 2) 10.1s + 0s | 0.5, 0.55 |
| 4D | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | Fixed SUM159 | Halo-NUP205 ss-YFP-KDEL | 488nm 560nm | 104x104x209/400 | 512x488x101 RAW 297x657x102 DSKW 297x648x100 DECON | (488nm, 560nm) (47.6ms ,47.6ms) (2, 2) 10.1s + 0s | 0.5, 0.55 |
| 5 | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | SUM159 | Halo-NUP107 or Halo-NUP205 | 560nm | 104x104x1045/2000 | 608x348x1 RAW Bleach 608x348x21 RAW Vol. 608x676x21 DSKW Vol. | 48.2ms-Bleach/Vol. 1000-Bleach; 6-Vol. 0.05s + 0s-Bleach; 1.05s + 15min-Vol. | 0.5, 0.55 |
| S3A | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | SUM159 | eGFP-NUP133 mCherry-Sec61β | 488nm 592nm | 104x104x209/400 | 768x512x250 RAW 768x1329x253 DSKW | (488nm, 592nm) (47.4ms ,47.4ms) (60, 60) 25s + 2min | 0.5, 0.55 |
| S4A | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Objective scan | NA | eGFP on coverslip | 488nm | 104x104x500 | 512x412x20 RAW | Variable exposure time (texp) 60 texp + 0s | 0.505, 0.6 |
| 2F, G; S7, S8 | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | Fixed SUM159 | Halo-NUP107 or Halo-NUP205 | 560nm 642nm | 104x104x104/200 | 700x512x301 RAW 1192x512x303 DSKW 1192x512x303 DECON | (560nm, 642nm) (200ms, 200ms) | 0.5, 0.55 |
| 3G,H | sCMOS (Hamamatsu, ORCA Flash 4.0 v2) | multiBessel lattice Dithered Sample scan | Fixed SUM159 | Halo-NUP107 or Halo-NUP205 ss-YFP-KDEL | 488nm 560nm | 104x104x104/200 | 700x512x400 RAW 1355x512x405 DSKW 1355x512x405 DECON | (488nm, 560nm) (200ms, 200ms) | 0.5, 0.55 |
Halo staining and cell imaging
Same day live-cell imaging
SUM 159 cells expressing eGFP-Nup133 together with Halo-Nup107 or Halo-Nup205 were plated overnight on 5 mm or 25 mm coverslips and grown in SUM 159 culture medium. The following day, cells were washed once with pre-warmed α-MEM and then incubated with 125 nM Janelia Fluor ® 549 HaloTag ligand dissolved in α-MEM for 15 min at 37°C. After 3 consecutive washes with α-MEM, the cells were imaged using Leibovitz’s L-15 Medium with lattice light-sheet or spinning disc microscopy.
Pulse-chase live-cell imaging
SUM 159 cells expressing Halo-Nup107 or Halo-Nup205 were plated in 24-well plates and grown in SUM159 culture medium. The following day, cells were washed once with pre-warmed α-MEM and then incubated with 125 nM Janelia Fluor ® 549 dissolved in α-MEM for 15 min at 37°C. After 3 consecutive washes with α-MEM, the cells were incubated with the SUM159 culture media for three additional days. The cells were then trypsinized and plated on 5 mm diameter coverslips (Bellco Glass), grown overnight in SUM medium, washed once with pre-warmed α-MEM (GIBCO) followed by incubation with 125 nM Janelia Fluor ® 646 HaloTag ligand in α-MEM for 15 min at 37°C. After three consecutive washes in α-MEM, the cells were imaged using lattice light-sheet microscopy. We inferred that the fluorescently labeled cells underwent normal cycles of division, from the similar increases in the numbers of cells exposed and unexposed to the fluorescent labels.
Fixed-cell imaging
SUM159 cells expressing Halo-Nup107 or Halo-Nup205 were plated and grown overnight in SUM159 culture media, washed once with pre-warmed α-MEM and then incubated with 125 nM Janelia Fluor ® 549 in α-MEM for 15 min at 37°C. After three consecutive washes in α-MEM, the cells were incubated with SUM159 culture media for additional two days. Cells were then trypsinized, plated and after overnight growth transfected with the ss-YFP-KDEL plasmid. After 8-10 hours of further growth, the cells were trypsinized and re-plated on cleaned 5 mm coverslips; following overnight growth, the cells were fixed at room temperature for 20 min with 4% PFA dissolved in PBS and imaged using lattice light-sheet microscopy using as medium phenol red free, Leibovitz’s L15 supplemented with 20% FBS and 20mM HEPES.
Pulse-chase fixed-cell imaging
For the experiments in (Fig. 3G,H), SUM159 cells expressing Halo-Nup107 or Halo-Nup205 were grown in SUM 159 culture medium. Confluent cells were fluorescently labeled with 125 nM Janelia Fluor ® X549 dissolved in α-MEM for 15 minutes at 37°C, then washed with pre-warmed medium three times for 10 minutes, then trypsinized and plated at 33% confluency, allow to grow for three days, then trypsinized, plated at 60% confluency and 12 hr later transfected and then grown for additional 24 hr. Cells were trypsinized, plated at 60% confluency and allowed to grow for 14-16 hr before fixation and imaging. Fixation at room temperature was done by incubation for 20 min with 4% paraformaldehyde dissolved in PBS, followed by incubation at room temperature with 30 mM glycine, pH 7.4 for 5 minutes at room temperature, washed three times with PBS and then imaged volumetrically using LLSM.
For the experiments in Figs. 2F,G, S11 and S12, SUM159 cells expressing Halo-Nup107 or Halo-Nup205 were grown in SUM 159 culture medium. On day one a 10-cm plate with cells at 33% confluency were incubated with 125 nM Janelia Fluor ® X549 dissolved in α-MEM for 15 minutes at 37°C, then washed with pre-warmed medium three times for 10 minutes. On day two, the cells were incubated with 125 nM Janelia Fluor X646 for 15 minutes at 37°C followed by the same wash. On day three, cells were trypsinized and plated at 33% confluency, and allowed to grow for four more days. On day 7, cells were plated at 33% confluence and allowed to grow for three days. On day 10, cells were plated at 33% confluency and allowed to grow for three days. Aliquots of cells on days 3, 7, 10 and 13 were trypsinized and plated at 60% confluency in 24-well plates containing 5 mm glass coverslips and allowed to grow for 14-16 hours to enrich for mitotic cells. These population of cells were fixed with 4% paraformaldehyde dissolved in PBS for 20 minutes at room temperature, followed by incubation with 30 mM glycine, pH 7.4 for 5 min at room temperature, washed three times with PBS and then imaged volumetrically by LLSM.
