

Abstract
Mucopolysaccharidoses comprise a group of rare metabolic diseases, in which the lysosomal degradation of glycosaminoglycans (GAGs) is impaired due to genetically inherited defects of lysosomal enzymes involved in GAG catabolism. The resulting intralysosomal accumulation of GAG-derived metabolites consequently manifests in neurological symptoms and also peripheral abnormalities in various tissues like liver, kidney, spleen and bone. As each GAG consists of differently sulfated disaccharide units, it needs a specific, but also partly overlapping set of lysosomal enzymes to accomplish their complete degradation. Recently, we identified and characterized the lysosomal enzyme arylsulfatase K (Arsk) exhibiting glucuronate-2-sulfatase activity as needed for the degradation of heparan sulfate (HS), chondroitin sulfate (CS) and dermatan sulfate (DS). In the present study, we investigated the physiological relevance of Arsk by means of a constitutive Arsk knockout mouse model. A complete lack of glucuronate desulfation was demonstrated by a specific enzyme activity assay. Arsk-deficient mice show, in an organ-specific manner, a moderate accumulation of HS and CS metabolites characterized by 2-O-sulfated glucuronate moieties at their non-reducing ends. Pathophysiological studies reflect a rather mild phenotype including behavioral changes. Interestingly, no prominent lysosomal storage pathology like bone abnormalities were detected. Our results from the Arsk mouse model suggest a new although mild form of mucopolysacharidose (MPS), which we designate MPS type IIB.
Introduction
Lysosomal sulfatases play a critical role in the degradation of macromolecules like sulfated glycosaminoglycans (GAGs), namely heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS) and keratan sulfate (KS), as well as a class of sulfolipids, also known as sulfatides. Sulfatide degradation is dependent on a single lysosomal sulfatase as reflected by the severe lysosomal storage disease (LSD) metachromatic leukodystrophy resulting from deficiency of arylsulfatase A [1]. In contrast, sulfated GAGs are degraded by a set of at least six lysosomal sulfatases, which act, along with a defined number of exoglycosidases and one acetyltransferase, in a concerted sequential degradation pathway proceeding at the non-reducing end (NRE) of the GAG chain [2,3]. Sugar composition and sulfation patterns of the different GAGs demand specific sets of degrading enzymes with some overlap for shared hydrolytic steps [4]. As a consequence, some enzyme defects (e.g. β-glucuronidase) affect the breakdown of various GAGs, whilst other enzymes are exclusively needed for a single GAG like HS glucosamine N-sulfatase and N-acetylglucosamine 6-sulfatase in HS degradation [3].
Deficiencies in as many as 12 GAG catabolizing enzymes are classified as a subgroup of LSDs called mucopolysacharidoses (MPSs) [5–7]. Biochemically, MPSs are characterized by an incomplete breakdown of one or several GAGs, which leads to a progressive accumulation of GAG metabolites inside the lysosomal compartment and to a pathophysiological increase in unusual GAG fragments in blood, urine and other body fluids [5]. The clinical course and severity vary considerably between different MPS disorders, as some diseases typically present in early childhood with severe somatic and neurological syndromes, whereas others manifest with mild clinical symptoms in adulthood or even remain unrecognized as MPS for a long time [5]. Interestingly, several MPSs do not or just mildly affect the central nervous system and thus enzyme replacement therapy (ERT) approaches have been developed. Indeed, ERT is currently available for MPS I, MPS II, MPS IVA, MPS VI and MPS VII [8].
Among the MPSs, the MPS III group (also called Sanfilippo syndrome) is of particular interest, as it comprises five distinct enzyme defects (subtypes IIIA–E), which all result in progressive HS storage [9]. While MPS IIIA–IIID (Sanfilippo A–D) are long known and well-described LSDs, which all have an early onset and severe neurological impairments in common, we just recently identified the first MPS IIIE patients suffering from a late-onset vision and hearing loss, which is caused by HS glucosamine-3-O-sulfatase deficiency (arylsulfatase G deficiency) [6,10].
Glucuronate-2-O-sulfation (GlcA2S) in HS has been reported for the first time in 1985 and shown to occur in CS/DS as well [11]. In mammals, 2-O-sulfation is exclusively catalyzed by the enzyme HS 2-O-sulfotransferase (Hs2st), which preferentially modifies iduronic acid (IdoA) but also shows limited activity towards glucuronic acid (GlcA) thus forming glucuronate-2-O-sulfate (GlcA2S) as a relatively rare modification [12]. A different enzyme, namely uronyl 2-O-sulfotransferase (Ust), is involved in the formation of 2-O-sulfated GlcA in CS/DS. As a consequence, lysosomal degradation of both HS and CS/DS requires a GlcA2S desulfating enzyme. Shaklee et al. [13] already described in 1985 such a specific hydrolyzing GlcA2S from GAG-derived disaccharides at low pH, as is expected for a lysosomal sulfatase. However, the molecular nature of this enzyme remained enigmatic until several new human sulfatase candidate genes were identified on the basis of the so-called sulfatase signature sequence CXPSR by genome analysis and cDNA library screening [14–16]. The arylsulfatase K (ARSK) gene was among these candidates and after cloning and expression in human cells, we demonstrated sulfatase activity of the recombinant human ARSK enzyme (rhARSK) against small arylsulfate pseudosubstrates, such as p-nitrocatechol sulfate and p-nitrophenyl sulfate at acidic pH [17]. A lysosomal localization of endogenous ARSK was initially suggested due to its appearance in subproteome analyses, in which the specific lysosomal sorting signal mannose 6-phosphate (M6P) was exploited to identify novel lysosomal hydrolases (reviewed in [18]). We confirmed the M6P-modification as well as the lysosomal localization of ARSK [17]. Recently, we demonstrated 2-O-sulfatase activity of rhARSK against two synthetic disaccharides, namely glucuronate-2-O-sulfate-N-acetylglucosamine (G2A0) and glucuronate-2-O-sulfate-N-sulfoglucosamine (G2S0), which are identical in structure to disaccharide units found in HS and also in CS/DS [19]. Since these synthetic substrates were not turned over by iduronate 2-sulfatase (IDS) and, vice versa, iduronate 2-sulfate containing disaccharide substrates of IDS were not turned over by rhARSK, we concluded that ARSK is the long sought lysosomal glucuronate-2-sulfatase (GDS) which, by analogy to the well-known IDS, we designated GDS [19].
In the present study, we generated an ARSK-deficient mouse model in order to evaluate the physiological relevance of lysosomal GDS activity and, after establishing a GDS-specific assay, confirmed the complete loss of glucuronate desulfation. Moreover, we identified and biochemically characterized GAG-derived storage material found at moderate levels particularly in kidney and brain of knockout mice. Ex vivo desulfation of this lysosomal storage material by rhARSK, but not by rhIDS, verified that ARSK is the only GDS in lysosomes. Pathophysiological studies of these Arsk-deficient mice reflect a rather mild storage pathology including behavioral changes.
Materials and methods
Generation of Arsk-deficient mice
The Arsk (arylsulfatase K) knockout/reporter mouse model (official name: Arsktm1b(KOMP)Wtsi, (http://www.informatics.jax.org/allele/MGI:5446134)) was generated in a C57BL/6NJ background by the Knockout Mouse Phenotyping Program (KOMP2) at The Jackson Laboratory using embryonic stem (ES) cells provided by the International Knockout Mouse Consortium. These mice are now available at KOMP/MMRC (see (https://www.mmrrc.org/catalog/sds.php?mmrrc_id=46694). The L1L2_Bact_P targeting cassette was inserted upstream of exon 3 of the Arsk gene on Chromosome 13. The cassette was composed of a FRT site followed by a lacZ reporter, a loxP site, the selectable marker neomycin phosphotransferase (neo), a second FRT site and a second loxP site (see Figure 1A). A third loxP site was inserted downstream of the targeted exon 3 so that exon 3 was flanked by loxP sites. The construct was introduced into C57BL/6N-derived JM8.N4 ES cells, and correctly targeted ES cells were injected into B6(Cg)-Tyrc-2J/J (Stock No. 58) blastocysts. The resulting chimeric males were bred to C57BL/6NJ (Stock No. 005304) females and then to B6N.Cg-Tg(Sox2-cre)1Amc/J mice to remove the floxed neo-coding sequence as well as the critical exon 3, whereas the lacZ reporter was left behind. The resulting offspring were bred to C57BL/6NJ mice to remove the cre-expressing transgene. Routine genotyping was performed in a multiplex PCR reaction using the following primer pairs: WT_fwd_5′-GAAGGCCTATCCAGCCTACC-3′; WT_rev_5′-TGGCTGGTTTAGAGTCTGTGG-3′ and Arsk_KO_fwd_5′-GCTACAGTCAACAGCAACTGA-3′; Arsk_KO_rev_5′-GTGTTGCCTTGAGTGTCCTG-3′. The mice were housed under standard conditions in a pathogen-free animal facility at Bielefeld University. The experiments were performed using littermates as controls. All procedures and experiments in mice were performed in accordance with local guidelines and were approved by local authorities. For perfusion, mice were deeply anesthetized with an intraperitoneal injection of 100 mg/kg bodyweight (bw) ketamine, 16 mg/kg bw xylazine and 2 mg/kg bw acepromazine and pre-perfused with 1% procaine in 0.1 M phosphate-buffered saline (PBS).
Figure 1. Generation and validation of Arsk-deficient mice.
