

Abstract
Patients with congenital heart disease (CHD) are one of the fastest growing populations in cardiology, and valvular pathology is at the center of many congenital lesions. Derangements in valvular embryology lead to several anomalies prone to dysfunction, each with hemodynamic effects that require appropriate surveillance and management. Surgical innovation has provided new treatments that have improved survival in this population, though has also contributed to esotericism in patients who already have unique anatomic and physiologic considerations. Conduit and prosthesis durability are often monitored collaboratively with general and specialized congenital-focused cardiologists. As such, general cardiologists must become familiar with valvular disease with CHD for appropriate care and referral practices. In this review, we summarize the embryology of the semilunar and atrioventricular (AV) valves as a foundation for understanding the origins of valvular CHD and describe the mechanisms that account for heterogeneity in disease. We then highlight the categories of pathology from the simple (e.g., bicuspid aortic valve, isolated pulmonic stenosis) to the more complex (e.g., Ebstein’s anomaly, AV valvular disease in single ventricle circulations) with details on natural history, diagnosis, and contemporary therapeutic approaches. Care for CHD patients requires collaborative effort between providers, both CHD-specialized and not, to achieve optimal patient outcomes.
Keywords: Congenital heart disease (CHD), valvular heart disease, valvular embryology
Overview
Congenital heart defects are the most common type of birth defects, and with recent advances leading to improved survival, there are now over two million infants, children, and adults with congenital heart disease (CHD) in the United States alone (1-3). Many patients suffer from primary congenital valvular lesions or prosthetic/conduit valvular dysfunction. Given the substantial variability within each phenotype of CHD, this review will provide a basic background in embryology to allow the reader to create an adaptable construct in their approach to these patients (Table 1).
Table 1. Congenital defects, embryogenesis, pathologic consequences, recommended diagnostic imaging, and referral to specialized care.
| Category | Congenital defect | Embryologic etiology | Pathologic consequences | Diagnostic imaging | Class I indication for referral to CHD specialist | ||||
|---|---|---|---|---|---|---|---|---|---|
| TTE | TEE | CMRI | CT | ||||||
| Semilunar valves | |||||||||
| Aortic valve | Bicuspid aortic valve | Abnormal fusion of conotruncal endocardial cushions | Primary valvular disease (regurgitation/stenosis), aortopathy | ++ | + | + | ⍬ | ||
| Pulmonic valve | Congenital pulmonic stenosis | Abnormal fusion of conotruncal endocardial cushions and/or dysgenesis of endocardial cushion tissue | Inadequate right ventricular output, right ventricular dysfunction, tricuspid regurgitation | ++ | †‡ | † | ⍵ | ||
| Pulmonic regurgitation | Abnormal fusion of conotruncal endocardial cushions and/or dysgenesis of endocardial cushion tissue; though often iatrogenic | Right ventricular dysfunction, tricuspid regurgitation, increased arrhythmogenicity | ++ | ++‡ | ⍵ | ||||
| Tetralogy of Fallot | Anterior deviation of outflow tract spiral septum | Right ventricular dysfunction, tricuspid regurgitation, increased arrhythmogenicity | ++ | ++‡ | |||||
| Aortic and pulmonary valves | Truncus arteriosus | Failed septation of the ventricular outflow tract | Cyanosis due to right and left InterChamber communication | ++ | ++ | + | ++ | ||
| Atrioventricular valves | |||||||||
| Tricuspid valve | Ebstein’s anomaly | Failed delamination of septal and posterior tricuspid leaflets | Right ventricular dysfunction, tricuspid regurgitation, increased arrhythmogenicity | ++ | § | +‡ | ++ | ||
| Mitral valve | Cleft mitral valve | Partial failed fusion of ventral and dorsal atrioventricular endocardial cushions | Mitral regurgitation | ++ | + | ⍬ | |||
| Parachute mitral valve | Abnormal fusion of primitive papillary muscles during delamination | Mitral regurgitation and/or stenosis | ++ | + | ⍬ | ||||
| Tricuspid and mitral valves | Common atrioventricular valves | Total failed fusion of ventral and dorsal atrioventricular endocardial cushions | Cyanosis due to right and left InterChamber communication, valvular regurgitation/stenosis | ++ | ¶ | ¶ | ++ | ||
Note that truncus arteriosus, cleft mitral valve, and parachute mitral valve are included in this table to provide the reader with further examples of applied embryology in congenital heart disease. ++, indicates a recommendation for initial diagnostic testing, central component of surveillance, or Class I indication as a component of management; +, indicates a recommended secondary or adjunct component of management once diagnosis is established; †, cardiac MRI or CT may be indicated for assessment of peripheral (nonvalvular) pulmonary stenosis; ‡, cardiac MRI may be used to assess right ventricular size and function; §, transesophageal echocardiography can be useful for procedural planning if transthoracic images are inadequate to assess the tricuspid valve; ¶, transthoracic echocardiogram recommended for Atrioventricular Septal Defect per guidelines, though further imaging may be recommended by a CHD specialist; ⍬, if present with additional congenital structural abnormalities; ⍵, if valvular lesion is graded as more than mild in nature. CHD, congenital heart disease; CMRI, cardiac MRI; CT, computed tomography; TEE, transesophageal echocardiography; TTE, transthoracic echocardiography. Reference: Stout KK, Daniels CJ, Aboulhosn JA, Bozkurt B, Broberg CS, Colman JM, et al. 2018 AHA/ACC Guideline for the Management of Adults With Congenital Heart Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019 Apr 2;139 (14):e698-800.
As the prevalence of CHD grows, it is imperative that providers in all practice settings recognize pathology promptly, coordinate appropriate surveillance, and collaborate with CHD-specialists for optimal treatment. As such, this review provides details on the diagnosis, natural history, and contemporary therapeutic approaches to valvular heart disease in CHD. The information used to write this review was collected via computerized literature searches of electronic databases including U.S. National Library of Medicine (MEDLINE), ScienceDirect, and Google Scholar as well as hand searches of the references of the retrieved literature and discussions with experts in CHD. We present the following article in accordance with the Narrative Review reporting checklist (available at http://dx.doi.org/10.21037/cdt-19-693-b).
Congenital disease of the semilunar valves
Embryogenesis of the semilunar valves
The aortic and pulmonary valves form within the primitive outflow tract. Early in gestation, this is a hollow tube termed the conotruncus (4,5). Neural crest tissue migrates into the tube and forms two major collections of endocardial cushion tissue. The collections develop opposite each other, rotating in a counterclockwise spiral through the entire conotruncus (6). Once formed, the cushions grow into the tube’s lumen and fuse at the midline to create distinct, balanced systemic (left ventricular outflow tract and aortic root) and pulmonary [right ventricular outflow tract (RVOT) and main pulmonary artery] outflow channels (5,7-11). Inferiorly, the cushions fuse with the interventricular septum.
Extensions from the newly formed conotruncal septum protrude to give rise to the right and left leaflets of both semilunar valves. At the same level, there are two smaller endocardial cushions that grow out perpendicularly to form the posterior aortic and anterior pulmonic leaflets (Figure 1). The valve leaflets are exposed to the cyclical loading and unloading of the fetal circulation, causing remodeling and elongation into the thin, recognizable valve leaflets seen at full development
Figure 1.

Spiral septum and semilunar valve embryogenesis. (A) Early in development, the precursors to the cardiac outflow tract (OFT) are the truncus arteriosus and the conus. Combined, these make a hollow tubal structure at one end of primitive cardiac mass. The boundary between the conus and truncus arteriosus is marked by an outer curvature of the OFT (the conotruncal curvature). (B) The conotruncal cushions and intercalated cushions develop within the OFT. These cushions occupy four quadrants of the OFT (shown in cross-section). The conotruncal cushions fuse to septate the OFT, as shown in C. (C) Fusion of the conotruncal cushions forms a spiral septum, the truncal part of which divides the OFT into aorta and pulmonary trunk, whereas the conal part separates the OFT into left and right ventricular. The aortic valve and pulmonic valve develop at the conotruncal junction. (D) The conotruncal (superior and inferior septal) cushions give rise to the right and left leaflets of each of the semilunar valves. In the aorta, these are the right and left coronary leaflets, while in the pulmonary valve, these are the right and left cusps. The right-posterior and the left-anterior intercalated cushions develop respectively into the posterior aortic (non-coronary cusp of the aortic valve) and the anterior pulmonic (anterior cusp of the pulmonic valve) leaflets. AL, anterior leaflet; APIC, anterior pulmonary intercalated cushion; AV, aortic valve; CA, coronary artery; CTC, conotruncal cushion; ICC, intercalated cushion; ISC, inferior septal cushion; LL, left leaflet; LCL, left coronary leaflet; LVOT, left ventricular outflow tract; NCL, non-coronary leaflet; PAIC, posterior aortic intercalated cushion; PV, pulmonary valve; RCL, right coronary leaflet; RL, right leaflet; RVOT, right ventricular outflow tract; SSC, superior septal cushion. Images reproduced with permission from Lin et al. Partitioning the heart: mechanisms of cardiac septation and valve development. Development. 2012 Sep 15;139(18):3277-99 and Martin et al. Embryonic Development of the Bicuspid Aortic Valve [Internet]. Vol. 2, Journal of Cardiovascular Development and Disease. 2015. p. 248-72.
Deviation from this process can lead to several types of CHD. Tetralogy of Fallot is a consequence of anterior deviation of the outflow tract’s spiral septum which diminishes the RVOT and pulmonary valve orifice. Malrotation of the spiral septum can lead to transposition of the great arteries. Aberration in arrangement of endocardial cushions may lead to primary valvular dysfunction with many phenotypes, the most common of which are bicuspid aortic valve (BAV) and pulmonary valve stenosis, discussed below (12).
BAV
BAV is the most common congenital heart defect, with a prevalence of 0.5–2% in the general population and a clear predominance in men with a gender ratio of ~3:1 (13). The pathology is rooted in the fusion of two adjacent conotruncal endocardial cushions (8). This leaves patients with two, often asymmetric valve cusps and predisposes them to accelerated aortic valvular disease as well as the commonly associated aortopathy (14).
Patients’ pathologies vary dependent on the degree and orientation of cusp fusion (15-17). The most common morphologic subtype of BAV encountered in surgical series is fusion of the right and left coronary cusps, which accounts for as many as 70–85% of patients. The next most prevalent is a fusion of the right and non-coronary cusps (10–15%), and then left and non-coronary fusion (<5%). There are still rarer subtypes with either two raphe or no raphe and two symmetrical cusps (18,19).
The morphologic subtype of BAV has proven to be predictive of patterns of valvular dysfunction and event-free survival. Fusion of the right and non-coronary cusps is associated with more rapid progression in valvular dysfunction. Series indicate that a higher percentage of patients with right-left cusp fusions experience moderate-severe aortic regurgitation than those with right-noncoronary or left-noncoronary cusp fusions. Patients with right-noncoronary or left-noncoronary cusp fusions are at higher risk for moderate-severe aortic stenosis. Morphologic subtype is also predictive of patterns of aortopathy. For instance, right-noncoronary cusp fusions tend to have root-sparing ascending aortic dilation, where right-left cusp fusions are associated with aneurysmal change in both the ascending aorta and aortic root (15,16,19).
Familial clustering is well-established in patients with BAV. Screening series show a prevalence of 9.1% in first degree relatives of index cases (20,21). Inheritance is widely accepted to be polygenic (22,23). Early data indicate that different morphologic BAV subtypes may have distinct genetic underpinning (12). Screening echocardiography is recommended for first degree relatives of an index case.
BAV occurs mostly in isolation, though it is associated with the presence of other CHD. The incidence of BAV is as high as 30% in patients with subvalvular or supravalvular stenosis, 30% in patients with ventricular septal defects, and 15–20% in patients with Sinus of Valsalva Aneurysms (14,24,25). The morphologic type of BAV also appears to correlate with coronary dominance, and presence of an anomalous coronary takeoff (26,27).