Correlative light-electron microscopy
Light microscopy
Photo Etched German Glass Coverslips (#1.5, 25mm, Electron Microscopy Science, Cat. 72265-50) were cleaned by sonication in 1M KOH for 15 min and then rinsed 3 times in distilled water. SUM159 or SVG-A cells were plated and allowed to reach 40% confluency after overnight incubation at 37°C in the appropriate medium.
Fluorescence microscopy
The cells were chemically fixed for 10 minutes at room temperature with 0.2% glutaraldehyde and 2.5% paraformaldehyde (PFA) dissolved in 0.1M PIPES pH 7.4 buffer. The cells were washed once with 0.1M PIPES buffer and then imaged in the same buffer using an inverted spinning disc confocal microscope (Yokogawa Electric) set up (3I, Denver, CO) equipped with a 1.2x magnification lens. Imaging was performed using a 512 x 512 pixels cooled EMCCD camera (QuantEM:512sc, Photometrics) (Cureton et al., 2012). A montage of 25 adjacent low-magnification bright field images mapping the gridded numbers was first acquired using a computer controlled piezo Z stage and a linear encoded X and Y platform (Applied Scientific Instruments, Eugene, OR) using a 10x, 0.45 NA air Plan Apochromat objective (Carl Zeiss MicroImaging, Inc. Thornwood, NY). The X and Y positions of the selected cells were marked using Slidebook 6.0 (Intelligent Imaging Innovations). The selected cells were imaged in 3D at 200 nm intervals in the Z direction and ~130 nm/pixel in the X/Y directions using a 100x, 1.4 NA Plan Apochromat oil immersion objective (Carl Zeiss Microimaging, Inc.). Excitation lasers at 488 and 561 nm (Coherent, Santa Clara, CA) were used with 525/50 and 607/36 emission filters respectively (Semrock, Rochester, NY).
Electron Microscopy
Fixation and Staining
Following fluorescence imaging, the samples were immediately incubated in 0.1M PIPES pH 7.4 buffer including 2.5% PFA and 2% glutaraldehyde at 4°C for 16 hours, then washed twice with a solution containing 0.1M PIPES pH 7.4 buffer. A 2% OsO4 aqueous solution dissolved in 0.1 M PIPES, was used to stain the cells for 1 hour at RT, followed by another 1 hour at RT incubation in a solution containing 0.1 M PIPES, pH 7.4 and 2.5% potassium ferrocyanide. The cells were then washed three times at five-minute intervals with ultrapure water. It was then followed by a 30 minute incubation at RT with a filtered (Whatman, 0.2 μm) freshly prepared solution of 1 % thiocarbohydrazide made by dissolving it at 60°C for 15 minutes. The cells were consecutive washed three times in 5 minute intervals, followed by a second OsO4 staining with 2% OsO4 in ultrapure water for 1 hour at RT. The cells were again washed three times in five minute intervals with ultrapure water, followed by two washes with 0.05M maleate buffer, pH 5.15 (Sigma-Aldrich), and finally incubated with 1% Uranyl Acetate dissolved in 0.05M maleate buffer pH 5.15 for 12 hours at 4°C.
Dehydration and Embedding
Samples placed on wet ice were washed two times for five minutes with ultrapure water, followed by dehydration using a graded series of ethanol solutions (30, 50, 70, 90%) each for three minutes, then three times in 100% absolute ethanol for 10 minutes ending with three washes with anhydrous acetone (Sigma-Aldrich) for 10 minutes at RT. A freshly prepared resin mixture containing methylhexahydrophthalic anhydride and cycloaliphatic epoxide resin (ERL 4221, Electron Microscopy Sciences) at a weight 1.27:1 ratio mixed with the catalyzing agent (Hishicolin PX-4ET, Nippon Chemical Industrial) at a 1:100 by volume was homogenized in a water bath sonicator at RT for 15 minutes, and the anhydrous samples were mixed with the resin at a 1:1 volumetric ratio and gently rocked on a plate rocker for 12 hours. The resin mixture was then removed by aspiration and replaced with an aliquot of newly prepared resin mixture and incubated for two hours; this step was repeated three more times. Finally, the glass cover slips with the attached cells were placed on the cut-off caps from 1.5 mL Eppendorf tubes containing freshly prepared resin, oriented with the cells towards the cap. The resin was allowed to polymerize for 12 hours at 100°C, after which the caps with the polymerized resin were immersed in boiling water for 5 minutes and then quickly transferred into liquid nitrogen leading to separation of the glass cover slip from the resin. The polymerized resin blocks were cut out from the caps and glued onto the top of aluminum pin mount stubs (Ted Pella) using conductive silver epoxy adhesive (EPO-TEK H20S, Electron Microscopy Sciences) with the glass side facing up. This face was coated with carbon (20 nm thickness) generated from a high purity carbon cord source (Electron Microscopy Sciences) using a Quorum Q150R ES sputter coater (Quorum Technologies).
FIB-SEM Imaging
A Zeiss Crossbeam 540 FIB-SEM (Carl Zeiss Microscopy, LLC) was used to perform all correlative fluorescence/electron microscopy experiments. Samples were loaded into the FIB-SEM and the stage tilted at 30 degrees to reveal the layout of the numbered grid embedded in the resin block. Bright-field images of the grid numbers were used as a guide for finding the cells on the surface of the block that had been subjected to high magnification 3D fluorescence imaging. For mapping the orientation of the cells, SEM was performed with an accelerating voltage of 8 kV and a probe current of 2 nA using the SE2 (Everhart-Thornley) detector. After mapping the cells, the stage was tilted to 54 degrees (normal to the FIB column) and a trench was etched into the resin block with the ion beam to expose a cross section for high-resolution SEM imaging. Datasets were acquired at 1.5 kV accelerating voltage at 400 pA probe current using a backscatter electron detector (ESB) with a voltage grid set to 808 V to filter out scattered secondary electrons. A dwell time of 3μs, line averaging of 8, and pixel size of 10 x 10 nm (X/Y) was used for all correlative datasets. FIB milling was performed with a 30 kV gallium ion beam in 10 nm steps to create isotropic 10 x 10 x 10 nm (XYZ) voxels.