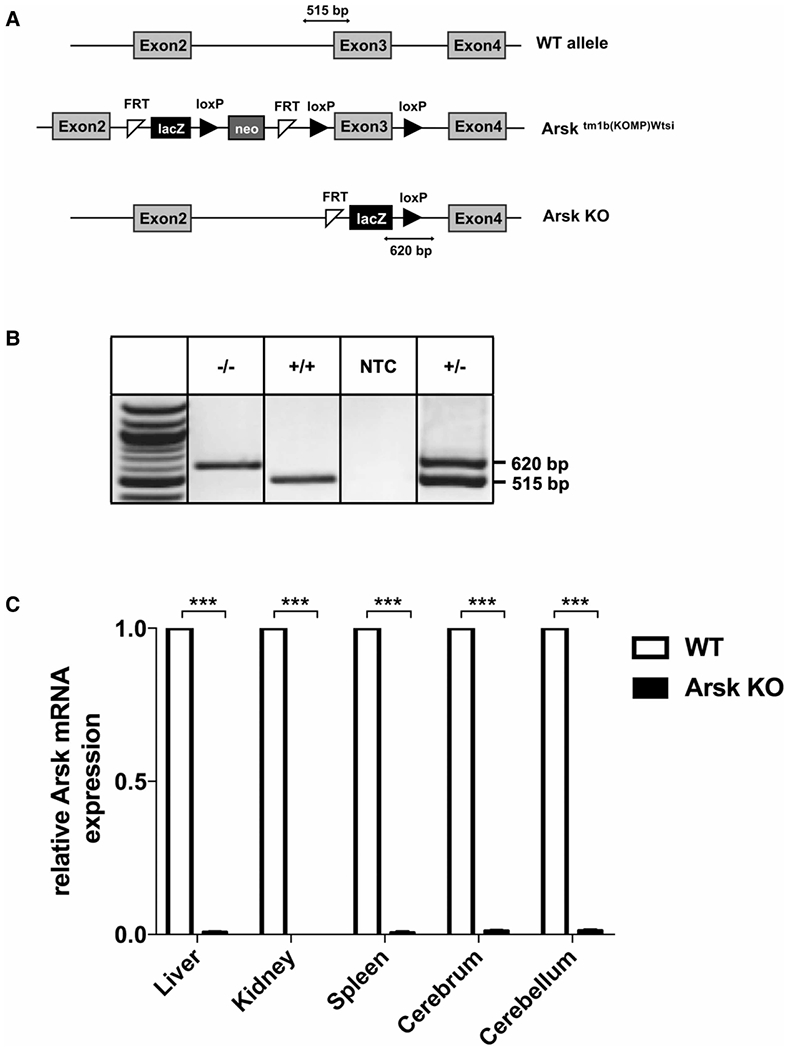
(A) Schematic representation of the Arsk gene locus in wildtype (WT, upper panel) and Arsk knockout mice (Arsk KO, lower panel). The constitutive Arsk knockout allele (knockout first strategy) was achieved by Cre recombinase expression in B6N(Cg)-Arsktm1b(KOMP)Wtsi/J mice resulting in the loxP-dependent deletion of exon 3 of the Arsk gene as well as the neomycin phosphotransferase gene (neo) while maintaining the FRT-loxP-flanked β-galactosidase (lacZ) reporter from the targeting cassette. PCR amplicons are indicated for the WT allele (515 bp) and the knockout allele (620 bp). (B) PCR-based genotyping of mice resulted in a 620-bp- and a 515-bp-product in knockout and wildtype, respectively. NTC, no template control. (C) The absence of functional Arsk transcript was verified in various tissues of Arsk knockout mice (Arsk KO) by SYBR-green-based quantitative (q)PCR using Gapdh as reference gene.
cDNA synthesis and qPCR
For total RNA preparation, 100 mg of fresh tissue was disrupted and homogenized using a rotor-stator homogenizer (Ultra-Turrax, IKA, Staufen, Germany). RNA was isolated using RNeasy Midi Kit (Qiagen, Hilden, Germany), and cDNA was subsequently synthesized using the iScript Kit from Bio-Rad (Hercules, CA, U.S.A.). Quantitative PCR (qPCR) was performed in a StepOnePlus Real-Time PCR cycler (Thermo Fisher Scientific, Waltham, MA, U.S.A.) using the KAPA SYBR Fast Universal Kit (PeqLab, VWR, Erlangen, Germany). Differences in gene expression were normalized to the housekeeping gene glycerinaldehyde 3-phosphate dehydrogenase (Gapdh). qPCR analysis was performed using the following primers: Arsk-qPCR-F_5′-TGGAGAAGCATGGCTATCAGAC-3′; Arsk-qPCR-R 5′-ATGGGTCTGCCTTCTTGTCG-3′; Gapdh-F_5′;-GCAGTGCCAGCCTCGTCCC-3′; Gapdh-R_5′-CAGGCGCCCAATACGGCCA-3′.
Lysosome-enriched fractions form mouse liver (tritosomes)
Lysosome-enriched fractions (tritosomes) from liver of 12-month-old mice were isolated with the non-ionic detergent tyloxapol (Sigma) by a combination of differential and isopycnic centrifugation steps as described previously [20]. Mice were treated by a single injection (intraperitoneal) of 0.75 mg of tyloxapol per g of body weight four days prior to lysosome isolation.
Disaccharide derivatization with AMAC
Derivatization of the disaccharide biomarkers G2A0 und G2S0 with AMAC was performed as described [21,22]. Lyophilized disaccharides (100 nmol) were reconstituted with 5 μl of a 100 mM AMAC solution in acetic acid/dimethyl sulfoxide (DMSO) (3 : 17, v/v) to which 5 μl of a freshly prepared solution of 1 M sodium cyanoborohydride in water was added. Derivatization was performed by incubating at 45°C for 4 h. Finally, 100 μl of 50% (v/v) DMSO was added to the samples and aliquots were used for RP-HPLC analysis to separate AMAC-labeled disaccharide from free AMAC label.
RP-HPLC separation of AMAC-labeled disaccharides
HPLC separation of disaccharides was performed by reversed-phase chromatography with an Ettan LC system (GE Healthcare) equipped with a Phenomenex Jupiter C18 RP column (300 μm, 4.6 mm diameter, 3.2 ml CV) as described before [23]. In brief, ammonium acetate (60 mM, pH 5.6) was used as binding buffer and saccharide elution was achieved using acetonitrile in this buffer. After injecting the samples (25–100 nmol), the different saccharides were eluted by a three-step linear acetonitrile gradient at a flow rate of 1.0 ml/min. The first gradient step ranged from 2% to 4% acetonitrile for 4 ml, followed by 4–15% acetonitrile for 28 ml and a third step with 60% acetonitrile for 8 ml. The labeled saccharides were detected by UV absorbance at 255 nm (absorbance maximum of AMAC), dried in a speedvac and analyzed by ESI-MS. Moreover, AMAC-labeled disaccharides were treated with either purified recombinant ARSK (20 ng) or lysosome-enriched fractions (50 μg protein in 250 mM ammonium acetate pH 4.6) for 24 h at 37°C. After digest, proteins were removed using a 10-kDA-centrifugation filter and the filtrate was dried and re-chromatographed as described before with the Jupiter C18 RP column and analysed by ESI-MS.
RP-UPLC/nano-ESI-MS
Accurate Mass RP-UPLC/nano-ESI-MS measurements were performed using a Q-IMS-TOF mass spectrometer Synapt G2Si (Waters GmbH, Manchester, U.K.) in resolution mode, interfaced to a nano-ESI ion source. Nitrogen served both as the nanoflow gas (0.4 bar) and the dry gas (20 l/h) for UPLC/nano-ESI-MS. Nitrogen is generated by a nitrogen generator NGM 11. The mass axis was externally calibrated with fragment ions of GluFib as calibration standard. The UPLC system is a M-Class from waters, equipped with an autosampler, nano-UPLC pump and a column oven. Water, containing 0.1% formic acid served as solvent A, acetonitrile, containing 0.1% formic acid was used as solvent B. After trapping of the sample for 5 min using 100% A at a flow rate of 5 μl/min, the gradient (5 min at 95% A, ramping in 30 min to 65% A, in 5 min to 15% A, holding 15% A for 5 min, ramping to 95% A in 1 min and keeping it at 95% A for 14 min) was started. The gradient separation took 1 h at a flow rate of 300 nl/min. During the gradient separation, mass spectra were acquired in a mass range from m/z 350 to 2000. The spray voltage was set to 1.6 kV, the source temperature to 80°C, the sampling cone to 30 V and the source offset to 80 V. During the UPLC/MS measurements, singly protonated LeuEnk served as an internal calibration standard.
Scan accumulation and data processing was performed with MassLynx 4.1 (Waters GmbH, Manchester, U.K.). The spectra shown were generated by the accumulation and averaging of several single spectra. The determination of exact masses was performed using centroided data.
GAG purification
Organs were removed from Arsk-deficient mice or WT mice at 12 months of age and were homogenized in ice-cold buffer containing 50 mM sodium acetate and 0.2 M NaCl (pH 6.0) using a Polytron homogenizer. The resulting homogenates were digested on a shaker overnight at 37°C with 0.1 mg/ml pronase and 0.1% Triton X-100. Tissue homogenates were filtered through a 0.45-μm membrane to remove insoluble debris. GAGs were purified from cell homogenates by anion-exchange chromatography as described previously [24]. Briefly, 0.5 ml of diethylaminoethyl resin was washed with 20 column volumes (CV) of 50 mM sodium acetate buffer (pH 6) containing 0.2 M NaCl and 0.1% Triton X-100. After loading the sample, the column was washed with 40 CVs of wash buffer, and GAGs were eluted with 5 CVs of buffer containing 50 mM sodium acetate and 2 M NaCl (pH 6). Samples were desalted by gel filtration (PD10), and subsequently used for enzymatic depolymerization of HS using 2 mU of each of heparin lyases I, II and III or depolymerization of CS/DS using 20 mU of chondroitinase ABC. To confirm Arsk as the glucuronate-2-O-sulfatase, purified samples were treated with recombinant human (rh)Arsk (15 ng), recombinant human (rh)IDS (8 ng) or buffer overnight prior to the enzymatic depolymerization. The digest of lysosomal storage material was determined as described below.
GAG quantification and NRE analysis
Enzymatically depolymerized GAG preparations were differentially mass labeled by reductive amination with [12C6]aniline as previously described [25]. Each sample was mixed with a known amount of [13C6]aniline labeled standard unsaturated HS and CS disaccharides, including N-acetylglucosamine-2-O-sulfate (G2A0) and N-sulfoglucosamine-2-O-sulfate (G2S0), the specific NRE biomarkers for Arsk deficiency. Samples were analyzed by liquid chromatography–mass spectrometry (LC/MS) using an LTQ Orbitrap Discovery electrospray ionization mass spectrometer (Thermo Scientific) equipped with quaternary HPLC pump (Surveyor MS Pump; Finnigan) and a C-18 reverse-phase microbore column as described previously [3]. Internal disaccharides and the NRE monosaccharide were quantified manually based on their unique mass signatures relative to internal standards and normalized by either tissue wet weight or protein concentration, determined by BCA assay (Pierce).