Aortopathy is the BAV’s best known and studied comorbidity. Ascending aortic dilation occurs at a younger age in patients with BAV (even those without valvular dysfunction), at a more rapid rate, and is associated with a higher risk of aortic dissection compared to patients with tricuspid aortic valves (27,28). Though turbulent valvular hemodynamics are known to create asymmetric shear stress within the ascending aorta, the presence of aortic dilation in BAV patients independent of valvular dysfunction or hypertension confirms the significance of an inherent abnormality in aortic tissue. BAV aortopathy is characterized histopathologically by cystic medial necrosis with increased levels of cellular apoptosis, elastic fragmentation and collagen deposition (29-31). These factors, in combination with higher rates of aortic anomalies such as Coarctation, substantially increase this population’s risk of complications in the proximal aorta (32,33).
Management of patients with BAV largely consists of appropriate surveillance, treatment of comorbid cardiovascular disease, and determining the timing for procedural intervention. Symptoms and diagnostic findings on physical exam, electrocardiogram (ECG), and chest roentgenography (CXR) should be comparable to patients with tricuspid aortic valve dysfunction (e.g., a crescendo decrescendo murmur at the right upper sternal border will be present in both bicuspid and tricuspid aortic valves with stenotic hemodynamics). Serial Doppler echocardiography assessment is recommended to monitor for progressing pathology at specified time intervals dependent on patients’ age, symptoms, and known valvular dysfunction. Computed tomographic or magnetic resonance angiography of the aorta is indicated in patients when the aortic sinuses, sinotubular junction, or ascending aorta cannot be fully assessed accurately by echocardiography and as a complementary imaging modality for a more thorough assessment of the entire aorta at specified time intervals (Figure 2) (34).
Figure 2.

Bicuspid aortic valve. (A,B,C,D) Transthoracic echocardiography study of a bicuspid aortic valve with no raphe and two symmetrical leaflets in parasternal short-axis (A) and in the apical three-chamber view (B,C,D). Note the presence of mixed valvular disease with central aortic regurgitation on color Doppler (C) and severe aortic stenosis assessed on continuous wave Doppler (D). (E) CT aortogram in the same patient, with valvular morphology redemonstrated on an oblique view.
Intervention on the aortic valve is indicated in patients with severe aortic stenosis and/or regurgitation who are experiencing symptoms and/or have impairment in left ventricular function. In the case of aortic regurgitation, intervention is indicated if there is progressive left ventricular dilatation as is the case with aortic regurgitation of alternate etiologies. For most patients with BAV the threshold for prophylactic aortic surgery is at a diameter of 5.5 cm, though lower thresholds exist for patients considered to be at increased risk for dissection, lower surgical risk, or with valvular dysfunction meeting criteria for surgery (35).
There are several considerations when determining the appropriate intervention for patients with BAV. They often require treatment at a younger age, frequently have mixed valvular disease, and commonly have other structural abnormalities. Those with isolated aortic stenosis without calcification or regurgitation can be considered for balloon valvuloplasty, though stenosis is likely to recur (36). Patients not amenable to valvuloplasty can be considered for surgical aortic valve repair, aortic valve replacement, the Ross Procedure, or more recently transcatheter aortic valve replacement (TAVR).
Aortic Valve repairs including suture plication, reconstruction with autologous pericardium (the Ozaki Procedure), and commissuroplasty have proven durable; however experience is confined to specialized centers (37-39). The Ross Procedure provides good hemodynamic performance with a pulmonic autograft in the aortic position, though can be limited by eventual dysfunction of the implanted homograft in the pulmonary position (40). The role of TAVR for BAV is being explored with great interest despite early concerns for issues with elliptical annuli and consequent paravalvular leak. Initial off-label observational data indicate technical difficulty with early TAVR technology in BAV. Newer generation transcatheter valves, improvement in pre-procedural imaging, and refined technique have led to improvements in practice such that more contemporary outcomes data show similar 30-day and one year mortality rates in BAV stenosis and tricuspid aortic valve stenosis in appropriately selected patients (41-44).
Subaortic and supravalvular aortic stenosis (SVAS)
While BAV is the most common structural congenital anomaly, there are several other important congenital lesions of the left ventricular outflow tract. Structural anomalies immediately inferior or superior to the aortic valve, while less common, require a unique approach. Amongst children recognized to have obstructive congenital left ventricular outflow tract lesions, 71–77% have aortic valvular stenosis, 10–14% have subvalvular stenosis, 8–14% have supravalvular stenosis, and 0–8% have multi-level obstruction (45-47). These lesions, like valvular stenosis, may be progressive in nature and not diagnosed until adulthood. As such, it is imperative that the general cardiologist recognize these pathologies and refer patients to specialized centers when appropriate.
Subvalvular or Subaortic stenosis (SAS) comes in many forms, with the most common being a thin, discrete, fibrous, circumferential membrane within the outflow tract that is attached to the ventricular septum. The second form is a muscular, diffuse, tunnel-like obstruction within the outflow tract, and is more rare (48). SAS is often found in patients with comorbid CHD, with the most common associations being with ventricular septal defects (23–37%), patent ductus arteriosus (34%), BAV (23%), coarctation of the aorta (23%), and pulmonic stenosis (9%) (48-50). It is associated with the Shone complex of left-sided obstructive lesions (51). The embryogenesis of SAS is unclear, and it has been found to recur after surgical removal. Current theories postulate that SAS develops and progresses in the context of particular geometric relationships between the ventricular septum, aorta, and mitral valve. The width of the aorto-mitral separation is twice normal in SAS patients, creating a steeper aorto-septal angle and an overriding aorta (52-54). This arrangement increases turbulence and shear stress within the outflow tract, causing progressive myocardial thickening, fibrosis, and scarring to result in the development of the structural anomaly.
SVAS is also heterogeneous in its nature. The majority of cases consist of a discrete thickening at the sinotubular junction, creating the so-called ‘hourglass’ deformity. A minority (25–40% of cases) have extension from the sinotubular junction to the more distal ascending aorta, aortic arch, and peripheral vasculature (55-57). The phenotype may be rooted in genetic disturbance of an elastin gene. Histologic specimens of the tunica media show minimal, disorganized elastin fibers, smooth muscle hypertrophy, and excessive collagen fibers. Williams-Beuren syndrome, a multisystem genetic syndrome with a deletion in the elastin gene, is associated with SVAS with a prevalence of ~70% in affected patients (58). Isolated SVAS can also be inherited in an autosomal dominant pattern or occur sporadically.
Clinically, SAS and SVAS may be difficult to distinguish from aortic stenosis or alternate fixed-obstructive left ventricular outflow tract lesions. ECG may show left ventricular hypertrophy. CXR may show cardiomegaly in advanced cases. Physical exam findings are similar for valvular aortic stenosis, SAS, and SVAS, though there are subtle differences. SAS more often causes aortic regurgitation due to valvular remodeling caused by subvalvular shear stress. If SVAS involves the more distal ascending aorta or brachiocephalic artery, patients may have unequal carotid pulses. Thorough imaging evaluation is key to appropriate diagnosis; two-dimensional (2D), three-dimensional (3D), and doppler transthoracic and transesophageal echocardiography are commonly used for defining both the anatomy of the obstruction and consequent pressure gradients. Cardiac MRI and CT imaging often serve as adjunct modalities to better characterize lesions following diagnosis. Cardiac catheterization may be used to characterize the magnitude and character of obstruction (particularly when multiple lesions are suspected), though is used less and less in contemporary practice.
Management of SAS and SVAS largely consists of surgical relief of obstruction (i.e., enlargement of the sinotubular junction and affected aorta for SVAS; discrete membrane excision in a focal SAS; myectomy or even Konno procedure for more diffuse SAS). Criteria for surgery include an obstruction causing a greater than 50 mmHg gradient, symptoms attributable to the obstruction, depression in ejection fraction, or coronary ischemia caused by the lesion (59). Following surgery, patients should continue with routine monitoring in cardiology specialty clinic. Surgical series in SAS have shown recurrent obstruction, progression in aortic regurgitation, increased risk of infectious endocarditis, and genetic predisposition to other valvular pathology (48,58,60-62).
Congenital pulmonary stenosis (PS)
Congenital PS is a common entity within CHD with an incidence of 0.5 per 1,000 live births (47). Though most often found as an isolated defect, valvular PS is associated with comorbid CHD—most commonly conotruncal defects such as tetralogy of Fallot, transposition of the great arteries, and double-outlet right ventricle. It is also associated with the genetic Noonan Syndrome (63). Valvular PS is the most common form of RVOT obstruction, though infundibular, supravalvular, and peripheral pulmonary arterial pathologies are well established (64).
Valvular PS presentations can vary from neonatal right ventricular (RV) failure to incidental discovery without symptoms depending on the severity of pathology. Heritability of congenital valvular PS is low at less than 4% amongst offspring, though is an autosomal dominant trait with variable penetrance in certain genetic syndromes (65). Cardiac physical exam findings classically include a systolic ejection murmur heard best at the second left intercostal space, possibly with a palpable thrill depending on intensity. The RVOT obstruction increases the time required for RV systole, leading to a split S2. Wider splitting of the S2 represents increased obstruction. Depending on the particulars of PV pathology, there may be an audible ejection click as the valve opens that decreases in intensity with inspiration. In these cases the proximity of the click to the normal S1 sound correlates to increasing severity.
ECG may show right axis deviation, right atrial enlargement, and/or RV hypertrophy dependent on the severity of the obstruction. CXR may show right heart enlargement and dilation of the pulmonary arteries. Echocardiography is crucial to verify diagnosis, where continuous wave Doppler profiles in pulmonic stenosis correlate well with invasive hemodynamics in the cardiac catheterization laboratory (Figure 3) (66,67).
Figure 3.

Pulmonic stenosis. Two cases of pulmonic stenosis are shown. (A,B,C) show a case of congenital pulmonic stenosis of the most common, dome-shaped phenotype in the parasternal short-axis view on a transthoracic echocardiogram. (A) Demonstrates that valve closed during diastole, (B) shows the valve doming due to the commissural fusion, and (C) reveals turbulent flow across the stenotic valve. (D,E,F) show a case of conduit stenosis, a known postoperative complication of surgical right ventricular to pulmonary arterial prostheses. (D) CT pulmonary angiogram showing the stenotic segment proximal to the pulmonary arterial bifurcation. (E) Parasternal short-axis view of the stenotic segment with flow acceleration demonstrated on color Doppler and (F) pulsed-wave Doppler.
Congenital valvular PS has three major morphologic subtypes: (I) the most common, dome-shaped phenotype with a central, narrowed effective orifice within three symmetric, mobile valve leaflets with fusion at the sites of the expected commissures; (II) the dysplastic phenotype with myxomatous thickening and poorly mobile leaflets without commissural fusion; and (III) uni- or bicuspid pulmonary valve phenotypes typical of tetralogy of Fallot (64,68). Similar to BAV, PS may be accompanied by an arteriopathy and post-stenotic pulmonary arterial dilation. Contributing factors are inherent medial abnormality of the vasculature and increased wall stress from eccentric, post-stenotic flow (69,70).
The natural history of congenital valvular PS is dependent on the severity of disease. Patients with mild stenosis with gradients less than 25 mmHg will likely never experience progression in disease or require intervention (71). Moderate or severe congenital PS often cause exertional intolerance in infancy or childhood, and many receive treatment before transitioning to adult cardiology practice. Greater than 20% of patients diagnosed with moderate PS who are initially asymptomatic will experience symptoms within 10 years of diagnosis and require intervention (58). Severe PS or pulmonary atresia presents early with cyanosis and right heart failure requiring prompt surgical evaluation to avoid irreversible RV remodeling and dysfunction.
Current guidelines recommend intervening on patients with moderate PS who experience associated symptoms and on all patients with severe valvular PS regardless of symptoms (59). In most cases, balloon pulmonary valvuloplasty is the preferred intervention. The procedure is minimally invasive and experienced centers have published generations of reassuring safety and efficacy data (72-77). A major limitation of balloon valvuloplasty, however, is its high rate of subsequent pulmonic regurgitation (PR). This can be progressive and lead to RV dysfunction (75,78-80). The presence of symptoms attributable to post-procedural PR is a Class I indication for valve replacement (59). Surgical valvotomy is an alternative to balloon valvuloplasty, but is typically reserved for those with dysplastic valvular leaflets, hypoplastic pulmonary annuli, or severe PR (81).
Pulmonic regurgitation in CHD
While mild PR is relatively common in normal individuals, there are special considerations within CHD (82). Isolated congenital PR is rare, most often associated with congenital absence of the pulmonary valve (83). PR is more often a component of mixed valvular disease in one of the aforementioned PS morphologic subtypes or as an iatrogenic consequence of prior percutaneous or surgical intervention (81,84-86).
