

Abstract
The poor prognosis for pancreatic ductal adenocarcinoma (PDAC) patients impels an improved understanding of disease biology, to facilitate the development of better therapies. PDAC typically features a remarkably dense stromal reaction, featuring and established by a prominent population of cancer-associated fibroblasts. Genetically engineered mouse models and increasingly sophisticated cell culture techniques have demonstrated important roles for fibroblasts in PDAC progression and therapy response, but these roles are complex, with strong evidence for both tumor-supportive and tumor-suppressive or homeostatic functions. Here we review the recent literature which has improved our understanding of heterogeneity in fibroblast fate and function in this disease including the existence of distinct fibroblast populations, and highlight important avenues for future study.
Introduction
Over fifty years ago, Michael Stoker’s lab demonstrated that normal, static fibroblasts could suppress the growth of polyoma-transformed cells, providing early evidence that components of normal tissue architecture can serve as barriers to tumorigenesis (1). About twenty years later, Mina Bissell’s lab showed that a local wounding reaction promoted Rous sarcoma virus-mediated tumor formation (2), illustrating a principle that has been bolstered across tumor types in the decades since: a fibro-inflammatory microenvironment plays critical roles in supporting tumor progression (3). These and additional, compelling studies suggest that a normal microenvironment prevents premalignant cells from progressing into overt cancer, while an abnormal or wound-repair-associated microenvironment can be tumor-promoting (4). Since these pioneering studies, extensive efforts have uncovered links between oncogenic signaling and induction of a wound-like reaction in the local tissue environment, including activation of resident fibroblasts. The interaction between transformed cells and cellular wound-healing mediators such as fibroblasts is of particular relevance to PDAC in light of its remarkably dense stroma, made up of a prominent population of cancer-associated fibroblasts (CAFs), as well as immune cells and a dense extracellular matrix derived in large part from the CAF pool. Inducible mouse models of PDAC driven by oncogenic KRAS have demonstrated the link between epithelial oncogenic signaling and fibroblast activation: extinction of oncogenic KRAS signaling leads to a rapid reduction in stromal fibroblast activation (5, 6). These results likely reflect regulation of myriad cytokines and growth factors by oncogenic KRAS signaling, such that fibroblast populations are sensing and responding to a wounded epithelium, yielding diverse and complex interactions. The increasingly appreciated complexity of the CAF component of the PDAC microenvironment, together with its apparent relevance to disease progression and therapy response, have motivated recent efforts to analyze CAF functional and transcriptional diversity.
Functional Diversity Among PDAC CAFs
Reacting to a disruption in tissue homeostasis, fibroblasts take on diverse functions which can support or suppress tumorigenesis. Epithelial cell responses to CAF functions are likely context-dependent with respect to stage of tumorigenesis, tumor genotype, and host tissue. In the pancreas, early responses to oncogenic signaling in the pancreatic epithelium include the activation and expansion of fibroblasts, such that low-grade premalignant pancreatic intraepithelial neoplasia (PanIN) lesions are already surrounded by areas of fibrosis (5). As these fibroblasts co-evolve with the advancing cancer, they take on diverse functions that can support or suppress tumor growth. An improved understanding of the precise mechanisms underlying stromal support for PDAC progression versus those that restrain tumor growth will be important for developing rational therapies targeting the tumor microenvironment.
Tumor-promoting functions of PDAC CAFs
In enacting a wound-healing response as they have evolved to do, CAFs exhibit diverse functions that effectively support pancreatic tumor growth. These tumor-promoting CAF functions include providing metabolic support to enable proliferation in the neoplastic compartment, which gains significance in the context of the hypovascular and nutrient-poor pancreatic tumor microenvironment (TME) (7–9). This stromal metabolic support includes direct and indirect provision of amino acids to support biomass production by PDAC cells. For example, CAFs secrete high levels of alanine, which PDAC cells preferentially use to fuel the tricarboxylic acid (TCA) cycle (10). Stroma-derived alanine ultimately supports lipid and non-essential amino acid biosynthesis, enabling proliferation despite low levels of serum nutrients (including glucose) that typically fuel these metabolic processes. CAFs also provide amino acids to PDAC cells indirectly by producing a dense, collagen-rich extracellular matrix (ECM). The amino acid composition of collagen is proline-rich (~25%), and recent work has demonstrated that collagen uptake and catabolism, with subsequent proline catabolism by PRODH1, supports PDAC cell proliferation under relevant conditions of nutrient challenge in vitro and promotes tumor progression in vivo (11). In addition to amino acids, CAFs secrete specific lipid species that PDAC cells take up and catabolize to reduce their need for de novo lipid synthesis (12). This lipid scavenging mechanism is desirable for PDAC cells in a hypoxic environment (13), as lipogenesis and lipid desaturation involve oxygen-dependent SCD1 activity. Paracrine transfer of metabolites from stromal fibroblasts to PDAC cells may also occur via CAF-derived exosomes, which were shown to harbor diverse metabolites including amino acids and TCA cycle intermediates (14) and to promote PDAC cell proliferation and chemoresistance (15). Beyond biomass production, CAF-derived metabolites may also support PDAC cell proliferation in the context of chemotherapy treatment: PDAC CAFs were recently shown to secrete deoxycytidine, protecting PDAC cells from gemcitabine toxicity (16). Metabolite release by stromal fibroblasts may reflect conserved mechanisms from a wound-healing reaction as a means for mesenchymal cells with a lower proliferative demand to provide energy and support proliferation within the regenerating epithelium. Alternatively, pancreatic cancer cells may evolve to stimulate these metabolic functions within their surrounding stroma via tumor-specific mechanisms.