Histological examination
Mouse organs were inspected by light- and electron microscopy as described previously in detail [26]. For perfusion, 8 to 13-month-old mice were deeply anaesthetized with an intraperitoneal injection of 100 mg/kg bodyweight (bw) ketamine, 16 mg/kg bw xylazine and 2 mg/kg bw acepromazine and pre-perfused with 1% procaine in 0.1 M PBS. Fixation was then performed with transcardial vascular perfusion with 6% glutaraldehyde in 0.1 M PBS. For electron-microscopy, tissues were additionally fixed with 2% osmium tetroxide before embedding in araldite. Ultrathin sections were stained with uranyl acetate and lead citrate and finally analyzed with a Zeiss EM 900 electron microscope (Zeiss).
Skeletal analysis
The whole skeletons of 6-month-old mice were first analyzed by contact radiography (35 kV, 2 s) (Faxitron XRay Corp., U.S.A.). For micro-computed tomography (μCT) analysis, the right femur of each mouse was fixed and placed into a radio-translucent sample holder. Scanning and analysis were performed as previously described [27] with a voxel resolution of 10 μm using a μCT 40 desktop cone-beam μCT (Scanco Medical, Switzerland) according to standard guidelines [28]. Trabecular bone was analyzed in the distal metaphysis in a volume situated 2500–500 μm proximal of the distal growth plate. Cortical bone was analyzed in a 1000 μm long volume situated in the middle of the diaphysis. The cortical bone evaluation was performed with a threshold of 300, whereas for trabecular bone a threshold of 250 was used. For bone histology, skeletons were fixed in 3.7% PBS-buffered formaldehyde for 48 h before they were transferred into 80% ethanol. The lumbar vertebral bodies L1 to L4 and the left tibia of each mouse were dehydrated in ascending alcohol concentrations, before they were embedded into methylmethacrylate. Sections of 4 μm thickness were cut in the sagittal plane using a Microtec rotation microtome (Tachno-Med GmbH, Germany). Sections were stained by von Kossa/van Gieson and toluidine blue staining procedures as described previously [27]. Histomorphometry was carried out according to the guidelines of the American Society for Bone and Mineral Research [29] using an OsteoMeasure system (Osteometrics Inc., U.S.A.). Statistical analysis was performed using unpaired, two-tailed Student’s test, and P-values below 0.05 were considered statistically significant.
Behavioral analysis
WT and ARSK-deficient mice (n = 13 and 14, all females) were subjected to a behavioral test battery at the age of 12 months. Mice were housed at standard laboratory conditions (12 h light/dark cycle, constant room temperature and humidity). Behavioral experiments were performed during the light phase of the activity cycle. Food and water were available ad libitum. All procedures were reviewed and approved by the ethical research committee of KU Leuven. Behavioral analysis was described before in detail with minor modifications regarding motor function (forelimb grip strength, rotarod test and home cage activity) [30], exploratory testing (open field locomotion and elevated plus maze) [31].
Sociability and preference for social novelty (SPSN) was evaluated in a three-chamber Plexiglas set-up in which chambers are connected by closable doors. The two side chambers (26 × 26 cm) contain cylindrical wired cages (diameter 10 cm, height 11 cm) in which a stranger mouse could be placed. First, an acclimation trial was performed during which mice were allowed to explore the central chamber (42 × 26 cm) for 5 min. During the 10 min sociability trial, mice could enter all chambers of which one side chamber contained a caged stranger mouse and the other an empty cage. The side of the social chamber was randomized. Preference for social novelty was subsequently measured by placing a second stranger mouse into the previously empty cage, and recording preferential exploration again for 10 min. Total path length, the average distance to the cages and time spent in proximity of the cages were measured.
Spatial learning and memory were tested in the Morris water maze with hidden escape platform. We used a 150 cm circular pool filled with water (constant temperature: 26°C) which was opacified with nontoxic white paint. A 15 cm round platform was hidden 1 cm beneath the surface of the water at a fixed position. Mice learned to navigate to the escape platform using different visuo-spatial cues present in the room. Daily trial blocks of four swimming trials (intertrial interval 15 min) were performed, starting randomly from each of four starting positions. Mice that failed to find the platform within 2 min were guided to the platform. They had to remain on the platform for 10 s before being rescued and returned to their home cage (maximum three attempts for completing 10 s). Two acquisition sessions of five trial blocks were each followed by 2 days of rest and a probe trial. During these probe trials, the platform was removed from the pool, and the swimming paths of the animals were recorded for 100 s using EthoVision video tracking equipment and software (Noldus, Wageningen, The Netherlands). Probe trial analysis was performed using animals that showed less than 15% immobility time to allow reliable assessment of spatial preferences (probe 1 final WT n =11, KO n = 14; probe 2 final WT n = 8, KO n = 11).
Passive avoidance learning was examined in a step-through box with a small illuminated compartment and a larger dark compartment containing a grid-floor. After 30 min dark adaptation, the mouse was placed in the light compartment for a training trial. After 5 s, the sliding door to the dark compartment was opened and step-through latency was manually recorded up to a 300-s cut-off. When all four paws were placed on the grid, a mild electric shock (0.3 mA, 1 s) was delivered with a constant current shocker (MED Associates Inc., St. Albans, Vermont, U.S.A.). Retention memory was tested 24 h later using the same procedure without shock delivery.
Statistical analysis.
Behavioral data are presented as mean + standard error of the mean (SEM). Shapiro—Wilk and Brown—Forsythe tests were used to determine normality and variance homogeneity. Performance of WT and ARSK-deficient mice was compared using Mann—Whitney rank sum tests (grip strength, SPSN acclimation, target entries probe trial 1), independent sample t-tests (open field, elevated plus maze, other water maze probe trial measures), one-sample t-tests (water maze probe trial coincidence analysis) and 2-way (rotarod, cage activity, water maze acquisition) or 3-way (SPSN sociability and social novelty) repeated-measures ANOVA.
Results
Generation and validation of an Arsk knockout mouse model
We recently demonstrated that the lysosomal sulfatase ARSK shows glucuronate 2-sulfatase (GDS) activity towards synthetic disaccharide substrates. Physiologically, a GlcA2S-specific GDS enzyme would represent the last missing link in the complex HS degradation pathway. To confirm the in vivo function of ARSK in the catabolism of both HS and CS and to evaluate the general physiological relevance of glucuronate 2-desulfation, we took advantage of a commercially available mouse line (B6N(Cg)-Arsktm1b(KOMP)Wtsi/J; JAX 018657), which we acquired from the KOMP2 at The Jackson Laboratory (now available at KOMP/MMRC). In this particular mouse genome, exon 3 of the Arsk gene, as well as an inserted neomycin phosphotransferase (neo) cassette were primarily flanked by loxP sites (Figure 1A, middle panel; for further information see (https://www.mmrrc.org/catalog/sds.php?mmrrc_id=46694). Upon breeding male mice carrying this cassette with females expressing Cre recombinase both exon 3 and neo were removed leaving a ß-galactosidase (lacZ)-cassette behind, which can be used as reporter gene for expression studies (Figure 1A, lower panel). The Cre transgenic element was removed by congenic breeding into C57Bl/6 background. For genotyping, we established a routine multiplex PCR protocol, in which the wildtype allele was detected as a 515-bp-fragment using a primer pair binding within intron 2 and exon 3, respectively, whereas the knockout allele was detected as a 620-bp-fragment resulting from primers binding the lacZ-cassette and the loxP site, respectively (see Figure 1A,B). As the deletion of exon 3 does not necessarily lead to a complete Arsk gene knockout, we validated transcriptional Arsk inactivation by quantitative (q)PCR analyses revealing virtual no residual Arsk mRNA in visceral organs and the CNS (Figure 1C).
To demonstrate that specifically glucuronate 2-sulfatase activity is absent from Arsk-deficient mice, we developed a specific enzyme assay employing our synthetic disaccharide substrates glucuronate-2-O-sulfate-N-acetylglucosamine (G2A0) and glucuronate-2-O-sulfate-N-sulfoglucosamine (G2S0) (for disaccharide code also see [32]). Both disaccharides were labeled with the fluorescent dye 2-aminoacridone (AMAC) at the reducing end of glucosamine, hence leaving the unaltered NRE of glucuronate-2-O-sulfate exposed for degradation by lysosomal enzymes. AMAC-labeled disaccharides were incubated in the presence or absence of highly lysosome-enriched fractions (tritosomes) derived from liver of 12-month-old tyloxapol-treated wildtype or Arsk knockout male mice and subsequently analyzed by reversed-phase nano-liquid chromatography/nano-electrospray ionization mass spectrometry (RP-nano-LC/nano-ESI-MS) of AMAC-containing fractions (Figure 2). Incubation of G2A0 as well as G2S0 with tritosomes from the mutant did not alter hydrophobicity or m/z values of the disaccharides, as shown for G2A0 in Figure 2A,D. In contrast, incubation of G2A0 with wildtype tritosomes shifted the AMAC-containing peak to a less hydrophilic product. This new AMAC peak was analyzed by mass spectrometry and exhibited a m/z value of 416.18, which corresponds to an AMAC-labeled N-acetylated hexosamine, resulting from the two-step degradation of G2A0 by GDS and, subsequently, ß-D-glucuronidase present in wildtype tritosomes (Figure 2B,D). To demonstrate that this specific GDS activity is mediated by Arsk, we again incubated the AMAC-labeled G2A0 disaccharide with knockout tritosomes, this time supplemented with recombinant human ARSK (rhARSK). Interestingly, three AMAC-labeled peaks were detected, which were analyzed by MS (Figure 2C,D). The first minor peak with a m/z value of 672.17 represented the undigested AMAC-labelled disaccharide, while the second minor peak with m/z 592.21 resulted from rhARSK-mediated desulfation of the disaccharide as the hydrolytic cleavage of a sulfate group accounts for a decrease in mass by 79.96 Da. The major peak (m/z 416.18) matches the product obtained after incubation with wildtype lysosomes, hence confirming the two-step conversion of the G2A0 disaccharide to the N-acetylated hexosamine monosaccharide. These data clearly demonstrate that liver from Arsk knockout mice is totally devoid of GDS activity and that rhARSK can reconstitute this activity.