Patients with severe PR are often asymptomatic at the time of diagnosis (87). Initial signs may be exertional intolerance due to dyspnea given patients’ inability to augment RV output (88). If RV failure develops, patients may experience hepatic congestion, ascites, and pedal edema. Right atrial and ventricular remodeling confer increased risk of arrhythmia, and patients may present with consequent lightheadedness or syncope. Cardiac physical exam findings depend on the mechanism of PR. Patients with congenital absence of the pulmonary valve will not have a P2, and instead a loud to-and-fro murmur over the second left intercostal space given unobstructed retrograde flow. The murmur is a lower-to-medium pitch, in contrast to the high-pitched Graham-Steell murmur associated with pulmonary hypertension and increased left-sided cardiac pressures. Similar to PS, ECG assessment may reveal right-axis deviation, RV hypertrophy, and right bundle branch block. CXR may show right heart enlargement.
Appropriate imaging diagnosis is crucial to assess the mechanism and severity of PR. Doppler and 2D echocardiography have historically been the cornerstones of evaluation. Severity is determined by PR jet width, duration, pressure half time, and increased density on continuous wave Doppler. In severe PR the color jet fills greater than 50% of the of the pulmonary valve annulus dimension and is of shorter duration due to rapid equalization of pressures in the pulmonary artery and right ventricle. Furthermore, color flow reversal can be seen in both the main pulmonary artery as well as pulmonary artery branches (Figure 4) (89). Cardiac MRI, however, has emerged as the preferred imaging modality for patients with PR, specifically for RV volume assessment. It has outperformed echocardiography in several studies in assessing RV remodeling and dysfunction (90-95). Nonetheless, limited availability of MRI machines curbs widespread use.
Figure 4.

Pulmonic regurgitation. Demonstrates a case of severe pulmonary regurgitation in a patient with tetralogy of Fallot on transthoracic echocardiography. (A) Parasternal short axis view of the pulmonary valve and main pulmonary artery with color Doppler showing a large regurgitant jet, meeting criteria for severe pathology as it covers greater than 50% of the pulmonary valve annulus. (B) Characteristic continuous-wave Doppler of severe pulmonary regurgitation with a dense regurgitant jet with steep deceleration and rapid termination. (C) 2D echocardiographic image showing right ventricular enlargement as a consequence of ongoing pulmonary regurgitation.
Patients with known PR of moderate or greater severity should undergo regular imaging evaluation and follow with a specialized CHD cardiologist (59). Indications for procedural interventions are dependent on the presence of symptoms and RV volumes and dysfunction. Increased arrhythmias have also been related to RV remodeling, and should be a factor when consideration valvular intervention. Patients with tetralogy of Fallot are the best studied CHD population on appropriate timing for pulmonary valve intervention in PR, though experts caution on extrapolation to those with different underlying pathology. In tetralogy of Fallot, pulmonary valve replacement (PVR) is recommended in patients with cardiac MRI-derived RV end-diastolic indexed volume of greater than 150 mL/m2 or RV end-systolic indexed volume >80 mL/m2, QRS duration of >180 msec or prolongation of >3.5 msec/year, residual shunt defects, concomitant RVOT obstruction (RV systolic pressure greater than two-thirds systemic pressure), symptoms of progressive exercise intolerance, or heart failure and sustained arrhythmia (59,96-100).
Surgical PVR is the most studied therapeutic approach for patients with PR in native RVOTs. Historically, the preferred surgical practice was placement of an aortic or pulmonary homograft in younger patients. More recently, there has been movement toward more traditional prostheses given high failure rates with homografts—possibly a consequence of younger patients’ immune response to donor tissue (101,102). Surgical outcomes data with this practice are encouraging, though younger age at intervention continues to be a risk factor for prosthesis failure requiring re-intervention (103-105).
Transcatheter PVR (TPVR)
TPVR has had an expanding role in various CHD. The procedure initially emerged in 2000 as a minimally invasive means to treat dysfunctional surgical RV to pulmonary arterial conduits (106). Twenty years later, it is usually the first line intervention depending on anatomy. In experienced centers, TPVR is now feasible in the native RVOT or failing conduits or bioprostheses (107-109). Mid-term outcomes data show decreases in RV volumes, stabilization of QRS duration, and favorable valvular hemodynamics with low periprocedural morbidity, mortality, and reintervention rates (110-114).
There are currently two FDA-approved, commercially available devices for TPVR: the Melody® valve made from bovine jugular vein (Medtronic, Minneapolis, MN, USA) and the Sapien XT bovine pericardial valve (Edwards Lifesciences, Irvine, CA, USA). The Melody® valve is limited by smaller diameters (18–22 mm), but is approved for use in both dysfunctional conduits and failing bioprostheses. It can be expanded to 24 mm at the price of mild central PR. The Sapien XT is available in diameters from 20 to 29 mm, though is only FDA-approved for use in failing conduits.
There are several important limitations to consider with TPVR. The early Melody® valve experience suffered from a ~20% rate of stent fracture and consequent prosthesis dysfunction. Fortunately, this has been mitigated by pre-stenting the intended landing zone (115,116). TPVR in native RVOTs and dilated pulmonary arteries is limited by available transcatheter valve sizes as under-sizing can result in poor apposition, paravalvular leak, and valve embolization. CHD patients also have widely variable RVOT anatomy, and delivery systems have limitations with tortuous anatomy. Perhaps most concerning is that native RVOTs and RV to PA conduits are closely juxtaposed to the aortic root and coronary arteries. The introduction of the rigid transcatheter valve stent struts may cause coronary artery or aortic root compression requiring urgent intervention (117-119). Series show that the risk of coronary artery compression may be as high as ~5%, and accordingly there is a Class I indication for balloon compression testing prior to valve deployment (58).
Congenital disease and the atrioventricular (AV) valves
Embryogenesis of the AV valves
AV valves form at the junction of the primitive atria and ventricles (termed AV canal) (6,7). At this level, large superior and inferior AV endocardial cushions grow into the lumen of the AV canal. They merge to create a septum separating the tricuspid and mitral orifices. The edges of this septum evolve to become the septal leaflets of their respective AV valve (the anterior mitral leaflet on the left and the septal tricuspid leaflet on the right). The confluence also serves as an anchor to merge with the atrial and ventricular septae (6,7,120). The mural AV valve leaflets are derived from smaller, lateral endocardial cushions (Figure 5).
Figure 5.
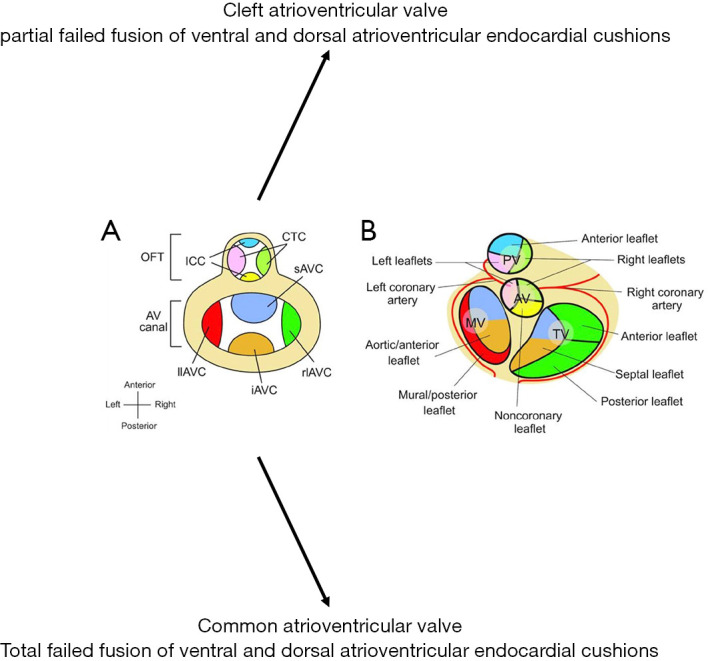
Endocardial cushions and heart valve leaflets. (A) Schematic of endocardial cushions in the atrioventricular (AV) canal and the outflow tract (OFT). The figure is a superior view of the heart with atria removed. The endocardial cushions are color coded to correspond to their derived valve leaflets illustrated in (B). (B) Schematic (superior view) of atrioventricular and semilunar valve leaflets that develop from the corresponding cushions color coded in (A). CTC, conotruncal cushions; ICC, intercalated cushions; sAVC, superior AV cushion; iAVC, inferior AV cushion; rlAVC, right lateral AV cushion; llAVC, left lateral AV cushion; PV, pulmonary valve; AV, aortic valve; TV, tricuspid valve; MV, mitral valve. Figure reproduced with permission from Lin et al. Partitioning the heart: mechanisms of cardiac septation and valve development. Development. 2012 Sep 15;139(18):3277-99.
AV valve leaflets form through a process known as delamination (121-123). In this process, fenestrations develop in the ventricular myocardium underlying the AV endocardial cushions. This allows the primitive valve tissue to mobilize, exposing it to the shear stress of repeated loading and unloading in the fetal circulation. This cyclical tension precipitates fibrous remodeling of the ventricular aspects of the valves and their remaining tethers to the myocardium (chordae tendineae). The anchoring trabeculae go on to become papillary muscles (Figure 6) (124,125).
Figure 6.

Delamination of atrioventricular valves. Once endocardial cushion tissue surrounds the atrioventricular (AV) orifices, valve leaflets are formed via a delamination process. The superficial endocardial cushion tissue differentiates into precursory valve tissue. As this occurs fenestrations develop within the underlying myocardium, allowing the primitive valve to mobilize. As myocardium disappears, the primitive valve leaflets are left attached to focal patches of resilient myocardium that will go on to become papillary muscles. The valve cusps are exposed to repeated loading and unloading in the primitive circulation, and their tissue remodels to accommodate the associated shear stress. Fibrous chordae tendineae develop to maintain the integrity of the valve cusp as it grows to accommodate the AV orifice. Normal development of AV valves is represented by the progression of (A,B,C,D) moving horizontally across the figure. † represents the failed delamination of the septal tricuspid leaflet seen in Ebstein anomaly. ‡ represents fusion of tethered trabeculae resulting in a single papillary muscle for the mitral valve. This phenotype is known as the ‘Parachute’ mitral valve.
The nature of this development is at the root of the variability appreciated in AV valve anatomy seen in patients with and without CHD. Maladaptive fusion of endocardial cushions may result in congenital mitral and/or tricuspid valve dysplasia with consequent stenosis or atresia. Total failure of AV endocardial cushion fusion can lead to profound AV canal defects that leave a common (neither identifiable as mitral or tricuspid) AV valve straddling the entire ventricular inlet (126).
Ebstein anomaly
Ebstein anomaly occurs when the tricuspid valve’s septal and posterior leaflets fail to delaminate from their underlying myocardium. These leaflets are often hypoplastic. The anterior leaflet is often enlarged, redundant, and ‘sail-like’. It can also contain fenestrations and be abnormally tethered to the RV myocardium (127-130). These derangements cause apical displacement of the tricuspid orifice’s fibromuscular skeleton, and consequently the tricuspid annulus. In extreme cases the valvular anatomy can be so apically displaced near the pulmonary valve that the anterior leaflet can cause obstruction of the RVOT (131).
The area between the traditional AV groove and the apically displaced tricuspid annulus is the so-called ‘atrialized’ right ventricle. This segment of myocardium is thinner, more distensible, and poorly contractile in comparison to normal RV myocardium. In fact, it balloons out maladaptively during atrial systole to create a passive reservoir and thus decreases forward flow (127,132). The derangement in the tricuspid orifice’s fibromuscular skeleton also diminishes its capability as an electrical barrier between the right atrium and ventricle, allowing for uninsulated muscular connections and associated accessory pathways (133). The remaining functional right ventricle is diminished in its effective geometry and function due to the impinging valve.
Ebstein anomaly is rare, with a reported incidence at ~1 per 20,000 live births (132,134,135). The genetic and environmental contributions to the phenotype remain ambiguous. It is widely accepted to be genetically heterogeneous, with several genes implicated in small subpopulations. Twinning, white race, and a family history of cardiovascular malformations have been associated with increased incidence. Maternal exposure to benzodiazepines or environmental exposure to paint varnish have been shown to increase risk (136). The classic association with in utero exposure to Lithium is present, though has been exaggerated by biased retrospective reports (137).