Correlation alignment
Fluorescence images obtained using spinning disc confocal microscopy were subjected to image deconvolution using the Lucy–Richardson algorithm (deconvlucy function in MATLAB) with experimentally measured PSF for 15 iterations. For purposes of correlation, the resulting images were compared with the raw fluorescent images to determine empirically the signal thresholds and dynamic ranges, so that the images showed spots with fluorescence signal from at least 8 copies of eGFP molecules. The resulting 3D fluorescent image stacks were used to align with the corresponding FIB-SEM dataset.
The sequential FIB-SEM images were registered using the Fiji plugin StackReg with Rigid Body transformation. For visualization, the registered stacks were filtered with Median 3D (x=3, y=3, z=3) and enhanced signals by adjusting gamma to 1.75 using the Amira software (Thermo Fisher Scientific).
The pre-processed stacks of fluorescent and FIB-SEM images described above were then imported into Amira and processed as follows. 3D volume renderings of the fluorescent and FIB-SEM images were created, thus revealing the cell shape. The fluorescent signal corresponding to the ER distributed throughout the cell volume was then used to orient and align the cells during prometaphase, telophase and interphase. For interphase cells we aligned the fluorescent signals of the nuclear envelope and Nup133. Displayed FIB-SEM images corresponding to sub-volumes from the entire 3D stacks were rendered to achieve clear representation of the ER and nuclear envelope contour (Lighting: Diffuse, Interpolation: cubic, Colormap: H:0.147, S:1, V:1 and ranging dependent on signal, Gamma: 5.3-8, Opacity:1).
QUANTIFICATION AND STATISTICAL ANALYSIS
Single molecule fluorescence calibration
The LLSM and TIRF microscopes were calibrated for single- or three-eGFP detection using bacterially generated eGFP adsorbed to a glass coverslip (Adell et al., 2017; Cocucci et al., 2012). The LLSM was similarly calibrated for single-molecule detection using Janelia Fluor ® 549 and 635 HaloTag ligands. The imaging conditions were adjusted to detect the fluorescence signal within a diffraction-limited spot with a signal-to-noise ratio of at least 3 (Adell et al., 2017). Briefly, the microscopes were adjusted to detect the fluorescence from single molecules within a diffraction limited spot from which we generated a calibration curved using different exposures and the same laser power used to image cells. A custom-made MATLAB (MathWorks) script automatically detected the 3D-fitted asymmetric Gaussian fluorescence intensity associated with a diffraction-limited spot; a T-test was used to identify valid spots whose 3D-fitted fluorescence signal was statistically higher than their local background before bleaching. The fluorescence intensity distribution was then fitted to a mixture-model Gaussian function (Aguet et al., 2013) such that the first Gaussian population corresponded to the signal elicited by a single molecule. The fluorescence signals detected in the cells were then converted to number of molecules taking into account the propagated error (square root of the squared sums of uncertainties from the 3D fitted fluorescence signal and from the single- molecule calibration curve). The TIRF microscope was calibrated for single-molecule eGFP as described above but using instead a 2D Gaussian fluorescence intensity-fitting algorithm (Aguet et al., 2013; Cocucci et al., 2012).
Automated detection of spots obtained using LLSM
The LLSM was adjusted to detect the fluorescence from single molecules, data collected using scan mode (sample on the coverslip moved along the s-axis) and post-processed as described (Aguet et al., 2016). Diffraction limited spots from fluorescence signals elicited by the tagged-Nup’s were detected through a least-squares minimization numerical fitting with a model of the microscope PSF approximated by a 3D Gaussian function (Aguet et al., 2016). Estimated fluorescence intensities associated with each spot were calculated from the corresponding amplitudes of the fitted 3D Gaussian function (Aguet et al., 2016). Showing the localized spots as a volume whose dimensions and intensity were the corresponding fitted 3D Gaussian functions facilitated their visualization in the volumetric images (Fig. 2 F,G, S7 and S8).
Statistical analysis
MATLAB (Mathworks, versions 2014a-2019b) was used for fitting Gaussian distributions (either to the mode of the distribution, or mixture-model fitting) and to determine the mean ± SD of the fits. The MATLAB functions were built using previously published functions (Aguet et al., 2013). The number of measurements used for each of the fits is noted in the figures and/or corresponding figure legends. Mann-Whitney and unpaired t-tests determined using GraphPad Prism 8 were used to evaluate the statistical significance of differences between the independent groups for the data in Fig. S3.
Supplementary Material
Video S1. 3D live cell LLSM imaging of Nup133 in a dividing cell, Related to Figure 2.
Time-lapse movie obtained using live cell 3D LLSM of genome-edited SVG-A dividing cells expressing eGFP-Nup133. The central region of the time-lapse movie shows an example of a cell undergoing cell division, from interphase through mitosis and ending with cytokinesis. The xy-projections show (1) loss of fluorescence signal from Nup133 in NPCs at the nuclear margin of the cell during interphase, (2) loss of this signal and its redistribution into the volume excluded from the chromosome mass and into bright spots associated with kinetochores during mitosis, (3) separation of the signals at the kinetochores into the daughter cells at the onset of anaphase and (4) post-mitotic reappearance of signal in both daughter cells corresponding to their newly formed nuclear pores. Time-lapse acquired every 123 s for three hours; each time point is a stack of 100 consecutive planes spaced one um.
Video S2. Live cell LLSM imaging of Nup107 subassemblies in the volume excluded from chromatin during metaphase. Related to Figure 3.
Time-lapse movie acquired in a single optical plane using live cell LLSM from a genome-edited SUM159 cell in metaphase expressing Halo-Nup133. Cells on a cover slip were briefly incubated with the fluorescent dyes Janelia Fluor ® 549 and Janelia Fluor ® 635 during the first and second days, respectively, and a cell undergoing mitosis imaged during the sixth day. The bright fluorescence spots in the volume excluded from the chromatin represent octameric subassemblies of Nup107 that had survived several cycles of cell division. The time-lapse movie shows that the two sets of octameric structures do not colocalize. Time-lapse acquired every 100 ms for 14 s; a single time point comprised two channels sequentially acquired every 50 ms using 47 ms exposures.