Figure 2. Lysosomes from Arsk-deficient mice do not convert G2A0.
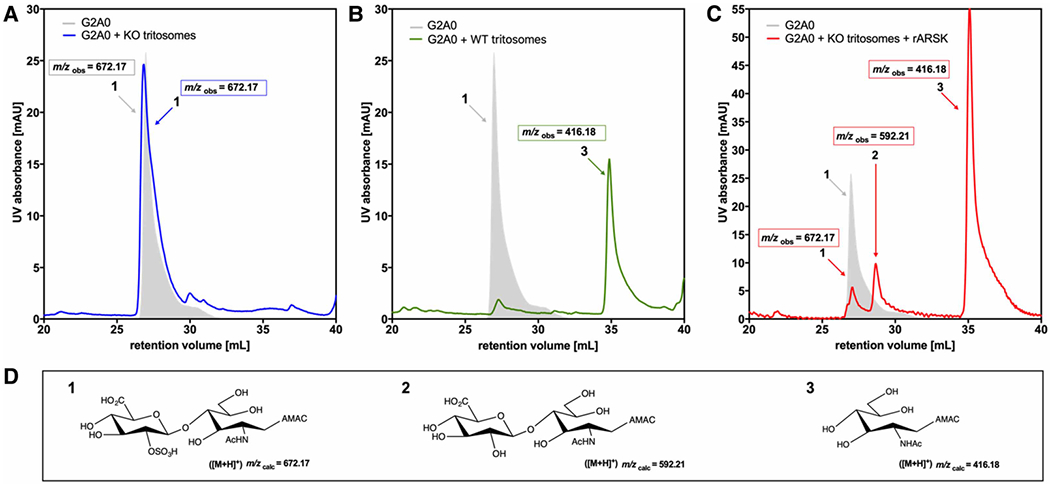
G2A0 (12.5 nmol) was pre-labeled with the fluorescent dye 2-aminoacridone (AMAC). Fifty micrograms of a lysosome-enriched fraction (tritosomes) were incubated with G2A0-substrate at 37°C for 24 h and subsequently analyzed by C18-reversed-phase-HPLC combined to nano-ESI-MS. For disaccharide code also see [32]. (A) G2A0 was treated with Arsk-deficient tritosomes (blue) resulting in unreacted (gray shaded) substrate 1 (m/z 672.17) or was treated with wildtype (WT) tritosomes. (B) resulting in the novel peak 3 (m/z 416.18). (C) Simultaneous treatment of G2A0 (672.17) with Arsk-deficient tritosomes and human recombinant (r)ARSK (20 ng) resulted in peak 2 (m/z 592.21) indicating the loss of a sulfate group and peak 3 (m/z 416.18) representing the AMAC-labelled hexosamine after desulfation and disaccharide cleavage. (D) Molecular structures and calculated m/z-ratios of the G2A0 disaccharide (1), its desulfated product (2) and the resulting AMAC-labelled monosaccharide product (3), which is due to tritosome-mediated glycosidase activity.
GDS-deficient mice show storage of GAGs
Urinal GAG excretion and lysosomal GAG storage are common hallmarks associated with deficiencies of lysosomal GAG degradation. In a first approach, we determined the total sulfated GAG content from urine of knockout and wildtype mice by the dimethylmethylene blue (DMMB) assay and observed slightly, but not significantly, elevated amounts in urine (Supplementary Figure S1).
As glucuronate-2-sulfate is a particular modification found in both HS and CS, we quantified more precisely total amounts of these GAGs in various tissues isolated from three 12-month-old male wildtype and three matched knockout mice by Glycan Reductive Isotope Labeling-liquid chromatography/mass spectrometry (GRIL-LC/MS). The data revealed significantly elevated amounts of total HS in brain, kidney and spleen of Arsk knockout mice (Figure 3A). The highest total amount of 400 μg HS/g wet weight was found in knockout kidney, which corresponds to a five-fold increase in total HS compared with WT mice. We also compared HS amounts derived from isolated liver lysosomes after tyloxapol treatment from wildtype and Arsk-deficient mice and observed a 2.5-fold increase in HS in knockout lysosomes (Figure 3B). Of note, while CS was also elevated in kidney (5x), lung (3x) and brain (2x) of knockout mice, showing clear statistical significance at least for kidney (Figure 3C, P = 0.04), liver homogenates and purified liver lysosomes from both genotypes exhibited comparable CS levels (Figure 3C,D). More detailed analysis of the sulfate composition of HS disaccharides (Figure 3E) isolated from purified liver lysosomes showed increased levels of 2-O-sulfated HS in knockout samples even though the 2-O-sulfafation of HS is clearly a sort of rare modification. In contrast, CS did not exhibit elevated 2-O-sulfation values (Figure 3F), which is in line with the unaffected total CS amounts in Arsk-deficient mice mentioned above.
Figure 3. Storage of HS and CS disaccharides in Arsk-deficient mice.

Glycosaminoglycans were extracted from brain, kidney, liver, lung and spleen or from isolated liver lysosomes (tritosomes) of 12-month-old WT or ARSK-deficient mice. The amount of heparan sulfate (HS) was determined in the indicated tissues (A) and tritosomes (B) by LC/MS after glycan reductive isotope labeling (GRIL). Similarly, the amount of chondroitin sulfate (CS) was determined in the various tissues (C) and liver lysosomes (D) (n = 3 per genotype, values represent mean ± SEM, *P < 0.05, **P < 0.01). (E,F) The composition of HS- and CS-derived disaccharides were evaluated regarding acetylation (N-Ac) and sulfation by GRIL-LC-MS indicating a minor increase in 2-O-sulfated HS disaccharides.
In all reported cases, the defect of a specific enzyme in the defined sequential degradation pathway, proceeding at the NRE of the GAG chains, leads to the accumulation of unique NRE saccharides that can be considered as biomarkers for the disease [3,4]. In case of Arsk deficiency, one would expect to find elevated amounts of 2-O-sulfated glucuronate-containing NREs (G2S0 and G2A0) deriving from HS and CS, respectively. For NRE-analysis, HS and CS from the brain and kidney of wildtype and knockout mice were isolated, depolymerized by bacterial lyases and the resulting disaccharides were chemically tagged with [12C6]aniline prior to LC/MS analysis. Quantification was enabled by adding [13C6]-labeled 2-O-sulfoglucuronate-containing disaccharides G2A0 and G2S0 as internal standard (Figure 4). In brain, G2S0 was 22-fold elevated in knockout mice (Figure 4A, left panel), whereas G2A0, which is hardly detectable in this tissue, showed an eightfold increase in knockout mice (Figure 4A, right panel). In kidney from knockout mice, disaccharides comprising 2-O-sulfated glucuronates at their NREs were highly elevated by a factor of eight (G2S0) and twelve (G2A0), respectively, in comparison with wildtype (Figure 4A). With regard to total amounts, the disulfated G2S0 (2078 pmol/g wet weight) exceeded the monosulfated G2A0 (1461 pmol/g). Isolated liver lysosomes from Arsk-deficient mice exhibited a 10-fold increase in G2S0-containing disaccharides (Figure 4B). G2A0 was not detected in liver lysosomes (data not shown). Interestingly, analysis of CS-derived NREs from brain and kidney of knockout mice (Figure 4D) did not exhibited elevated G2a0 amounts and G2a4 was not detectable at all (data not shown). Of note, we cannot exclude that other, even higher sulfated disaccharides (e.g. with additional glucosamine 6-O-sulfation) escaped our analysis, although additional unidentified peaks were not observed.
Figure 4. Arsk-deficient mice accumulate 2-O-sulfated non-reducing end (NRE) biomarkers.
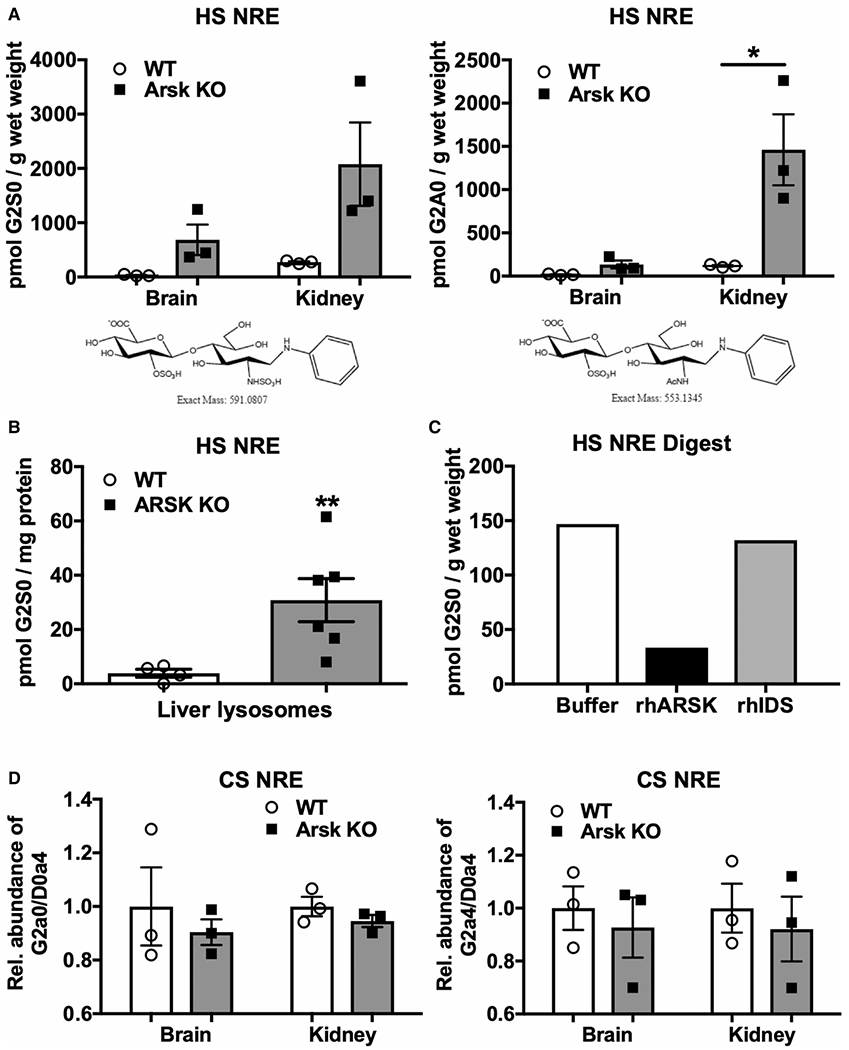
HS NREs derived from brain and kidney (A) and isolated liver lysosomes (B) of Arsk KO or WT mice were obtained by GAG-depolymerization using bacterial lyases. Quantification of HS NREs from tissues or liver lysosomes was achieved by GRIL-LC/MS with known amounts of [13C6]aniline-labeled internal standards including the specific G2S0 (A left panel; B) and G2A0 (A right panel) disaccharides. (n = 3 for each genotype, values represent mean ± SEM, *P < 0.05, **P < 0.01). (C) Kidney-derived 2-O-sulfat HS NREs were incubated with recombinant human (rh)ARSK (15 ng), rhIDS (8 ng) or with buffer overnight and quantified as described above. (D) Quantification of CS-derived NREs from brain and kidney of WT and Arsk KO mice.