Clinical presentations with Ebstein anomaly vary widely dependent on the severity of displacement and dysplasia of the tricuspid valve leaflets, the RV substrate, and comorbid CHD (138-140). Interatrial communications [i.e., patent foramen ovales (PFOs), atrial septal defects (ASDs)] are present in 79% to 94% of patients, leaving them vulnerable to right-to-left shunting, paradoxical emboli, and right heart volume overload (141-143). Clinical severity often correlates with age at presentation. Fetal, neonatal, and infant patients are more likely to have additional CHD diagnoses and suffer more dire manifestations with cyanosis, right heart failure, and a high mortality rate (144). Children, adolescents, and adults can present with dyspnea, fatigue, ascites, edema, exertional intolerance, or cyanosis in a time course correlating with the severity of their disease; however a large portion come to attention with atrial arrhythmias (145-147).
Physical examination also correlates with severity of disease. The characteristic murmur is one of tricuspid regurgitation (TR) during systole at the lower left sternal border with a possible added mid-diastolic rumble caused by turbulent blood flow across the dysmorphic tricuspid annulus. Jugular venous pressure can be elevated, though commonly will be blunted due to the increased compliance of the enlarged right atrium. S1 and S2 are often widely split, and accompanied by an S3 and/or S4 in what is termed a “triple” or “quadruple” rhythm (141). Manifestations of cyanosis may be present depending on the degree of right-to-left shunting. Features of right heart failure including hepatomegaly, ascites, and edema are signs of decompensation. ECG may be notable for enlarged ‘Himalayan’ P waves representing right atrial enlargement. First degree AV block and right bundle branch block are commonly seen. Atypical S waves may be present due to abnormal depolarization in the atrialized right ventricle. The classic CXR findings are a globular cardiac silhouette with a narrow pulmonary artery (132).
Doppler and 2D echocardiography are used for initial diagnosis, assessing valvular hemodynamics, elucidating presence of interatrial shunting, and ongoing surveillance for disease progression. The cardinal echocardiographic feature of Ebstein anomaly is apical displacement of the septal tricuspid leaflet by greater than or equal to 8 mm/m2 body surface area in the apical four chamber view (130,148,149). Quantification of TR by Doppler is often challenging due to the valve orifice’s anatomic complexity and the presence of fenestrations. Jet vectors are variable dependent on patient anatomy. Complicating things further still, patients’ pulmonary pressures are typically normal (except for those with comorbid anatomic pulmonic stenosis or left heart disease). This, along with the RV myopathy, makes for a low velocity TR jet that can be difficult to ascertain with Doppler imaging (Figure 7).
Figure 7.

Ebstein anomaly. Demonstrates a case of Ebstein anomaly on transthoracic echocardiography. (A) Apical four-chamber view showing right ventricular dilation and apical displacement of the tricuspid annulus due to restriction of the septal leaflet and a larger, redundant anterior leaflet. (B) Tricuspid regurgitation due to the dysmorphic valve is visualized on color Doppler. (C) Demonstrates the severity of valve abnormality in this case with the septal tricuspid valve leaflet closely juxtaposed to the pulmonary valve. (D) Demonstrates the classic ‘Sail’ morphology of the redundant anterior tricuspid leaflet in Ebstein anomaly. ATVL, anterior tricuspid valve leaflet; PV, pulmonary valve; STVL, septal tricuspid valve leaflet.
There are also challenges with transthoracic echocardiography in patients with severely enlarged right-sided heart chambers. Cardiac MRI serves a complementary role with its ability to better quantify parameters of right atrial and ventricular volumes and function, but can also better elucidate the anatomy of the posterior tricuspid leaflet and fenestrations within the tricuspid valve (150,151). Transesophageal echocardiography may also be used when transthoracic imaging is unable to fully characterize tricuspid valve morphology and function (59).
Adult patients diagnosed with Ebstein anomaly should be referred to a cardiologist with subspecialty training in CHD. Guidelines recommend surveillance imaging, evaluation for arrhythmias, and exercise testing at regular intervals dependent on the severity of their disease. Surgical intervention is indicated in patients with heart failure symptoms, objective evidence of worsening exercise capacity, or progressive RV systolic dysfunction by echocardiography or Cardiac MRI. Waiting beyond these developments confers a poorer prognosis. Current guidelines also state that surgical repair may be beneficial in patients with progressive RV enlargement, systemic desaturation felt to be caused by right-to-left atrial shunting, paradoxical embolism, and/or atrial tachyarrhythmias (59,152-155).
Surgical techniques have evolved with time, though all have generally consisted of a tricuspid valve repair or replacement, closure of the interatrial communication, plication of the atrialized right ventricle, selective right atrioplasty, and disruption of any accessory electrophysiologic pathways not amenable to catheter ablation. Earlier surgeries attempted to create a competent monocuspid valve from the enlarged anterior leaflet (156,157). The more contemporary repair is a “cone reconstruction,” where all three tricuspid leaflets are mobilized and then sewn together to create a neo-valve or “cone”. The atrialized right ventricle is then plicated to create a compact neo-annulus, and the cone is sewn in with careful attention to avoid injury to the right coronary artery (158). Outcome data of the monocuspid repair has favorable long term survival rates of 86% and freedom from reoperation in 70% of patients 15 years postoperatively (154). Should valve repair prove too difficult, valve replacement is performed with a preference toward bioprostheses to avoid thromboembolic complications (159,160). In severe cases, patients may undergo a bidirectional Glenn procedure in addition to valve intervention to facilitate ventricular unloading or even complete Fontan palliation to bypass the right heart entirely (161).
Congenital mitral valve disease
The stochastic nature of AV valve development yields substantial variability in both the valvular and subvalvular apparatus. Accordingly, derangements in embryogenesis can lead to a wide spectrum of congenital pathology. Partially failed fusion of endocardial cushions can lead to cleft valve leaflets. Abnormal fusion of myocardial trabeculae or derangements in delamination may create dysfunctional subvalvular apparatus (148).
Congenital mitral valvular disease can exist in isolation, but is often recognized in patients with comorbid CHD. Mitral valve clefting is often a component of a partial AV canal defect. A parachute mitral valve, where all chordae are attached to a solitary central papillary muscle (Figure 8), and supramitral ring are associated with the classic Shone complex of structural left-sided obstructive lesions. Mitral atresia is almost exclusively associated with hypoplasia of the left-sided chambers or transposition of the great vessels and a common ventricle. Several types of congenital mitral dysplasia have also been described, including the hammock mitral valve (where the papillary muscles are directly connected to the valve leaflets without interposed chordae) and the double orifice mitral valve with an interposed fibrous connection. An echocardiography series showed that approximately 0.5% of patients referred to a major pediatric cardiology practice had some type of congenital malformation of the mitral valve (51,162-164).
Figure 8.

Parachute mitral valve. Demonstrates a case of a ‘Parachute’ Mitral Valve, in which all anchoring chordae tendineae supporting the mitral valve arise from a single papillary muscle. This is a consequence of abnormal fusion of myocardial trabeculae during delamination. (A,B) Apical four-chamber view using 2D echocardiography to show (A) the single anchoring papillary muscle and (B) color flow Doppler showing mitral regurgitation from the maladaptive tethering apparatus. (C) Cardiac MRI image showing the ‘Parachute’ mitral valve.
Doppler and 2D echocardiography are used for initial diagnosis, assessing valvular anatomy, and valvular hemodynamics. 3D echocardiography has proven increasingly useful as an adjunct modality (165). Clinical presentations, diagnostic testing, and physical exam findings vary depending on the type of mitral valvular pathology and the degree of hemodynamic disturbance. Mitral valve cleft, which most often affects the anterior mitral leaflet, leads to mitral regurgitation. The parachute mitral valve, supramitral membrane, and double-orifice mitral valve more often cause stenotic physiology. The hammock mitral valve, with its thickened and less mobile subvalvular apparatus, leads to mixed valvular disease due to restriction of valve leaflets (164).
Mitral valve repair is preferable to mitral valve replacement when possible, though congenital anomalies often represent complex cases where repair may not be feasible. Given the importance of a successful primary repair, guidelines recommend that patients follow at a medical center where such abnormalities can be followed and future interventions considered (59).
AV valves in the univentricular heart
An extensive discussion of CHD that results in a univentricular circulation is beyond the scope of this review. Briefly, a univentricular circulation is a surgical endpoint created via multiple staged procedures in patients with complex CHD. In these cases, patients have a single effective ventricle (right, left, or indeterminate) capable of providing systemic cardiac output. In a series of surgeries, the vena cavae are anastomosed to the pulmonary arterial circulation without an interposed subpulmonic ventricle. Thus, systemic venous return flows passively into the pulmonary arterial circulation. Pulmonary venous return flows back into the heart, through the AV valve(s), and into the effective ventricle to supply blood to the systemic circulation (166-168).
In this physiology, transpulmonary flow is driven solely by the gradient between systemic venous pressure and pulmonary venous pressure. Even modest AV valvular dysfunction can drive increases in pulmonary venous pressure, causing pulmonary and venous congestion. Worse still is that AV valve regurgitation will lead to ventricular remodeling, which in turn leads to annular dilation and maladaptive valve tethering, causing worse regurgitation (169). This remodeling has been shown to increase the risk of arrhythmias, failure of the univentricular circuit, and need for repeat surgery (170). Retrospective cohorts have consistently found that the presence of AV valve dysfunction is associated with significant increases in morbidity and mortality (171-174).
Patients with a common (single, merged) AV valve or a morphologically tricuspid valve are more likely to suffer from AV valve dysfunction (171,175-179). Series indicate that prolapse and dysplasia are often the principal mechanisms (179), though leaflet cleft or restriction can also contribute. Annular dilation leading to secondary valvular regurgitation is also a common phenomenon, though is infrequently the only pathologic mechanism (171,180).
Unfortunately, data to guide appropriate surveillance and timing for surgical management of AV valves in a univentricular circulation is scant. Guidelines recommend surveillance echocardiography annually, and cardiac MRI every two to three years depending on the presence of symptoms or higher risk pathology (59). Studies are largely retrospective surgical and autopsy series from specialized centers with substantial variability in patient phenotype and procedural technique. Also, a significant percentage of patients in these series underwent AV valve surgery before or at the time of their surgical conversion to univentricular physiology (181). Despite this, AV valve repair and replacement are amongst the most common redo-surgeries in this population (182). Postoperative ventricular function and residual AV valve dysfunction appear to be major predictors for requiring repeat intervention, and survival (174).
Conclusions
There is considerable heterogeneity and complexity in the valvular pathology associated with CHD. Early recognition of congenital valvular heart disease, and prompt referral to specialized centers is crucial. Multimodality imaging is central to elucidating complex valvular anatomy and dysfunction in several types of CHD. Management of valvular disease in CHD should involve a multidisciplinary approach involving teams of congenital cardiac surgeons, interventional cardiologists, multimodality imagers, and CHD-focused cardiologists to provide optimal care to this complex group of patients.
Acknowledgments
We would like to acknowledge Brianna Core for her administrative support.
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editors (Deborah H. Kwon and Bo Xu) for the series “Heart Valve Disease” published in Cardiovascular Diagnosis and Therapy. The article has undergone external peer review.
Reporting Checklist: The authors have completed the Narrative Review reporting checklist. Available at http://dx.doi.org/10.21037/cdt-19-693-b).
Conflicts of Interest: Both authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/cdt-19-693-b). The series “Heart Valve Disease” was commissioned by the editorial office without any funding or sponsorship. The authors have no other conflicts of interest to declare.
