

Abstract
Glutamate-ammonia ligase (glutamine synthetase; Glul) is enriched in astrocytes and serves as the primary enzyme for ammonia detoxification and glutamate inactivation in the brain. Loss of astroglial Glul is reported in hippocampi of epileptic patients, but the mechanism by which Glul deficiency might cause disease remains elusive. Here we created a novel mouse model by selectively deleting Glul in the hippocampus and neocortex. The Glul deficient mice were born without any apparent malformations and behaved unremarkably until postnatal week three. There were reductions in tissue levels of aspartate, glutamate, glutamine and GABA and in mRNA encoding glutamate receptor subunits GRIA1 and GRIN2A as well as in the glutamate transporter proteins EAAT1 and EAAT2. Adult Glul-deficient mice developed progressive neurodegeneration and spontaneous seizures which increased in frequency with age. Importantly, progressive astrogliosis occurred before neurodegeneration and was first noted in astrocytes along cerebral blood vessels. The responses to CO2-provocation were attenuated at four weeks of age and dilated microvessels were observed histologically in sclerotic areas of cKO. Thus, the abnormal glutamate metabolism observed in this model appeared to cause epilepsy by first inducing gliopathy and disrupting the neurovascular coupling.
Keywords: cerebrovascular dysfunction, epilepsy, Glul, neurodegeneration, metabolism, gliopathy
1. Introduction
Glutamate-ammonia ligase (glutamine synthetase; Glul) is enriched in astrocytes (Martinez-Hernandez et al., 1977) and plays a critical role in the conversion of glutamate to glutamine and thereby also in the removal of neurotoxic ammonia in the central nervous system (CNS). Because treatment of rodents with the Glul inhibitor methionine sulfoximine induces convulsive seizures (Eid et al., 2016) and because Glul is deficient in parts of the hippocampus in patients with mesial temporal lobe epilepsy (MTLE) (Eid et al., 2004; van der Hel et al., 2005), a role for Glul in epileptogenesis has been proposed. Consistent with this idea, the few described patients suffering from congenital Glul deficiency, all had neonatal onset, severe epileptic encephalopathy (Haberle et al., 2012; Spodenkiewicz et al., 2016).
The exact role of Glul in epilepsy, however, has been difficult to establish, and the mechanism by which Glul deficiency might cause disease is poorly understood. First, MTLE patients with secondary Glul deficiency may suffer from the consequences of the Glul deficiency as well as from effects of the primary insult (which is unknown in most cases). Second, the patients with congenital (primary) Glul deficiency were not only suffering from the cerebral consequences of Glul insufficiency, but also from the consequences of multiple organ failure with secondary effects on the brain (Spodenkiewicz et al., 2016). Third, methionine sulfoximine has several effects other than inhibiting Glul. It decreases tissue glutathione (Shaw and Bains, 2002), increases astrocyte glycogen (Bernard-Helary et al., 2002) and excites neurons via a Glul-independent mechanism (Kam and Nicoll, 2007). To separate the consequences of a primary Glul defect in theCNS from those of other events, a genetic approach is urgently needed; however, attempts to resolve this issue have been met with limited success. Mice completely deficient in Glul die already at embryonic day E3 (He et al., 2007) and mice with deficiency limited to the CNS die three days after birth (He et al., 2010).
Because cortical structures are important for seizure generation and propagation(Eid et al., 2008; Blumenfeld et al., 2009) and because intact brain stem functions are required for survival, we selectively deleted Glul in the cerebral cortex (i.e. neocortex and hippocampus) of mice by taking advantage of the Emx1-IRES Cre line, which directs Cre-mediated gene deletion in the cortex while sparing critical brain stem regions (Gorski et al.,2002). Using this approach, we demonstrate here that loss of cortical Glul causes abnormal glutamate signaling and induces astrogliosis and vascular impairment, ultimately culminating in spontaneous recurrent (epileptic) seizures and neuron loss.
2. Materials and methods
2.1. Materials
N,N’ methylene bisacrylamide was from Promega (Madison, WI, USA). Molecular mass markers for SDS polyacrylamide gel electrophoresis (SDS PAGE) and nitrocellulose sheets (0.22 µm pores, 100 % nitrocellulose) were from GE Healthcare (Buckinghamshire, UK). Paraformaldehyde was from Electron Microscopy Sciences (Hatfield, PA, USA), and glutaraldehyde was from TAAB (Reading, UK). Sodium dodecyl sulfate (SDS) of high purity (>99 % C12 alkyl sulfate) was from Millipore (Carrigtwohill, C. Cork, Ireland). Electrophoresis equipment was from Hoefer Scientific Instruments (San Francisco, CA, USA). Permount mounting medium, ProLong anti-fade DAPI media, DyNAzyme II DNA Polymerase were from ThermoFisher Scientific (Waltham, Massachusetts, USA). All other reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA).
2.2. Animals
All animal studies were carried out in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) and the United States Animal Welfare Act. Formal approval to conduct the experiments was obtained from the animal subjects review board of the Norwegian Governmental Institute of Public Health (Oslo, Norway) and the Institutional Animal Care and Use Committee at the Yale School of Medicine. Care was taken to avoid suffering and minimize the number of the experimental animals. The mice were housed in individually ventilated (IVC) cages at constant temperature (22 ± 0.7°C) and humidity (56 ± 6%), and were fed with Harlan Teklad 2018 (Harlan Laboratories Inc., Indianapolis, IN, USA) with the assess to water ad libitum. Biopsies from ear or tail were collected for determining genotype.