Beyond metabolic support, CAFs support PDAC progression through paracrine activation of mitogenic and pro-survival signaling pathways. In-depth phosphoproteomic analyses demonstrated the existence of reciprocal signaling networks between PDAC cells and CAFs (17). In this study, the authors demonstrated that pancreatic epithelial cells harboring mutant KRAS signal via secreted factors to rewire intracellular signaling within pancreatic CAFs, increasing stromal production of growth factors such as IGF1 and GAS6. These stimulated CAFs in turn signal to the KRAS-mutant epithelium to widely regulate the phosphoproteome, stimulating phosphorylation of targets including IGF1R, AXL/TYRO3, and AKT. CAF-mediated reciprocal signaling altered tumor cell behavior, increasing mitochondrial membrane polarization, superoxide production, and spare respiratory capacity, with potential implications for stroma-driven liabilities for therapeutic intervention. A subsequent phosphoproteomic analysis of CAF/PDAC cell interaction focused on phosphotyrosine identified CAF-derived LIF as a key paracrine regulator of STAT3 signaling in the epithelial compartment (18). Genetic inhibition of the LIF receptor (Lifr) in pancreatic epithelial cells significantly prolonged survival in a genetically engineered mouse model of PDAC, while pharmacologic inhibition of LIF with a monoclonal antibody together with chemotherapy extended survival and yielded a more differentiated tumor histology compared to chemotherapy alone, highlighting a role for stroma-derived LIF signaling in both PDAC progression and chemoresistance. PDAC CAFs influence signaling and phenotype in PDAC cells not only through protein-based paracrine networks, but also through lipids. We recently demonstrated that PDAC CAFs secrete high levels of lysophosphatidylcholines (LPCs) (12). As described above, these CAF-derived lipids can be taken up by PDAC cells and incorporated into the cancer cell lipidome; however, LPCs can also be hydrolyzed by the secreted enzyme autotaxin, highly expressed by PDAC cells, to generate lysophosphatidic acid and stimulate mitogenic signaling events such as AKT phosphorylation. Genetic or pharmacologic autotaxin inhibition suppressed PDAC growth in vivo, pointing to another node of tumor-stroma interaction that may serve as a therapeutic target within the complex TME.
In addition to these paracrine interactions with cancer cells, PDAC CAFs can also support tumor progression through growth-permissive modulation of the immune microenvironment. A subset of pancreatic CAFs express the membrane protein fibroblast activation protein (FAP), and in a subcutaneous mouse model of PDAC, depletion of FAP+ cells fostered immune-mediated tumor regressions in response to vaccination against the tumor-associated antigen mesothelin (19). Mechanistically, CAFs have been shown to restrict anti-tumor T cell responses in two ways, and additional mechanisms may certainly be relevant. First, FAP+ CAFs are the principal repository for chemokine CXCL12 in the pancreatic TME, and CXCL12 promotes spatial exclusion of T cells, as pharmacologic inhibition of the interaction of CXCL12 with its receptor CXCR4 promoted T cell accumulation in tumor centers and fostered efficacy of immune checkpoint blockade (20). Second, the dense and rigid ECM produced by CAFs (including FAP+ CAFs (21)) has been shown to restrict T cell motility in the lung TME (22), and high ECM density was subsequently shown to restrict T cell proximity to cancer cells in PDAC (23, 24). A recent report identified a population of PDAC CAFs expressing components of the MHC II complex; these antigen-presenting CAFs (apCAFs) were able to present antigen to T cells in vitro but did so in the absence of costimulatory molecule expression, suggesting an additional potential interaction between PDAC CAFs and intratumoral T cells (25). Immune modulation by PDAC CAFs extends beyond T cells. For example, a subset of CAFs serve as the predominant source of IL6 in PDAC (26); in fact, PDAC cells stimulate IL6 release by pancreatic fibroblasts in a paracrine manner (27). CAF-derived IL6 was reported to promote differentiation of monocyte precursors into myeloid-derived suppressor cells (MDSCs) and promote immune suppression (28, 29). In addition, through secretion of thymic stromal lymphopoietin (TSLP), CAFs in PDAC (and in breast cancer) can polarize dendritic cells to promote pro-tumorigenic TH2 responses instead of potentially tumor-suppressive TH1 responses (30). PDAC CAFs were recently shown to produce high levels of matrix protein βig-h3, which may act directly on tumor-specific CD8+ T cells and on macrophages (31). Together, these studies provide evidence for modulation of the innate and adaptive immune microenvironment by PDAC CAFs and the dense ECM they produce. Targeting these mechanisms may hold promise in evoking efficacy of immune checkpoint blockade, which is otherwise ineffective in PDAC (32) (Figure 1).
Figure 1.