References
- 1.Gilboa SM, Devine OJ, Kucik JE, et al. Congenital Heart Defects in the United States. Circulation 2016;134:101-9. 10.1161/CIRCULATIONAHA.115.019307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilboa SM, Salemi JL, Nembhard WN, et al. Mortality Resulting From Congenital Heart Disease Among Children and Adults in the United States, 1999 to 2006. Circulation 2010;122:2254-63. 10.1161/CIRCULATIONAHA.110.947002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.CDC. Data and Statistics on Congenital Heart Defects | CDC [Internet]. Centers for Disease Control and Prevention 2019 [cited 2019 Sep 20]. Available online: https://www.cdc.gov/ncbddd/heartdefects/data.html
- 4.Milos NC, Nordstrom DB, Ongaro I, et al. Variations in structure of the outflow tract of the human embryonic heart: A new hypothesis for generating bicuspid aortic semilunar valves. Ann Anat 2017;211:88-103. 10.1016/j.aanat.2016.12.004 [DOI] [PubMed] [Google Scholar]
- 5.Radu-Ioniţă F, Bontaş E, Goleanu V, et al. Heart Embryology: Overview. In: Right Heart Pathology. Springer, Cham; 2018:3-24. [Google Scholar]
- 6.Lin CJ, Lin CY, Chen CH, et al. Partitioning the heart: mechanisms of cardiac septation and valve development. Development 2012;139:3277-99. 10.1242/dev.063495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Combs MD, Yutzey KE. Heart Valve Development: Regulatory networks in development and disease. Circ Res 2009;105:408-21. 10.1161/CIRCRESAHA.109.201566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin PS, Kloesel B, Norris R, et al. Embryonic Development of the Bicuspid Aortic Valve. J Cardiovasc Dev Dis 2015;2:248-72. 10.3390/jcdd2040248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Restivo A, Piacentini G, Placidi S, et al. Cardiac outflow tract: a review of some embryogenetic aspects of the conotruncal region of the heart. Anat Rec A Discov Mol Cell Evol Biol 2006;288:936-43. 10.1002/ar.a.20367 [DOI] [PubMed] [Google Scholar]
- 10.Enriquez A, Bradfield J, Shivkumar K, et al. Anatomy of the Outflow Tract Region [Internet]. Cardiac Mapping 2019:61-9. Available online: 10.1002/9781119152637.ch6 10.1002/9781119152637.ch6 [DOI]
- 11.Vanmierop LH, Wiglesworth FW. Pathogenesis of transposition Complexes. Am J Cardiol 1963;12:226-32. 10.1016/0002-9149(63)90312-6 [DOI] [PubMed] [Google Scholar]
- 12.Fernández B, Durán AC, Fernández-Gallego T, et al. Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. J Am Coll Cardiol 2009;54:2312-8. 10.1016/j.jacc.2009.07.044 [DOI] [PubMed] [Google Scholar]
- 13.Masri A, Svensson LG, Griffin BP, et al. Contemporary natural history of bicuspid aortic valve disease: a systematic review. Heart 2017;103:1323-30. 10.1136/heartjnl-2016-309916 [DOI] [PubMed] [Google Scholar]
- 14.Braverman AC. The bicuspid aortic valve and associated aortic disease. Valvular heart disease A companion to Braunwald’s heart disease. 4th edn. Elsevier Saunders; 2014:179-98. [Google Scholar]
- 15.Fernandes SM, Khairy P, Sanders SP, et al. Bicuspid Aortic Valve Morphology and Interventions in the Young. J Am Coll Cardiol 2007;49:2211-4. 10.1016/j.jacc.2007.01.090 [DOI] [PubMed] [Google Scholar]
- 16.Kang JW, Song HG, Yang DH, et al. Association Between Bicuspid Aortic Valve Phenotype and Patterns of Valvular Dysfunction and Bicuspid Aortopathy. JACC Cardiovasc Imaging 2013;6:150-61. 10.1016/j.jcmg.2012.11.007 [DOI] [PubMed] [Google Scholar]
- 17.Sievers HH, Schmidtke C. A classification system for the bicuspid aortic valve from 304 surgical specimens. J Thorac Cardiovasc Surg 2007;133:1226-33. 10.1016/j.jtcvs.2007.01.039 [DOI] [PubMed] [Google Scholar]
- 18.Sabet HY, Edwards WD, Tazelaar HD, et al. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc 1999;74:14-26. 10.4065/74.1.14 [DOI] [PubMed] [Google Scholar]
- 19.Asher CR, Griffin BP. Manual of Valvular Heart Disease. Lippincott Williams & Wilkins; 2017:550. [Google Scholar]
- 20.Cripe L, Andelfinger G, Martin LJ, et al. Bicuspid aortic valve is heritable. J Am Coll Cardiol 2004;44:138-43. 10.1016/j.jacc.2004.03.050 [DOI] [PubMed] [Google Scholar]
- 21.Huntington K, Hunter AG, Chan KL. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol 1997;30:1809-12. 10.1016/S0735-1097(97)00372-0 [DOI] [PubMed] [Google Scholar]
- 22.Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature 2005;437:270-4. 10.1038/nature03940 [DOI] [PubMed] [Google Scholar]
- 23.Martin LJ, Ramachandran V, Cripe LH, et al. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum Genet 2007;121:275-84. 10.1007/s00439-006-0316-9 [DOI] [PubMed] [Google Scholar]
- 24.Oppenheimer-Dekker A, Gittenberger-de Groot AC, Bartelings MM, et al. Abnormal architecture of the ventricles in hearts with an overriding aortic valve and a perimembranous ventricular septal defect (“Eisenmenger VSD”). Int J Cardiol 1985;9:341-55. 10.1016/0167-5273(85)90032-4 [DOI] [PubMed] [Google Scholar]
- 25.Neumayer U, Stone S, Somerville J. Small ventricular septal defects in adults. Eur Heart J 1998;19:1573-82. 10.1053/euhj.1998.1083 [DOI] [PubMed] [Google Scholar]
- 26.Koenraadt WMC, Tokmaji G, DeRuiter MC, et al. Coronary anatomy as related to bicuspid aortic valve morphology. Heart 2016;102:943-9. 10.1136/heartjnl-2015-308629 [DOI] [PubMed] [Google Scholar]
- 27.Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med 2014;370:1920-9. 10.1056/NEJMra1207059 [DOI] [PubMed] [Google Scholar]
- 28.Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol 2010;55:2789-800. 10.1016/j.jacc.2009.12.068 [DOI] [PubMed] [Google Scholar]
- 29.Tadros TM, Klein MD, Shapira OM. Ascending aortic dilatation associated with bicuspid aortic valve: pathophysiology, molecular biology, and clinical implications. Circulation 2009;119:880-90. 10.1161/CIRCULATIONAHA.108.795401 [DOI] [PubMed] [Google Scholar]
- 30.Warnes CA. Bicuspid aortic valve and coarctation: two villains part of a diffuse problem. Heart 2003;89:965-6. 10.1136/heart.89.9.965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikonomidis JS, Jones JA, Barbour JR, et al. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with bicuspid or tricuspid aortic valves. J Thorac Cardiovasc Surg 2007;133:1028-36. 10.1016/j.jtcvs.2006.10.083 [DOI] [PubMed] [Google Scholar]
- 32.Becker AE, Becker MJ, Edwards JE. Anomalies associated with coarctation of aorta: particular reference to infancy. Circulation 1970;41:1067-75. 10.1161/01.CIR.41.6.1067 [DOI] [PubMed] [Google Scholar]
- 33.Oliver JM, Alonso-Gonzalez R, Gonzalez AE, et al. Risk of aortic root or ascending aorta complications in patients with bicuspid aortic valve with and without coarctation of the aorta. Am J Cardiol 2009;104:1001-6. 10.1016/j.amjcard.2009.05.045 [DOI] [PubMed] [Google Scholar]
- 34.Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014;63:e57-185. 10.1016/j.jacc.2014.02.536 [DOI] [PubMed] [Google Scholar]
- 35.Hiratzka LF, Creager MA, Isselbacher EM, et al. Surgery for Aortic Dilatation in Patients With Bicuspid Aortic Valves: A Statement of Clarification From the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2016;67:724-31. 10.1016/j.jacc.2015.11.006 [DOI] [PubMed] [Google Scholar]
- 36.Satou GM, Perry SB, Lock JE, et al. Repeat balloon dilation of congenital valvar aortic stenosis: immediate results and midterm outcome. Catheter Cardiovasc Interv 1999;47:47-51. [DOI] [PubMed] [Google Scholar]
- 37.Pettersson GB, Crucean AC, Savage R, et al. Toward predictable repair of regurgitant aortic valves: a systematic morphology-directed approach to bicommissural repair. J Am Coll Cardiol 2008;52:40-9. 10.1016/j.jacc.2008.01.073 [DOI] [PubMed] [Google Scholar]
- 38.Ozaki S, Kawase I, Yamashita H, et al. Reconstruction of Bicuspid Aortic Valve With Autologous Pericardium. Circ J 2014;78:1144-51. 10.1253/circj.CJ-13-1335 [DOI] [PubMed] [Google Scholar]
- 39.Zeeshan A, Idrees JJ, Johnston DR, et al. Durability of Aortic Valve Cusp Repair With and Without Annular Support. Ann Thorac Surg 2018;105:739-48. 10.1016/j.athoracsur.2017.09.019 [DOI] [PubMed] [Google Scholar]
- 40.Liu T, Xie M, Lv Q, et al. Bicuspid Aortic Valve: An Update in Morphology, Genetics, Biomarker, Complications, Imaging Diagnosis and Treatment. Front Physiol 2019;9:1921. 10.3389/fphys.2018.01921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makkar RR, Yoon SH, Leon MB, et al. Association Between Transcatheter Aortic Valve Replacement for Bicuspid vs Tricuspid Aortic Stenosis and Mortality or Stroke. JAMA 2019;321:2193-202. 10.1001/jama.2019.7108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoon SH, Lefèvre T, Ahn JM, et al. Transcatheter Aortic Valve Replacement With Early- and New-Generation Devices in Bicuspid Aortic Valve Stenosis. J Am Coll Cardiol 2016;68:1195-205. 10.1016/j.jacc.2016.06.041 [DOI] [PubMed] [Google Scholar]
- 43.Mylotte D, Lefevre T, Søndergaard L, et al. Transcatheter aortic valve replacement in bicuspid aortic valve disease. J Am Coll Cardiol 2014;64:2330-9. 10.1016/j.jacc.2014.09.039 [DOI] [PubMed] [Google Scholar]
- 44.Yoon SH, Bleiziffer S, De Backer O, et al. Outcomes in Transcatheter Aortic Valve Replacement for Bicuspid Versus Tricuspid Aortic Valve Stenosis. J Am Coll Cardiol 2017;69:2579-89. 10.1016/j.jacc.2017.03.017 [DOI] [PubMed] [Google Scholar]
- 45.Kitchiner D, Jackson M, Malaiya N, et al. Incidence and prognosis of obstruction of the left ventricular outflow tract in Liverpool (1960-91): a study of 313 patients. Br Heart J 1994;71:588-95. 10.1136/hrt.71.6.588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu CW, Hwang B, Lee BC, et al. Aortic stenosis in children: 19-year experience. Zhonghua Yi Xue Za Zhi 1997;59:107-13. [PubMed] [Google Scholar]
- 47.Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002;39:1890-900. 10.1016/S0735-1097(02)01886-7 [DOI] [PubMed] [Google Scholar]
- 48.Brauner R, Laks H, Drinkwater DC, Jr, et al. Benefits of early surgical repair in fixed subaortic stenosis. J Am Coll Cardiol 1997;30:1835-42. 10.1016/S0735-1097(97)00410-5 [DOI] [PubMed] [Google Scholar]
- 49.Leichter DA, Sullivan I, Gersony WM. “Acquired” discrete subvalvular aortic stenosis: Natural history and hemodynamics. J Am Coll Cardiol 1989;14:1539-44. 10.1016/0735-1097(89)90395-1 [DOI] [PubMed] [Google Scholar]