As Figure 1A shows, to generate the conditional Glul KO mouse line (Glulf/f), the targeting vector was prepared by recombineering as described by Lee and co-workers (Lee et al., 2001). Briefly, approximately 12 kb of Glul genomic fragment containing the entire Glul was retrieved from the bacterial artificial chromosome (BAC) clone, RP24– 326N10 (obtained from the BACPAC Resources Center at the Children’s Hospital Oakland Research Institute, Oakland, CA) by gap repair. The 5’LoxP site as inserted in intron 1 at position 628, nucleotides 5’ of exon 2, and the second 3’Lox sequence together with the Frt-PGKneo-Frt selectable marker was inserted 140 nucleotides 5’ of exon 7. Thus, loxP sites were inserted on each flank of the DNA encoding exon 2–6. This vector containing approximately 4kb and 2.9kb of 5’-long and 3’-short arms, respectively, was then linearized by NotI digestion, purified and then electroporated into ES cells, which was derived from F1(129sv/C57BL6j) blastocyst. ES cells were cultured in the presence of G418 and Ganciclovir after electroporation according to Wurst and Joyner (Wurst and Joyner, 1999) and drug resistant colonies were picked and cultured in 96-well plates. Drug resistant ES clones were screened by nested long-range PCR using primers specific to genomic sequences outside the homology arms and LoxP sites to identify targeted ES clones. Targeted clones were expanded and screened again to confirm their identity prior to the generation of chimeric animals by aggregation with CD1 morula. Chimeric males were then bred with ROSA26-Flpe female (Soriano, 1997) to remove the PGKneo cassette to generate the final Glul floxed allele. Two chimeric founder lines (1F7 and 1B4) were produced, and backcrossed more than ten generations into the C57BL/6 background. Both were crossed with Cre-lines as described. The Flox-Glul mice were first tested by crossing with a general deletor strain (Htrp1-Cre: Jackson Laboratories, Stock No. 004302; RRID: IMSR_JAX:004302) mediating deletion in all cells including germ cells and a global astrocyte deletor (hGFAP-Cre: Jackson Laboratories, Stock No. 004600; RRID: IMSR_JAX:004600). Heterozygote Glul mice of two lines were obtained and they were fertile, but homozygote knockout mice were not obtained (data not shown) in agreement with data from others using another Flox-Glul mouse line (He et al., 2007; He et al., 2010). We then tested the Emx1-IRES-Cre line (Jackson Laboratories, Stock No. 005628; Gorski et al., 2002; RRID: IMSR_JAX:005628)}) by crossing it with a reporter line [Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo; Jackson Laboratories, Stock No. 007676; RRID: IMSR_JAX: 007676). The resultant litters had Cre expressed in neurons, astrocytes and oligodendrocytes, (data not shown) consistent with data from others (Gorski et al., 2002). Crossing the female or male Emx1-IRES-Cre line with the male or female Flox-Glul mice resulted in live mice lacking Glul in the cerebral cortex (Fig. 1B). The breeding scheme to generate cortical Glul knockouts was as follows:
Fig. 1.
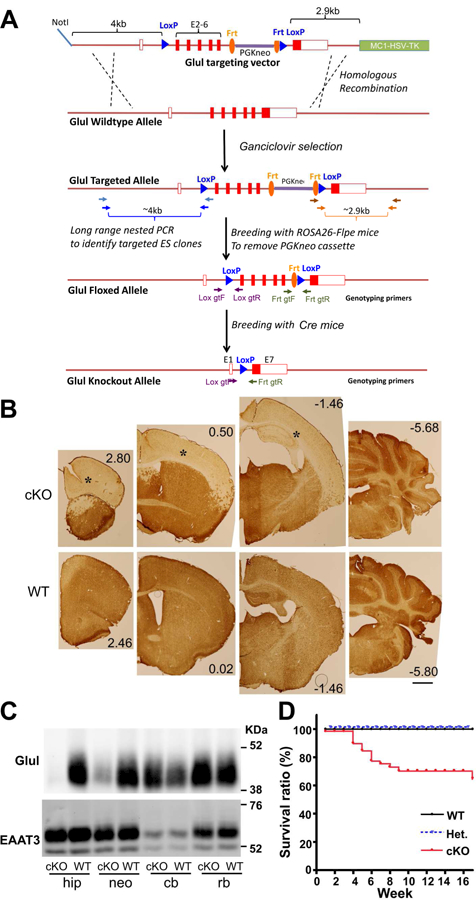
Selective deletion of the Glul gene in the cerebral cortex was achieved by crossing Emx1-Cre mice with Glul-flox mice. (A) Construction of Glul flox mice (see Methods for details). (B) The resulting Glul conditional knockout mice (cKO) lacked Glul protein in the neocortex and the hippocampus. Sections through different parts of brains from cKO and wild-type (WT) mice were labeled with antibodies to Glul. Note that there is hardly any staining in the cerebral cortex (*) of the cKO mice. And as the Emx1-Cre driver is not active in the cerebellum (cb), Glul expression is preserved. Scale bar 1 mm. (C) Immunoblots confirmed that Glul labeling was virtually absent in the hippocampus and neocortex of the cKO mice. Identical blots were prepared from fresh brain tissue from the hippocampus (hip), neocortex (neo), cerebellum (cb) and rest of the brain (rb) from cKO and WT mice, and probed with antibodies to Glul or EAAT3 as indicated. (D) Mortality of the cKOs during 16-week-observation period (n = 97). Note that the mortality was highest between 4 weeks and 9 weeks with few deaths before and after.
1st generation: heterozygote Cre-mice ([Emx1-Cre+/0]) were crossed with homozygote Flox-Glul mice ([Glulf/f]). This crossing gave two possible genotype combinations, namely [Emx1-Cre+/0;Glulf/w] and [Glulf/w].
