

Abstract
Background
Stimulation of 4-1BB with agonistic antibodies is a promising strategy for improving the therapeutic efficacy of immune checkpoint inhibitors (ICIs) or for overcoming resistance to ICIs. However, dose-dependent hepatotoxicity was observed in clinical trials with monoclonal anti-4-1BB agonistic antibodies due to the activation of 4-1BB signaling in liver resident Kupffer cells.
Methods
To avoid this on-target liver toxicity, we developed a novel bispecific antibody (4-1BB×PD-L1 bispecific antibody, termed “ABL503”) uniquely designed to activate 4-1BB signaling only in the context of PD-L1, while also blocking PD-1/PD-L1 signaling.
Results
Functional evaluation using effector cells expressing both 4-1BB and PD-1 revealed superior biological activity of ABL503 compared with the combination of each monoclonal antibody. ABL503 also augmented T-cell activation in in vitro assays and further enhanced the anti-PD-L1-mediated reinvigoration of tumor-infiltrating CD8+ T cells from patients with cancer. Furthermore, in humanized PD-L1/4-1BB transgenic mice challenged with huPD-L1-expressing tumor cells, ABL503 induced superior anti-tumor activity and maintained an anti-tumor response against tumor rechallenge. ABL503 was well tolerated, with normal liver function in monkeys.
Conclusion
The novel anti-4-1BB×PD-L1 bispecific antibody may exert a strong anti-tumor therapeutic efficacy with a low risk of liver toxicity through the restriction of 4-1BB stimulation in tumors.
Keywords: costimulatory and inhibitory T-cell receptors, T-lymphocytes, immunotherapy, adaptive immunity
Introducion
Cancer treatment has been revolutionized by T-cell-directed immunotherapies, such as immune checkpoint inhibitors (ICIs) targeting the PD-1/PD-L1 pathway. However, a substantial proportion of patients with cancer do not respond to ICIs, and many patients ultimately develop ICI resistance through various mechanisms.1 These problems highlight the unmet need for developing novel immunotherapeutic strategies with improved efficacy. Given that anti-tumor responses do not always occur despite enhanced T-cell responses on ICI therapy,2 3 the targeting of co-stimulatory receptors (eg, 4-1BB, GITR, and OX-40) appears to be a promising therapeutic option for overcoming a non-response to immunotherapies and further enhancing the function of exhausted tumor-specific T cells, eliciting significant anti-tumor responses.4 5 To this end, several agents that target co-stimulatory receptors are in an early phase of clinical investigation.4
In the development of effective cancer immunotherapies, including ICIs, agonistic antibodies, and combination therapies,6 various techniques are used for antibody optimization.7 A bispecific antibody (BsAb) is engineered to bind two different targets through the physical linkage of two antigen binding sites and the dual targeting concepts have been used in a wide depending on the target molecules and mechanisms.8 Considering that the co-stimulatory receptors and ligands complex must be structurally clustered to deliver strong co-stimulation signals, a BsAb can be used to induce a supercluster of targets (eg, co-stimulatory receptors) without FcγR-mediated clustering, and to restrict off-target effects by designing the cross-linkage of two different molecules where one targets specific locations or cell types.4 Thus, a BsAb has unique features compared with combination therapy in terms of specificity and efficacy.
The receptor 4-1BB (CD137 or TNFRSF9) is a uniquely compelling target for cancer immunotherapy. Agonistic 4-1BB antibodies have exhibited potent anti-tumor efficacy in various preclinical models and human CD8+ tumor-infiltrating lymphocytes (TILs).9–11 Targeting 4-1BB is appealing due to the prominent 4-1BB expression on highly exhausted PD-1high CD8+ TILs, which contribute to cancer progression, and because 4-1BB signaling induces clonal expansion of CD8+ TILs, which exhibit tumor reactivity without terminal differentiation.11 12 Urelumab was the first agonistic antibody to be developed that induced potent activation of 4-1BB-mediated signaling, but its clinical development has been slowed by two cases of severe hepatotoxicity leading to patient mortality.13 This hepatotoxicity may be caused by the activation of 4-1BB signaling on liver myeloid cells and subsequent induction of interleukin-27 production.14 Further studies have demonstrated that urelumab dose is the most important factor influencing the development of hepatotoxicity.15 However, whether a urelumab with a relatively low dose to avoid hepatoxicity can trigger a sufficient anti-tumor response remains questionable. Utolimumab is another anti-4-1BB antibody that exhibits milder hepatotoxicity, although a phase I trial revealed suboptimal efficacy.16 Thus, the development of a 4-1BB agonistic antibody with minimal hepatotoxicity but sufficient anti-tumor efficacy is still needed.
Various candidate biomarkers have been investigated for predicting the response to ICIs. In particular, PD-L1 expression on tumor tissue appears to predict the response to anti-PD-1/PD-L1 therapy,17 which can be explained by PD-L1 expression being potently induced on IFN-γ production in accordance with the notion that an optimal anti-tumor response on checkpoint blockade relies on a pre-existing anti-tumor response.18–21 As 4-1BB is prominently expressed on CD8+ TILs from the tumor microenvironment and further upregulated after PD-1 blockade, 4-1BB and PD-L1 could be mechanistically beneficial partners for inducing anti-tumor response.11
In the present study, we describe the development of a novel tumor-targeting anti-4-1BB×PD-L1 BsAb for cancer treatment. Considering that the trimeric receptor and superclustering of trimer receptor–ligand complexes are critical for optimal induction of 4-1BB signaling, and that a BsAb is a suitable tool for superclustering of trimeric receptor–ligand complexes, we designed the 4-1BB portion of BsAb to be conditionaly clustered and activated in the context of PD-L1 binding in the tumor microenvironment.4 22 23 Thus, this BsAb activates 4-1BB signaling in a manner dependent on PD-L1 engagement, thereby restricting the 4-1BB stimulatory activity within the tumor microenvironment and simultaneously blocking PD-1/PD-L1 signaling. We tested this antibody using various in vitro functional assays including functional restoration assays using TILs from patients with cancer and in vivo animal models. Our results show that the 4-1BB×PD-L1 BsAb significantly potentiates anti-tumor responses only when PD-L1 is engaged, as indicated by further enhancement of the anti-PD-L1-mediated reinvigoration of exhausted tumor-infiltrating CD8+ T cells while minimizing the risk of peripheral toxicity.
Materials and methods
Expression and purification of recombinant antibodies
To produce recombinant antibodies, the constructed vectors were transiently expressed in ExpiCHO-S cells (Thermo Fisher) using (ExpiFectamine CHO Kit (Thermo), cultured in ExpiCHO Expression medium (Thermo) under the conditions of 30°C to 37°C for 7 to 15 days in a CO2 incubator equipped with a rotating shaker. Plasmid DNA (250 µg) and ExpiFectamin CHO Reagent (800 µL) were mixed with Opti-MEM I medium (20 mL final volume) and allowed to stand at room temperature for 5 min. The mixed solution was added to 6×106 ExpiCHO cells cultured in ExpiCHO Expression Medium and gently mixed in a shaker incubator at 37°C with a humidified atmosphere of 8% CO2 in air. At 18 hours post-transfection, 1.5 mL of ExpiFectamin CHO Transfection Enhancer 1 and 60 mL of ExpiFectamin CHO Transfection Feed were added to each flask. Each antibody was purified from the cell culture supernatant by recombinant Protein A affinity chromatography (Hitrap Mabselect Sure; GE Healthcare) and gel filtration chromatography with a HiLoad 26/200 Superdex200 prep grade column (GE Healthcare). SDS-PAGE (NuPage 4%–12% Bis–Tris gel) and size exclusion HPLC (Agilent) analysis with SE-HPLC column (SWXL SE-HPLC column; TOSOH) were performed to detect and confirm the size and purity of each BsAb. Purified proteins were concentrated in PBS by ultrafiltration using a Amicon Ultra 15 30K device (Merck).