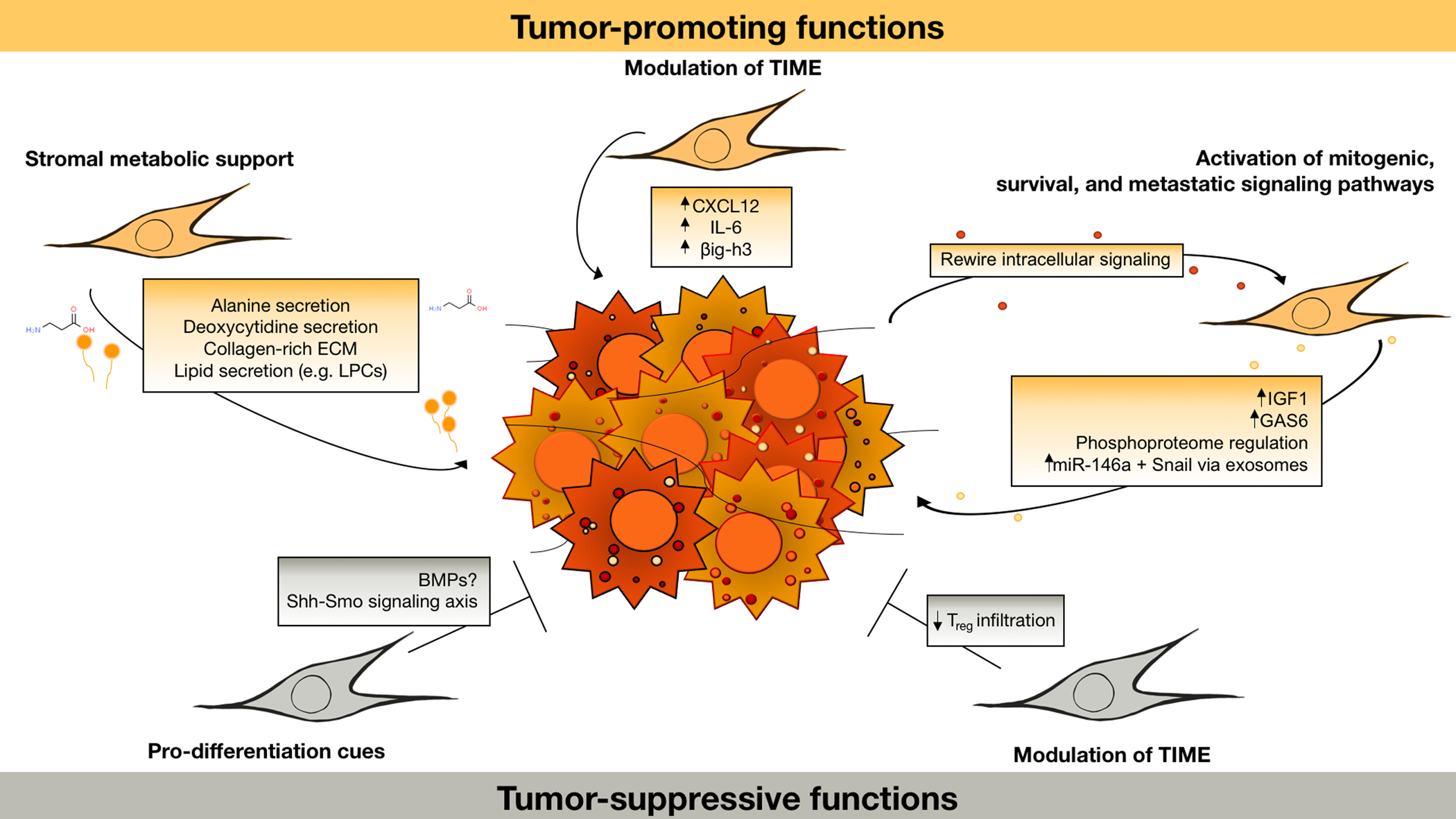
Summary of tumor-promoting and potential tumor-suppressive functions of PDAC CAFs. TIME: tumor immune microenvironment. CAFs can promote tumor progression via paracrine regulation of oncogenic signal transduction, including via IGF1/GAS6 and LIF signaling and reciprocal regulation of the PDAC phosphoproteome (17, 18). Through the release of exosomes and metabolites such as deoxycytidine (14–16), CAFs also regulate cancer cell metabolism and drug sensitivity. Metabolic regulation through secretion of lipids (12) and direct or indirect provision of amino acids (10, 11) enables proliferation within the nutrient-poor tumor microenvironment. CAFs also orchestrate growth-permissive regulation of the immune microenvironment through secretion of cytokines and other immune-modulatory factors such as CXCL12 (20), IL6 (26, 27), and βig-h3 (31). While tumor-suppressive CAF functions remain poorly understood, results from various CAF depletion models suggest that a Shh-dependent, aSMA-positive subset of CAFs promote a more differentiated and less aggressive tumor phenotype (33–35). This may be mediated by release of differentiation cues such as BMPs (38). CAF interactions with the immune system can be tumor-suppressive in part, through suppressing Treg infiltration (33), though interactions between CAFs and the anti-tumor immune response are complex and an important area for further study.
Evidence for tumor-suppressive CAF function
While the studies described above and others have documented tumor-promoting functions of PDAC CAFs, CAF ablation studies in genetically engineered mouse models of PDAC yielded the unexpected result that these cells can suppress tumor progression. In three papers published in 2014, the authors used genetic or pharmacologic approaches to ablate CAFs during pancreatic tumorigenesis. While results varied somewhat, likely due to the different systems used, all three studies found that CAF ablation made matters worse, resulting in poorly differentiated tumors and shortened survival (33–35).
Two of these studies targeted CAFs dependent on the Shh-Smo signaling axis (34, 35). Rhim et al. ablated Shh-dependent CAFs during PDAC progression in two ways: they conditionally deleted Shh in pancreatic epithelial cells by crossing a Shh-floxed allele into the established Pdx1-Cre;KrasLSL-G12D/+;Trp53fl/+;Rosa26LSL-YFP/+ (PKCY) model of pancreatic cancer, thus preventing the accumulation of a Shh-dependent stroma at all stages of tumorigenesis. ShhPKCY mice developed tumors earlier than controls and succumbed to the disease significantly faster, with a greater frequency of metastasis. Examining young mice at 8 weeks of age, loss of Shh-dependent stroma resulted in a higher frequency of PanIN lesions as well as acinar-to-ductal metaplasia (ADM) compared to PKCY mice. ShhPKCY tumors were poorly differentiated with significantly reduced levels of αSMA-positive CAFs and leukocytes including F4/80+ monocytes. Consistent with a prior study linking the Shh-driven stroma to poor perfusion and drug delivery (36), genetic Shh inhibition increased vascular density and perfusion in these tumors and also increased cancer cell proliferation, perhaps due to increased nutrient availability. In the same study, KrasLSL-G12D/+;Trp53LSL-R172H/+;Pdx1-Cre (KPC) mice (37) were treated with Smo inhibitor IPI-926 beginning at 8 weeks of age, prior to PDAC formation (though ADM and PanINs are present at this time). As in the genetic inhibition model, long-term IPI-926 treatment yielded poorly differentiated tumors and shortened survival—here, mice met endpoint criteria with significantly smaller tumors than those found in vehicle controls at endpoint. Interestingly, these inhibitor-treated mice succumbed with small tumors but with severe weight loss, raising the possibility of an inverse relationship between Shh-driven stroma and the characteristic tissue wasting commonly seen in pancreatic cancer patients. Lee et al. had very similar findings: conditional deletion of Shh from pancreatic epithelial cells in the Ptf1a-Cre;KrasLSL-G12D/+ (KC) model enhanced epithelial cell proliferation as well as PanIN and PDAC formation. Extending these findings to two additional genetically engineered mouse models, the authors found that pharmacologic inhibition of Shh-Smo signaling reduced the density of Gli1+ (Shh-responsive) stroma while increasing epithelial cell proliferation and PanIN and PDAC formation. Pharmacologic Shh-Smo pathway activation, however, increased Gli1+ stromal density, and reduced epithelial cell proliferation and neoplastic progression. It is important to note that the reported results of Shh inhibition in PDAC are difficult to reconcile, owing to our incomplete understanding of the mechanisms and consequences of the Shh pathway in this setting. While the tumor-suppressive functions of Shh-dependent CAFs remain unknown, similar findings in bladder cancer suggest that Shh-driven stromal elements are a source of urothelial differentiation factors, and BMPs in particular (38). Shh-dependent stroma may similarly suppress tumor growth in the pancreas in part through provision of pro-differentiation cues.