- 50.Choi JY, Sullivan ID. Fixed subaortic stenosis: anatomical spectrum and nature of progression. Br Heart J 1991;65:280-6. 10.1136/hrt.65.5.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shone JD, Sellers RD, Anderson RC, et al. The developmental complex of “parachute mitral valve,” supravalvular ring of left atrium, subaortic stenosis, and coarctation of aorta. Am J Cardiol 1963;11:714-25. 10.1016/0002-9149(63)90098-5 [DOI] [PubMed] [Google Scholar]
- 52.Barkhordarian R, Wen-Hong D, Li W, et al. Geometry of the left ventricular outflow tract in fixed subaortic stenosis and intact ventricular septum: an echocardiographic study in children and adults. J Thorac Cardiovasc Surg 2007;133:196-203. 10.1016/j.jtcvs.2006.09.010 [DOI] [PubMed] [Google Scholar]
- 53.Kleinert S, Geva T. Echocardiographic morphometry and geometry of the left ventricular outflow tract in fixed subaortic stenosis. J Am Coll Cardiol 1993;22:1501-8. 10.1016/0735-1097(93)90563-G [DOI] [PubMed] [Google Scholar]
- 54.Rosenquist GC, Clark EB, McAllister HA, et al. Increased mitral-aortic separation in discrete subaortic stenosis. Circulation 1979;60:70-4. 10.1161/01.CIR.60.1.70 [DOI] [PubMed] [Google Scholar]
- 55.Flaker G, Teske D, Kilman J, et al. Supravalvular aortic stenosis. A 20-year clinical perspective and experience with patch aortoplasty. Am J Cardiol 1983;51:256-60. 10.1016/S0002-9149(83)80045-9 [DOI] [PubMed] [Google Scholar]
- 56.Keane JF, Fellows KE, LaFarge CG, et al. The surgical management of discrete and diffuse supravalvar aortic stenosis. Circulation 1976;54:112-7. 10.1161/01.CIR.54.1.112 [DOI] [PubMed] [Google Scholar]
- 57.Aboulhosn J, Child JS. Left ventricular outflow obstruction: subaortic stenosis, bicuspid aortic valve, supravalvar aortic stenosis, and coarctation of the aorta. Circulation 2006;114:2412-22. 10.1161/CIRCULATIONAHA.105.592089 [DOI] [PubMed] [Google Scholar]
- 58.Bruno E, Rossi N, Thüer O, et al. Cardiovascular findings, and clinical course, in patients with Williams syndrome. Cardiol Young 2003;13:532-6. 10.1017/S1047951103001124 [DOI] [PubMed] [Google Scholar]
- 59.Stout KK, Daniels CJ, Aboulhosn JA, et al. 2018 AHA/ACC Guideline for the Management of Adults With Congenital Heart Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019;139:e698-800. [DOI] [PubMed] [Google Scholar]
- 60.Wright GB, Keane JF, Nadas AS, et al. Fixed subaortic stenosis in the young: medical and surgical course in 83 patients. Am J Cardiol 1983;52:830-5. 10.1016/0002-9149(83)90423-X [DOI] [PubMed] [Google Scholar]
- 61.Jacobs JP, Palatianos GM, Cintron JR, et al. Transaortic resection of the subaortic membrane. Treatment for subvalvular aortic stenosis. Chest 1994;106:46-51. 10.1378/chest.106.1.46 [DOI] [PubMed] [Google Scholar]
- 62.Firpo C, Maitre Azcárate MJ, Quero Jiménez M, et al. Discrete subaortic stenosis (DSS) in childhood: a congenital or acquired disease? Follow-up in 65 patients. Eur Heart J 1990;11:1033-40. 10.1093/oxfordjournals.eurheartj.a059631 [DOI] [PubMed] [Google Scholar]
- 63.Colquitt JL, Noonan JA. Cardiac findings in Noonan syndrome on long-term follow-up. Congenit Heart Dis 2014;9:144-50. 10.1111/chd.12102 [DOI] [PubMed] [Google Scholar]
- 64.Bashore TM. Adult congenital heart disease: right ventricular outflow tract lesions. Circulation 2007;115:1933-47. 10.1161/CIRCULATIONAHA.105.592345 [DOI] [PubMed] [Google Scholar]
- 65.Driscoll DJ, Michels VV, Gersony WM, et al. Occurrence risk for congenital heart defects in relatives of patients with aortic stenosis, pulmonary stenosis, or ventricular septal defect. Circulation 1993;87:I114-20. [PubMed] [Google Scholar]
- 66.Johnson GL, Kwan OL, Handshoe S, et al. Accuracy of combined two-dimensional echocardiography and continuous wave Doppler recordings in the estimation of pressure gradient in right ventricular outlet obstruction. J Am Coll Cardiol 1984;3:1013-8. 10.1016/S0735-1097(84)80361-7 [DOI] [PubMed] [Google Scholar]
- 67.Aldousany AW, DiSessa TG, Dubois R, et al. Doppler estimation of pressure gradient in pulmonary stenosis: maximal instantaneous vs peak-to-peak, vs mean catheter gradient. Pediatr Cardiol 1989;10:145-9. 10.1007/BF02081678 [DOI] [PubMed] [Google Scholar]
- 68.Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to develop guidelines on the management of adults with congenital heart disease). Circulation 2008;118:e714-833. [DOI] [PubMed] [Google Scholar]
- 69.Fenster BE, Schroeder JD, Hertzberg JR, et al. 4-Dimensional cardiac magnetic resonance in a patient with bicuspid pulmonic valve: characterization of post-stenotic flow. J Am Coll Cardiol 2012;59:e49. 10.1016/j.jacc.2011.09.087 [DOI] [PubMed] [Google Scholar]
- 70.Vedanthan R, Sanz J, Halperin J. Bicuspid pulmonic valve. J Am Coll Cardiol 2009;54:e5. 10.1016/j.jacc.2009.05.027 [DOI] [PubMed] [Google Scholar]
- 71.Hayes CJ, Gersony WM, Driscoll DJ, et al. Second natural history study of congenital heart defects. Results of treatment of patients with pulmonary valvar stenosis. Circulation 1993;87:I28-37. [PubMed] [Google Scholar]
- 72.Kan JS, White RI, Jr, Mitchell SE, et al. Percutaneous balloon valvuloplasty: a new method for treating congenital pulmonary-valve stenosis. N Engl J Med 1982;307:540-2. 10.1056/NEJM198208263070907 [DOI] [PubMed] [Google Scholar]
- 73.Chen CR, Cheng TO, Huang T, et al. Percutaneous balloon valvuloplasty for pulmonic stenosis in adolescents and adults. N Engl J Med 1996;335:21-5. 10.1056/NEJM199607043350104 [DOI] [PubMed] [Google Scholar]
- 74.Kaul UA, Singh B, Tyagi S, et al. Long-term results after balloon pulmonary valvuloplasty in adults. Am Heart J 1993;126:1152-5. 10.1016/0002-8703(93)90668-Y [DOI] [PubMed] [Google Scholar]
- 75.Taggart NW, Cetta F, Cabalka AK, et al. Outcomes for balloon pulmonary valvuloplasty in adults: comparison with a concurrent pediatric cohort. Catheter Cardiovasc Interv 2013;82:811-5. 10.1002/ccd.24973 [DOI] [PubMed] [Google Scholar]
- 76.Wang SZ, Ou-Yang WB, Hu SS, et al. First-in-Human Percutaneous Balloon Pulmonary Valvuloplasty Under Echocardiographic Guidance Only. Congenit Heart Dis 2016;11:716-20. 10.1111/chd.12380 [DOI] [PubMed] [Google Scholar]
- 77.Merino-Ingelmo R, Santos-de Soto J, Coserria-Sánchez F, et al. Long-term Results of Percutaneous Balloon Valvuloplasty in Pulmonary Valve Stenosis in the Pediatric Population. Rev Esp Cardiol (Engl Ed) 2014;67:374-9. 10.1016/j.rec.2013.08.020 [DOI] [PubMed] [Google Scholar]
- 78.Fawzy ME, Hassan W, Fadel BM, et al. Long-term results (up to 17 years) of pulmonary balloon valvuloplasty in adults and its effects on concomitant severe infundibular stenosis and tricuspid regurgitation. Am Heart J 2007;153:433-8. 10.1016/j.ahj.2006.11.021 [DOI] [PubMed] [Google Scholar]
- 79.Garty Y, Veldtman G, Lee K, et al. Late outcomes after pulmonary valve balloon dilatation in neonates, infants and children. J Invasive Cardiol 2005;17:318-22. [PubMed] [Google Scholar]
- 80.Harrild DM, Powell AJ, Trang TX, et al. Long-Term Pulmonary Regurgitation Following Balloon Valvuloplasty for Pulmonary Stenosis. J Am Coll Cardiol 2010;55:1041-7. 10.1016/j.jacc.2010.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fathallah M, Krasuski RA. Pulmonic Valve Disease: Review of Pathology and Current Treatment Options. Curr Cardiol Rep 2017;19:108. 10.1007/s11886-017-0922-2 [DOI] [PubMed] [Google Scholar]
- 82.Yoshida K, Yoshikawa J, Shakudo M, et al. Color Doppler evaluation of valvular regurgitation in normal subjects. Circulation 1988;78:840-7. 10.1161/01.CIR.78.4.840 [DOI] [PubMed] [Google Scholar]
- 83.Zucker N, Rozin I, Levitas A, et al. Clinical presentation, natural history, and outcome of patients with the absent pulmonary valve syndrome. Cardiol Young 2004;14:402-8. 10.1017/S1047951104004093 [DOI] [PubMed] [Google Scholar]
- 84.Earing MG, Connolly HM, Dearani JA, et al. Long-term follow-up of patients after surgical treatment for isolated pulmonary valve stenosis. Mayo Clin Proc 2005;80:871-6. 10.4065/80.7.871 [DOI] [PubMed] [Google Scholar]
- 85.Rebergen SA, Chin JG, Ottenkamp J, et al. Pulmonary regurgitation in the late postoperative follow-up of tetralogy of Fallot. Volumetric quantitation by nuclear magnetic resonance velocity mapping. Circulation 1993;88:2257-66. 10.1161/01.CIR.88.5.2257 [DOI] [PubMed] [Google Scholar]
- 86.McCrindle BW, Kan JS. Long-term results after balloon pulmonary valvuloplasty. Circulation 1991;83:1915-22. 10.1161/01.CIR.83.6.1915 [DOI] [PubMed] [Google Scholar]
- 87.Shimazaki Y, Blackstone EH, Kirklin JW. The natural history of isolated congenital pulmonary valve incompetence: surgical implications. Thorac Cardiovasc Surg 1984;32:257-9. 10.1055/s-2007-1023399 [DOI] [PubMed] [Google Scholar]
- 88.Frigiola A, Giardini A, Taylor A, et al. Echocardiographic assessment of diastolic biventricular properties in patients operated for severe pulmonary regurgitation and association with exercise capacity. Eur Heart J Cardiovasc Imaging 2012;13:697-702. 10.1093/ehjci/jes002 [DOI] [PubMed] [Google Scholar]
- 89.Zoghbi WA, Adams D, Bonow RO, et al. Recommendations for noninvasive evaluation of native valvular regurgitation: a report from the American Society of Echocardiography developed in collaboration with the Society for Cardiovascular Magnetic Resonance. J Am Soc Echocardiogr 2017;30:303-71. 10.1016/j.echo.2017.01.007 [DOI] [PubMed] [Google Scholar]
- 90.Samyn MM, Powell AJ, Garg R, et al. Range of ventricular dimensions and function by steady-state free precession cine MRI in repaired tetralogy of Fallot: Right ventricular outflow tract patch vs. conduit repair. J Magn Reson Imaging 2007;26:934-40. 10.1002/jmri.21094 [DOI] [PubMed] [Google Scholar]
- 91.Mercer-Rosa L, Yang W, Kutty S, et al. Quantifying pulmonary regurgitation and right ventricular function in surgically repaired tetralogy of Fallot: a comparative analysis of echocardiography and magnetic resonance imaging. Circ Cardiovasc Imaging 2012;5:637-43. 10.1161/CIRCIMAGING.112.972588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zdradzinski M, Elkin R, Flamm S, et al. Incremental value of cardiac magnetic resonance for assessing pulmonic valve regurgitation. J Heart Valve Dis 2015;24:502-7. [PubMed] [Google Scholar]
- 93.Crean AM, Maredia N, Ballard G, et al. 3D Echo systematically underestimates right ventricular volumes compared to cardiovascular magnetic resonance in adult congenital heart disease patients with moderate or severe RV dilatation. J Cardiovasc Magn Reson 2011;13:78. 10.1186/1532-429X-13-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rajiah P, Nazarian J, Vogelius E, et al. CT and MRI of pulmonary valvular abnormalities. Clin Radiol 2014;69:630-8. 10.1016/j.crad.2014.01.019 [DOI] [PubMed] [Google Scholar]