Surface plasmon resonance (SPR)
SPR experiments were performed on a Biacore T200 instrument using a Protein A-immobilized chip (Cytiva) at 25°C. HBS-EP buffer (Cytiva) was diluted 10-fold with distilled water and then filtered using a 0.22 µm membrane filter (Corning). The filtered HBS-EP buffer was then used as the running buffer. The ABL503 candidate was diluted with the HBS-EP buffer containing 500 mM NaCl (Sigma) and 2% (w/v) bovine serum albumin (BSA, Gibco). The diluted ABL503 candidate was captured on the Protein A-immobilized chip surface at a flow rate of 30 µL/min for 60 s and then the chip surface washed using the running buffer for 30 s. The histidine-tagged human PD-L1 (R&D Systems) was diluted serially with running buffer from 500 nM to 31.25 nM. The histidine-tagged human 4-1BB (R&D Systems) was serially diluted with the running buffer from 250 nM to 15.625 nM. Diluted recombinant proteins were individually injected over the chip surface for 60 s at a flow rate of 30 µL/min. At the end of the sample injection, the running buffer was passed over the sensor surface for 180 s to monitor the dissociation. The sensor chip was regenerated with 10 mM glycine–HCl pH 1.5 (Cytiva) at a flow rate of 30 µL/min for 30 s. The kinetic analysis was performed in a 1:1 binding model (for PD-L1) and bivalent analyte model (for 4-1BB) using BiaEvaluation software (Cytiva).
Target binding study by ELISA
The binding of ABL503 to PD-L1 or 4-1BB proteins (human or cynomolgus monkey) and B7 or TNF receptor superfamily proteins was analyzed by ELISA. All recombinant proteins were purchased from Sino Biological (human PD-L1: 10084-H08H, monkey PD-L1: 90251-C08H, human 4-1BB: 10041-H02H, monkey 4-1BB: 90847-K02H, human B7-H3: 11188-H08H, human B7-H4: 10738-H08H, human OX40: 10481-H08H, human GITR: 10643-H08H). Each well of the Nunc Maxisorp plate was coated with 0.5 µg/mL of proteins overnight at 4°C. After washing, the plates were blocked for 1 hour with the blocking buffer containing 1% BSA (Gibco) and then incubated with serially diluted ABL503 for 1 hour at 37°C. The wells were washed and incubated for 1 hour at 37°C with horseradish peroxidase (HRP)–conjugated goat anti-human IgG Fc or F(ab′)2 secondary antibody (Pierce), followed by the addition of HRP chromogenic substrate 3,3′,5,5′-tetramethylbenzidine (TMB; Sigma). Absorbance at 450 nm was measured using a microplate reader (Molecular Devices). For the dual-antigen capture ELISA, human PD-L1 (10084-H02H; Sino Biological) was coated and ABL503 binding detected using human 4-1BB-His (10041-H08H; Sino Biological) and HRP-conjugated anti-His antibody (Roche).
Reporter cell line assays
To confirm the 4-1BB stimulation by ABL503, various cancer cell lines that differentially expressed PD-L1 (Panc-1, NCI-N87, MDA-MB-231, HCC1954, or RKO) were seeded at 2.5×104 cells per well in 96-well white plates (Costar) and incubated overnight in a CO2 incubator at 37°C. The next day, GloResponse NFκB-Luc2/h4-1BB Jurkat cells (Promega) in assay medium (RPMI1640+1% FBS) were added at 2.5×104 cells per well with pre-plated cancer cells or without cancer cells. Serially diluted ABL503 was incubated with co-cultured cells or only 4-1BB Jurkat cells for 6 hours in a CO2 incubator at 37°C. After 6-hour incubation, an equal volume of Bio-Glo luciferase assay reagent (Promega) was added and the luciferase activity analyzed by measuring luminescence using the PHERAstar FSX microplate reader (BMG Labtech). The PD-1/PD-L1 blockade bioassay (Promega) and PD-1+4-1BB combination bioassay (Promega) were performed according to the manufacturer’s instructions.
Cytotoxicity assays
ADCC activity was determined using ADCC reporter bioassay kit (Promega) following the protocol provided by the supplier. For CDC assays, target cells were prepared at a density of 1.0×105 cells/mL and plated 100 µL to each well in the 96-well plate. Serially diluted antibodies were then added to the wells and 25 µL of twofold diluted human complement serum (Quidel) was added to the well followed by incubation for 5 hours at 37°C. Rituximab was used as a positive control. The detection of luminescence was performed by a plate reader (PHERAstar) following the addition of CytoTox-Glo reagent (Promega).
Toxicity study in cynomolgus monkeys
Each group included one male and one female cynomolgus monkey. The groups were administered a slow intravenous injection of ABL503 at a dose of 0, 15, or 75 mg/kg once weekly, four times (days 1, 8, 15, and 22). Necropsy was performed on day 29. Safety and toxicity were assessed based on standard parameters. This monkey study was conducted in accordance with the Safety and Quality Assurance guidelines in the Guideline for Experiments Document of WuXi AppTec.
Isolation of peripheral blood mononuclear cells (PBMCs) and tumor single-cell suspension
PBMCs were isolated from whole blood by standard Ficoll-Paque (GE Healthcare, Uppsala, Sweden) density gradient centrifugation. Purified lymphocytes were cryopreserved until use. Tumor single-cell suspensions were generated as described previously.24 Tumor tissues were obtained from patients with pathologically confirmed HCC who underwent surgical resection at Asan Medical Center (Seoul, Korea). To purify single-cell suspensions from tissue samples, fresh tumor tissues obtained from a prospective cohort of 79 patients with pathologically confirmed HCC who underwent surgical resection at Asan Medical Center (Seoul, Korea) were immediately cut into small pieces (2–4 mm) after removing fat and necrotic components. Up to 1 g of tumor tissue was then transferred to a gentle MACS C-Tube (Miltenyi Biotec, Bergisch Gladbach, Germany) containing 200 µL of enzyme H, 100 µL of enzyme R, and 25 µL of enzyme A (Miltenyi Biotec) pre-mixed in 4.7 mL of RPMI. The C-tubes were then placed on a gentle MACS Octo Dissociator (Miltenyi Biotec) and program 37°C_h_TDK_3 run for 1 hour. After the cell suspension was passed through a 70 µm cell strainer, the cells were washed once. Cells in 20 µL of media were stained with 20 µL of AOPI staining solution (Nexcelcom Bioscience, Lawrence, MA) and the number and viability of mononuclear cells assessed using a Cellometer Auto 2000 (Nexcelcom). The cells were then washed once and cryopreserved.