Video S3. Volume imaging of a cell during prometaphase. Related to Figure 5.
Subvolume of a gene-edited SVG-A cell expressing eGFP-Nup133 during pro-metaphase obtained by confocal spinning disc confocal microscopy and FIB-SEM. The movie first shows the overall segmentation (yellow) of this volume obtained by FIB-SEM highlighting the fenestrated ER sheets, then the distribution of fluorescence spots of eGFP-Nup133 (green) within the volume excluded by chromosome mass, finally focusing on one location showing colocalization of the fluorescence signal with a region of highly fenestrated ER sheet.
Video S4. Volume imaging of a cell during interphase. Related to Figure 6.
Subvolume of a genome-edited SVG-A cell expressing eGFP-Nup133 and transiently expressing mCherry-Sec61β during pro-metaphase obtained by confocal spinning disc confocal microscopy and FIB-SEM. The movie first shows sequential single plane views obtained with FIB-SEM, then the overall segmentation (yellow) highlighting nuclear pores on the nuclear envelope, then the distribution of fluorescence spots of eGFP-Nup133 (green) within the nuclear envelope and the intranuclear tubular connection between to sites of nuclear envelope, finally focusing on one location showing colocalization of the eGFP-Nup133 fluorescence signal (green) with the nuclear envelope. The movie ends by comparing the distribution of eGFP-Nup133 (green) with the location of the ER marked by mCherry-Sec61β (purple).
Video S5. Volume imaging of a cell during telophase. Related to Figures 5, 6 and S6.
Subvolumes of two daughter genome-edited SVG-A cells expressing eGFP-Nup133 during telophase obtained by confocal spinning disc confocal microscopy and FIB-SEM. The movie first shows the overall segmentation (yellow) of this volume obtained by FIB-SEM, then the distribution of fluorescence spots of eGFP-Nup133 (green) with the developing nuclear envelope, finally focusing on several locations showing colocalization of highly fenestrated nuclear envelope with the fluorescence signal. Most of the double-membrane holes are uniform and ~ 80 nm in diameter.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit monoclonal antibody specific for Nup133 | Abcam | ab155990 |
| Donkey anti-rabbit IgG HRP-conjugated secondary antibody | VWR | 95017-556 |
| ECO transfection reagent | Gift from Zheng-Rong Lu and Andrew Schilb Case (Western Reserve University) | N/A |
| goat anti-rabbit secondary antibody conjugated to horseradish peroxidase | Thermo Fisher | 65-6120 |
| rabbit monoclonal antibody specific for actin | Bethyl Laboratories | A300-491A |
| Rabbit polyclonal antibody to β-actin | Abcam | 8227 |
| Rabbit polyclonal antibody to Nup133 | Abcam | 233086 |
| rabbit monoclonal antibody specific for Nup133 | Abcam | ab155990 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| 2-(N-morpholino) ethanesulfonic acid sodium salt | Sigma-Aldrich | M5057 |
| aprotinin | Sigma-Aldrich | A6106 |
| BSA | Sigma-Aldrich | A9418 |
| catalyzing agent | Nippon Chemical Industrial | Hishicolin PX-4ET |
| cycloaliphatic epoxide resin | Electron Microscopy Sciences | ERL 4221 |
| disuccinimidyl suberate | Thermo Fisher | A39267 |
| DMEM/F-12/GlutaMAX | Life Technologies | 10565-042 |
| EDTA | Sigma-Aldrich | E9884 |
| fetal bovine serum | Atlanta Biologicals | S11150 |
| Geneticin | Life Technologies | 11811031 |
| glutaraldehyde | Electron Microscopy Science | 16220 |
| Halt protease inhibitor | Thermo Fisher | 78430 |
| HEPES | Mediatech | 25-060-CI |
| Hoechst 33342 | Thermo Fisher | R37605 |
| Hydrocortisone | Sigma-Aldrich | H4001 |
| Insulin | Sigma-Aldrich | I9278 |
| Janelia Fluor ® 549 HaloTag ligand | https://www.janelia.org/open-science/janelia-fluor-dyes | NA |
| Janelia Fluor ® 635 HaloTag ligand | https://www.janelia.org/open-science/janelia-fluor-dyes | NA |
| Janelia Fluor ® 646 HaloTag ligand | https://www.janelia.org/open-science/janelia-fluor-dyes | NA |
| Janelia Fluor ® x549 HaloTag ligand | https://www.janelia.org/open-science/janelia-fluor-dyes | NA |
| Janelia Fluor ® x646 HaloTag ligand | https://www.janelia.org/open-science/janelia-fluor-dyes | NA |
| Leibovitz’s L15 media | Thermo Fisher | 11415064 |
| leupeptin | Thermo Fisher | 78435 |
| methylhexahydrophthalic anhydride | J&K Scientific | 25550-51-0 |
| Minimum Essential Medium | Corning | 10-010-CV |
| NaCl | Sigma-Aldrich | S9888 |
| NaF | Sigma-Aldrich | 201154 |
| NaVO3 | Sigma-Aldrich | 72060 |
| Nocodazole | Sigma-Aldrich | M1404 |
| non-fat dry milk | Sigma-Aldrich | M7409 |
| NP-40 | Sigma-Aldrich | 492016 |
| OsO4 | Electron Microscopy Sciences | 19100 |
| Paraformaldehyde | Sigma-Aldrich | 158127 |
| PBS | Sigma-Aldrich | P3813 |
| penicillin and streptomycin | VWR International | 45000-652 |
| PIPES | Sigma-Aldrich | P6757 |
| PMSF | Thermo Fisher | 36978 |
| potassium ferrocyanide | Sigma-Aldrich | 455989 |
| protease inhibitor cocktail | Roche | 4693116001 |
| SDS | Sigma-Aldrich | L3771 |
| sodium deoxycholate | Sigma-Aldrich | 30970 |
| thiocarbohydrazide | Electron Microscopy Sciences | 21900 |