- 95.Jonas SN, Kligerman SJ, Burke AP, et al. Pulmonary Valve Anatomy and Abnormalities: A Pictorial Essay of Radiography, Computed Tomography (CT), and Magnetic Resonance Imaging (MRI). J Thorac Imaging 2016;31:W4-12. 10.1097/RTI.0000000000000182 [DOI] [PubMed] [Google Scholar]
- 96.Lee C, Kim YM, Lee CH, et al. Outcomes of pulmonary valve replacement in 170 patients with chronic pulmonary regurgitation after relief of right ventricular outflow tract obstruction: implications for optimal timing of pulmonary valve replacement. J Am Coll Cardiol 2012;60:1005-14. 10.1016/j.jacc.2012.03.077 [DOI] [PubMed] [Google Scholar]
- 97.Ammash NM, Dearani JA, Burkhart HM, et al. Pulmonary regurgitation after tetralogy of Fallot repair: clinical features, sequelae, and timing of pulmonary valve replacement. Congenit Heart Dis 2007;2:386-403. 10.1111/j.1747-0803.2007.00131.x [DOI] [PubMed] [Google Scholar]
- 98.Oosterhof T, van Straten A, Vliegen HW, et al. Preoperative Thresholds for Pulmonary Valve Replacement in Patients With Corrected Tetralogy of Fallot Using Cardiovascular Magnetic Resonance. Circulation 2007;116:545-51. 10.1161/CIRCULATIONAHA.106.659664 [DOI] [PubMed] [Google Scholar]
- 99.Therrien J, Siu SC, Harris L, et al. Impact of pulmonary valve replacement on arrhythmia propensity late after repair of tetralogy of Fallot. Circulation 2001;103:2489-94. 10.1161/01.CIR.103.20.2489 [DOI] [PubMed] [Google Scholar]
- 100.Buechel ERV, Dave HH, Kellenberger CJ, et al. Remodelling of the right ventricle after early pulmonary valve replacement in children with repaired tetralogy of Fallot: assessment by cardiovascular magnetic resonance. Eur Heart J 2005;26:2721-7. 10.1093/eurheartj/ehi581 [DOI] [PubMed] [Google Scholar]
- 101.Caldarone CA, McCrindle BW, Van Arsdell GS, et al. Independent factors associated with longevity of prosthetic pulmonary valves and valved conduits. J Thorac Cardiovasc Surg 2000;120:1022-30; discussion 1031. 10.1067/mtc.2000.110684 [DOI] [PubMed] [Google Scholar]
- 102.Baskett RJ, Ross DB, Nanton MA, et al. Factors in the early failure of cryopreserved homograft pulmonary valves in children: preserved immunogenicity? J Thorac Cardiovasc Surg 1996;112:1170-8; discussion 1178-9. 10.1016/S0022-5223(96)70130-7 [DOI] [PubMed] [Google Scholar]
- 103.McKenzie ED, Khan MS, Dietzman TW, et al. Surgical pulmonary valve replacement: a benchmark for outcomes comparisons. J Thorac Cardiovasc Surg 2014;148:1450-3. 10.1016/j.jtcvs.2014.02.060 [DOI] [PubMed] [Google Scholar]
- 104.Shinkawa T, Anagnostopoulos PV, Johnson NC, et al. Performance of bovine pericardial valves in the pulmonary position. Ann Thorac Surg 2010;90:1295-300. 10.1016/j.athoracsur.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen PC, Sager MS, Zurakowski D, et al. Younger age and valve oversizing are predictors of structural valve deterioration after pulmonary valve replacement in patients with tetralogy of Fallot. J Thorac Cardiovasc Surg 2012;143:352-60. 10.1016/j.jtcvs.2011.10.079 [DOI] [PubMed] [Google Scholar]
- 106.Bonhoeffer P, Boudjemline Y, Saliba Z, et al. Percutaneous replacement of pulmonary valve in a right-ventricle to pulmonary-artery prosthetic conduit with valve dysfunction. Lancet 2000;356:1403-5. 10.1016/S0140-6736(00)02844-0 [DOI] [PubMed] [Google Scholar]
- 107.Boudjemline Y, Brugada G, Van-Aerschot I, et al. Outcomes and safety of transcatheter pulmonary valve replacement in patients with large patched right ventricular outflow tracts. Arch Cardiovasc Dis 2012;105:404-13. 10.1016/j.acvd.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 108.Gillespie MJ, Rome JJ, Levi DS, et al. Melody valve implant within failed bioprosthetic valves in the pulmonary position: a multicenter experience. Circ Cardiovasc Interv 2012;5:862-70. 10.1161/CIRCINTERVENTIONS.112.972216 [DOI] [PubMed] [Google Scholar]
- 109.Cabalka AK, Asnes JD, Balzer DT, et al. Transcatheter pulmonary valve replacement using the melody valve for treatment of dysfunctional surgical bioprostheses: A multicenter study. J Thorac Cardiovasc Surg 2018;155:1712-24.e1. 10.1016/j.jtcvs.2017.10.143 [DOI] [PubMed] [Google Scholar]
- 110.Coats L, Khambadkone S, Derrick G, et al. Physiological and Clinical Consequences of Relief of Right Ventricular Outflow Tract Obstruction Late After Repair of Congenital Heart Defects. Circulation 2006;113:2037-44. 10.1161/CIRCULATIONAHA.105.591438 [DOI] [PubMed] [Google Scholar]
- 111.Lurz P, Nordmeyer J, Giardini A, et al. Early versus late functional outcome after successful percutaneous pulmonary valve implantation: are the acute effects of altered right ventricular loading all we can expect? J Am Coll Cardiol 2011;57:724-31. 10.1016/j.jacc.2010.07.056 [DOI] [PubMed] [Google Scholar]
- 112.Cheatham JP, Hellenbrand WE, Zahn EM, et al. Clinical and hemodynamic outcomes up to 7 years after transcatheter pulmonary valve replacement in the US melody valve investigational device exemption trial. Circulation 2015;131:1960-70. 10.1161/CIRCULATIONAHA.114.013588 [DOI] [PubMed] [Google Scholar]
- 113.Plymen CM, Bolger AP, Lurz P, et al. Electrical remodeling following percutaneous pulmonary valve implantation. Am J Cardiol 2011;107:309-14. 10.1016/j.amjcard.2010.09.017 [DOI] [PubMed] [Google Scholar]
- 114.Morray BH, McElhinney DB, Boudjemline Y, et al. Multicenter Experience Evaluating Transcatheter Pulmonary Valve Replacement in Bovine Jugular Vein (Contegra) Right Ventricle to Pulmonary Artery Conduits. Circ Cardiovasc Interv 2017;10:e004914. 10.1161/CIRCINTERVENTIONS.116.004914 [DOI] [PubMed] [Google Scholar]
- 115.Lurz P, Coats L, Khambadkone S, et al. Percutaneous pulmonary valve implantation: impact of evolving technology and learning curve on clinical outcome. Circulation 2008;117:1964-72. 10.1161/CIRCULATIONAHA.107.735779 [DOI] [PubMed] [Google Scholar]
- 116.McElhinney DB, Hellenbrand WE, Zahn EM, et al. Short- and medium-term outcomes after transcatheter pulmonary valve placement in the expanded multicenter US melody valve trial. Circulation 2010;122:507-16. 10.1161/CIRCULATIONAHA.109.921692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lindsay I, Aboulhosn J, Salem M, et al. Aortic root compression during transcatheter pulmonary valve replacement. Catheter Cardiovasc Interv 2016;88:814-21. 10.1002/ccd.26547 [DOI] [PubMed] [Google Scholar]
- 118.Fraisse A, Assaidi A, Mauri L, et al. Coronary artery compression during intention to treat right ventricle outflow with percutaneous pulmonary valve implantation: incidence, diagnosis, and outcome. Catheter Cardiovasc Interv 2014;83:E260-8. 10.1002/ccd.25471 [DOI] [PubMed] [Google Scholar]
- 119.Morray BH, McElhinney DB, Cheatham JP, et al. Risk of coronary artery compression among patients referred for transcatheter pulmonary valve implantation: a multicenter experience. Circ Cardiovasc Interv 2013;6:535-42. 10.1161/CIRCINTERVENTIONS.113.000202 [DOI] [PubMed] [Google Scholar]
- 120.Webb S, Qayyum SR, Anderson RH, et al. Septation and separation within the outflow tract of the developing heart. J Anat 2003;202:327-42. 10.1046/j.1469-7580.2003.00168.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oosthoek PW, Wenink AC, Vrolijk BC, et al. Development of the atrioventricular valve tension apparatus in the human heart. Anat Embryol (Berl) 1998;198:317-29. 10.1007/s004290050187 [DOI] [PubMed] [Google Scholar]
- 122.de la Cruz MV, Markwald RR. Embryological Development of the Ventricular Inlets. Septation and Atrioventricular Valve Apparatus [Internet]. Living Morphogenesis of the Heart; 1998:131-55. Available online: 10.1007/978-1-4612-1788-6_6 10.1007/978-1-4612-1788-6_6 [DOI]
- 123.Tzifa A, Gauvreau K, Geggel RL. Factors associated with development of atrial septal restriction in patients with tricuspid atresia involving the right-sided atrioventricular valve. Am Heart J 2007;154:1235-41. 10.1016/j.ahj.2007.07.037 [DOI] [PubMed] [Google Scholar]
- 124.Moorjani N, Rana BS, Wells FC. Anatomy and Physiology of the Mitral Valve. In: Moorjani N, Rana BS, Wells FC. editors. Operative Mitral and Tricuspid Valve Surgery. London: Springer London; 2018:1-20. [Google Scholar]
- 125.Kanani M, Moorman AFM, Cook AC, et al. Development of the atrioventricular valves: clinicomorphological correlations. Ann Thorac Surg 2005;79:1797-804. 10.1016/j.athoracsur.2004.06.122 [DOI] [PubMed] [Google Scholar]
- 126.Annabi MR, Makaryus AN. Embryology, Atrioventricular Septum. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2019. [PubMed] [Google Scholar]
- 127.Warnes CA. Adult congenital heart disease importance of the right ventricle. J Am Coll Cardiol 2009;54:1903-10. 10.1016/j.jacc.2009.06.048 [DOI] [PubMed] [Google Scholar]
- 128.Anderson KR, Lie JT. The right ventricular myocardium in Ebstein’s anomaly: a morphometric histopathologic study. Mayo Clin Proc 1979;54:181-4. [PubMed] [Google Scholar]
- 129.Attenhofer Jost CH, Connolly HM, Warnes CA, et al. Noncompacted myocardium in ebstein’s anomaly: initial description in three patients. J Am Soc Echocardiogr 2004;17:677-80. 10.1016/j.echo.2004.02.013 [DOI] [PubMed] [Google Scholar]
- 130.Edwards WD. Embryology and pathologic features of Ebstein’s anomaly. Prog Pediatr Cardiol 1993;2:5-15. 10.1016/1058-9813(93)90042-X [DOI] [Google Scholar]
- 131.Zuberbuhler JR, Allwork SP, Anderson RH. The spectrum of Ebstein’s anomaly of the tricuspid valve. J Thorac Cardiovasc Surg 1979;77:202-11. 10.1016/S0022-5223(19)40958-6 [DOI] [PubMed] [Google Scholar]
- 132.Attenhofer Jost CH, Connolly HM, Dearani JA, et al. Ebstein’s Anomaly. Circulation 2007;115:277-85. 10.1161/CIRCULATIONAHA.106.619338 [DOI] [PubMed] [Google Scholar]
- 133.Ho SY, Goltz D, McCarthy K, et al. The atrioventricular junctions in Ebstein malformation. Heart 2000;83:444-9. 10.1136/heart.83.4.444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pradat P, Francannet C, Harris JA, et al. The epidemiology of cardiovascular defects, part I: a study based on data from three large registries of congenital malformations. Pediatr Cardiol 2003;24:195-221. 10.1007/s00246-002-9401-6 [DOI] [PubMed] [Google Scholar]
- 135.Lupo PJ, Langlois PH, Mitchell LE. Epidemiology of Ebstein anomaly: prevalence and patterns in Texas, 1999-2005. Am J Med Genet A 2011;155A:1007-14. 10.1002/ajmg.a.33883 [DOI] [PubMed] [Google Scholar]