2nd generation: the [Emx1-Cre+/0;Glulf/w] mice were crossed with homozygote Flox-Glul mice ([Glulf/f]). This crossing gave four possible genotype combinations: [Emx1-Cre+/0;Glulf/f], [Emx1-Cre+/0;Glulf/w], [Glulf/f] and [Glulf/w].
The [Emx1-Cre+/0;Glulf/f] mice are hereafter referred to as cortical knockouts (cKO), while the [Emx1-Cre+/0;Glulf/w] are referred to as conditional heterozygotes and the mice lacking Cre ([Glulf/f] and [Glulf/w]) are referred to as wildtype (WT).
The phenotypes of the two Flox-Glul founder lines were similar (data not shown). Most of our study here was based on the knockout generated from the 1F7 founder line.
2.3. Genotyping
Biopsies were digested (95°C, 1 h) with alkaline lysis solution (25 mM NaOH and 0.2 mM EDTA) and chilled on ice before addition of neutralizing solution (40 mM Tris-HCl). The primers to detect the GS-flox genotype were GS lox gtF (5’-agtcagcagtgtgctccttg-3’) and GS lox gtR (5’-gctcagctcttggaacaacc-3’). The cycle is described as following: 1. 94°C 3 min; 2. 94°C 30 sec; 3. 55°C 30 sec; 4. 72°C 30 sec; Repeat 2–4 for 35 cycles; 5. 72°C, 4 min. The expected PCR products were 346 bp for the wild-type allele and 440 bp for the flox allele. The primers to detect the Emx1-Cre (Gorski et al., 2002) are oIMR1084 (5’- GCG GTC TGG CAG TAA AAA CTA TC-3’) and oIMR1085 (5’-GTG AAA CAG CAT TGC TGT CAC TT-3’). The cycle is described as following: 1. 94°C 3 min; 2. 94°C 30 sec; 3. 51.7°C 1 min; 4. 72°C 1 min; steps 2–4 were repeated 35 times; 5. 72°C, 2 min. The expected PCR product was about 100 bp (data not shown).
2.4. Antibodies
Affinity purified anti-peptide antibodies to sheep anti-EAAT2 (GLT-1; Ab#8; Li et al., 2012; RRID: AB_2714090), rabbit anti-EAAT1 (GLAST; Ab#314; Holmseth et al., 2009; RRID: AB_2314561) and rabbit anti-EAAT3 (EAAC1; Ab#371 Holmseth et al., 2012a; RRID: AB_2714048) were from the same batch as previously described. Because antibody batches may differ from each other (Danbolt et al., 2016a), we identify antibody batches by the unique identification number (“Ab#”) they are given by our electronic laboratory information system (software provided by Science Linker AS; Oslo, Norway).
Rabbit anti-VGLUT1 (Cat. No. 135303; RRID: AB_887875) was gifts from Henrik Martens (Synaptic System GmbH, Goettingen, Germany). Rabbit anti-GLUT1 (Cat. no. ab14683; RRID: AB_301408) were from Abcam. Rabbit anti-glutamine synthetase (Cat. No G2781; RRID: AB_259853), and mouse monoclonal anti-GFAP (Cat no. G3893; RRID: AB_477010) were from Sigma (St. Louis, MO, USA). IRDye 680RD Donkey anti-Rabbit IgG (H+L) (Cat. no. P/N 926–68073; RRID: AB_10954442) and IRDye 800CW Donkey anti-mouse IgG (H+L) (Cat. no. P/N 926–32212; RRID: AB_621847) were from Li-Cor Bioscience UK Ltd, and Alexa Fluor 680 AffiniPure Donkey anti-sheep IgG (H+L) (Cat. no. 713–625-147; RRID: AB_2340753) was from Jackson ImmunoResearch. Alexa fluor goat anti-mouse 488 (Cat. no A11029; RRID: AB_2534088), goat anti-rat 488 (Cat. no A11006; RRID: AB_2534074), goat anti-rabbit 555 (Cat. no A21429; RRID: AB_2535850) antibodies were purchased from Molecular probes (Eugene, OR, USA).
2.5. Tissue preparation for electrophoresis and immunoblotting
The tissue wet weight was recorded before the tissue was homogenized in 10–20 volumes of 1% SDS in 10 mM sodium phosphate buffer (NaPi) with pH 7.4 (NaPi). As brain tissue contains close to 10 % protein (Lowry, 1953; Lowry et al., 1954), the wet weight gave a first approximation of the protein content. The total protein concentrations of the extracts were subsequently determined by Lowry assay (Lowry et al., 1951), and then diluted to the same concentrations in SDS-sample buffer (Laemmli, 1970), and loaded onto SDS-polyacrylamide gel (Laemmli, 1970) using Hamilton syringes (Hamilton Robotics, NV, USA) to ensure correct loading. After electrophoresis and immunoblotting, Ponceau-S stain or total REVERT protein staining (Li-Cor, Lincoln, NE, USA) of the membranes in combination with Coomassie blue stain of the gels were used to check that the blots indeed had received equal amounts of protein after transfer. Identical blots were prepared in parallel and probed with different primary antibodies. Because the expression of housekeeping proteins can alter in diseased state (Ferguson et al., 2005) and glutamine synthetase deficiency in murine cortex causes progressive neurodegeneration, house keeping proteins might not be optimal as useful references in the present study, and were only used when stated.