| Thymidine | Sigma-Aldrich | T9250 |
| Tris-HCl | Sigma-Aldrich | 10812846001 |
| Triton X-100 | Sigma-Aldrich | T8787 |
| Tween 20 | Sigma-Aldrich | P9416 |
| Uranyl Acetate | Electron Microscopy Sciences | 22400 |
| Critical commercial assays | ||
| BCA assay | Pierce | 23225 |
| ECL substrate | Pierce | 32134 |
| Gibson Assembly Cloning Kit | New England Biolabs | E5510S |
| GoTaq Polymerase | Promega | M3001 |
| Deposited data | ||
| Experimental models: cell lines | ||
| SUM159 human breast carcinoma cells | Forozan et al., 1999; J. Brugge (Harvard Medical School) | N/A |
| SUM157 eGFP-Nup133 | This paper | NA |
| SUM157 eGFP-Nup133/Halo-Nup107 | This paper | NA |
| SUM157 eGFP-Nup133/Halo-Nup205 | This paper | NA |
| SUM157 Halo-Nup107 | This paper | NA |
| SUM157 Halo-Nup133 | This paper | NA |
| SUM157 Halo-Nup205 | This paper | NA |
| SVG-A eGFP-Nup133 | This paper | NA |
| SVG-A Halo-Nup107 | This paper | NA |
| SVG-A Halo-Nup133 | This paper | NA |
| SVG-A Halo-Nup205 | This paper | NA |
| SVG-A human fetal astroglial cells | Chou et al., 2016b; W. J. Atwood (Brown University) | N/A |
| Experimental models: organisms/strains | ||
| Oligonucleotides | ||
| Nup133 up F1: gaattcgagctcggtacccCAACTGGCCAGTTTTACCAAGC | This paper | NA |
| Nup133 up R1: GACTCCAAGGAGCAGCGACT | This paper | NA |
| eGFP-Nup133 F2: AGTCGCTGCTCCTTGGAGTCATGGTGAGCAAGGGCGAGGAGC | This paper | NA |
| eGFP-Nup133 R2: GGAACCACCAGAACCACCAG | This paper | NA |
| Halo-Nup133 F2: AGTCGCTGCTCCTTGGAGTCATGGCAGAAATCGGTACTGGCTT | This paper | NA |
| Halo-Nup133 R2: GGAACCACCAGAACCACCAG | This paper | NA |
| Nup133 down F3: GGTTCTGGTGGTTCTGGTGGTTCCTTCCCAGCCGCCCCTTCTCC | This paper | NA |
| Nup133 down R3: gtcgactctagaggatccccAACATGGTGTGTGTTGGGGGTG | This paper | NA |
| Nup107 up F1: gaattcgagctcggtacccCAAAAATGTGACTGACTCCTTC | This paper | NA |
| Nup107 up R1: GGCTAAAGCCTTTTCCACAC | This paper | NA |
| Halo-Nup107 F2: GTGTGGAAAAGGCTTTAGCCATGGCAGAAATCGGTACTGG | This paper | NA |
| Halo-Nup107 R2: GGAACCACCAGAACCACCAGAACC | This paper | NA |
| Nup107 down F3: GGTTCTGGTGGTTCTGGTGGTTCCGACAGGTCAGTACTGATGGTG | This paper | NA |
| Nup107 down R3: gtcgactctagaggatccccCTTTAGAACAATGGCAGTCAC | This paper | NA |
| Nup205 up F1: gaattcgagctcggtacccCGTTATTGATGCTGAGTGTTTG | This paper | NA |
| Nup205 up R1: CTTAGAGGCGCACTAACAGAG | This paper | NA |
| Halo-Nup205 F2: CTCTGTTAGTGCGCCTCTAAGATGGCAGAAATCGGTACTGG | This paper | NA |
| Halo-Nup205 R2: GGAACCACCAGAACCACCAGAACC | This paper | NA |
| Nup205 down F3: GGTTCTGGTGGTTCTGGTGGTTCCGCGACGCCTTTGGCGGTAAA | This paper | NA |
| Nup205 down R3: gtcgactctagaggatccccCCTACTATGCGACAGCAGTG | This paper | NA |
| Recombinant DNA | ||
| mCherry-Sec61β | Lu et al., 2009; Gift from Gia Voeltz | Addgene; Plasmid #49155 |
| ss-YFP–KDEL | Valm et al., 2017; J. Lippincott-Schwartz (HHMI) | N/A |
| NLS-RFP | Gift from S. Liu and D. Pellman (Harvard Medical School) | N/A |
| Software and algorithms | ||
| 3D Gaussian fitting for 3D point source detection | Aguet et al., 2016 | https://github.com/francois-a/llsmtools |
| Amira | Thermo Fisher | Versions 5.6 – 2020.1 |
| FIJI | Schneider et al., 2012 | https://imagej.net/Fiji |
| Imaris | Bitplane | Versions 8-9 |
| MATLAB | Mathworks | 2015 - 2020 |
| Other | ||
| 8-well chambered coverglass slides | Thermo Fisher | 155411 |
Highlights.
Incomplete mitotic NPC dissociation yields octameric inner and outer ring subassemblies
Octameric inner and outer ring subassemblies localize to fenestrated mitotic ER
Octameric inner and outer ring subassemblies template post-mitotic NPC formation
NPC’s inherit octameric inner and outer ring subassemblies
ACKNOWLEDGEMENTS
We thank Joan Brugge for generously providing the parental SUM159 cells, Walter J. Atwood for the parental SVG-A cells, Luke D. Lavis and Jonathan B. Grimm for the Janelia Fluor® HaloTag ligands, David Shiwei Liu and David Pellman for providing the NLS-RFP lentivirus construct, Jennifer Lippincott-Schwartz for the ss-YFP-KDEL expression plasmid, Justin H. Houser for maintaining the spinning disc confocal microscope and for operating the FIB-SEM, Stephen C. Harrison for discussions and editorial help, and members of our laboratory for help and encouragement. The research was supported by National Institutes of Health R01 GM075252, National Institute of General Medical Sciences Maximizing Investigators’ Research Award GM130386, NIH Harvard Virology Program training Grant T32 AI07245 with a postdoctoral fellowship to Alex.J.B.Kreutzberger, NNF16OC0022166 Novo Nordisk Foundation / Danish Technical University grants to Tom Kirchhausen and a Biogen Sponsored Research Agreement to Tom Kirchhausen.
Footnotes
DECLARATION OF INTERESTS
The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AND MATERIALS AVAILABILITY
All data needed to evaluate the conclusions in this paper are available in the main text and the supplementary materials. The datasets used and/or analyzed during the current study are available from the corresponding authors S.U. and T.K. upon request.