In vitro functional assay using PBMCs
PBMCs (3×104) were co-cultured with HCC1954 (1×104) in the presence of 5 µg/mL of human anti-CD3 antibody (clone UCHT1, BioLegend, 300438) and incubated with indicated doses of purified antibodies. The concentrations of IFN-γ in cell supernatants were measured using human IFN-γ Quantikine ELISA kit (R&D 637 Systems, SIF50) following 3 days of incubation at 37°C in 5% CO2.
In vitro T-cell restoration assay using tumor single-cell suspensions
The tumor single-cell suspensions (2×105 cells) were seeded to wells at duplicated or triplicated containing soluble anti-CD3 antibody (1 ng/mL, OKT-3; eBioscience) and either 10 µg/mL indicated antibodies, including isotype control (MOPC-21; Biolegend), PD-L1, 4-1BB (1A10), and urelumab. To estimate the cytokine production of CD8+ TILs, the cells were incubated for a total of 36 hours. After 24 hours of incubation, we added brefeldin A (GolgiPlug; BD Biosciences) and monensin (GolgiStop; BD Biosciences), and then the cells were incubated for an additional 12 hours. To measure CD8+ TIL proliferation, the tumor single-cell suspensions were labeled with CellTrace Violet (CTV; Invitrogen) before seeding and cultured for 72 hours. The mitotic index was calculated according to mitotic events using the absolute number of precursor cells and the number of cells in each mitotic division. Representative FACS plots showing gating strategy is shown in online supplemental figure S4.
jitc-2021-002428supp001.pdf (2MB, pdf)
In vivo mouse study to evaluate therapeutic efficacy
Male C57BL/6-Cd274tm1(CD274)Tnfrsf9tm1(TNFRSF9) (humanized hPD-L1/h4-1BB) mice generated by Bcgen (Beijing Biocytogen Co., Ltd) were subcutaneously injected with human PD-L1-expressing MC38 (MC38hPD-L1) colon carcinoma tumor cells (5×105). When the tumor size reached ~120 mm3, tumor-bearing animals were randomly enrolled into different study groups. Groups of seven animals each were intravenously administered 7.5 mg/kg hIgG1 or 0.4, 2, or 10 mg/kg ABL503, at a frequency of one dose every 3 days, four times. Tumor volume and survival were monitored and recorded twice per week. The mice were sacrificed once the tumor volume reached 3000 mm3 or, if the tumor resolved, after a survival duration of 61 days.
To further assess the dose dependency of the effect, humanized female B-hPD-L1/h4-1BB mice were subcutaneously injected with MC38hPD-L1 tumor cells, as described previously. Once the mean tumor size reached ~120 mm3, tumor-bearing animals were randomly enrolled into different study groups. Groups of seven animals each were intravenously administered 6 mg/kg hIgG1, 3 mg/kg B6 (anti-PD-L1 antibody), 3 mg/kg 1A10 (anti-4-1BB antibody), 3 mg/kg B6+3 mg/kg 1A10, or 4 mg/kg ABL503, at a frequency of one dose every 3 days, four times. Twice per week, tumor volume was measured—calculated as (length×width×width of tumor)/2. Survival data were assessed using Kaplan-Meier estimator plots. The mice were sacrificed when the tumor volume reached 2500 mm3 or, if the tumor resolved, after a survival duration of 42 days.
Complete regression (CR) was exhibited by a total of nine mice, including two from the 2 mg/kg group and seven from the 10 mg/kg group. To evaluate the maintained immune response after tumor clearance, the mice in CR were re-challenged with subcutaneous injection of MC38hPD-L1 tumor cells (5×105) at 40 days after primary tumor injection. Five previously untreated mice were used as controls. The protocol and any amendments or procedures involving the care and use of animals in this study were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Biocytogen (Wakefield, MA, USA).
Immunohistochemistry and PD-L1 scoring
Tumor tissues from in vivo mouse study were stained with anti-PD-L1 antibody (Dako, Clone 28-8) and visualized with images on ×20 objective scale using a microscope. PD-L1 expression was determined by combined positive score (CPS), which is the total number of tumor cells and immune cells (including lymphocytes and macrophages)stained with PD-L1 divided by the total number of viable tumor cells, and multiplied by 100. Five spots were counted and then final CPS was defined by multiplying the mean CPS from five spots by the ratio of the PD-L1 positive area to the total area, as indicated by manufacturer (Dako).
Cytokine release assay
PBMCs from healthy donors (2×105 cells) in RPMI-1640 medium with 10% FBS in 96-well flat-bottom cell culture plates were treated with 1, 5, 10, or 50 µg/mL of ABL503. Human IgG1 or IgG4 was used as a negative control, and 10 µg/mL of plate-bound anti-CD3 stimulation was used as a positive control. After 48 hours of incubation, the levels of cytokines IFN-γ, TNF-α, IL-10, IL-6, IL-1β, and IL-17A in the culture medium were measured by the LEGENDplex Human Inflammation Panel 1 kit according to the manufacturer’s protocol. Fluorescence signals were measured by BD LSR II flow cytometry (BD Biosciences) and analyzed by FlowJo V.10 (Tristar).
Statistical analysis
Statistical analyses were performed using GraphPad Prism Software V.9 (GraphPad) and R V.3.6.2. The EC50 values were determined using a nonlinear regression curve. To compare data between multiple groups, we performed unpaired one-way ANOVA or mixed-effects model with Tukey’s multiple comparison test using lme4 and emmeans package in R (formula: mitotic index ~~treatment+(1+treatment|subject)) or GraphPad Prism Software. Survival analysis was performed using the log-rank (Mantel-Cox) test. P values were determined by two-sided tests and p <0.05 was considered significant.
Results
Development of a novel tumor-targeting 4-1BB×PD-L1 bispecific antibody (ABL503)
The 4-1BB×PD-L1 bispecific antibody (BsAb), ABL503, was constructed from the anti-huPD-L1 monoclonal huIgG1 antibody and a single-chain variable fragment (scFv) targeting hu4-1BB (1A10), which is fused at the C terminus of the anti-huPD-L1 antibody, with a single amino acid substitution (N299A) on the Fc region (figure 1A). By nature of the dual binding of BsAb, ABL503 was designed to deliver the 4-1BB activation signal depending on PD-L1 engagement (figure 1B). Binding affinity analysis by surface plasmon resonance (SPR) revealed that ABL503 bound to recombinant hu4-1BB (KD: 1.380±0.137×10−8 M) and huPD-L1 (KD: 3.072±0.088×10−9 M) with high affinity (figure 1C; online supplemental table S1). Moreover, dual-antigen capture ELISA showed that ABL503 simultaneously bound to both 4-1BB and PD-L1 (figure 1D). We also confirmed that ABL503 exhibited negligible binding activities to other members of the TNF receptor superfamily and B7 family, including human OX40, GITR, B7-H3, and B7-H4 (online supplemental figure S1).
Figure 1.