Using an entirely different approach, Özdemir et al. had similar findings upon PDAC CAF depletion. Here, an αSMA-tk allele was crossed into the highly aggressive Ptf1a-Cre;KrasLSL-G12D/+;Tgfbr2flox/flox (PKT) model of PDAC to enable depletion of αSMA-expressing proliferative cells, including CAFs, with ganciclovir. Ganciclovir treatment early or late in PDAC development yielded poorly differentiated tumors and shortened survival, consistent with the studies described above. Depletion of αSMA+ cells also remodeled the immune microenvironment, including reduced F4/80+ monocytes as seen with Shh inhibition, as well as increased Treg infiltration. This depletion strategy made otherwise resistant PKT tumors responsive to anti-CTLA4 immune checkpoint blockade, consistent with prior findings (20). These results are bolstered by patient outcome data: PDAC patients with higher αSMA scores or higher tumor stromal densities have improved overall survival (33, 39). Together, these studies suggest that Shh-dependent, αSMA-positive CAFs—or a sub-population of CAFs meeting these criteria—function through unknown mechanisms to restrain PDAC growth.
Evidence for PDAC CAF Heterogeneity
The divergent results of CAF manipulation in PDAC models underscores the need to better understand these cells and their functions and reconcile these confounding results. To that end, it may be that any individual CAF can be both tumor-supportive and tumor-suppressive, by nature of its diverse functions related to its evolutionary role in wound healing. However, increasing evidence suggests heterogeneity among PDAC CAFs (Figure 2), consistent with the notion that the complex tumor microenvironment includes CAF subtypes that support and others that suppress tumor growth. Moving towards an improved understanding of the distinct functions of these CAF populations will be critical for the potential development of rational, stroma-targeted therapies.
Figure 2.
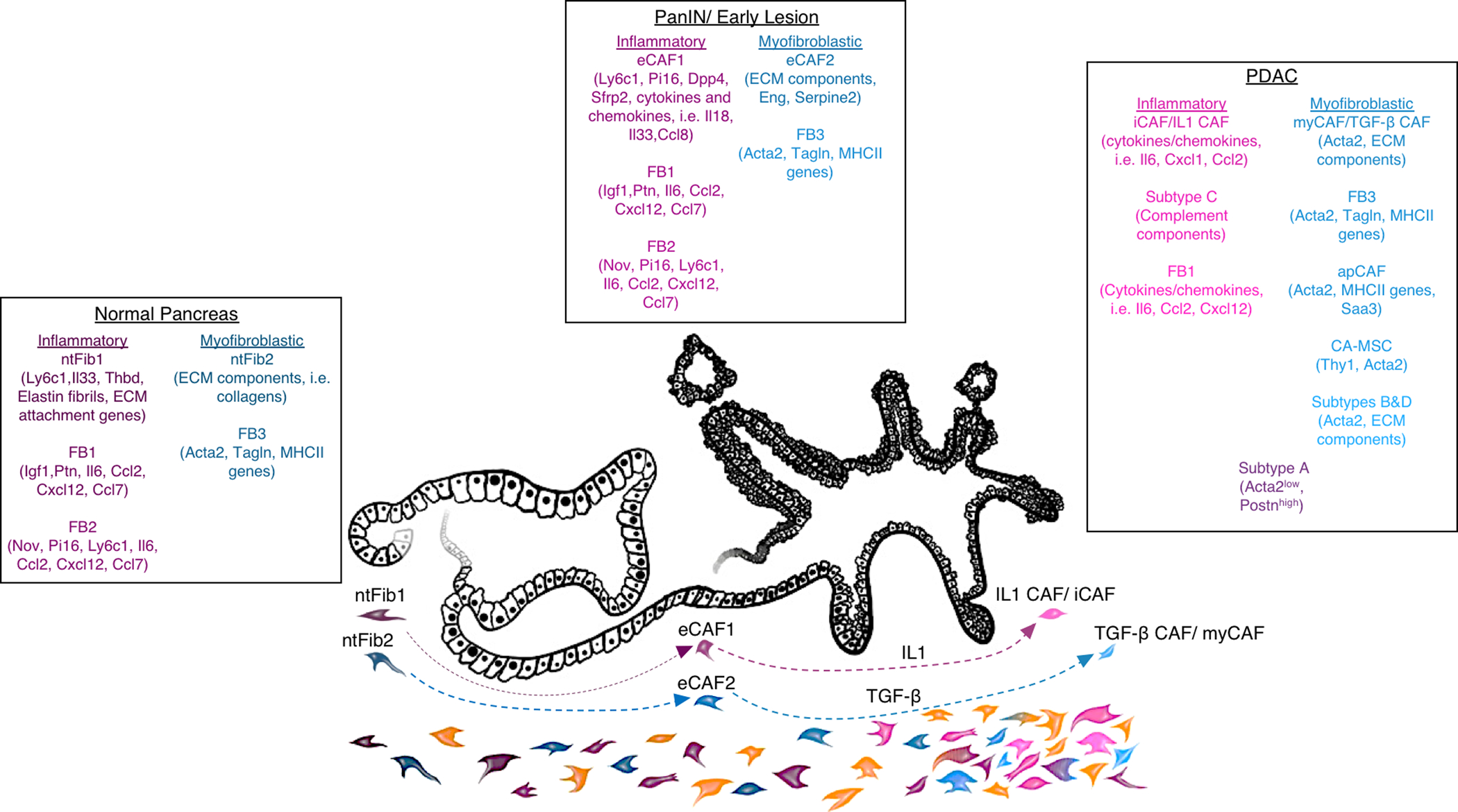
Overview of PDAC CAF subtypes identified from transcriptional profiling, including overlap of key markers for each population. In normal pancreas tissue, early or PanIN lesions, and PDAC, fibroblast transcriptional programs generally fall into two categories: inflammatory signatures including cytokines and other immune-modulatory molecules (indicated in shades of pink), and myofibroblastic signatures including classical markers of activated fibroblasts (Acta2, Tagln), ECM components and remodeling factors, and in some myofibroblastic sub-populations, genes encoding the MHC II complex (indicated in shades of blue). Single-cell RNA-seq has identified 2 (ntFib1 and ntFib2) (46) or 3 (FB1, FB2, and FB3) (44) fibroblast populations in normal pancreas, and computational modeling suggests that these tissue-resident fibroblast populations likely give rise to CAFs (46). Based on transcriptional similarities, it seems that these fibroblasts in normal pancreas tissue give rise to inflammatory (eCAF1 in (46), FB1/FB2 in (44)) or myofibroblastic (eCAF2 in (46), FB3 in (44)) fibroblasts in early lesions (per analysis in (46) and indicated by the dashed arrows). Depending on analysis method, model used, and perhaps other factors, 2–4 CAF populations are found in established PDAC, though boundaries seem non-discrete (for example, subtype A in (45)) and sub-populations exist within these CAF designations. An inflammatory population of CAFs (FB1 in (44), IL1 CAFs in (46), iCAF in (26), subtype C in (45)) seems to arise from tissue-resident inflammatory fibroblasts (46) as a result of IL1 signaling (46, 47). Myofibroblastic CAFs (FB3 in (44), TGF-β CAFs in (46), myCAF in (26), subtypes B and D in (45), and sub-populations of myofibroblastic CAFs including antigen-presenting CAFs or apCAFs (25) and cancer-associated mesenchymal stem cells or CA-MSC (55, 56)) seem to arise from tissue-resident myofibroblastic cells (46) as a result of TGF-β signaling (46, 47). Additional populations not fitted to these categories may also be present in some PDAC cases perhaps dependent on genotype or stage, indicated here in orange. Key genes expressed by these defined fibroblast or CAF subtypes are listed.