REFERENCES
- Adell MAY, Migliano SM, Upadhyayula S, Bykov YS, Sprenger S, Pakdel M, Vogel GF, Jih G, Skillern W, Behrouzi R, et al. (2017). Recruitment dynamics of ESCRT-III and Vps4 to endosomes and implications for reverse membrane budding. Elife 6, e31652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguet F, Antonescu CN, Mettlen M, Schmid SL, and Danuser G (2013). Advances in Analysis of Low Signal-to-Noise Images Link Dynamin and AP2 to the Functions of an Endocytic Checkpoint. Dev Cell 26, 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguet F, Upadhyayula S, Gaudin R, Chou Y-Y, Cocucci E, He K, Chen B-C, Mosaliganti K, Pasham M, Skillern W, et al. (2016). Membrane dynamics of dividing cells imaged by lattice light-sheet microscopy. Mol Biol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M, Zimmerli CE, Rantos V, Wilfling F, Ronchi P, Fung HKH, Lee C-W, Hagen W, Turonova B, Karius K, et al. (2020). In-cell architecture of the nuclear pore and snapshots of its turnover. Nature 586, 796–800. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, and Hetzer MW (2007). Nuclear envelope formation by chromatin-mediated reorganization of the endoplasmic reticulum. Nat Cell Biol 9, 1160–1166. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Vargas JD, Hsiao JP, and Hetzer MW (2009). Recruitment of functionally distinct membrane proteins to chromatin mediates nuclear envelope formation in vivo. J Cell Biol 186, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appen, von A, Kosinski J, Sparks L, Ori A, DiGuilio AL, Vollmer B, Mackmull M-T, Banterle N, Parca L, Kastritis P, et al. (2015). In situ structural analysis of the human nuclear pore complex. Nature 526, 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appen, von A, LaJoie D, Johnson IE, Trnka MJ, Pick SM, Burlingame AL, Ullman KS, and Frost A (2020). LEM2 phase separation promotes ESCRT-mediated nuclear envelope reformation. Nature 582, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudouin J, Gerlich D, Daigle N, Eils R, and Ellenberg J (2002). Nuclear envelope breakdown proceeds by microtubule-induced tearing of the lamina. 108, 83–96. [DOI] [PubMed] [Google Scholar]
- Belgareh N, Rabut G, Baï SW, van Overbeek M, Beaudouin J, Daigle N, Zatsepina OV, Pasteau F, Labas V, Fromont-Racine M, et al. (2001). An evolutionarily conserved NPC subcomplex, which redistributes in part to kinetochores in mammalian cells. J Cell Biol 154, 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilir Ş, Kojidani T, Mori C, Osakada H, Kobayashi S, Koujin T, Hiraoka Y, and Haraguchi T (2019). Roles of Nup133, Nup153 and membrane fenestrations in assembly of the nuclear pore complex at the end of mitosis. Genes Cells 24, 338–353. [DOI] [PubMed] [Google Scholar]
- Boucrot E, and Kirchhausen T (2007). Endosomal recycling controls plasma membrane area during mitosis. Proc Natl Acad Sci USA 104, 7939–7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui KH, Appen, von A, DiGuilio AL, Ori A, Sparks L, Mackmull M-T, Bock T, Hagen W, Andres-Pons A, Glavy JS, et al. (2013). Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155, 1233–1243. [DOI] [PubMed] [Google Scholar]
- Cai E, Marchuk K, Beemiller P, Beppler C, Rubashkin MG, Weaver VM, Gérard A, Liu T-L, Chen B-C, Betzig E, et al. (2017). Visualizing dynamic microvillar search and stabilization during ligand detection by T cells. Science 356, eaal3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B-C, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, Liu Z, et al. (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. 346, 1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y-Y, Cuevas C, Carocci M, Stubbs SH, Ma M, Cureton DK, Chao L, Evesson F, He K, Yang PL, et al. (2016a). Identification and characterization of a novel broad spectrum virus entry inhibitor. 90, 4494–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y-Y, Krupp A, Kaynor C, Gaudin R, Ma M, Cahir-McFarland E, and Kirchhausen T (2016b). Inhibition of JCPyV infection mediated by targeted viral genome editing using CRISPR/Cas9. Sci. Rep. 6, 36921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocucci E, Aguet F, Boulant S, and Kirchhausen T (2012). The first five seconds in the life of a clathrin-coated pit. 150, 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman VC, and Wu J-Q (2014). Every laboratory with a fluorescence microscope should consider counting molecules. Mol Biol Cell 25, 1545–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cureton DK, Harbison CE, Cocucci E, Parrish CR, and Kirchhausen T (2012). Limited transferrin receptor clustering allows rapid diffusion of canine parvovirus into clathrin endocytic structures. 86, 5330–5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle N, Beaudouin J, Hartnell L, Imreh G, Hallberg E, Lippincott-Schwartz J, and Ellenberg J (2001). Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J Cell Biol 154, 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dultz E, Zanin E, Wurzenberger C, Braun M, Rabut G, Sironi L, and Ellenberg J (2008). Systematic kinetic analysis of mitotic dis- and reassembly of the nuclear pore in living cells. J Cell Biol 180, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg J, Siggia ED, Moreira JE, Smith CL, Presley JF, Worman HJ, and Lippincott-Schwartz J (1997). Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J Cell Biol 138, 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forozan F, Veldman R, Ammerman CA, Parsa NZ, Kallioniemi A, Kallioniemi OP, and Ethier SP (1999). Molecular cytogenetic analysis of 11 new breast cancer cell lines. Br. J. Cancer 81, 1328–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz C, Walczak R, Yavuz S, Santarella R, Gentzel M, Askjaer P, Galy V, Hetzer M, Mattaj IW, and Antonin W (2007). MEL-28/ELYS is required for the recruitment of nucleoporins to chromatin and postmitotic nuclear pore complex assembly. EMBO Rep 8, 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Laylin LK, Riel-Mehan M, Chen B-C, Lord SJ, Goddard TD, Ferrin TE, Nicholson-Dykstra SM, Higgs H, Johnson GT, Betzig E, et al. (2017). Actin-based protrusions of migrating neutrophils are intrinsically lamellar and facilitate direction changes. Elife 6, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Meth 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Meth 12, 244–50–3pfollowing250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, et al. (2017). A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat Meth 14, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li D, Zhang S, Yang Y, Liu J-J, Wang X, Liu C, Milkie DE, Moore RP, Tulu US, et al. (2018). Visualizing Intracellular Organelle and Cytoskeletal Interactions at Nanoscale Resolution on Millisecond Timescales. Cell 175, 1430–1442.e17. [DOI] [PubMed] [Google Scholar]