Briefly, the blots were first rinsed in PBS (10 mM NaPi pH7.4 and 135 mM NaCl) and then were blocked (1 hour) with 0.05 % (w/v) casein in PBS before incubating with primary antibodies in bovine serum albumin (BSA; 30 mg/ml) in PBST (PBS with 1 ml/liter Tween 20 and 0.5 mg/ml NaN3) overnight, room temperature. The membrane was rinsed (4×10 min) with PBST before incubation (1 hour) in secondary antibody solution (1:20000). Rinse membrane several times with PBST and then PBS to remove residual Tween 20 before scanning. The blots were examined for immunofluorescence using an infrared scanner (Licor Odyssey system, LI-COR Biotechnology-UK Ltd, Cambridge, UK). Densitometric data were extracted from the images using the Gel analyzer tool included in our electronic laboratory information system (software provided by Science Linker AS; Oslo, Norway). The results are presented as percent of control (average ± SEM where n represents the number of pairs of littermates). Unpaired student’s T-test was used to compare the means of two groups of data. P-values are given when there were statistically significant differences (p < 0.05) between the data from the Emx1-Glul knockout mice and the control mice.
2.6. Immunocytochemistry
Mice were transcardinally perfused with 4% formaldehyde in 0.1M NaPi with or without 0.05% glutaraldehyde (Danbolt et al., 1998). Briefly, they were deeply anesthesized by intraperitoneal injection with ZFR cocktail (at least 0.1 ml per 10 g body weight). ZRF is of a mixture of zolazepam (3.3 mg/ml; CAS 31352–82-6), tiletamine (3.3 mg/ml; CAS 14176–49-9), xylazine (0.5 mg/ml; CAS 7361–61-7) and Fentanyl (2.6 µg/ml; CAS 437–38-7). After cessation of all reflexes, the mice were perfused first with 0.1 M NaPi pH7.4 to wash out blood and then immediately followed by 4 % formaldehyde in 0.1 M NaPi with or without 0.05 % glutaraldehyde, as stated, for five minutes. The relevant tissues were collected and immersed in fixative for about 2–4 hour at room temperature. Sections (40 µm thick) were cut from the fixed unfrozen tissue using a Vibratome 1000 plus® (Vibratome, Bannockburn, UK).
Diaminobenzidine-peroxidase labeling was performed as described (Lehre et al., 1995). Briefly, the sections were pre-incubated in 1 % hydrogen peroxide in 0.1 M NaPi, rinsed and treated (30 min, room temperature) in 1 M ethanolamine in 0.1 M NaPi with 0.5 % (v/v) Triton X-100 and rinsed (× 3) in PBS (0.135 M NaCl,10 mM NaPi pH 7.4 with 0.5 % (v/v) Triton X-100) prior to incubation (1 h, room temperature) in blocking solution (10 % (v/v) newborn calf serum (NCS) in TBST (TBS with 0.5 % (v/v) Triton X-100), with 0.1 mg/mL NaN3). Next, sections were incubated (over night, room temperature) with primary antibodies diluted (at concentrations as stated) in blocking solution. After rinsing (×5) in TBST with 1 % (v/v) NCS, the sections were incubated (1 h, room temperature) with secondary antibodies diluted (1:300) in TBST with 1 % (v/v) NCS, and, after rinsed as above, incubated (1 h, room temperature) with streptavidin-biotinylated horseradish peroxidase complex, diluted (1:300) in TBST with 1 % (v/v) NCS. The rinsing steps were repeated and sections were then rinsed (×3) in PBS prior to the color development step with diaminobenzidine-peroxidase.
Immunofluorescent labeling was done as previously described (Zhou et al., 2012). Briefly, The sections were rinsed (3 × 5 min) in TBST (TBS with 0.5 % Triton X-100), treated with 1 M ethanolamine in 0.1 M NaPi pH 7.4 for 30 min, washed in TBST, incubated (1 hour) in TBST containing 10 % newborn calf serum and 3 % bovine serum albumin followed by incubation with primary antibodies and finally with secondary antibodies (Alexa fluor goat anti-mouse 488, Molecular probes; Eugene, OR, USA) diluted 1:1000 dilution. After being washed 3 times with TBST, the sections were then mounted with ProLong Gold antiFade mounting medium with DAPI (ThermoFisher Cat. No. P36935, Waltham, MA, USA) or Fluoroshield with DAPI histology mounting medium (Sigma, F6057) and examined using a Zeiss Axioplan 2 microscope equipped with a Zeiss LSM 510 meta confocal scanner head (Zeiss, Jena, Germany).
2.7. Histological staining
2.7.1. Nissl staining
Sections were mounted onto SuperFrost Plus slides (ThermoFisher, Waltham, MA, USA), air dried and incubated (3 min) in Walter’s cresyl violet solution on a heating plate. Excess staining solution was removed, the stain was differentiated in tap water (3 min) and the sections were dehydrated in graded ethanol, cleared in xylene and mounted with Permount (Cat. no: 12377369, Fisher Scientific).
2.7.2. NeuroSilver staining
Vibratome sections were stained using the FD NeuroSilver kit according to the manufacturer’s instructions (FD NeuroTechnolologies Inc, Columbia, USA). Briefly, sections were rinsed in MilliQ water (2 × 5 min), transferred into a mixture containing equal volumes of Solution A and B (2 × 10 min) and incubated in a mixture of equal volumes of Solution A and B with Solution E, and then in a mixture of Solution C and Solution F (2 × 2 min). After incubation in a mixture of Solution D and Solution F for 5 min, the sections were rinsed in MilliQ water (2 × 3 min) and incubated in Solution G (2 × 5 min). The sections were finally mounted on SuperFrost slides, air dried, cleared in xylene and coverslipped with Permount.
2.8. Amino acid measurements
Mice were decapitated, and the heads immediately plunged into liquid nitrogen for 5 second. The neocortex and hippocampi were dissected out on ice, weighed, and homogenized in 10 volumes of perchloric acid, 2% (vol/vol) containing α-amino adipate, 1 mmol/L. Protein was removed by centrifugation, and the supernatant was neutralized with KOH, 10 mol/L. Amino acids were quantified by HPLC and fluorescence detection after pre-column derivatization with o-phthaldialdehyde, as described (Dahlberg et al., 2014).