To further confirm that the storage material in Arsk-deficient mice reflects the physiological substrate of Arsk, we isolated GAGs from kidney of knockout mice and treated them in parallel either with buffer, recombinant human ARSK (rhARSK) or recombinant human iduronate-2-sulfatase (rhIDS), respectively (Figure 4C). For analysis, we depolymerized the GAGs with appropriate bacterial lyases and quantified the amount of resulting NREs by GRIL-LC/MS. After incubation with rhARSK, G2S0-NRE was reduced by 80% in comparison with the buffer control, while after incubation with rhIDS only a very low reduction, if any, of G2S0 was observed.
In summary, these results clearly show that Arsk-deficient mice accumulate significant amounts of HS, which exhibits glucuronate-2-O-sulfated NREs, particularly in brain and kidney. Moreover, recombinant ARSK/GDS efficiently degrades the glucuronate-2-O-sulfated NREs of the accumulated HS. In conclusion, lysosomal storage of 2-O-sulfated HS is the hallmark of a new type of mucopolysaccharidosis caused by ARSK/GDS-deficiency.
Histological analysis of Arsk-deficient mice
Mucopolysaccharidoses are often characterized by a storage pathology, in which the extent and the type of vacuolation may vary in a tissue- or even cell type-specific manner. In fact, a given MPS may also exhibit different types of storage material in terms of electron-lucent to electron-dense material with either floccular, fibrillogranular or zebra-body like appearance in different tissues reflecting differences in secondary storage subsequent to the primary storage process [33,34]. As the biochemical analysis of knockout mice exhibited considerable amounts of accumulated GAGs in kidney, brain and, to a lesser extent, also in other organs, we examined various tissues including kidney, liver, lung, trachea, retina and brain for storage pathology by light and electron microscopy of 8- to 12-month-old mice. We did not detect storage vacuolation in any inspected tissue that correlated with accumulated GAGs as expected from our HS and CS quantitation. The kidney of 8- and 12-month-old Arsk-deficient mice exhibited the moderate but significant appearance of supra-nuclear dense bodies in the thick ascending limbs at the inner stripe of the outer medulla upon toluidine blue-staining (Figure 5B,D) as well as in some intermediate tubules of the inner medulla (Figure 5F), whereas these structures were absent from wildtype specimens (Figure 5A,C and E). In electron microscopy, these dense bodies found their equivalent in electron-dense structures indicating lipofuscin-like material. These structures were exclusively observed in Arsk-deficient mice (Figure 5G,H) and not in wildtype mice.
Figure 5. Histopathology of kidney in 12-month-old Arsk-deficient mice.
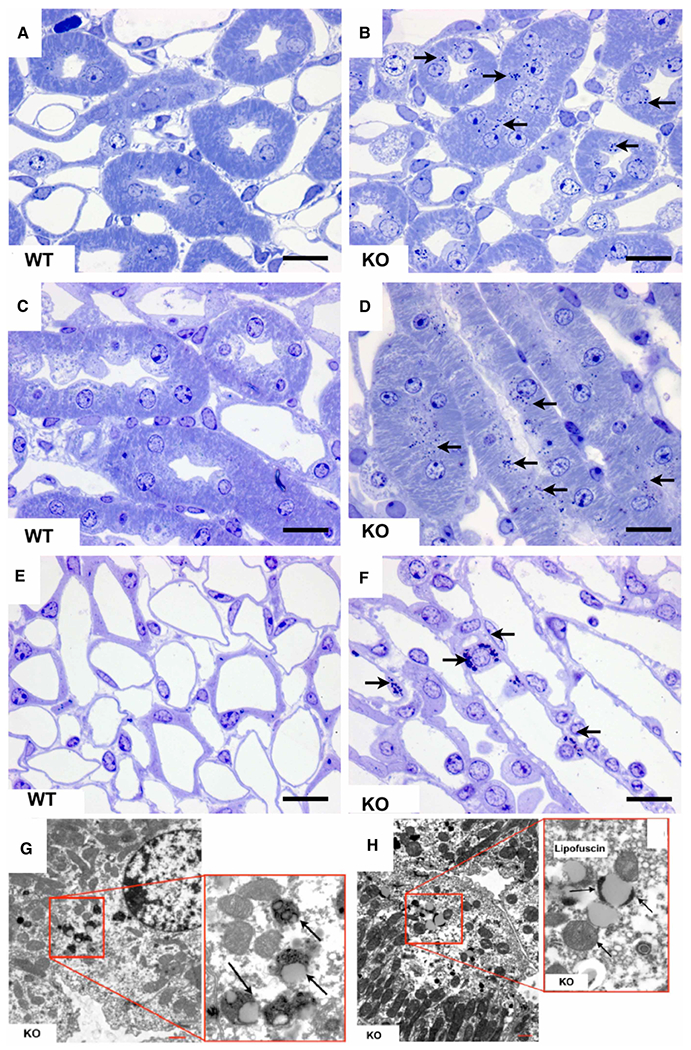
Toluidine blue-stainings of semi-thin sections of Arsk-deficient mice kidneys exhibit dense bodies (arrows) in the thick ascending limbs of the inner stripe of the outer medulla (B,D) as well as in intermediate tubules of the inner medulla (F); the respective WT controls are shown on the left (panels A, C and E). In electron microscopy (G,H), the equivalent of these dense bodies (arrows) appeared as lipofuscin-related material exclusively in the kidney of Arsk-deficient mice. Scale bars: 20 μm (light microscopy); 5 μm (electron microscopy).
In many MPS mouse models with neuronal participation, the accumulation of lysosomal storage material is often accompanied by an expansion of the endosomal/lysosomal compartment mediated by inflammation processes and by secondary storage of lipids in the CNS of affected mice. However, the lysosomal marker Lamp1, the macrophage marker F4/80 and the microglia marker CD68 did not accumulate in 3- to 18-month-old Arsk-deficient compared with wildtype mice. Also, other common markers indicative of pathophysiological alterations in the CNS like glial fibrillary acidic protein (GFAP, astrocyte marker), myelin basic protein (MBP, oligodendrocyte marker), calbindin (Purkinje cell marker) and neuronal nuclear protein (NeuN) did not show any apparent differences between both genotypes. Retina degeneration is considered as a sensitive pathological hallmark in several types of MPS III including the explicit late-onset arylsulfatase G-deficiency MPS IIIE. However, immunofluorescence analyses of the retina regarding photoreceptor and microglia cell numbers as well as retina thickness were inconspicuous in 12- and 24-month-old male Arsk-deficient mice (data not shown).
Bone formation and bone remodeling
A defining hallmark of many types of MPS is skeletal dysplasia due to bone growth impairments, which results from growth plate destruction, failure in ossification and imbalance of bone growth. Clinical features related to skeletal dysplasia may include dwarfism, rigidity or laxity of joints, tracheal obstructions, kyphoscoliosis and many other complications. To analyze a possible influence of Arsk deficiency on the skeleton, we first applied μCT to compare femoral bones from 24-week-old wildtype and Arsk-deficient mice. We did not detect a significant difference between the two groups in terms of femur length or midshaft diameter (Figure 6A). Similarly, trabecular bone mass was comparable in wildtype and Arsk-deficient mice, and there was no statistically significant difference in the bone volume per tissue volume (BV/TV). We did observe, however, a reduced bone mineral density in Arsk-deficient animals, yet the difference from wildtype controls was rather moderate (Figure 6B). With respect to cortical bone, we observed similar cortical thickness in wildtype and Arsk-deficient mice (Figure 6C), thereby supporting the overall conclusion that Arsk deficiency does not impact bone mass in either the trabecular or the cortical compartment. Consistently, there was no significant difference between the two groups, when we subjected the femora to three-point-bending assays (Figure 6D).
Figure 6. Arsk-deficient mice exhibit no obvious skeletal phenotype.