CAF marker expression as a basis for stromal heterogeneity
Among the challenges in studying CAFs and manipulating them in their host tissues is a lack of specific markers. CAFs are often defined by the absence of markers for other established cell populations including EpCAM to mark epithelial cells (including normal pancreatic epithelial cells and most PDAC cells), CD45 to mark leukocytes, and CD31 to mark endothelial cells. The remaining population includes numerically minor cell types in the PDAC microenvironment such as pericytes, but is mostly comprised of CAFs. Markers used to identify CAFs, including some commonly thought to be pan-CAF markers, have indicated heterogeneity in expression that likely signifies distinct subpopulations.
Commonly used PDAC CAF markers to date include αSMA/ACTA2, FAP, PDGFRα, FSP1, and PDPN. While αSMA was long thought to be a pan-CAF marker, recent investigation of CAF subpopulations in human and mouse PDAC showed that a minority of CAFs in PDAC stroma are in fact αSMA-negative, but have the characteristic morphology of activated fibroblasts and express other fibroblast markers such as PDGFRβ (26). These αSMA-negative CAFs are found in KPC tumors in mice, and in human PDAC. These cells comprise a minority of PDAC CAFs and are spatially restricted, mostly localized to stromal regions lacking juxtacrine interactions with tumor cells. Consistent with these results, a recent analysis of CAFs in the Kras+/LSLG12Vgeo;Trp53lox/lox;Rosa26+/LSLEYFP;Elas-tTA/tetO-Cre (KPeCY) genetically engineered mouse model of PDAC found that ~75% of PDAC CAFs express αSMA; of those, about half also express PDGFRα (40). An additional 9% of PDAC CAFs in this model express PDGFRα but not αSMA, while 16% express neither marker. PDGFRα also marks only a subset of PDAC CAFs in KPC tumors at a frequency similar to findings in the KPeCY model (~45%), and the limited overlap between αSMA+ and PDGFRα+ CAFs is further supported by recent single-cell RNA-seq analysis demonstrating that PDGFRA expression is enriched among ACTA2-negative CAFs in KPC tumors and human PDAC (25). FSP1 has received less attention as a PDAC CAF marker, and its utility is limited somewhat given that FSP1 may also mark tumor cells that have undergone EMT and in some cases lost expression of E-cadherin (and perhaps other epithelial markers) (41). However, genetic lineage-labeling efforts have enabled FSP1 expression analysis on stromal cells of non-epithelial origin, which showed FSP1 expression on a subset of PDAC CAFs and minimal overlap between FSP1 and αSMA expression (42). The spatial orientation, transcriptional profile, and functions of this FSP1+ CAF population are currently unknown. The cell surface glycoprotein FAP was identified on reactive fibroblasts in human cancers, and depletion of FAP+ cells fostered immunological control of PDAC growth in a subcutaneous model and in KPC mice (19, 20). The marker profile of FAP+ stromal cells changes over the course of stepwise tumorigenesis: FAP+ cells associated with pre-malignant PanIN lesions are αSMA−, but FAP+ CAFs in PDAC are αSMA+, with FAP expressed on 92% of αSMA+ CAFs (20). PDPN has emerged more recently as a robust marker for PDAC CAFs. Single-cell RNA-seq results from KPC tumors and human PDAC suggest that PDPN is a pan-CAF marker (25), and its expression on the cell surface makes PDPN useful for CAF quantification and isolation by FACS. Interestingly, high PDPN expression in CAFs correlates with worse prognosis in PDAC (43). Importantly, all of these markers are expressed by additional cell types beyond the tumor microenvironment: for example, PDPN is expressed on lymphatic endothelial cells, αSMA is expressed by pericytes, and FAP is expressed on mesenchymal cells in skeletal muscle and bone. These are important considerations when designing models to target and manipulate these cell populations in vivo. While markers pervasively expressed on PDAC CAFs will be helpful for isolation and characterization of this cell population as a whole, the restriction of other markers to CAF subsets is an important indicator of underlying transcriptional and functional heterogeneity, an important topic for ongoing and future investigation.