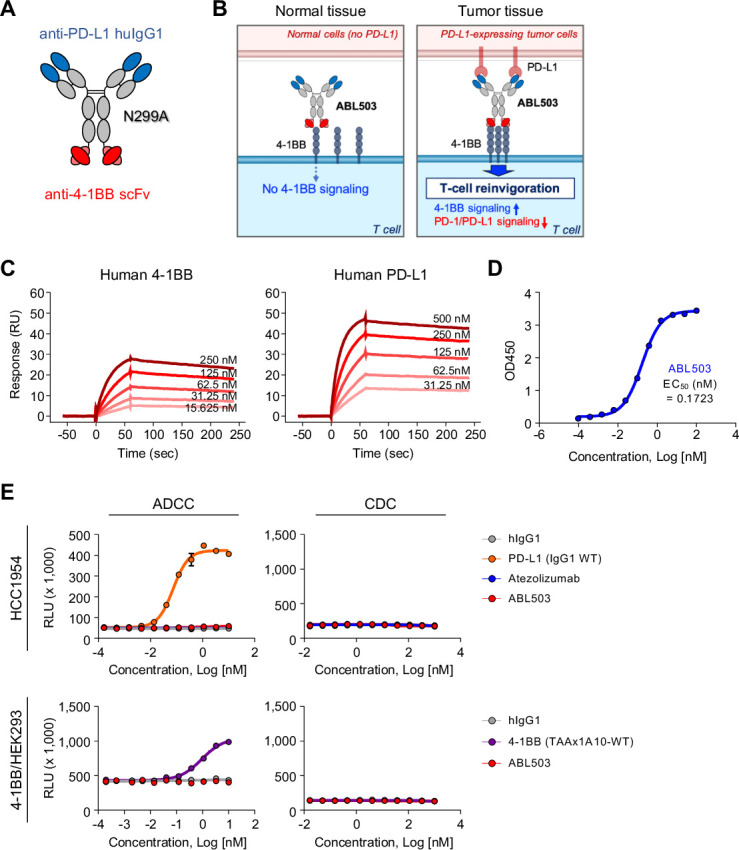
Novel tumor-targeting anti-PD-L1×4-1BB bispecific antibody (ABL503) simultaneously binds unique sites on its targets. (A) ABL503 was designed to target PD-L1 and 4-1BB at the F(ab)2 and Fc portions, respectively, which prevents the Fc portion from binding FcγR. (B) Schematic illustrating the mechanism of action of ABL503. (C) Surface plasmon resonance confirmed the binding of ABL503 to the targets 4-1BB (left) and PD-L1 (right) at the indicated concentrations. (D) Dual-antigen capture ELISA showed that ABL503 simultaneously bound 4-1BB and PD-L1 at the indicated antibody concentrations. The EC50 of ABL503 indicates the mean. (E) The capacity of inducing antibody-dependent cell-mediated cytotoxicity (ADCC) was estimated by ADCC reporter bioassay during ABL503 treatment. The complement-dependent cytotoxicity (CDC) efficacy of ABL503 was also measured by CDC assay. All experiments were performed by duplicated or triplicated samples.
jitc-2021-002428supp002.pdf (61.3KB, pdf)
Since the 4-1BB×PD-L1 BsAb was designed to eliminate Fc-effector function via a single amino acid substitution (N299A) on the FcγR-binding domain of Fc region, we evaluated binding affinity of ABL503 to Fcγ receptors and investigated whether ABL503 induces antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). The results of the binding affinity of ABL503 to Fcγ receptors demonstrated that ABL503 did not exhibit any significant binding activity to FcγRIIa (H167), FcγRIIa (R167), FcγRIIb (H167), FcγRIIIa (F176), FcγRIIIa (V176), and FcγRIIIb while maintaining binding activity to FcγRI and the neonatal Fc receptor, FcRn (online supplemental table S2). In vitro functional assays confirmed that ABL503 did not induce ADCC against PD-L1-expressing cells (HCC1954) or 4-1BB-overexpressed HEK293 cells like atezolizumab (figure 1E), whereas wildtype anti-PD-L1 monoclonal antibody (mAb) (the same clone as in ABL503) or BsAb consisted of wildtype anti-4-1BB (1A10-WT) and undisclosed tumor associated antigen (TAA) elicited significant ADCC activity. Moreover, ABL503 did not mediate any CDC effects against HCC1954 cells and 4-1BB-overexpressed HEK293 cells (figure 1E) while rituximab induced significant CDC activity against CD20-expressing cells (Raji and Daudi) (online supplemental figure S2).
ABL503 is a conditional 4-1BB agonist depending on PD-L1 engagement that can simultaneously block PD-1/PD-L1 signaling
ABL503 possesses unique properties such that the 4-1BB activation signal is induced only on PD-L1 binding. Therefore, we investigated the T-cell stimulatory activity of ABL503 using 4-1BB and/or PD-1/PD-L1 signaling reporter assays. In a luciferase reporter NF-κB-Luc2/4-1BB Jurkat cell line, ABL503 did not activate NF-κB pathway without PD-L1 engagement, whereas urelumab induced strong 4-1BB signaling (figure 2A). To confirm that the 4-1BB stimulation activity of ABL503 was dependent on PD-L1 engagement, we co-cultured the luciferase reporter Jurkat cell line with various cancer cell lines that differentially expressed PD-L1 (PD-L1low (Panc-1 and NCI-N87), PD-L1intermediate (HCC1954 and MDA-MB-231), or PD-L1high (RKO)) in the presence of antibody combinations of anti-PD-L1 and anti-4-1BB (figure 2B, C). Urelumab activated the NF-κB pathway regardless of PD-L1 expression (figure 2C). Intriguingly, ABL503 induced significant 4-1BB signaling only when PD-L1 was engaged, whereas the NF-κB pathway was not activated by anti-4-1BB (1A10, same clone as in ABL503) mono-treatment or combination treatment (figure 2C). Moreover, compared with urelumab, ABL503 yielded a significantly stronger intensity of NF-κB signaling when co-cultured with PD-L1intermediate and PD-L1high tumor cell lines (figure 2C).
Figure 2.