2.9. EEG
Mice were anesthetized with 0.25–2% isoflurane (Baxter, Deerfield, Ill.) in O2, placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) and implanted with stainless-steel screw electrodes (Plastics One, Roanoke, Va, USA) that were positioned in the epidural space, over the parietal cortex bilaterally. The female ends of the electrodes were inserted in a pedestal (Plastics One, Roanoke, VA; USA) which was cemented onto the skull with UV light cured acrylated urethane adhesive (Loctite 3106 Light Cure Adhesive, Henkel Corp., Rocky Hill, CT, USA) to form a headcap. The mice were placed individually in custom-made Plexiglas cages. A 6-channel cable was connected to the electrode pedestal on one end and to a commutator (Plastics One) on the other. A second cable connected the commutator to the digital EEG recording unit (CEEGraph Vision LTM, Natus/Bio-Logic Systems Corp., San Carlos, CA, USA). Digital cameras with infrared light detection capability were used to record animal behavior (two cages per camera). The digital video signal was encoded and synchronized to the digital EEG signals. Seizures were identified by visual inspection of the EEG record. The Racine criteria (Racine et al., 1973) were modified and used to classify the seizures from the video records, as follows: Stage 1, immobilization, eye blinking, twitching of vibrissae, and mouth movements; Stage 2, head nodding, often accompanied by severe facial clonus; Stage 3, forelimb clonus; Stage 4, rearing, Stage 5, rearing, falling, and generalized convulsions.
2.10. fMRI imaging and data analysis
For imaging experiments, animals were first anesthetized using 3% isoflurane in a mixture of O2 and N2O (30/70%), then placed in a custom built frame where they were freely breathing through a nose cone. During the scanning procedure, isoflurane was turned off and anesthesia was maintained using a subcutaneous infusion of dexmedetomidine (Zoetis): 2µL + 4µL/h per gram of body weight, at 0.05 mg/mL concentration. Body temperature was monitored using a rectal probe, and maintained within 35–37ºC using warm water pumped through the bed.
MRI data were obtained on a modified 9.4T system with Varian spectrometer and custom built 1H 12mm surface coil. Images were acquired over 8 contiguous coronal slices (thickness = 1mm), covering the parenchyma between the olfactory bulb and cerebellum, with an in-plane field of view of 1.28 × 1.28 cm. Resting-state (task-free) images were obtained using an echoplanar imaging (EPI) sequence (TR/TE= 1000/13 ms in a 32 × 32 matrix, for an in-plane resolution of 250µm) for 6 minutes (360 repetitions), and repeated three times per animal. The cerebrovascular reactivity (CVR; repeated twice per animal) functional scans lasted for 9 minutes with 10% CO2 added to the breathing gas mixture between minutes 3 and 6 of the acquisition. Lastly, a high-resolution spin-echo anatomical image (32 slices at 0.25 mm thickness, 128 × 128 for an in-plane resolution of 100µm, 4 averages) was used for co-registration and volumetric analysis.
Using AFNI (https://afni.nimh.nih.gov/), functional images of CO2 challenge were corrected for motion and spatially smoothed using a Gaussian filter (FWHM = 1 mm). Parametric maps of CVR were generated from a voxel-level linear model and expressed as the t-statistical value of the contrast between baseline (Frames 1 to 180) and CO2 challenge (Frames 210 to 360).
Anatomical images were used to generate a non-linear transform from each animal subject’s native space to a common standard space. The transform was then applied to all parametric maps of CVR, which were then Fisher transformed. Group averages and contrasts were calculated with a voxel-level linear model for each modality with an adjusted threshold of p < 0.05 corrected for multiple comparisons. Finally, brain volume was estimated by manually drawing an intracranial mask over the anatomical images (in native space) by an experimenter blinded to group conditions.
2.11. Open field test (OF)
The 15-min OF tests were carried out using 4–15 weeks old (male and female) under a light intensity of 100 ± 10 Lux. The tests were performed between 4.0–0.5 hours before the start of dark cycle. First, animals were placed in the testing room in their home cage for at least 10 min to acclimate. The test started by gently removing each mouse from their home cage and immediately placing them in the center of the test chamber (chamber dimensions: L50 × W50 × H21.5 cm). A series of 12.5 × 12.5 cm zones were defined in the test chamber. The outer zone consists of 12 blocks while the center/inner zone consists of 4 blocks. The entire session was videotaped for later analysis. All the animals were first-time for OFs. After the tests, the animals were housed in a new cage, and the number of feces produced during the sessions was counted. 70 % ethanol was used to clean the chamber between sessions. All OF videos were then analyzed by one individual in a double-blind manner. Horizontal activities were measured by total number of crossings (during each min and in 15 min) and vertical activities were measured by the total number of rearing (to the walls). For accessing anxiety behavior, both total number of crossings to the center and the number of fecal boli produced were counted
2.13. Experimental Design and statistical analysis
All experiments were performed at least three times unless stated otherwise. Statistical significance between two independent samples was assessed by Student’s t test. Statistical analyses and plotting graphs were performed with GraphPad Prism4 (GraphPad Software). Differences between groups were judged to be significant when P values were smaller than 0.05. *, ** and not significant represent P<0.05, P<0.01 and not significant, respectively. Error bars always indicate the standard error of the mean (± SEM) except when stated (Table 1). No samples or animals were excluded from the analysis. For HPLC analysis, data were obtained in a blind manner in which the experimenters did not know the mouse genotypes.
Table 1.