Transcriptional heterogeneity among PDAC CAFs
Diverging results from CAF ablation studies together with marker expression patterns suggest that PDAC CAFs are heterogeneous, perhaps including subtypes that support and others that suppress tumor growth. To address this possibility, several groups have performed gene expression profiling on primary CAFs from genetically engineered PDAC mouse models or from patient samples, and the results of these studies all suggest the existence of multiple subpopulations of CAFs with distinct transcriptional programs. Single-cell RNA-seq analyses of KPC tumors and human PDAC highlighted the existence of three CAF populations on the basis of gene expression (25). These include a population that transcriptionally resemble classically activated myofibroblasts, with high levels of expression of αSMA, ECM components, and contractility factors and were termed myCAFs. A second population of CAFs lacks αSMA expression but has a transcriptional profile rich in cytokines and chemokines such as IL6 and CXCL12, named inflammatory CAFs or iCAFs. A third population expresses the genes that comprise the MHC II complex, including the invariant chain CD74, but in the absence of co-stimulatory molecules typically expressed by antigen-presenting cells; this population was termed antigen-presenting CAFs or apCAFs, and were able to present antigen to T cells in vitro. Interestingly, the gene Saa3 was identified as a marker of the apCAF population. Consistent with a presumed immune-modulatory function for these cells, a recent study found that PDAC in the context of a Saa3-null stroma had increased levels of intratumoral macrophages, including CD11c+ CD206− (potentially tumoricidal) macrophages (40).
An independent single-cell RNA-seq study on multiple genetically engineered PDAC mouse models resulted in similar findings. While three fibroblast populations were found in normal pancreas tissue and early lesions (named FB1, FB2, and FB3), only two of these populations—FB1 and FB3—were found in advanced PDAC (44). These results were consistent across three mouse models, all driven by mutant Kras but also by Ink4a loss, p53 mutation (R172H), or p53 loss. FB1 had a transcriptional profile consistent with the previously described iCAF phenotype, rich in expression of cytokines and chemokines including Il6, Cxcl12, and Ccl2. FB3 had features of myCAFs including expression of Acta2 and numerous myofibroblast-associated contractility factors such as Tagln, but also expressed MHC II components as seen among apCAFs. Analysis of low-passage primary human PDAC CAFs in culture showed the existence of similar subtypes (45), which suggests stability of at least some transcriptional heterogeneity as this was maintained in culture in the absence of relevant signaling gradients in the tumor microenvironment. RNA-seq analysis of 16 primary CAF samples led to the identification of 4 subtypes, named A-D. Subtypes B and D had hallmarks of myCAFs in their expression of ACTA2 and numerous ECM components. Subtype C somewhat resembled the iCAF phenotype in expressing inflammatory mediators such as complement components. Subtype A had features of both iCAFs and myCAFs: ACTA2 expression was low, while expression of myofibroblastic genes such as POSTN was high, though this may reflect heterogeneity within this subtype given that bulk RNA-seq was performed. These results together support the existence of three CAF subtypes, albeit with non-discrete boundaries.
A more recent single-cell RNA-seq analysis of fibroblasts in normal pancreas and PDAC provided insights into fibroblast evolution during tumor progression, and also identified a CAF marker of prognostic significance in immunotherapy clinical trials (46). The authors performed sequencing on stromal populations isolated from normal mouse pancreas, as well as early lesions and established tumors in Pdx1Cre/+;LSL-KrasG12D/+;p16/p19flox/flox (KPP) mice. These analyses identified 2 major CAF populations in early lesions, one characterized by ECM component production aligned with the myCAF subtype and another positive for immune-modulatory transcripts similar to the iCAF subtype; 2 similar populations were identified in established tumors, suggesting an evolutionary relationship. This study also identified 2 fibroblast populations in normal pancreas tissue which likely give rise to these myCAF and iCAF populations based on differentiation trajectories developed from expression profiles as well as pseudo-time analysis. This argues for the development of PDAC CAFs from pre-existing fibroblast populations in normal tissue. Normal pancreas also harbored a mesothelial cell population which expressed genes associated with the apCAF subtype. Importantly, human PDAC samples revealed similar fibroblast evolution from tissue-resident fibroblasts, through an activated fibroblastic state in early lesions with ECM and immune-modulatory gene expression, to 2 CAF populations in established tumors similar to the myCAF and iCAF subtypes. Human PDAC CAFs with an ECM-rich, myofibroblastic transcriptional profile, here called TGFb CAFs, comprise the majority of CAFs and express the marker LRRC15. This novel CAF marker and its associated transcriptional signature predict a poor response to immunotherapy, impelling future studies to characterize the functional relationship between this CAF population and the anti-tumor immune response. Though these CAF heterogeneity studies to date have substantial similarities, key differences (for example, the precise cadre of cytokines and chemokines expressed by pro-inflammatory CAF subpopulations) are noted and will be important to understand. These differences are likely in part technical, reflecting distinct sequencing methods or depth and different tissue dissociation protocols; and in part biological, reflecting a distinct stromal milieu across different mouse models and stages of tumorigenesis.