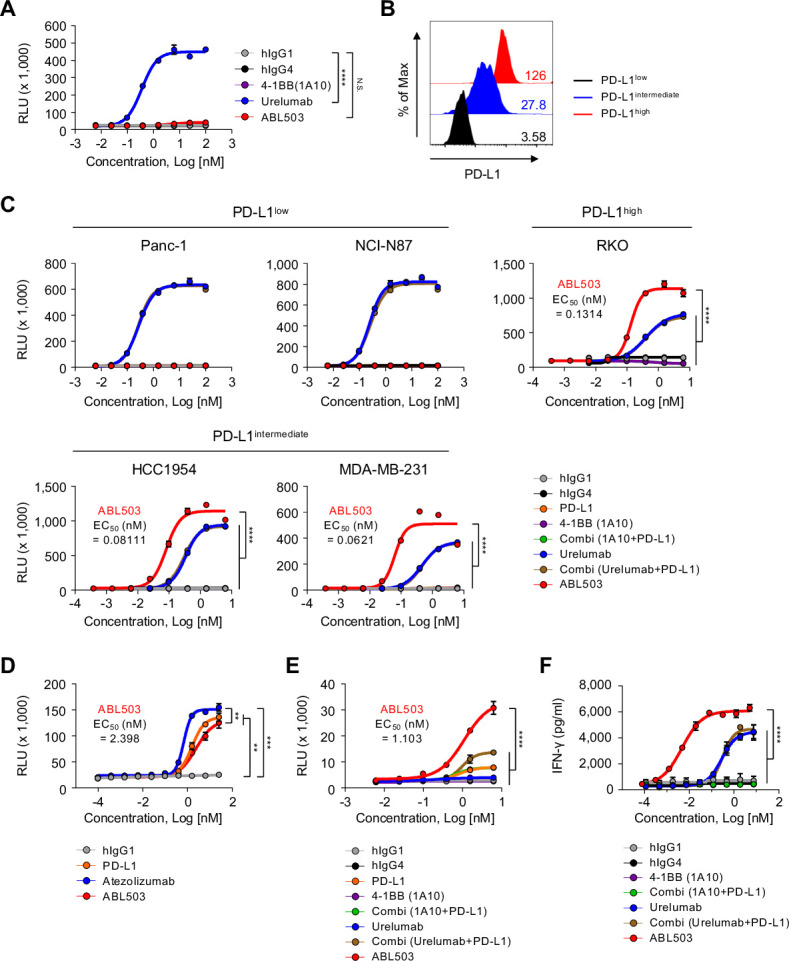
ABL503 induces 4-1BB signaling only in the context of PD-L1 engagement, which also blocks PD-1/PD-L1 signaling. (A) The luciferase reporter NF-kB-Luc2/4-1BB Jurkat cell line was cultured in the presence of the indicated treatments without target cells. Luciferase activity was measured by relative luminescence units (RLU). (B) Mean fluorescence intensity (MFI) shows the basal expression of PD-L1 in the Panc-1, HCC1954, and RKO cell lines. (C) Induction of 4-1BB signaling was observed when Panc-1, NCI-N87, MDA-MB-231, HCC1954, and RKO cells were co-cultured with reporter Jurkat cells and certain antibodies. (D) A PD-1/PD-L1 blockade bioassay (Promega) was performed to estimate the ability of ABL503 to block PD-1/PD-L1 signaling. (E,) A PD-L1/4-1BB combination bioassay (Promega) was performed, confirming the bispecific effect of ABL503. The EC50 in C, D, and E is derived from ABL503 group. (F) Human peripheral blood mononuclear cells and HCC1954 cells expressing PD-L1 were cocultured with the indicated antibodies. IFN-γ production was assessed by ELISA using cultured supernatant. All samples were duplicated or triplicated and the figures show the representative of three or four independent experiments. Significance was estimated by an unpaired one-way ANOVA with Tukey’s multiple comparisons test using the area under the curve (AUC). The EC50 of ABL503 indicates the mean. Data are presented as mean±SEM. N.S., not significant. **p<0.01; ****p<0.0001.
PD-L1 is a tumor targeting antigen and is also a clinically proven immune checkpoint for cancer immunotherapy. Therefore, we next investigated whether ABL503 can effectively inhibit the PD-1/PD-L1 signaling pathway. A PD-1/PD-L1 blockade bioassay revealed that ABL503 efficiently inhibited PD-1/PD-L1 signaling similar to that observed with treatment using atezolizumab or anti-PD-L1 blocking antibody alone (figure 2D). Next, we performed a PD-L1/4-1BB combination bioassay that measures the combined immunomodulatory effects of the PD-1/PD-L1 and 4-1BB signaling pathways. This revealed that ABL503 induced significantly greater NF-κB pathway enhancement compared with anti-PD-L1 or anti-4-1BB alone and their combination (figure 2E). Notably, ABL503 resulted in significantly higher luciferase activity than combination treatment with anti-PD-L1 and urelumab (figure 2E). The combination of anti-PD-L1 and anti-4-1BB (1A10) yielded stimulatory activity similar to that of anti-PD-L1 only, which was significantly lower than the potency of ABL 503 (figure 2E). Furthermore, we examined the effects of ABL503 in terms of augmented T-cell effector function using human PBMCs co-cultured with PD-L1-expressing HCC 1954. ABL503 induced robust IFN-γ production, which was significantly superior to treatment with urelumab alone or combination treatment with urelumab and anti-PD-L1 (figure 2F). In addition, anti-4-1BB (the same clone as in ABL503) alone failed to induce IFN-γ production (figure 2F).
Overall, our results suggest that ABL503 exerted 4-1BB stimulation activity only in the context of PD-L1 engagement. Moreover, ABL503 exhibited additive (or synergistic) effects of blockade of PD-L1 pathway and stimulation of 4-1BB pathway simultaneously.
ABL503 is well tolerated and does not induce non-specific production of pro-inflammatory cytokines
Next, we assessed the safety of ABL503 in cynomolgus monkeys (figure 3A). We first examined whether ABL503 was cross-reactive to monkey 4-1BB and PD-L1 by SPR analysis and ELISA, and found that ABL503 bound to monkey 4-1BB (KD: 0.759±0.138×10−8 M) and monkey PD-L1 (KD: 6.016±0.148×10−9 M) with comparable affinity with human protein (figure 3B and online supplemental table S1). The treatment of ABL503 was well tolerated in the monkeys repeatedly injected with two doses of ABL503 (15 mg/kg and 75 mg/kg), respectively, with no observed treatment-related mortality, clinical signs, or changes in body weight, food consumption, hematology, or coagulation (data not shown). Moreover, all monkeys treated with 15 mg/kg of ABL503 exhibited levels of ALT and AST, clinical chemistry parameters in a normal range associated with the normal liver function (figure 3C). Similarly, monkeys treated with high-dose (75 mg/kg) ABL503 exhibited overall good tolerance, with normal ALT and AST levels (figure 3C), while one monkey showed minimal increase in ALT and AST at D29 post-treatment.
Figure 3.

Treatment with ABL503 is well tolerated in cynomolgus monkeys. (A) Total 4 monkeys were injected with the indicated concentration of ABL503 four times at 7-day intervals. (B) The binding of ABL503 to targets, human (red) and monkey (blue) 4-1BB and PD-L1, was observed by single-antigen captured ELISA at indicated concentration. (C) Clinical parameters including AST and ALT were measured.
To examine the non-specific production of various inflammatory cytokines such as IFN-γ, TNF-α, IL-10, IL-6, IL-1β, and IL-17A by ABL503, PBMCs from healthy donors (n=6) were treated with ABL503 at various concentrations and the levels of cytokines were measured by cytometric bead array assay. The results confirmed that ABL503 did not induce non-specific production of pro-inflammatory cytokines across different concentrations (online supplemental figure S3).
ABL503 further augments effector function of exhausted tumor-infiltrating CD8+ T cells compared with anti-PD-L1
We next investigated whether ABL503 could reinvigorate effector functions of tumor-infiltrating T cells (TILs) from patients with cancer which were manifested by exhaustion and functional impairment in the tumor microenvironment. We performed a series of in vitro functional restoration assays using tumor single-cell suspensions from patients with hepatocellular carcinoma (n=19), to evaluate the effects of ABL503 on the potential reinvigoration of tumor-infiltrating exhausted CD8+ T cells. We found that CD8+ TILs exhibited significantly enhanced proliferation following treatment with ABL503, but not by anti-4-1BB (1A10) alone (figure 4A). Notably, the level of ABL503-mediated enhanced proliferation of CD8+ TILs was much higher than that by single PD-L1 blockade or combination treatment of anti-PD-L1 with anti-4-1BB (1A10) (figure 4A).