- 136.Correa-Villaseñor A, Ferencz C, Neill CA, et al. Ebstein’s malformation of the tricuspid valve: genetic and environmental factors. The Baltimore-Washington Infant Study Group. Teratology 1994;50:137-47. 10.1002/tera.1420500208 [DOI] [PubMed] [Google Scholar]
- 137.Cohen LS, Friedman JM, Jefferson JW, et al. A reevaluation of risk of in utero exposure to lithium. JAMA 1994;271:146-50. 10.1001/jama.1994.03510260078033 [DOI] [PubMed] [Google Scholar]
- 138.Celermajer DS, Bull C, Till JA, et al. Ebstein’s anomaly: presentation and outcome from fetus to adult. J Am Coll Cardiol 1994;23:170-6. 10.1016/0735-1097(94)90516-9 [DOI] [PubMed] [Google Scholar]
- 139.Seward JB, Tajik AJ, Feist DJ, et al. Ebstein’s anomaly in an 85-year-old man. Mayo Clin Proc 1979;54:193-6. [PubMed] [Google Scholar]
- 140.Giuliani ER, Fuster V, Brandenburg RO, et al. Ebstein’s anomaly: the clinical features and natural history of Ebstein's anomaly of the tricuspid valve. Mayo Clin Proc 1979;54:163-73. [PubMed] [Google Scholar]
- 141.Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000;342:334-42. 10.1056/NEJM200002033420507 [DOI] [PubMed] [Google Scholar]
- 142.Attenhofer Jost CH, Connolly HM, O’Leary PW, et al. Left heart lesions in patients with Ebstein anomaly. Mayo Clin Proc 2005;80:361-8. 10.4065/80.3.361 [DOI] [PubMed] [Google Scholar]
- 143.Danielson GK, Driscoll DJ, Mair DD, et al. Operative treatment of Ebstein’s anomaly. J Thorac Cardiovasc Surg 1992;104:1195-202. 10.1016/S0022-5223(19)34605-7 [DOI] [PubMed] [Google Scholar]
- 144.Celermajer DS, Cullen S, Sullivan ID, et al. Outcome in neonates with Ebstein’s anomaly. J Am Coll Cardiol 1992;19:1041-6. 10.1016/0735-1097(92)90291-T [DOI] [PubMed] [Google Scholar]
- 145.Cappato R, Schlüter M, Weiss C, et al. Radiofrequency current catheter ablation of accessory atrioventricular pathways in Ebstein’s anomaly. Circulation 1996;94:376-83. 10.1161/01.CIR.94.3.376 [DOI] [PubMed] [Google Scholar]
- 146.Lev M, Gibson S, Miller RA. Ebstein’s disease with Wolff-Parkinson-White syndrome; report of a case with a histopathologic study of possible conduction pathways. Am Heart J 1955;49:724-41. 10.1016/0002-8703(55)90218-0 [DOI] [PubMed] [Google Scholar]
- 147.Watson H. Natural history of Ebstein’s anomaly of tricuspid valve in childhood and adolescence. An international co-operative study of 505 cases. Br Heart J 1974;36:417-27. 10.1136/hrt.36.5.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Feneck R, Kneeshaw J, Ranucci M. Core Topics in Transesophageal Echocardiography. Cambridge University Press; 2010:416. [Google Scholar]
- 149.Shiina A, Seward JB, Edwards WD, et al. Two-dimensional echocardiographic spectrum of ebstein’s anomaly: Detailed anatomic assessment. J Am Coll Cardiol 1984;3:356-70. 10.1016/S0735-1097(84)80020-0 [DOI] [PubMed] [Google Scholar]
- 150.Attenhofer Jost CH, Edmister WD, Julsrud PR, et al. Prospective comparison of echocardiography versus cardiac magnetic resonance imaging in patients with Ebstein’s anomaly. Int J Cardiovasc Imaging 2012;28:1147-59. 10.1007/s10554-011-9923-1 [DOI] [PubMed] [Google Scholar]
- 151.Bharucha T, Anderson RH, Lim ZS, et al. Multiplanar review of three-dimensional echocardiography gives new insights into the morphology of Ebstein’s malformation. Cardiol Young 2010;20:49-53. 10.1017/S1047951109991971 [DOI] [PubMed] [Google Scholar]
- 152.Badiu CC, Schreiber C, Hörer J, et al. Early timing of surgical intervention in patients with Ebstein’s anomaly predicts superior long-term outcome. Eur J Cardiothorac Surg 2010;37:186-92. 10.1016/j.ejcts.2009.06.052 [DOI] [PubMed] [Google Scholar]
- 153.Chen JM, Mosca RS, Altmann K, et al. Early and medium-term results for repair of Ebstein anomaly. J Thorac Cardiovasc Surg 2004;127:990-8; discussion 998-9. 10.1016/j.jtcvs.2003.11.037 [DOI] [PubMed] [Google Scholar]
- 154.Brown ML, Dearani JA, Danielson GK, et al. The outcomes of operations for 539 patients with Ebstein anomaly. J Thorac Cardiovasc Surg 2008;135:1120-36, 1136.e1-7. [DOI] [PubMed]
- 155.Sarris GE, Giannopoulos NM, Tsoutsinos AJ, et al. Results of surgery for Ebstein anomaly: a multicenter study from the European Congenital Heart Surgeons Association. J Thorac Cardiovasc Surg 2006;132:50-7. 10.1016/j.jtcvs.2005.10.062 [DOI] [PubMed] [Google Scholar]
- 156.Danielson GK, Fuster V. Surgical Repair of Ebsteinʼs Anomaly. Ann Surg 1982;196:499-504. 10.1097/00000658-198210000-00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Carpentier A, Adams DH, Filsoufi F. Carpentier’s Reconstructive Valve Surgery: From Valve Analysis to Valve Reconstruction. Saunders/Elsevier; 2010:354. [Google Scholar]
- 158.da Silva JP, Baumgratz JF, da Fonseca L, et al. The cone reconstruction of the tricuspid valve in Ebstein’s anomaly. The operation: early and midterm results. J Thorac Cardiovasc Surg 2007;133:215-23. 10.1016/j.jtcvs.2006.09.018 [DOI] [PubMed] [Google Scholar]
- 159.Kiziltan HT, Theodoro DA, Warnes CA, et al. Late results of bioprosthetic tricuspid valve replacement in Ebstein’s anomaly. Ann Thorac Surg 1998;66:1539-45. 10.1016/S0003-4975(98)00961-8 [DOI] [PubMed] [Google Scholar]
- 160.Holst KA, Connolly HM, Dearani JA. Ebstein’s Anomaly. Methodist Debakey Cardiovasc J 2019;15:138-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Raju V, Dearani JA, Burkhart HM, et al. Right ventricular unloading for heart failure related to Ebstein malformation. Ann Thorac Surg 2014;98:167-73; discussion 173-4. 10.1016/j.athoracsur.2014.03.009 [DOI] [PubMed] [Google Scholar]
- 162.Banerjee A, Kohl T, Silverman NH. Echocardiographic evaluation of congenital mitral valve anomalies in children. Am J Cardiol 1995;76:1284-91. 10.1016/S0002-9149(99)80357-9 [DOI] [PubMed] [Google Scholar]
- 163.Aslam S, Khairy P, Shohoudi A, et al. Shone Complex: An Under-recognized Congenital Heart Disease With Substantial Morbidity in Adulthood. Can J Cardiol 2017;33:253-9. 10.1016/j.cjca.2016.09.005 [DOI] [PubMed] [Google Scholar]
- 164.Séguéla PE, Houyel L, Acar P. Congenital malformations of the mitral valve. Arch Cardiovasc Dis 2011;104:465-79. 10.1016/j.acvd.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 165.Ziani AB, Latcu DG, Abadir S, et al. Assessment of proximal isovelocity surface area (PISA) shape using three-dimensional echocardiography in a paediatric population with mitral regurgitation or ventricular shunt. Arch Cardiovasc Dis 2009;102:185-91. 10.1016/j.acvd.2008.12.008 [DOI] [PubMed] [Google Scholar]
- 166.Frescura C, Thiene G. The new concept of univentricular heart. Front Pediatr 2014;2:62. 10.3389/fped.2014.00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gewillig M. The Fontan circulation. Heart 2005;91:839-46. 10.1136/hrt.2004.051789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fontan F, Baudet E. Surgical repair of tricuspid atresia. Thorax 1971;26:240-8. 10.1136/thx.26.3.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Veldtman GR, Opotowsky AR, Wittekind SG, et al. Cardiovascular adaptation to the Fontan circulation. Congenit Heart Dis 2017;12:699-710. 10.1111/chd.12526 [DOI] [PubMed] [Google Scholar]
- 170.d’Udekem Y, Xu MY, Galati JC, et al. Predictors of survival after single-ventricle palliation: the impact of right ventricular dominance. J Am Coll Cardiol 2012;59:1178-85. 10.1016/j.jacc.2011.11.049 [DOI] [PubMed] [Google Scholar]
- 171.Honjo O, Mertens L, Van Arsdell GS. Atrioventricular Valve Repair in Patients With Single-ventricle Physiology: Mechanisms, Techniques of Repair, and Clinical Outcomes. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2011;14:75-84. 10.1053/j.pcsu.2011.02.001 [DOI] [PubMed] [Google Scholar]
- 172.Barber G, Helton JG, Aglira BA, et al. The significance of tricuspid regurgitation in hypoplastic left-heart syndrome. Am Heart J 1988;116:1563-7. 10.1016/0002-8703(88)90744-2 [DOI] [PubMed] [Google Scholar]
- 173.Imai Y, Takanashi Y, Hoshino S, et al. Modified Fontan procedure in ninety-nine cases of atrioventricular valve regurgitation. J Thorac Cardiovasc Surg 1997;113:262-8; discussion 269. 10.1016/S0022-5223(97)70322-2 [DOI] [PubMed] [Google Scholar]
- 174.Ohye RG, Gomez CA, Goldberg CS, et al. Tricuspid valve repair in hypoplastic left heart syndrome. J Thorac Cardiovasc Surg 2004;127:465-72. 10.1016/j.jtcvs.2003.07.053 [DOI] [PubMed] [Google Scholar]
- 175.King G, Ayer J, Celermajer D, et al. Atrioventricular Valve Failure in Fontan Palliation. J Am Coll Cardiol 2019;73:810-22. 10.1016/j.jacc.2018.12.025 [DOI] [PubMed] [Google Scholar]
- 176.Liu VJH, Yong MS, d’Udekem Y, et al. Outcomes of atrioventricular valve operation in patients with Fontan circulation. Ann Thorac Surg 2015;99:1632-8. 10.1016/j.athoracsur.2015.01.035 [DOI] [PubMed] [Google Scholar]
- 177.Mavroudis C, Stewart RD, Backer CL, et al. Atrioventricular valve procedures with repeat fontan operations: influence of valve pathology, ventricular function, and arrhythmias on outcome. Ann Thorac Surg 2005;80:29-36; discussion 36. 10.1016/j.athoracsur.2005.01.071 [DOI] [PubMed] [Google Scholar]
- 178.Laux D, Vergnat M, Lambert V, et al. Atrio-ventricular valve regurgitation in univentricular hearts: outcomes after repair. Interact Cardiovasc Thorac Surg 2015;20:622-9; discussion 629-30. 10.1093/icvts/ivv011 [DOI] [PubMed] [Google Scholar]
- 179.Honjo O, Atlin CR, Mertens L, et al. Atrioventricular valve repair in patients with functional single-ventricle physiology: Impact of ventricular and valve function and morphology on survival and reintervention. J Thorac Cardiovasc Surg 2011;142:326-35.e2. 10.1016/j.jtcvs.2010.11.060 [DOI] [PubMed] [Google Scholar]
- 180.Chauvaud S, Fuzellier JF, Houel R, et al. Reconstructive surgery in congenital mitral valve insufficiency (Carpentier’s techniques): long-term results. J Thorac Cardiovasc Surg 1998;115:84-92; discussion 92-3. 10.1016/S0022-5223(98)99001-8 [DOI] [PubMed] [Google Scholar]
- 181.Cetta F, Driscoll DJ. Bad Atrioventricular Valve, Bad Fontan: Stop Creating Bad Fontans. J Am Coll Cardiol 2019;73:823-5. 10.1016/j.jacc.2018.12.026 [DOI] [PubMed] [Google Scholar]
- 182.Pundi KN, Johnson JN, Dearani JA, et al. 40-Year Follow-Up After the Fontan Operation: Long-Term Outcomes of 1,052 Patients. J Am Coll Cardiol 2015;66:1700-10. 10.1016/j.jacc.2015.07.065 [DOI] [PubMed] [Google Scholar]