Understanding the mechanisms that drive transcriptional heterogeneity will become increasingly important as the functional differences of these subpopulations come to light. Several recent studies provide evidence that growth factor or cytokine signaling gradients set up by cancer cells play key roles in shaping CAF heterogeneity. For example, PDAC cells were recently found to be sources of two critical factors—IL1 and TGF-β—which together regulate CAF transcriptional programs in a paracrine manner (47). IL1 signaling through IL1R on CAFs was shown to promote the iCAF transcriptional program via NF-κB and subsequent induction of LIF, leading to autocrine activation of JAK/STAT signaling. TGF-β signaling instead promoted the myCAF phenotype, consistent with its role as a key regulator of fibrogenic gene expression and the myofibroblast state (48, 49). Though both come from tumor cells, this work suggests the relevance of signaling gradients within the tumor microenvironment, such that CAFs in close proximity to PDAC cells experience dominant TGF-β signaling and take on a myCAF phenotype, whereas CAFs at a greater distance from the nearest PDAC cell are more strongly influenced by IL1/IL1R signaling and are more likely to take on an iCAF phenotype. An independent analysis of fibroblast evolution further supported a role for TGF-β and IL1 signaling in driving the myCAF and iCAF fates, respectively (46). Interestingly, TGF-β signaling not only promoted a myCAF phenotype, but appeared to actively suppress the iCAF fate, suggesting a degree of mutual exclusivity consistent with single-cell RNA-seq results in patient samples (25).
A recent study suggests that, in addition to spatial context with respect to tumor cell proximity, tumor genotype plays an important role in the regulation of CAF heterogeneity (50). Specifically, p53 status determined the outcome of paracrine signaling to neighboring CAFs: by comparing CAFs isolated from KPC tumors (p53-mutant) versus KPflC tumors (p53-null), the authors found that PDAC cells harboring mutant p53 exert a dominant function in shaping neighboring CAFs to promote a pro-metastatic and chemoresistant microenvironment. Cancer cell-intrinsic NF-κB activity was again implicated as a regulator of CAF heterogeneity: p53-mutant PDAC cells had higher levels of NF-κB pathway activity than p53-null cells, and secreted higher levels of NF-κB target gene TNFα. TNFα in turn stimulated CAFs to secrete HSPG2 or perlecan, an ECM component that contributed both to the heightened pro-metastatic and chemoresistance phenotypes associated with p53-mutant cancer cells. Interestingly, p53 status was recently linked to local and systemic regulation of neutrophils and metastatic capacity in breast cancer (51), raising the possibility that distinct immune microenvironments may also indirectly regulate PDAC CAFs adjacent to cancer cells of different underlying mutational landscapes. As cancer cells of distinct genotypes coexist within the tumor microenvironment, the genetic aberrations within PDAC cells may represent important drivers of CAF heterogeneity.
CAF-intrinsic features also contribute to heterogeneity in transcriptional program and perhaps CAF function. For example, differentiation of tissue-resident pancreatic stellate cells (PSCs) from a quiescent state to an activated, myofibroblast-like phenotype is thought to give rise to the majority of PDAC CAFs (52). However, activated PSCs exhibit substantial transcriptional plasticity: ligands for the nuclear receptors RAR or VDR broadly suppress the activation-associated transcriptional program in these cells, highlighting the existence of a spectrum of activation states and fibroinflammatory potential for PSC-derived CAFs (53, 54). CAF cell of origin may also contribute to heterogeneity. For example, mesenchymal stem cells were recently identified as a numerically minor subpopulation of primary patient-derived CAFs, and were found to be a critical source of the cytokine GM-CSF (55). These cells promoted PDAC cell proliferation, invasion, and metastatic potential, and raise the possibility of a broader contribution of the bone marrow niche or other tissues to the PDAC CAF pool. Mesenchymal stem cells were also described in murine PDAC, as tumor-promoting regulators of macrophage function (56). These studies lay the foundation of our understanding of CAF complexity in the PDAC microenvironment, and impel further investigation of the origins and functions of CAF subtypes.
Conclusions and Future Directions
The work discussed above has much improved our understanding of CAF heterogeneity in PDAC, particularly with respect to distinct transcriptional programs within this stromal population. To bring these results to bear for PDAC patients, important future efforts will be needed to address knowledge gaps brought to light be these prior studies. The transcriptional heterogeneity among PDAC CAFs raises the possibility that, in addition to signaling gradients within the tumor microenvironment, CAFs have distinct cellular origins, with relevance for disease progression and for the development of model systems to study these cells. Further, the identification of tumor-suppressive or homeostatic CAF populations and their key mechanisms will be crucial in efforts to design rational stroma-targeted therapies that leave these tumor-suppressive mechanisms in place. Addressing outstanding questions will be greatly facilitated by the development of robust and specific new models to manipulate CAFs or their cellular precursors in vivo, enabling investigation of the previously described CAF subtypes to uncover their functions within the appropriate tissue context. These improved models, together with our enhanced understanding of CAF complexity and function, have the potential to foster the development of effective therapies for this highly recalcitrant cancer.
Statement of Significance.
While the abundant stromal reaction associated with pancreatic cancer has long been appreciated, the functions of the cancer-associated fibroblastic cells that establish this stromal reaction remain unclear. An improved understanding of the transcriptional and functional heterogeneity of pancreatic CAFs, as well as their tumor-supportive versus tumor-suppressive capacity, may facilitate the development of effective therapies for this disease.
Footnotes
Conflict of interest disclosure statement: The authors declare no potential conflicts of interest.