Figure 4.

ABL503 activates in vitro T-cell activation and potentially reinvigorates the function of CD8+ human tumor-infiltrating lymphocytes (TILs). (A, C, D) Human tumor single-cell suspensions from patients with hepatocellular carcinoma (HCC) (n=10 for A; n=14 for C and D) were incubated with anti-CD3 and specific antibodies for 72 hours. CellTrace Violet (CTV) staining was performed to measure the proliferative response of CD8+ TILs. (B, C, D) To analyze the cytokine production of CD8+ TILs, tumor single-cell suspensions from patients with HCC (n=12 for B, C, and D) were incubated with anti-CD3 and specific antibodies for 36 hours, with additional treatment with GolgiStop and GolgiPlug during the last 12 hours of incubation. Then the intracellular cytokines were stained. All experiments were performed in duplicated or triplicated samples. Significance was calculated using mixed-effects model with Tukey’s multiple comparison test. N.S., not significant. *p<0.05; **p<0.01; ***p<0.001.
In addition to the enhanced proliferation, ABL503 treatment significantly increased the production of IFN-γ and TNF-α by CD8+ TILs, whereas treatment with anti-4-1BB (1A10) alone did not (figure 4B). In particular, IFN-γ production was remarkably upregulated by ABL503 stimulation compared with single PD-L1 blockade or combination treatment of anti-PD-L1 with anti-4-1BB (1A10), similar to the results of the proliferation assay (figure 4B). These findings suggest that ABL503 could further enhance anti-PD-L1-mediated T-cell reinvigoration. Comparing the activities of ABL503 versus the combination of urelumab and anti-PD-L1, it was found that ABL503 induced similar functional CD8+ TIL responses, including proliferation and cytokine production, compared with the combination of urelumab plus anti-PD-L1 (figure 4C, D).
ABL503 induces tumor regression in vivo in humanized mice
Finally, we evaluated the anti-tumor efficacy of ABL503 in vivo using humanized huPD-L1/hu4-1BB transgenic mice established with syngeneic MC38 tumor cells expressing human PD-L1 (MC38hPD-L1). Starting from the day 6 post-MC38 inoculation, tumor-bearing humanized transgenic mice (n=7/each group) were treated four times (at 3-day intervals) with ABL503, anti-PD-L1, anti-4-1BB, or the combination of anti-4-1BB plus anti-PD-L1, and then monitored tumor progression and survival (figure 5A). Monotherapy with anti-4-1BB did not attenuate tumor progression in terms of tumor growth and survival, whereas monotherapy with anti-PD-L1 and combined therapy with anti-4-1BB plus anti-PD-L1 induced therapeutic effects (figure 5B). Importantly, ABL503 treatment yielded almost complete tumor regression, compared with single treatment with anti-PD-L1 and combined treatment with anti-PD-L1 and anti-4-1BB, resulting in extended survival of up to 39 days (figure 5B).
Figure 5.

ABL503 treatment in vivo exerts anti-tumor effects, and recovered mice maintains an anti-tumor response after complete tumor regression. (A) Humanized transgenic mice expressing human PD-L1 and 4-1BB (n=7 per group) were subcutaneously inoculated with MC38hPD-L1 tumor cells. At 6 days after tumor cell injection, when the mean tumor size reached approximately 120 mm3, tumor-bearing mice were randomly allocated to different groups and treated with the indicated antibodies four times at a 3-day interval. (B, C) Tumor volume and survival rate were assessed for the mice described in (A). Tumor growth is presented as mean±SEM, and the significance was calculated by mixed-effects model with Tukey’s multiple comparison test (B and C). N.S., not significant. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. The survival rate was analyzed by the log-rank (Mantel-Cox) test for survival curve comparisons with the control group. N.S., not significant. *p<0.05; **p<0.01; ***p<0.001. (D) At 40 days after primary tumor injection, the recovered group described in C (n=9, including two mice from the 2 mg/kg group, and seven mice from the 10 mg/kg group) was rechallenged using the same tumor cells. Tumor size was measured, and the significance of tumor growth was estimated by mixed-effects model. ****p<0.0001.
To evaluate the anti-tumor effect of ABL503 in relation to different amounts of PD-L1-expressing cells, we measured tumor growth after ABL503 treatment of various tumor cell mixtures including different proportions of MC38hPD-L1 and parent MC38 cells (online supplemental figure S5). At the end of the study, the tumor volume was larger with the mixture including 10% MC38hPD-L1 cells (MC38hPD-L1 and MC38 cells at a ratio of 1:9, CPS of 3), compared with in the mixtures including 100% MC38hPD-L1 cells and 50% MC38hPD-L1 cells (CPS of 16). However, ABL503 induced a significant anti-tumor response in all groups, indicating that ABL503 induces anti-tumor activity even in cancer with a low PD-L1 CPS (low percentage of PD-L1+ tumors) (online supplemental figure S5).
Next, we investigated whether the therapeutic efficacy of ABL503 was dose dependent. The in vivo anti-tumor activity of ABL503 increased in a dose-dependent manner (n=7/each group, figure 5C), with a dose of 2 mg/kg or 0.4 mg/kg exhibiting partial therapeutic efficacy, whereas a dose of 10 mg/kg yielded complete tumor regression. Finally, we examined whether the mice that achieved complete regression with ABL503 treatment exhibited anti-tumor responses against tumor rechallenge. These nine cured mice (two mice from the 2 mg/kg group and seven from the 10 mg/kg group; figure 5C) were re-challenged with MC38hPD-L1 cells at 40 days after their first ABL503 treatment. Naive mice (n=5) were included as a control group. Notably, no tumor growth was observed in mice previously cured by ABL503 (figure 5D).
Overall, these results indicate that ABL503 induced significant anti-tumor activity in vivo, which was vastly superior to anti-PD-L1 or anti-4-1BB monotherapy. Moreover, the ABL503-induced anti-tumor response was also protective against tumor re-challenge.
Discussion
Despite potent T-cell activation and the anti-tumor efficacy of 4-1BB agonists, the development of clinical agents targeting 4-1BB has been delayed significantly by the on-target off-tumor toxicities caused by non-specific (over) activation of T cells. In the present study, we generated a novel class of BsAb that simultaneously binds human 4-1BB and PD-L1. This BsAb, termed ABL503, potently enhances anti-tumor responses through the agonistic effect of 4-1BB, which is triggered only in the presence of PD-L1 engagement, together with the additional or synergistic T-cell reinvigoration by PD-L1 blockade. ABL503 induces functional restoration of CD8 TILs, in terms of proliferation and cytokine production, that is at least similar to combination therapy with anti-PD-L1 and urelumab, without toxicity. These features are promising for clinical development of ABL503 with the expectation of minimizing systemic immune activation-related toxicities while preserving the capacity to activate tumor-specific T cells.