Total amino acids in the cerebral cortex (including hippocampus) from cortical Glul knockouts and wildtype littermates
| Cortex | Cerebellum | |||
|---|---|---|---|---|
| WT (Cre-; GSf/?) | cKO (Cre+; GSf/f) | WT (Cre-; GSf/?) | cKO (Cre+; GSf/f) | |
| Asp | 2.86 ± 0.52 | 0.86 ± 0.19* | 2.24 ± 0.36 | 2.38 ± 0.31 |
| Glu | 9.45 ± 1.76 | 4.82 ±0.52* | 8.17 ± 0.33 | 8.58 ± 0.59 |
| Gln | 4.39 ± 0.24 | 1.05 ± 0.25* | 4.81 ± 0.30 | 4.90 ± 1.17 |
| Tau | 10.20 ± 2.87 | 12.43 ± 0.89 | 6.09 ± 0.13 | 7.46 ± 0.79 |
| Ala | 0.84 ± 0.16 | 0.93 ± 0.10 | 0.25 ± 0.17 | 0.30 ± 0.12 |
| GABA | 1.55 ± 0.48 | 0.82 ± 0.18* | 1.08 ± 0.36 | 1.08 ± 0.03 |
Amino acid concentrations (nmol per mg wet weight of whole tissue) were determined by means of HPLC in samples from three conditional Glul cortical knockouts (cKO) and two wildtype littermates. The mice, all females, were about 8 weeks old. The cortex samples comprised the entire neocortex and hippocampus. The numbers represent mean ± SD
3. Results
3.1. Mice with cortical deletion of Glul are viable
Our model of cortical Glul deficiency was created by first tagging the Glul gene with flanking LoxP sites to permit cyclization recombinase (Cre)-mediated deletion of exons 2–6 (Fig. 1A). These animals are hereafter referred to as Glul-flox mice. We then crossed the Glul-flox mice with Emx1-IRES-Cre mice, in which Cre-expression is primarily restricted to telencephalic regions, in particular parts that give rise to the neocortex and hippocampus (Gorski et al., 2002).
Breeding of our Glul-flox mice with the Emx1-Cre mice resulted in conditional knockout (cKO) mice where the Glul protein was abolished in most of the neocortex and hippocampus (Fig. 1BC). Because Glul is predominantly an astroglial enzyme, it seems reasonable to assume that the Emx1-Cre driven deletion mostly affects astrocyte function.
The glutamate transporters EAAT3 (Fig. 1C) and EAAT2 (data not shown) were still present in the affected area, as these genes had not been floxed and therefore could not be excised by Cre. The cortical expression levels of Glul protein in heterozygous mice were approximately half of those in wild-type (WT) mice (52%, n = 2 pairs of mice). Further, we observed that Glul protein was absent from the cKO cortex at birth (data not shown), consistent with the reported start of Cre-expression at E10.5 (Gorski et al., 2002). Genotyping at 3 weeks of age from a breeding scheme (Cre+/0; Glulf/w × Cre0/0; Glulf/f) revealed a typical Mendelian distribution (2WT: 1Het : 1cKO) of 47.8 % wild-type (WT; Cre0/0; Glulf/f or f/wt; n=199), ~27.2 % heterozygous (Cre+/0;Glulf/w; n=113) and ~25.0% cKO mice (Cre+/0; Glulf/f; n=104). Thus, the deletion did not appear to increase prenatal mortality. Increased mortality was first noted after three weeks (Fig 1D).
3.2. Cortical deletion of Glul results in late-onset epileptic seizures
Despite deletion of Glul before birth, neither heterozygous nor homozygous cKO mice could be distinguished from their WT littermates with respect to overall appearance during the first two postnatal weeks. At postnatal week 3, the cKO mice began to exhibit behavioral abnormalities such as hypoactivity and periodic running fits when subjected to mild stimulation such as removing the cage lid or gentle knocking on the cage. To further assess the behavioral phenotype, we subjected the mice to the open field test which measures general locomotive activities. The mice (4–15 weeks old cKO and WT littermates) were observed for 15 min each. This was sufficient to reveal alterations in the locomotive activities (Fig. 2). One subgroup of the cKO mice (cKO1, 9 pairs of WT and cKO) were almost inactive during the test (Fig. 2AC) while another subset (cKO2, 6 pairs) showed hypoactivity interrupted by bursts of sudden wild running (Fig. 2BC). The cKO mice exhibited reduced vertical activity (Fig. 2D) and fewer entries to the center of the chamber (Fig. 2E), while the time spent in the center by the cKO mice was comparable to that of WT littermates (Fig. 2F). During these sessions, fewer fecal boli were produced by the cKO mice, suggestive of reduced anxiety (Fig. 2G).
Fig.2.