References
- 1.Stoker MG, Shearer M, O’Neill C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J Cell Sci. 1966;1(3):297–310. Epub 1966/09/01. [DOI] [PubMed] [Google Scholar]
- 2.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science. 1985;230(4726):676–8. Epub 1985/11/08. doi: 10.1126/science.2996144. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21(3):309–22. Epub 2012/03/24. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 4.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–9. Epub 2011/03/09. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122(2):639–53. Epub 2012/01/11. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–70. Epub 2012/05/01. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75(3):544–53. Epub 2015/02/04. doi: 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–29. Epub 2012/03/24. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–20. Epub 2012/04/03. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479–83. Epub 2016/08/12. doi: 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun. 2017;8:16031. Epub 2017/07/08. doi: 10.1038/ncomms16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019;9(5):617–27. Epub 2019/03/07. doi: 10.1158/2159-8290.CD-18-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A. 2013;110(22):8882–7. Epub 2013/05/15. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5:e10250. Epub 2016/02/28. doi: 10.7554/eLife.10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36(13):1770–8. Epub 2016/09/27. doi: 10.1038/onc.2016.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalin S, Sullivan MR, Lau AN, Grauman-Boss B, Mueller HS, Kreidl E, et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res. 2019; 79(22):5723–5733. Epub 2019/09/06. doi: 10.1158/0008-5472.CAN-19-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tape CJ, Ling S, Dimitriadi M, McMahon KM, Worboys JD, Leong HS, et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell. 2016;165(4):910–20. Epub 2016/04/19. doi: 10.1016/j.cell.2016.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature. 2019;569(7754):131–5. Epub 2019/04/19. doi: 10.1038/s41586-019-1130-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330(6005):827–30. Epub 2010/11/06. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 20.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–7. Epub 2013/11/28. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015;75(14):2800–10. Epub 2015/05/17. doi: 10.1158/0008-5472.CAN-14-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910. Epub 2012/02/02. doi: 10.1172/JCI45817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–60. Epub 2016/07/05. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, et al. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res. 2014;20(13):3422–33. Epub 2014/04/26. doi: 10.1158/1078-0432.CCR-13-2972. [DOI] [PubMed] [Google Scholar]
- 25.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019;9(8):1102–23. Epub 2019/06/15. doi: 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–96. Epub 2017/02/25. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013;73(20):6359–74. Epub 2013/10/08. doi: 10.1158/0008-5472.CAN-13-1558-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73(10):3007–18. Epub 2013/03/22. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang H, Hegde S, DeNardo DG. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother. 2017;66(8):1037–48. Epub 2017/04/30. doi: 10.1007/s00262-017-2003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208(3):469–78. Epub 2011/02/23. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goehrig D, Nigri J, Samain R, Wu Z, Cappello P, Gabiane G, et al. Stromal protein betaig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut. 2019;68(4):693–707. Epub 2018/11/12. doi: 10.1136/gutjnl-2018-317570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foley K, Kim V, Jaffee E, Zheng L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016;381(1):244–51. Epub 2016/01/03. doi: 10.1016/j.canlet.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34. Epub 2014/05/27. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–47. Epub 2014/05/27. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A. 2014;111(30):E3091–100. Epub 2014/07/16. doi: 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–61. Epub 2009/05/23. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. Epub 2005/05/17. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 38.Shin K, Lim A, Zhao C, Sahoo D, Pan Y, Spiekerkoetter E, et al. Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell. 2014;26(4):521–33. Epub 2014/10/15. doi: 10.1016/j.ccell.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Torphy RJ, Wang Z, True-Yasaki A, Volmar KE, Rashid N, Yeh B, et al. Stromal Content Is Correlated With Tissue Site, Contrast Retention, and Survival in Pancreatic Adenocarcinoma. JCO Precis Oncol. 2018;2018. Epub 2018/12/07. doi: 10.1200/PO.17.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Djurec M, Grana O, Lee A, Troule K, Espinet E, Cabras L, et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc Natl Acad Sci U S A. 2018;115(6):E1147–E56. Epub 2018/01/21. doi: 10.1073/pnas.1717802115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–61. Epub 2012/01/24. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, LeBleu VS, Carstens JL, Sugimoto H, Zheng X, Malasi S, et al. Dual reporter genetic mouse models of pancreatic cancer identify an epithelial-to-mesenchymal transition-independent metastasis program. EMBO Mol Med. 2018;10(10). Epub 2018/08/19. doi: 10.15252/emmm.201809085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirayama K, Kono H, Nakata Y, Akazawa Y, Wakana H, Fukushima H, et al. Expression of podoplanin in stromal fibroblasts plays a pivotal role in the prognosis of patients with pancreatic cancer. Surg Today. 2018;48(1):110–8. Epub 2017/07/14. doi: 10.1007/s00595-017-1559-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, et al. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight. 2019;5. Epub 2019/07/25. doi: 10.1172/jci.insight.129212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neuzillet C, Tijeras-Raballand A, Ragulan C, Cros J, Patil Y, Martinet M, et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J Pathol. 2019;248(1):51–65. Epub 2018/12/24. doi: 10.1002/path.5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2019. Epub 2019/11/09. doi: 10.1158/2159-8290.CD-19-0644. [DOI] [PubMed] [Google Scholar]
- 47.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019;9(2):282–301. Epub 2018/10/28. doi: 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans RA, Tian YC, Steadman R, Phillips AO. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res. 2003;282(2):90–100. Epub 2003/01/18. doi: 10.1016/s0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- 49.Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257(1):180–9. Epub 2000/06/15. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- 50.Vennin C, Melenec P, Rouet R, Nobis M, Cazet AS, Murphy KJ, et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat Commun. 2019;10(1):3637. Epub 2019/08/14. doi: 10.1038/s41467-019-10968-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wellenstein MD, Coffelt SB, Duits DEM, van Miltenburg MH, Slagter M, de Rink I, et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature. 2019;572(7770):538–42. Epub 2019/08/02. doi: 10.1038/s41586-019-1450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pothula SP, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016;381(1):194–200. Epub 2015/11/17. doi: 10.1016/j.canlet.2015.10.035. [DOI] [PubMed] [Google Scholar]
- 53.Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology. 2011;141(4):1486–97, 97 e1–14. Epub 2011/06/28. doi: 10.1053/j.gastro.2011.06.047. [DOI] [PubMed] [Google Scholar]
- 54.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159(1):80–93. Epub 2014/09/27. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waghray M, Yalamanchili M, Dziubinski M, Zeinali M, Erkkinen M, Yang H, et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov. 2016;6(8):886–99. Epub 2016/05/18. doi: 10.1158/2159-8290.CD-15-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mathew E, Brannon AL, Del Vecchio A, Garcia PE, Penny MK, Kane KT, et al. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia. 2016;18(3):142–51. Epub 2016/03/20. doi: 10.1016/j.neo.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]