The structural and functional hallmark of our novel BsAb is that 4-1BB co-stimulation is achieved only in the context of PD-L1 engagement. In contrast to urelumab, which delivered strong 4-1BB signaling regardless of PD-L1 engagement, the activation signal of ABL503 was dependent on the engagement of PD-L1 by generation of 4-1BB hyperclustering. More importantly, the simple combination of anti-4-1BB (the same clone as ABL503) plus anti-PD-L1 (the same clone as ABL503) did not achieve the same degree of functional activation and reinvigoration of ABL503. This strongly supports a greater therapeutic potential of ABL503 through the mechanism of PD-L1 engagement-specific 4-1BB activation. These results expectedly ensure tumor-localized co-stimulation of 4-1BB activation and the minimization of off-target effects. Furthermore, the unique structure of ABL503, which precludes the binding of FcγR, is expected to prevent 4-1BB cross-linking via FcγRIIb, which has been suggested to be one of the mechanisms by which 4-1BB agonist causes hepatotoxicity.14 The toxicity analysis of urelumab did not predict severe hepatotoxicity in humans due to the low binding affinity of urelumab to the monkey 4-1BB receptor. Although ABL503 shows strong binding affinity to monkey 4-1BB, our present results indicated that ABL503 was well tolerated in monkeys, supporting that ABL503 can be safely tolerated due to the restriction of 4-1BB stimulatory activity to tumors. In addition, the results of the in vitro assay system indicating the absence of non-specific production of pro-inflammatory cytokines by ABL503 suggest favorable safety profiles for ABL503.
Epitope study revealed that ABL503 binds to distinct residues of 4-1BB located in the cysteine-rich domain (CRD)−4 region (data not shown), whereas urelumab and utolimumab binds to CRD-1 and CRD-3 regions of hu4-1BB, respectively.25 Considering that 4-1BBL binds along the entire length of the receptor CRD-2 region and the A2 motif of CRD-3,25 ABL503 may not compete with endogenous 4-1BB ligands for binding hu4-1BB and has a unique binding site compared with urelumab and utomilumab.4 With its unique 4-1BB binding sites compared with previously reported agents, ABL503 was shown to reinvigorate exhausted tumor-infiltrating CD8+ T cells from patients with cancer and to substantially potentiate the function of CD8+ TILs and anti-tumor activity in mouse model. In particular, the stimulatory and reinvigoration activities of ABL503 were at least comparable with those of the combination of PD-L1 and urelumab, a known superagonist of 4-1BB.
Although other groups have reported the development of tumor-targeted anti-4-1BB agents, including agents that target FAP, HER2, EGFR, or, similarly, PD-L1,26–29 our BsAb has a mechanistic synergism that supports tumor-targeting activity and PD-L1 blockade. We previously reported that 4-1BB is prominently expressed on CD8+ TILs in tumors with a high degree of pre-existing T-cell activation,5 which is closely interconnected with high levels of PD-L1 expression in the tumor.24 Thus, active anti-tumor T-cell responses may underlie the co-expression of 4-1BB and PD-L1, and ABL503 mechanistically fits into this immunological context to maximize the anti-tumor response. Notably, functional PD-1 blockade further upregulated 4-1BB expression on CD8+ TILs.5 Therefore, the efficacy of our anti-PD-L1 portion in ABL503 in reinvigorating TILs suggests that the activity of the anti-PD-L1 portion of ABL503 supports TIL activation, even in tumors with CD8+ TILs that are not sufficiently activated to express 4-1BB.
Although ABL503 was originally designed to engage PD-L1 expressed on tumor cells, we expect to expand the indications for this BsAb. On immune cells, PD-L1 expression preferentially reflects the IFN-γ response, but can also indicate the presence of a sufficient anti-tumor response in the tumor microenvironment, where ABL503 is expected to be efficacious.30 This tumor microenvironment involves dynamic mobilization of CD8+ TILs; therefore, it is possible that the BsAb activates CD8+ TILs adjacent to PD-L1-expressing immune cells, which reach tumor cells to elicit the tumor killing activity.31 Taken together, these aspects suggest that the indications for the BsAb may not necessarily be restricted to patients expressing PD-L1 on tumor cells.32
Despite its short-term administration schedule, ABL-503 treatment induced a sustained anti-tumor response even after re-challenge with tumor cells. Metabolic restriction repressed by tumor microenvironment has been shown to contribute to the non-responsiveness of PD-1 blockades33 and 4-1BB signaling can reverse the repression of T-cell mitochondrial activity.34 35 Therefore, it is possible that metabolic reprogramming of exhausted T cells through 4-1BB signaling may, at least in part, explain the formation of durable anti-tumor response of ABL503. Exact mechanisms by which ABL503 induces the immunological memory remains an interesting issue to be elucidated.
In summary, our study presents a novel anti-4-1BB×PD-L1 BsAb based on Fc-engineered huIgG1 targeting PD-L1 and a scFv targeting 4-1BB. This BsAb is able to induce 4-1BB stimulation only when PD-L1 is engaged, which may restrict the 4-1BB stimulatory activity in tumors. Using the in vitro functional assay system with tumor single-cell suspensions from patients with cancer, we provide the first demonstration that 4-1BB co-stimulation by the anti-4-1BB×PD-L1 BsAb further enhances the function of exhausted CD8+ TILs and anti-PD-L1-mediated CD8+ TIL reinvigoration. We performed a T-cell restoration assay by using single-cell suspensions from tumor tissues, not using isolated CD8 TILs. This assay has the advantage of showing how the antibodies work in complex tumor microenvironments, but also has the limitation that the presently demonstrated effects of ABL503 in reinvigorating exhausted CD8 TILs were likely caused by direct stimulation of CD8 TILs and also indirect reinvigoration of CD8 TILs. In addition, given that ABL503 induces 4-1BB signaling in the context of PD-L1 engagement, tumor single-cell suspensions would be more appropriate for evaluating the effects of ABL503 due to the presence of PD-L1 sources. Importantly, 4-1BB has been shown to be exclusively expressed on PD-1high CD8+ TILs,5 which harbors the highest degree of T-cell activation, as well as a proliferative and reinvigoration potential in therapeutic interventions involving ICIs. Therefore, the in situ activation of 4-1BB by the BsAb in tumors specifies its target to the tumor-infiltrating effector T cells and also cooperates with anti-tumor activity by blocking the activity of the PD-1/PD-L1 pathway. In conclusion, the anti-4-1BB×PD-L1 BsAb, ABL503, represents a promising immunotherapeutic agent against human cancers.
Footnotes
Contributors: Conception and design: SJ, EP, ES, LF, WJ, ZW, S-HP, JJ. Antibody production: YK. In vitro study: SJ, EP, H-DK, ES, HK, JJ, YK, UJ, Y-GS, MJ, SH, WJ. Animal experiment: YH, HL. Analysis and interpretation of data: SJ, EP, HK, JJ, LF, WJ, ZW, E-CS. Writing and review of the manuscript: SJ, EP, H-DK, ZW, E-CS, S-HP, JJ.
Funding: This work was supported by the National Research Foundation of Korea (NRF) grant, funded by the Korea government (MSIT) (NRF-2019R1A2C2005176 to S-HP) and by the grant from ABL Bio Inc., Republic of Korea.