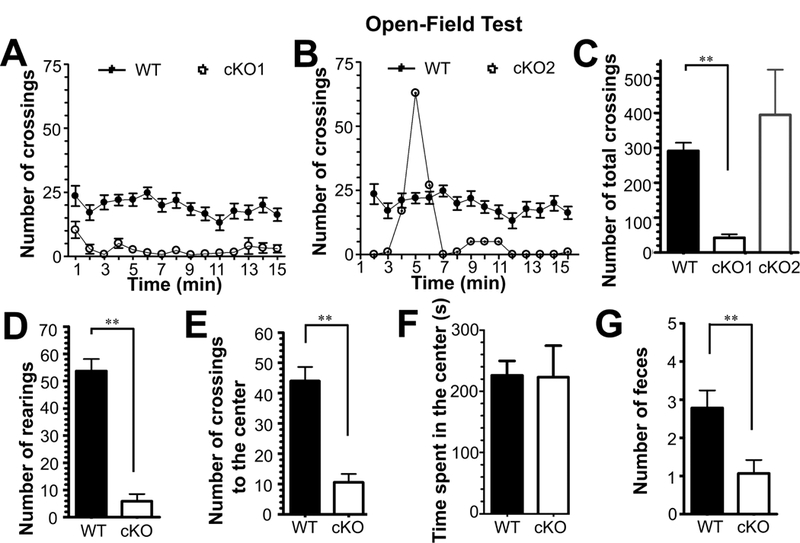
Glul conditional knockout (cKO) mice exhibited altered locomotive activities when subjected to the 15-min open-field test. A total of 15 pairs of WT and cKO mice (21 – 103 days old) were tested (10 pairs of males and 5 pairs of females). (A) Most of the cKO mice (cKO1; 9 pairs) were clearly hypoactive during the entire test period. (B) The remainder (cKO2; 6 pairs) had one or more sudden bursts of activity during the test period. The number of total crossings of all groups of cKO1 and cKO2 were represented in Panel (C). (D) The cKO mice were less active vertically (lower number of rearings), (E) they had fewer entries to the center, (F) but spent comparable time in the center. (G) The cKO also produced a lower number of feces during the test session. **p<0.01
Besides periodic running fits, facial automatisms with occasional falling and forelimb clonus were observed in the cKO mice older than 6 weeks, suggestive of seizures. Continuous video-intracranial EEG recordings of mice aged 4 – 28 weeks demonstrated spontaneous recurrent seizures in 4 of 7 cKO mice, but not in any of the heterozygous (n=9) or in the WT (n=11) mice (Fig. 3). The frequency, duration and behavioral severity of the seizures varied among animals, with a tendency for the seizures to become more frequent as the animals aged.
Fig. 3.
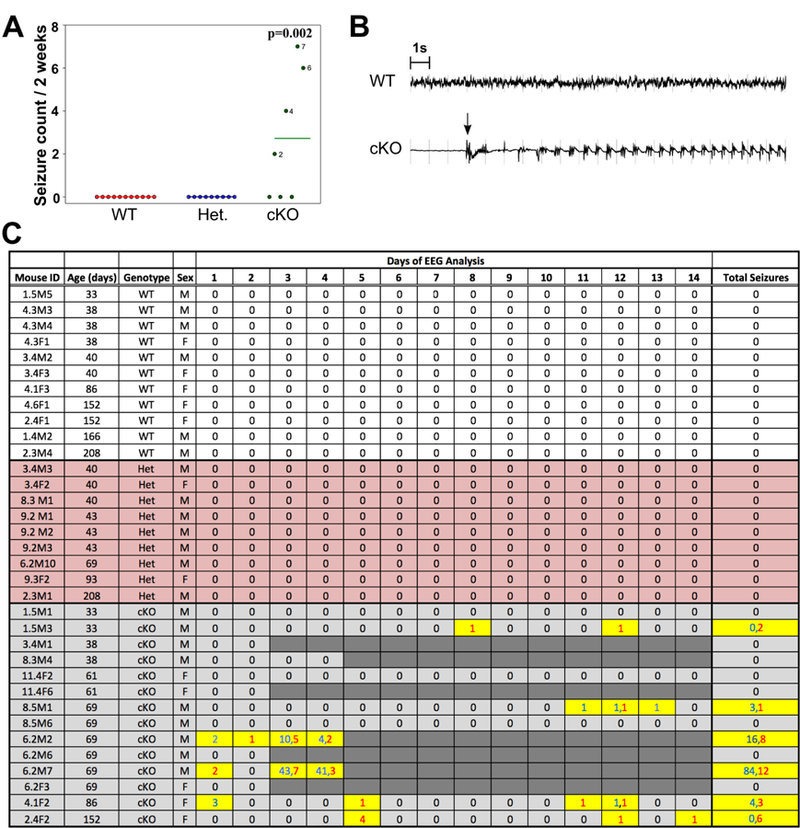
(A) The bars indicate the average number of seizures from the animals in c after exclusion of the 7 cKO that died during recordings. No seizures were observed in the wildtype (WT) or heterozygous Glul knockout (Het) mice, whereas the cKO mice had an average of 2.71 ± 1.13 seizures during the 2-week period (p =0.002). Twelve of 19 (63.2%) seizures were severe (Racine Grade 4–5). (B) Representative EEG tracing of a WT mouse and representative EEG tracing of a cKO mouse are shown. The arrow indicates the start of the seizure. The severity of the seizure was not apparent by EEG, and examination of the concurrent video record was necessary for severity staging. (C) Daily seizure counts and severity assessments over a 14-day recording period. Wildtype (WT; n = 11: 6 males, 5 females), heterozygous Glul knockout (Het; n = 9: 7 males, 2 females), and homozygous Glul knockout (cKO; n = 14: 9 males, 5 females) mice were monitored by continuous video-intracranial EEG analysis for two-weeks. The number of seizures per day is provided. Mild (Racine Grade 1–3) and severe (Racine Grade 4–5) seizures are indicated by blue and red numbers respectively. Seven of the 14 cKO animals died at different time points during the recording (indicated by dark gray shading). These animals are excluded from the plots in A. Abbreviations: F, female; M, male.
3.3. Cortical deletion of Glul causes progressive neurodegeneration and ultimately hippocampal sclerosis
Examination of Nissl-stained brain sections from 6 weeks old Glul cKO mice did not reveal any major abnormalities in the neocortical and hippocampal cytoarchitecture. However, focal lesions (small islands of neuronal loss) in the neocortex were occasionally found at this age, but only in some of the mice (data not shown). As the mice aged, more obvious neuropathological changes started in the hippocampus, as evidenced by selective degeneration of CA1 and CA3 pyramidal neurons and dentate granule cells. The hippocampal pathology occurred bilaterally and was readily apparent in the third postnatal month (Fig. 4AB). As the mice aged further, the neocortex became progressively affected in multiple areas, particularly the retrosplenial, motor, somatosensory, parietal association and visual cortices (Fig. 4C-E). The degree and pattern of neocortical neurodegeneration varied among animals at similar ages, but generally started in the most superficial layer of the cortex (data not shown). Neuro-Silver staining at 11 weeks of age (Fig. 4BD) confirmed that large numbers of neurons were degenerating in the cKO mice, but not in the conditional heterozygotes (Emx1-Cre+/0 ;Glulf/w) and the WT (Cre negative) mice.