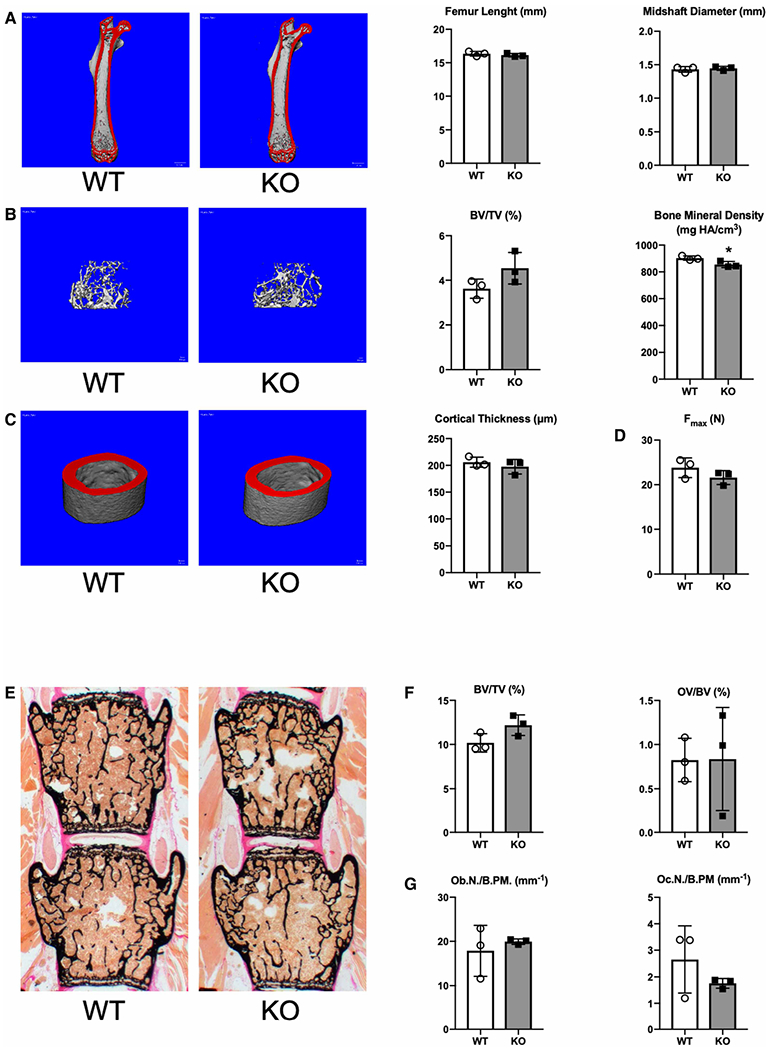
(A) Representative μCT images of femora from 24-week-old wildtype and Arsk-deficient mice. Mineralized bone appears in red. Quantification of the femoral length and midshaft diameter is given on the right. (B) Representative μCT images of trabecular bone from the same mice. Quantification of the bone volume per tissue volume (BV/TV) and the bone mineral density is given on the right. (C) Representative μCT images of cortical bone from the same mice. Quantification of the cortical thickness is given on the right. (D) Quantification of the maximal force (Fmax) to failure in three-point-bending assays of the femoral bones. (E) Representative Kossa-stained sections of vertebral bodies (L3 and L4) from 24-week-old wildtype and Arsk KO mice. Mineralized bone appears in black. (F) Quantification of bone volume per tissue volume (BV/TV) and osteoid volume per bone volume (OV/BV). (G) Quantification of osteoblast (Ob.N./B.Pm.) and osteoclast number (Oc.N./B.Pm.) per bone perimeter. All data represent mean ± SD (n = 3). The asterisk indicates a statistically significant difference (*P < 0.05) between the two groups.
To obtain cellular data regarding skeletal remodeling we additionally analyzed undecalcified spine sections from the 24-week-old wildtype and Arsk-deficient mice (Figure 6E). Similar to the μCT results, the trabecular bone volume was not significantly different between the two groups, and there was also no change in the osteoid volume per bone volume (OV/BV) indicating intact bone matrix mineralization (Figure 6F). Likewise, neither the osteoblast number nor the osteoclast number was affected by Arsk deficiency (Figure 6G). Taken together, these data demonstrate that Arsk does not play a major physiological role in bone remodeling in mouse.
Behavioral analysis
Behavioral assessment of 12-month-old female mice revealed no differences in grip strength, rotarod performance and home cage activity profiles, indicating that basic motor functions were intact in Arsk-deficient mice (data not shown). Both genotypes also showed comparable open field and elevated plus-maze exploration. This was corroborated by similar activity levels in the acclimation phase of the sociability/preference for social novelty (SPSN) test, which also showed no changes in subsequent phases. However, although there were no overall differences in preferential exploration during the ‘sociability’ phase (Figure 7A,B; proximity: P = 0.31; approaches: P = 0.15), we observed differences between wildtype and knockout mice dependent on the time interval (significant Time × Chamber × Genotype interactions; proximity: P < 0.05; approaches: P < 0.05). During the first time interval (0–2 min), knockout mice were on average closer to the empty cage than wildtype mice (Figure 7A, right panel) and also made more approaches towards it (Figure 7B, right panel), while being further away from the social cage (all P < 0.05). During the second 2-min interval (2–4 min), knockout mice showed increased approaches to the social cage (Figure 7B, right panel; P < 0.05). During the last interval (8–10 min), Arsk-deficient mice again made more approaches towards the empty cage compared with wildtype mice (Figure 7B, right panel; P < 0.05). These differences indicate a delayed and altered sociability process in knockout mice.
Figure 7. Behavioral alterations in 12-month-old Arsk-deficient mice.
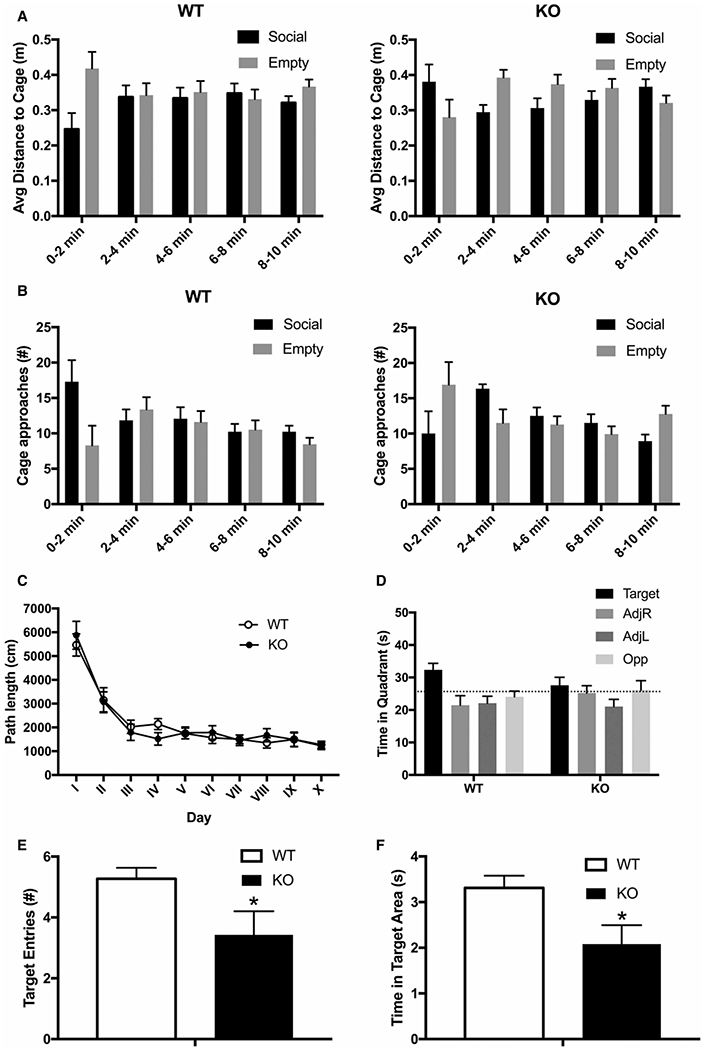
Social behavior of 12-month-old Arsk knockout (KO) mice and wildtype mice (WT) was assessed in the Sociability and Preference for Social Novelty test (SPSN). Knockout mice (right panel) showed time-dependent changes regarding overall proximity (A) as well as approach behavior (B) towards a caged conspecific in comparison with an empty cage, suggesting changes in sociability. Spatial learning and memory was tested in a Morris water maze set-up (C–F). Total path length during acquisition (C) and direct comparison of time spent in the target quadrant during the first probe trial (D) showed no significant changes. However, in contrast with WT mice, KO mice did not show target quadrant exploration above the coincidence level. This was corroborated by decreased numbers of target area entries (E) and average time spent in the target area (F), suggesting impaired spatial memory. Changes did not persist into the second probe trial. All data are represented as mean ± SEM (wildtype n = 13 females, knockout n = 14 females).
Spatial learning was tested during water maze acquisition, where mice learned to locate the hidden platform as indicated by the decreases in path length (P < 0.001) to reach the platform. There were no significant differences in overall or time-dependent performance between wildtype and knockout mice (Figure 7C; P = 0.99 and P = 0.79). During the first probe trial, knockout mice appeared to show a reduced preference for the target quadrant, but direct comparison was not significant (Figure 7D). However, wildtype mice spent significantly more time in the target quadrant than the 25% coincidence level (P < 0.01), while Arsk-deficient mice did not (P = 0.31). Furthermore, knockout mice made less target area entries (Figure 7E; P < 0.05) and spent less time in the target area (Figure 7F; P < 0.05) in comparison with wildtype mice. These results suggest impaired spatial reference memory in Arsk-deficient mice. During a second probe trial, both genotypes showed a clear preference for the target quadrant in comparison with other quadrants without significant differences in time spent in the target quadrant, target area entries and time spent in the target area.
In the passive avoidance test, both genotypes showed similar step-through latencies and both exhibited significant fear memory 24 h later. Arsk-deficient mice appeared slower to enter the dark compartment, but this effect did not reach significance (data not shown). To conclude, despite the absence of major patho-histological findings in the CNS, Arsk-deficient mice apparently showed behavioral changes albeit to a much lesser extent than all other mucopolysaccharidoses mouse models.