- Güttinger S, Laurell E, and Kutay U (2009). Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat. Rev. Mol. Cell Biol. 10, 178–191. [DOI] [PubMed] [Google Scholar]
- Hampoelz B, Andres-Pons A, Kastritis P, and Beck M (2019). Structure and Assembly of the Nuclear Pore Complex. Annu Rev Biophys 48, 515–536. [DOI] [PubMed] [Google Scholar]
- He K, Marsland lii R, Upadhyayula S, Song E, Dang S, Capraro BR, Wang W, Skillern W, Gaudin R, Ma M, et al. (2017). Dynamics of phosphoinositide conversion in clathrin-mediated endocytic traffic. Nature 552, 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Zhang Y, Zhu X, Zeng C, Wang Q, Zhou Q, Tao Q, Liu M, Lei J, Yan C, et al. (2020). Structure of the cytoplasmic ring of the Xenopus laevis nuclear pore complex by cryo-electron microscopy single particle analysis. Cell Res 30, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley K, Knockenhauer KE, Kabachinski G, and Schwartz TU (2015). Atomic structure of the Y complex of the nuclear pore. Nat. Struct. Mol. Biol. 22, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Fernandez-Martinez J, Nudelman I, Shi Y, Zhang W, Raveh B, Herricks T, Slaughter BD, Hogan JA, Upla P, et al. (2018). Integrative structure and functional anatomy of a nuclear pore complex. Nature 555, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski J, Mosalaganti S, Appen, von A, Teimer R, DiGuilio AL, Wan W, Bui KH, Hagen WJH, Briggs JAG, Glavy JS, et al. (2016). Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science 352, 363–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Golchoubian B, Belevich I, Jokitalo E, and Schlaitz A-L (2019). REEP3 and REEP4 determine the tubular morphology of the endoplasmic reticulum during mitosis. Mol Biol Cell 30, 1377–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaJoie D, and Ullman KS (2017). Coordinated events of nuclear assembly. Curr Opin Cell Biol 46, 39–45. [DOI] [PubMed] [Google Scholar]
- Lepore A, Taylor H, Landgraf D, Okumus B, Jaramillo-Riveri S, McLaren L, Bakshi S, Paulsson J, and Karoui ME (2019). Quantification of very low-abundant proteins in bacteria using the HaloTag and epi-fluorescence microscopy. Sci. Rep. 9, 7902–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DH, and Hoelz A (2019). The Structure of the Nuclear Pore Complex (An Update). Annu Rev Biochem 88, annurev–biochem–062917–011901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DH, Stuwe T, Schilbach S, Rundlet EJ, Perriches T, Mobbs G, Fan Y, Thierbach K, Huber FM, Collins LN, et al. (2016). Architecture of the symmetric core of the nuclear pore. Science 352, aaf1015–aaf1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, and Pellman D (2020). The coordination of nuclear envelope assembly and chromosome segregation in metazoans. Nucleus 11, 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loïodice I, Alves A, Rabut G, Van Overbeek M, Ellenberg J, Sibarita J-B, and Doye V (2004). The entire Nup107–160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol Biol Cell 15, 3333–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- Lu L, Ladinsky MS, and Kirchhausen T (2009). Cisternal organization of the endoplasmic reticulum during mitosis. Mol Biol Cell 20, 3471–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Ladinsky MS, and Kirchhausen T (2011). Formation of the postmitotic nuclear envelope from extended ER cisternae precedes nuclear pore assembly. J Cell Biol 194, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meselson M, and Stahl FW (1958). THE REPLICATION OF DNA IN ESCHERICHIA COLI. Proc Natl Acad Sci USA 44, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, and Ellenberg J (2017). Mechanisms of nuclear pore complex assembly: Two different ways of building one molecular machine. FEBS Lett 287, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, Bui KH, Schorb M, Hossain MJ, Politi AZ, Koch B, Eltsov M, Beck M, and Ellenberg J (2016). Nuclear pore assembly proceeds by an inside-out extrusion of the nuclear envelope. Elife 5, e19071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, Steyer AM, Schorb M, Hériché J-K, Hossain MJ, Sethi S, Kueblbeck M, Schwab Y, Beck M, and Ellenberg J (2018). Postmitotic nuclear pore assembly proceeds by radial dilation of small membrane openings. Nat. Struct. Mol. Biol. 25, 21–28. [DOI] [PubMed] [Google Scholar]
- Otsuka S, Szymborska A, and Ellenberg J (2014). Imaging the assembly, structure, and function of the nuclear pore inside cells. Methods Cell Biol 122, 219–238. [DOI] [PubMed] [Google Scholar]
- Poteryaev D, Squirrell JM, Campbell JM, White JG, and Spang A (2005). Involvement of the actin cytoskeleton and homotypic membrane fusion in ER dynamics in Caenorhabditis elegans. Mol Biol Cell 16, 2139–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhka M, Joensuu M, Vihinen H, Belevich I, and Jokitalo E (2012). Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Mol Biol Cell 23, 2424–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhka M, Vihinen H, Joensuu M, and Jokitalo E (2007). Endoplasmic reticulum remains continuous and undergoes sheet-to-tubule transformation during cell division in mammalian cells. J Cell Biol 179, 895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasala BA, Orjalo AV, Shen Z, Briggs S, and Forbes DJ (2006). ELYS is a dual nucleoporin/kinetochore protein required for nuclear pore assembly and proper cell division. Proc Natl Acad Sci USA 103, 17801–17806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz TU (2016). The Structure Inventory of the Nuclear Pore Complex. J Mol Biol 428, 1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuwe T, Correia AR, Lin DH, Paduch M, Lu VT, Kossiakoff AA, and Hoelz A (2015). Nuclear pores. Architecture of the nuclear pore complex coat. 347, 1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevathasan JV, Kahnwald M, Cieśliński K, Hoess P, Peneti SK, Reitberger M, Heid D, Kasuba KC, Hoerner SJ, Li Y, et al. (2019). Nuclear pores as versatile reference standards for quantitative superresolution microscopy. Nat Meth 16, 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbert S, Platani M, Boue S, and Mattaj IW (2006). Direct membrane protein-DNA interactions required early in nuclear envelope assembly. J Cell Biol 173, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungricht R, and Kutay U (2017). Mechanisms and functions of nuclear envelope remodelling. 18, 229–245. [DOI] [PubMed] [Google Scholar]