Competing interests: EP, ES, HK, UJ, JJ, YK, Y-GS, YH, HL, and JJung are employees of ABL Bio, Inc., and LF, WJ, and ZW are employees of I-Mab Biopharma which develop antibodies presented in this manuscript.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available on reasonable request. The data that support the findings of this study are available from the corresponding authors S-HP or JJ on reasonable request.
Ethics statements
Patient consent for publication
Not required.
Ethics approval
All included patients provided written informed consent and this study was approved by the Institutional Review Board of Asan Medical Center.
References
- 1.Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 2020;20:25–39. 10.1038/s41577-019-0218-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang AC, Postow MA, Orlowski RJ, et al. T-Cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017;545:60–5. 10.1038/nature22079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim KH, Cho J, Ku BM, et al. The first-week proliferative response of peripheral blood PD-1+CD8+ T cells predicts the response to anti-PD-1 therapy in solid tumors. Clin Cancer Res 2019;25:2144–54. 10.1158/1078-0432.CCR-18-1449 [DOI] [PubMed] [Google Scholar]
- 4.Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov 2018;17:509–27. 10.1038/nrd.2018.75 [DOI] [PubMed] [Google Scholar]
- 5.Kim Hyung‐Don, Park S, Jeong S, et al. 4‐1BB delineates distinct activation status of exhausted tumor‐infiltrating CD8+ T cells in hepatocellular carcinoma. Hepatology 2020;71:955–71. 10.1002/hep.30881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayat Mokhtari R, Homayouni TS, Baluch N, et al. Combination therapy in combating cancer. Oncotarget 2017;8:38022–43. 10.18632/oncotarget.16723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qi X, Li F, Wu Y, et al. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcγR affinity. Nat Commun 2019;10:2141. 10.1038/s41467-019-10088-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Labrijn AF, Janmaat ML, Reichert JM, et al. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov 2019;18:585–608. 10.1038/s41573-019-0028-1 [DOI] [PubMed] [Google Scholar]
- 9.Melero I, Shuford WW, Newby SA, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med 1997;3:682–5. 10.1038/nm0697-682 [DOI] [PubMed] [Google Scholar]
- 10.Buchan SL, Dou L, Remer M, et al. Antibodies to costimulatory receptor 4-1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity 2018;49:958–70. 10.1016/j.immuni.2018.09.014 [DOI] [PubMed] [Google Scholar]
- 11.Kim H-D, Park S, Jeong S, et al. 4-1BB delineates distinct activation status of exhausted tumor-infiltrating CD8+ T cells in hepatocellular carcinoma. Hepatology 2020;71:955-971. 10.1002/hep.30881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakellariou-Thompson D, Forget M-A, Creasy C, et al. 4-1BB agonist focuses CD8+ tumor-infiltrating T-cell growth into a distinct repertoire capable of tumor recognition in pancreatic cancer. Clin Cancer Res 2017;23:7263–75. 10.1158/1078-0432.CCR-17-0831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melero I, Hirschhorn-Cymerman D, Morales-Kastresana A, et al. Agonist antibodies to TNFR molecules that costimulate T and NK cells. Clin Cancer Res 2013;19:1044–53. 10.1158/1078-0432.CCR-12-2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartkowiak T, Jaiswal AR, Ager CR, et al. Activation of 4-1BB on liver myeloid cells triggers hepatitis via an interleukin-27-dependent pathway. Clin Cancer Res 2018;24:1138–51. 10.1158/1078-0432.CCR-17-1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segal NH, Logan TF, Hodi FS, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res 2017;23:1929–36. 10.1158/1078-0432.CCR-16-1272 [DOI] [PubMed] [Google Scholar]
- 16.Segal NH, He AR, Doi T, et al. Phase I study of single-agent utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin Cancer Res 2018;24:1816–23. 10.1158/1078-0432.CCR-17-1922 [DOI] [PubMed] [Google Scholar]
- 17.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19:133–50. 10.1038/s41568-019-0116-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, Crabill GA, Pritchard TS, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer 2019;7:305. 10.1186/s40425-019-0770-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji R-R, Chasalow SD, Wang L, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2012;61:1019–31. 10.1007/s00262-011-1172-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Teng F, Kong L, et al. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther 2016;9:5023–39. 10.2147/OTT.S105862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. 10.1172/JCI91190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Won E-Y, Cha K, Byun J-S, et al. The structure of the trimer of human 4-1BB ligand is unique among members of the tumor necrosis factor superfamily. J Biol Chem 2010;285:9202–10. 10.1074/jbc.M109.084442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wyzgol A, Müller N, Fick A, et al. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J Immunol 2009;183:1851–61. 10.4049/jimmunol.0802597 [DOI] [PubMed] [Google Scholar]
- 24.Kim H-D, Song G-W, Park S, et al. Association between expression level of PD1 by tumor-infiltrating CD8+ T cells and features of hepatocellular carcinoma. Gastroenterology 2018;155:1936–50. 10.1053/j.gastro.2018.08.030 [DOI] [PubMed] [Google Scholar]
- 25.Chin SM, Kimberlin CR, Roe-Zurz Z, et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat Commun 2018;9:4679. 10.1038/s41467-018-07136-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claus C, Ferrara C, Xu W, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med 2019;11. 10.1126/scitranslmed.aav5989. [Epub ahead of print: 12 06 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Compte M, Harwood SL, Muñoz IG, et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat Commun 2018;9:4809. 10.1038/s41467-018-07195-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinner MJ, Aiba RSB, Jaquin TJ, et al. Tumor-localized costimulatory T-cell engagement by the 4-1BB/HER2 bispecific antibody-anticalin fusion PRS-343. Clin Cancer Res 2019;25:5878–89. 10.1158/1078-0432.CCR-18-3654 [DOI] [PubMed] [Google Scholar]
- 29.Lakins MA, Koers A, Giambalvo R, et al. FS222, a CD137/PD-L1 tetravalent bispecific antibody, exhibits low toxicity and antitumor activity in colorectal cancer models. Clin Cancer Res 2020;26:4154–67. 10.1158/1078-0432.CCR-19-2958 [DOI] [PubMed] [Google Scholar]
- 30.Kowanetz M, Zou W, Gettinger SN, et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1). Proc Natl Acad Sci U S A 2018;115:E10119–26. 10.1073/pnas.1802166115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peranzoni E, Lemoine J, Vimeux L, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 2018;115:E4041–50. 10.1073/pnas.1720948115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee H-H, Wang Y-N, Xia W, et al. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell 2019;36:168–78. 10.1016/j.ccell.2019.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delgoffe GM. Filling the tank: keeping antitumor T cells metabolically fit for the long haul. Cancer Immunol Res 2016;4:1001–6. 10.1158/2326-6066.CIR-16-0244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menk AV, Scharping NE, Rivadeneira DB, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med 2018;215:1091–100. 10.1084/jem.20171068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawalekar OU, O'Connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 2016;44:380–90. 10.1016/j.immuni.2016.01.021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-002428supp001.pdf (2MB, pdf)
jitc-2021-002428supp002.pdf (61.3KB, pdf)
Data Availability Statement
Data are available on reasonable request. The data that support the findings of this study are available from the corresponding authors S-HP or JJ on reasonable request.