Discussion
Thirty-five years ago, Shaklee et al. [13] proved the existence of lysosome-associated enzymatic activity against glucuronate-2-O-sulfate residues derived from GAGs. In 2017, we demonstrated that this 2-sulfoglucuronate sulfatase activity is mediated by lysosomal arylsulfatase K (ARSK), which was renamed as GDS in conformity with iduronate-2-sulfatase (IDS) acting at the epimeric iduronate-2-sulfate [19]. In the present study, we questioned the physiological relevance of GDS by the characterization of a mouse model, in which the Arsk gene was constitutively inactivated as demonstrated at the genomic and transcriptional level. We confirmed GDS-deficiency in this mouse model using a novel GDS-specific enzyme assay based on HPLC/MS analysis of 2-aminoacridone (AMAC)-labeled disaccharides. Moreover, total GAG quantification and detailed analyses of GAG-derived NREs by GRIL-LC-MS confirmed that HS with 2-O-sulfoglucuronate NREs accounts for storage in selected tissues, primarily in brain and kidney. The total amount of accumulated GAGs in GDS-deficient mice was rather low, which is also reflected by the relative absence of storage pathology in light and electron microscopy analyses of various tissues. In kidney, some abnormal structures of unknown composition presented with marginal electron-dense bodies but all tissues analyzed so far were devoid of massive lysosomal burden typically seen in most HS-associated MPS types. Although neuro-pathology or bone formation changes were absent from the knockout mice, we observed signs of impaired social behavior and spatial memory in Arsk-deficient mice.
The group of mucopolysaccharidoses comprises seven types of LSDs which all have in common an inherited deficiency of a specific lysosomal enzyme involved in the degradation of GAGs. These enzyme dysfunctions lead to lysosomal storage of a defined set of GAGs or rather their specific accumulating metabolites and, as a consequence of the entire pathological cascade, often to secondary storage of unrelated material like lipids and polypeptides [9,33,35]. The lysosomal burden depends on the deficient enzyme as well as on the occurrence and accruing amount of its GAG substrate and hence may result in cellular dysfunction or even extensive cell death in affected organs and tissues [35–37]. Lysosomal accumulation and associated urinary excretion of HSHS is the hallmark of various types of MPS (I, II, III (subtypes A–E) and VII) many of which often present with severe phenotypes in human as well as in cohorts of naturally occurring or engineered animal models [38–40].
From the biochemical point of view, iduronate-2-sulfatase (IDS) deficiency (Hunter syndrome, MPS II) appears to be the most related disease to GDS-deficiency as both enzymes, GDS and IDS, react on the sulfate group in the C2 position of the uronic acids, either glucuronate (GlcA) or its epimer iduronate (IdoA) [41]. In general, uronic acids and their 2-O-sulfated forms can be found in HS/heparin (IdoA and GlcA), chondroitin sulfate (GlcA) and dermatan sulfate (IdoA) [41]. Hence, IDS-deficiency in Hunter syndrome leads to an accumulation of HS and DS in many organs causing a broad but variable set of symptoms including skeletal abnormalities, hepato-splenomegaly and progressive mental retardation [42]. Hunter syndrome is classified in a mild/attenuated form with short stature, joint stiffness and a severe form, which in addition to the aforementioned symptoms is characterized by an early onset of neurological degeneration and behavior disturbances [42]. However, so far no MPS-like patient with GAG accumulation was diagnosed as GDS-deficient, which might be attributed to the fact that the amount of 2-O-sulfated GlcA is low in HS [11,43]. For most naturally occurring HS species it is estimated that as little as 1% of the GAG-forming disaccharide units contain GlcA2S moieties [44,45]. This relative low abundance of GlcA2S is basically ascribed to an enzymatic complex consisting of the C5-epimerase, which converts GlcA into IdoA, and chain specific 2-O-sulfotransferases (Hs2st or Ust) responsible for the 2-O-sulfation of HS or CS/DS. These enzymes preferentially act on IdoA rather than GlcA moieties [46–48]. Thus 2-O-sulfated GlcA is a more rare modification, which is also reflected by the very modest phenotype in the GDS mouse model. The observed behavioral abnormalities in the GDS/Arsk-deficient mouse model might be ascribed to a higher GlcA2S degree of HS or CS/DS in the cerebrum as previously shown for human adult cerebral cortex, which exhibited significantly more GlcA2S (up to 10% of HS-derived disaccharides) compared with other adult tissues (0–2.5%), whereas the neonatal cortex was totally devoid of GlcA2S-containing disaccharides [45].
CS, or more precisely the CS D subform, is a physiological substrate of GDS. In our GAG analyses of the GDS mouse model, total CS was significantly elevated in kidney, whereas the amount of GlcA2S-containing NREs from CS/DS in knockout mice did not differ from controls suggesting a more general inhibition of CS catabolism in the knockout by accumulating GAG metabolites. 2-O-sulfation in CS (as well as DS) depends on the activity of uronyl 2-O-sulfotransferase (UST) [49,50]. The epimerization of GlcA into IdoA by the activity of the C5-epimerases DS-epi1 and DS-epi2 leads to the conversion of CS into DS, which gives rise to tissue-specific heterogeneity in the CS/DS composition [51]. On the other hand, CS catabolism, particularly the first steps of its breakdown, is less well understood than HS degradation. It has been shown that some hyaluronidases exhibit endolytic activity against CS chains [52].
In 1985, Shaklee et al. wondered whether ‘due to the low amount of GlcA2S in HS and CS, a defect in the GDS would not manifest itself in any clinical symptoms’ [13]. Here, we tested this possibility in a reverse genetic approach and can now draw the conclusion from our findings in the mouse model that GDS-deficiency in human most likely causes a late-onset form of MPS (referred to as type IIB), which so far either remained unnoticed or at least could not be classified. Interestingly, patients suffering from a rare form of late-onset MPS were identified (Type IIIE, in OMIM also listed as Usher syndrome type IV, USH4), whose causative genetic defect lies in the arylsulfatase G (ARSG) gene [10]. The ARSG gene product N-sulfoglucosamine 3-O-sulfatase is involved in the sequential degradation of HS and, in this context, is responsible for the removal of the rare 3-O-sulfate group at the glucosamine residue (GlcNS3S) [6]. As the GlcA2S modification is even more rare than the ARSG substrate GlcNS3S, one might expect an even milder phenotype in patients with GDS-deficiency as compared with patients with ARSG-dependent MPS IIIE/USH4. We believe that our GDS mouse model will help to identify patients with mild and late-onset MPS-like symptoms.
Supplementary Material
Acknowledgements
The authors thank Christiane Grebe, Marion Knufinke, Dagmar Niemeier, Mai-Britt Ilse and Björn Kowalewski for excellent technical assistance. The expert retinal analysis of our mice, performed in the group of Udo Bartsch (Department of Ophthalmology, University Medical Center Hamburg-Eppendorf), is gratefully acknowledged.
Funding
This work was supported by the German Research Foundation (DFG) with grant DI 575/10-1 to T.D., grant LU 1173/4-1 to T.L. and grant INST 215/484-1 FUGG to T.S., and also by a grant from the CURE Sanfilippo Foundation (to J.D.E.). Bastian Ramms was supported by a PhD fellowship from the German Academic Exchange Service (DAAD, 91558745). G.J.B. was supported by the National Institute of General Medical Sciences (P41GM103390) from the National Institutes of Health.
Abbreviations
- Arsk
official gene symbol for murine arylsulfatase K
- CS/DS
chondroitin sulfate/dermatan sulfate
- GAG
glycosaminoglycan
- GlcA
glucuronate
- GRIL-LC/MS
glycan reductive isotope labeling-liquid chromatography/mass spectrometry
- HPLC/MS
high-performance liquid chromatography–mass spectrometry
- HS
heparan sulfate
- IdoA
iduronate
- MPS
mucopolysaccharidosis
- nano-ESI
nano electrospray ionization
- NRE
non-reducing end
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- RP
reversed phase
- UPLC
ultra-performance liquid chromatography
- WT
wildtype.