- Urh M, and Rosenberg M (2012). HaloTag, a Platform Technology for Protein Analysis. Curr Chem Genomics 6, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, and Lippincott-Schwartz J (2017). Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri M, Schink KO, Campsteijn C, Wegner CS, Schultz SW, Christ L, Thoresen SB, Brech A, Raiborg C, and Stenmark H (2015). Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522, 231–235. [DOI] [PubMed] [Google Scholar]
- Wandke C, and Kutay U (2013). Enclosing chromatin: reassembly of the nucleus after open mitosis. 152, 1222–1225. [DOI] [PubMed] [Google Scholar]
- Wang S, Romano FB, Field CM, Mitchison TJ, and Rapoport TA (2013). Multiple mechanisms determine ER network morphology during the cell cycle in Xenopus egg extracts. J Cell Biol 203, 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weberruss M, and Antonin W (2016). Perforating the nuclear boundary - how nuclear pore complexes assemble. J Cell Sci 129, 4439–4447. [DOI] [PubMed] [Google Scholar]
- Wheatley DN, and Gray LH (1988). Mitosis and protein synthesis. 2. Synthesis of protein and RNA in synchronous HeLa S-3 cell populations entering and leaving M-phase of the cell cycle. Cytobios 55, 191–204. [PubMed] [Google Scholar]
- Yang L, Guan T, and Gerace L (1997). Integral membrane proteins of the nuclear envelope are dispersed throughout the endoplasmic reticulum during mitosis. J Cell Biol 137, 1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li S, Zeng C, Huang G, Zhu X, Wang Q, Wang K, Zhou Q, Yan C, Zhang W, et al. (2020). Molecular architecture of the luminal ring of the Xenopus laevis nuclear pore complex. Cell Res 30, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccolo M, Alves A, Galy V, Bolhy S, Formstecher E, Racine V, Sibarita J-B, Fukagawa T, Shiekhattar R, Yen T, et al. (2007). The human Nup107–160 nuclear pore subcomplex contributes to proper kinetochore functions. Embo J 26, 1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. 3D live cell LLSM imaging of Nup133 in a dividing cell, Related to Figure 2.
Time-lapse movie obtained using live cell 3D LLSM of genome-edited SVG-A dividing cells expressing eGFP-Nup133. The central region of the time-lapse movie shows an example of a cell undergoing cell division, from interphase through mitosis and ending with cytokinesis. The xy-projections show (1) loss of fluorescence signal from Nup133 in NPCs at the nuclear margin of the cell during interphase, (2) loss of this signal and its redistribution into the volume excluded from the chromosome mass and into bright spots associated with kinetochores during mitosis, (3) separation of the signals at the kinetochores into the daughter cells at the onset of anaphase and (4) post-mitotic reappearance of signal in both daughter cells corresponding to their newly formed nuclear pores. Time-lapse acquired every 123 s for three hours; each time point is a stack of 100 consecutive planes spaced one um.
Video S2. Live cell LLSM imaging of Nup107 subassemblies in the volume excluded from chromatin during metaphase. Related to Figure 3.
Time-lapse movie acquired in a single optical plane using live cell LLSM from a genome-edited SUM159 cell in metaphase expressing Halo-Nup133. Cells on a cover slip were briefly incubated with the fluorescent dyes Janelia Fluor ® 549 and Janelia Fluor ® 635 during the first and second days, respectively, and a cell undergoing mitosis imaged during the sixth day. The bright fluorescence spots in the volume excluded from the chromatin represent octameric subassemblies of Nup107 that had survived several cycles of cell division. The time-lapse movie shows that the two sets of octameric structures do not colocalize. Time-lapse acquired every 100 ms for 14 s; a single time point comprised two channels sequentially acquired every 50 ms using 47 ms exposures.
Video S3. Volume imaging of a cell during prometaphase. Related to Figure 5.
Subvolume of a gene-edited SVG-A cell expressing eGFP-Nup133 during pro-metaphase obtained by confocal spinning disc confocal microscopy and FIB-SEM. The movie first shows the overall segmentation (yellow) of this volume obtained by FIB-SEM highlighting the fenestrated ER sheets, then the distribution of fluorescence spots of eGFP-Nup133 (green) within the volume excluded by chromosome mass, finally focusing on one location showing colocalization of the fluorescence signal with a region of highly fenestrated ER sheet.
Video S4. Volume imaging of a cell during interphase. Related to Figure 6.
Subvolume of a genome-edited SVG-A cell expressing eGFP-Nup133 and transiently expressing mCherry-Sec61β during pro-metaphase obtained by confocal spinning disc confocal microscopy and FIB-SEM. The movie first shows sequential single plane views obtained with FIB-SEM, then the overall segmentation (yellow) highlighting nuclear pores on the nuclear envelope, then the distribution of fluorescence spots of eGFP-Nup133 (green) within the nuclear envelope and the intranuclear tubular connection between to sites of nuclear envelope, finally focusing on one location showing colocalization of the eGFP-Nup133 fluorescence signal (green) with the nuclear envelope. The movie ends by comparing the distribution of eGFP-Nup133 (green) with the location of the ER marked by mCherry-Sec61β (purple).
Video S5. Volume imaging of a cell during telophase. Related to Figures 5, 6 and S6.
Subvolumes of two daughter genome-edited SVG-A cells expressing eGFP-Nup133 during telophase obtained by confocal spinning disc confocal microscopy and FIB-SEM. The movie first shows the overall segmentation (yellow) of this volume obtained by FIB-SEM, then the distribution of fluorescence spots of eGFP-Nup133 (green) with the developing nuclear envelope, finally focusing on several locations showing colocalization of highly fenestrated nuclear envelope with the fluorescence signal. Most of the double-membrane holes are uniform and ~ 80 nm in diameter.
Data Availability Statement
This study did not generate any unique large-scale datasets or code.