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.von Figura K, Gieselmann V and Jaeken J (2001) Metachromatic leukodystrophy. In The Metabolic & Molecular Bases of Inherited Diseases (Scriver CR et al. ed), pp. 3694–3724, McGraw-Hill, New York [Google Scholar]
- 2.Neufeld EF, Lim TW and Shapiro LJ (1975) Inherited disorders of lysosomal metabolism. Annu. Rev. Biochem 44, 357–376 10.1146/annurev.bi.44.070175.002041 [DOI] [PubMed] [Google Scholar]
- 3.Lawrence R, Brown JR, Al-Mafraji K, Lamanna WC, Beitel JR, Boons GJ et al. (2012) Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol 8, 197–204 10.1038/nchembio.766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence R, Brown JR, Lorey F, Dickson PI, Crawford BE and Esko JD (2014) Glycan-based biomarkers for mucopolysaccharidoses. Mol. Genet. Metab 111, 73–83 10.1016/j.ymgme.2013.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neufeld EF and Muenzer J (2001) The mucopolysaccharidoses. In The Metabolic & Molecular Bases of Inherited Diseases (Scriver CR et al. eds), pp. 3421–3452, McGraw-Hill, New York. Chapter 136 [Google Scholar]
- 6.Kowalewski B, Lamanna WC, Lawrence R, Damme M, Stroobants S, Padva M et al. (2012) Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice. Proc. Natl. Acad. Sci. U.S.A 109, 10310–10315 10.1073/pnas.1202071109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cimaz R and La Torre F (2014) Mucopolysaccharidoses. Curr. Rheumatol. Rep 16, 389 10.1007/s11926-013-0389-0 [DOI] [PubMed] [Google Scholar]
- 8.Beck M (2017) Treatment strategies for lysosomal storage disorders. Dev. Med. Child Neurol 60, 13–18 10.1111/dmcn.13600 [DOI] [PubMed] [Google Scholar]
- 9.Jakobkiewicz-Banecka J, Gabig-Ciminska M, Kloska A, Malinowska M, Piotrowska E, Banecka-Majkutewicz Z et al. (2016) Glycosaminoglycans and mucopolysaccharidosis type III. Front. Biosci. (Landmark Ed) 21, 1393–1409 10.2741/4463 [DOI] [PubMed] [Google Scholar]
- 10.Khateb S, Kowalewski B, Bedoni N, Damme M, Pollack N, Saada A et al. (2018) A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet. Med 20, 1004–1012 10.1038/gim.2017.227 [DOI] [PubMed] [Google Scholar]
- 11.Bienkowski MJ and Conrad HE (1985) Structural characterization of the oligosaccharides formed by depolymerization of heparin with nitrous acid. J. Biol. Chem 260, 356–365 [PubMed] [Google Scholar]
- 12.Liu C, Sheng J, Krahn JM, Perera L, Xu Y, Hsieh PH et al. (2014) Molecular mechanism of substrate specificity for heparan sulfate 2-O-sulfotransferase. J. Biol. Chem 289, 13407–13418 10.1074/jbc.M113.530535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaklee PN, Glaser JH and Conrad HE (1985) A sulfatase specific for glucuronic acid 2-sulfate residues in glycosaminoglycans. J. Biol. Chem 260, 9146–9149 [PubMed] [Google Scholar]
- 14.Sardiello M, Annunziata I, Roma G and Ballabio A (2005) Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum. Mol. Genet 14, 3203–3217 10.1093/hmg/ddi351 [DOI] [PubMed] [Google Scholar]
- 15.Obaya AJ (2006) Molecular cloning and initial characterization of three novel human sulfatases. Gene 372, 110–117 10.1016/j.gene.2005.12.023 [DOI] [PubMed] [Google Scholar]
- 16.Dierks T, Lecca MR, Schlotterhose P, Schmidt B and von Figura K (1999) Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J 18, 2084–2091 10.1093/emboj/18.8.2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiegmann EM, Westendorf E, Kalus I, Pringle TH, Lubke T and Dierks T (2013) Arylsulfatase K, a novel lysosomal sulfatase. J. Biol. Chem 288, 30019–30028 10.1074/jbc.M113.499541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lubke T, Lobel P and Sleat DE (2009) Proteomics of the lysosome. Biochim. Biophys. Acta 1793, 625–635 10.1016/j.bbamcr.2008.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhamale OP, Lawrence R, Wiegmann EM, Shah BA, Al-Mafraji K, Lamanna WC et al. (2017) Arylsulfatase K is the lysosomal 2-Sulfoglucuronate sulfatase. ACS Chem. Biol 12, 367–373 10.1021/acschembio.6b01033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wattiaux R, Wibo M and Baudhuin P (1963) [Effect of the injection of Triton WR 1339 on the hepatic lysosomes of the rat. Arch. Int. Physiol. Biochim 71, 140–142 [PubMed] [Google Scholar]
- 21.Kitagawa H, Kinoshita A and Sugahara K (1995) Microanalysis of glycosaminoglycan-derived disaccharides labeled with the fluorophore 2-aminoacridone by capillary electrophoresis and high-performance liquid chromatography. Anal. Biochem 232, 114–121 10.1006/abio.1995.9952 [DOI] [PubMed] [Google Scholar]
- 22.Jackson P (1994) High-resolution polyacrylamide gel electrophoresis of fluorophore-labeled reducing saccharides. Methods Enzymol. 230, 250–265 10.1016/0076-6879(94)30017-8 [DOI] [PubMed] [Google Scholar]
- 23.Ambrosius M, Kleesiek K and Gotting C (2008) Quantitative determination of the glycosaminoglycan delta-disaccharide composition of serum, platelets and granulocytes by reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1201, 54–60 10.1016/j.chroma.2008.06.007 [DOI] [PubMed] [Google Scholar]
- 24.Esko JD (2001) Special considerations for proteoglycans and glycosaminoglycans and their purification. Curr. Protoc. Mol. Biol Chapter 17, Unit17 12 10.1002/0471142727.mb1702s22 [DOI] [PubMed] [Google Scholar]
- 25.Lawrence R, Olson SK, Steele RE, Wang L, Warrior R, Cummings RD et al. (2008) Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem 283, 33674–33684 10.1074/jbc.M804288200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stinchi S, Lullmann-Rauch R, Hartmann D, Coenen R, Beccari T, Orlacchio A et al. (1999) Targeted disruption of the lysosomal alpha-mannosidase gene results in mice resembling a mild form of human alpha-mannosidosis. Hum. Mol. Genet 8, 1365–1372 10.1093/hmg/8.8.1365 [DOI] [PubMed] [Google Scholar]
- 27.Yorgan TA, Peters S, Jeschke A, Benisch P, Jakob F, Amling M et al. (2015) The anti-osteoanabolic function of sclerostin is blunted in mice carrying a high bone mass mutation of Lrp5. J. Bone Miner Res. 30,1175–1183 10.1002/jbmr.2461 [DOI] [PubMed] [Google Scholar]
- 28.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ and Muller R (2010) Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res 25, 1468–1486 10.1002/jbmr.141 [DOI] [PubMed] [Google Scholar]
- 29.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H et al. (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR histomorphometry nomenclature committee. J. Bone Miner. Res 28, 2–17 10.1002/jbmr.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf H, Damme M, Stroobants S, D’Hooge R, Beck HC, Hermans-Borgmeyer I et al. (2016) A mouse model for fucosidosis recapitulates storage pathology and neurological features of the milder form of the human disease. Dis. Model. Mech 9, 1015–1028 10.1242/dmm.025122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stroobants S, Leroy T, Eckhardt M, Aerts JM, Berckmans D and D’Hooge R (2008) Early signs of neurolipidosis-related behavioural alterations in a murine model of metachromatic leukodystrophy. Behav. Brain Res. 189, 306–316 10.1016/j.bbr.2008.01.008 [DOI] [PubMed] [Google Scholar]
- 32.Lawrence R, Lu H, Rosenberg RD, Esko JD and Zhang L (2008) Disaccharide structure code for the easy representation of constituent oligosaccharides from glycosaminoglycans. Nat. Methods 5, 291–292 10.1038/nmeth0408-291 [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson FL, Holley RJ, Langford-Smith KJ, Badrinath S, Liao A, Langford-Smith A et al. (2012) Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One 7, e35787 10.1371/journal.pone.0035787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scriver CR (1995) The Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, Health Professions Division, New York [Google Scholar]
- 35.Vitner EB, Platt FM and Futerman AH (2010) Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem 285, 20423–20427 10.1074/jbc.R110.134452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Futerman AH and van Meer G (2004) The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol 5, 554–565 10.1038/nrm1423 [DOI] [PubMed] [Google Scholar]
- 37.Cox TM and Cachon-Gonzalez MB (2012) The cellular pathology of lysosomal diseases. J. Pathol 226, 241–254 10.1002/path.3021 [DOI] [PubMed] [Google Scholar]
- 38.Fedele AO (2015) Sanfilippo syndrome: causes, consequences, and treatments. Appl. Clin. Genet 8, 269–281 10.2147/TACG.S57672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Viana GM, Priestman DA, Platt FM, Khan S, Tomatsu S and Pshezhetsky AV (2020) Brain pathology in mucopolysaccharidoses (MPS) patients with neurological forms. J. Clin. Med 9, 396 10.3390/jcm9020396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamanna WC, Lawrence R, Sarrazin S and Esko JD (2011) Secondary storage of dermatan sulfate in Sanfilippo disease. J. Biol. Chem 286, 6955–6962 10.1074/jbc.M110.192062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanson SR, Best MD and Wong CH (2004) Sulfatases: structure, mechanism, biological activity, inhibition, and synthetic utility. Angew. Chem. Int. Ed. Engl 43, 5736–5763 10.1002/anie.200300632 [DOI] [PubMed] [Google Scholar]
- 42.Schwartz IV, Ribeiro MG, Mota JG, Toralles MB, Correia P, Horovitz D et al. (2007) A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr. 96, 63–70 10.1111/j.1651-2227.2007.00212.x [DOI] [PubMed] [Google Scholar]
- 43.Gill VL, Wang Q, Shi X and Zaia J (2012) Mass spectrometric method for determining the uronic acid epimerization in heparan sulfate disaccharides generated using nitrous acid. Anal. Chem 84, 7539–7546 10.1021/ac3016054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smeds E, Feta A and Kusche-Gullberg M (2010) Target selection of heparan sulfate hexuronic acid 2-O-sulfotransferase. Glycobiology 20, 1274–1282 10.1093/glycob/cwq089 [DOI] [PubMed] [Google Scholar]
- 45.Lindahl B, Eriksson L and Lindahl U (1995) Structure of heparan sulphate from human brain, with special regard to Alzheimer’s disease. Biochem. J 306 (Pt 1), 177–184 10.1042/bj3060177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rong J, Habuchi H, Kimata K, Lindahl U and Kusche-Gullberg M (2001) Substrate specificity of the heparan sulfate hexuronic acid 2-O-sulfotransferase. Biochemistry 40, 5548–5555 10.1021/bi002926p [DOI] [PubMed] [Google Scholar]
- 47.Bai X and Esko JD (1996) An animal cell mutant defective in heparan sulfate hexuronic acid 2-O-sulfation. J. Biol. Chem 271, 17711–17717 10.1074/jbc.271.30.17711 [DOI] [PubMed] [Google Scholar]
- 48.Pinhal MA, Smith B, Olson S, Aikawa J, Kimata K and Esko JD (2001) Enzyme interactions in heparan sulfate biosynthesis: uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc. Natl. Acad. Sci. U.S.A 98, 12984–12989 10.1073/pnas.241175798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi M, Sugumaran G, Liu J, Shworak NW, Silbert JE and Rosenberg RD (1999) Molecular cloning and characterization of a human uronyl 2-sulfotransferase that sulfates iduronyl and glucuronyl residues in dermatan/chondroitin sulfate. J. Biol. Chem 274, 10474–10480 10.1074/jbc.274.15.10474 [DOI] [PubMed] [Google Scholar]
- 50.Malmstrom A, Bartolini B, Thelin MA, Pacheco B and Maccarana M (2012) Iduronic acid in chondroitin/dermatan sulfate: biosynthesis and biological function. J. Histochem. Cytochem 60, 916–925 10.1369/0022155412459857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pacheco B, Malmstrom A and Maccarana M (2009) Two dermatan sulfate epimerases form iduronic acid domains in dermatan sulfate. J. Biol. Chem 284, 9788–9795 10.1074/jbc.M809339200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stern R and Jedrzejas MJ (2006) Hyaluronidases: their genomics, structures, and mechanisms of action. Chem. Rev 106, 818–839 10.1021/cr050247k [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.