Fig. 4.
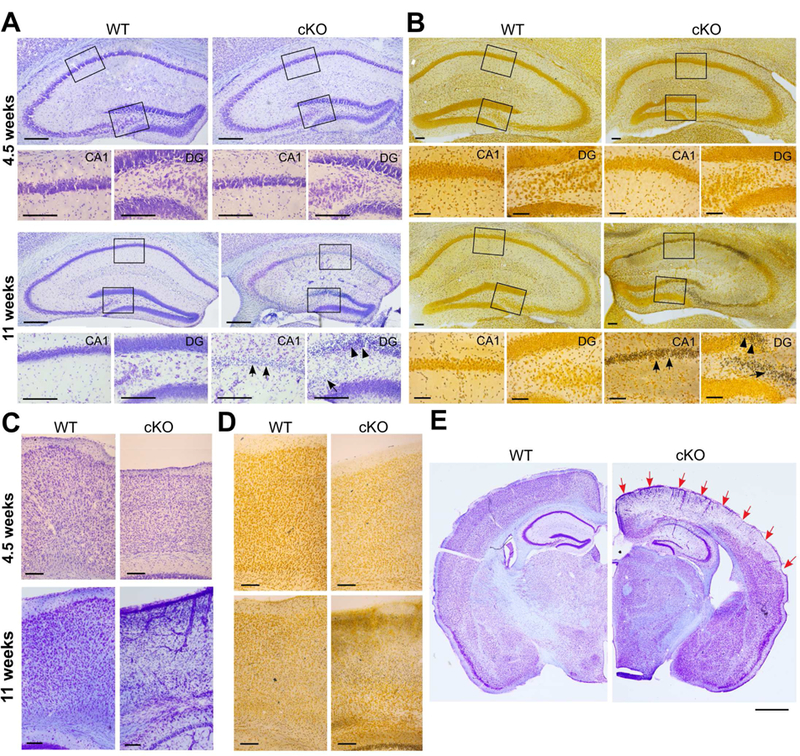
Mice lacking Glul in the cerebral cortex had progressive neurodegeneration. Nissl (A) and NeuroSilver (B) staining of hippocampus of WT and cKO mice showed massive neuronal degenerations (arrows) in CA1, in CA3, in part of the granule cell layer and in the hilus of the dentate gyrus (DG) at 11 weeks of age, but not at 4.5 weeks. (C) Nissl and (D) Neurosilver staining of motor cortex of Glul wildtype and conditional knockouts (cKO) showing massive neuronal degenerations in upper layer of cortex at 9 weeks of age. No neuron degenerations were seen at 4–5 weeks of age. (E) Selective degeneration in some areas of cortex indicated by red arrows. Scale bar: 50 µm (A-D); 1 mm (E).
3.4. Astrocytes lacking Glul become reactive prior to neuronal loss and epilepsy
Considering that Glul is an astrocytic protein, we assessed for reactive astrogliosis by examining the expression of glial fibrillary acid protein (GFAP). Western blotting revealed that the protein was hardly increased in cKO mice versus WT littermates at 2 weeks of age (Fig. 5B), but showed significant increases at 4 weeks of age, consistent with progressive astrogliosis. The increase in GFAP was confirmed by Taqman RT-PCR (data not shown) and immunocytochemistry (Fig. 5A).
Fig. 5.
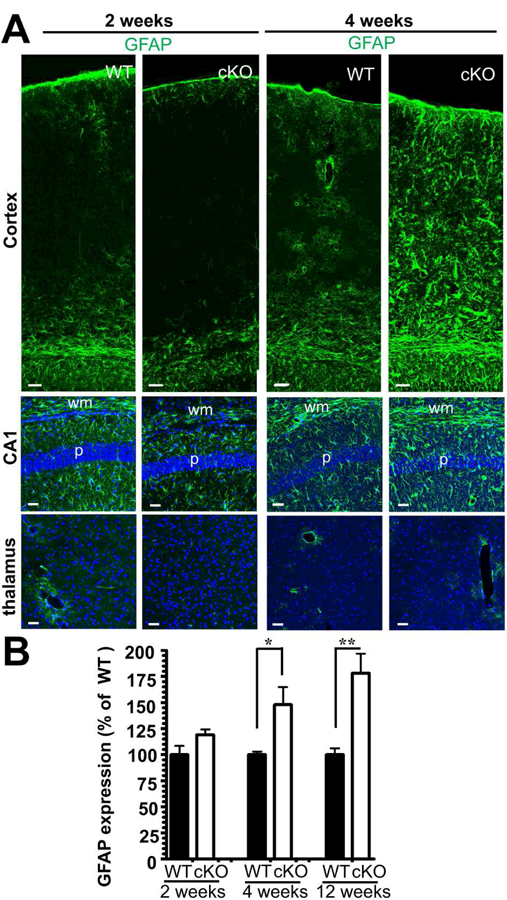
Lack of cortical Glul leads to astrogliosis preceding neurodegeneration. (A) Sections from the neocortex of WT and cKO mice (littermates processed in parallel; ages as indicated) were probed with antibodies to GFAP (green; labels astrocytes). Astrogliosis, as indicated by increased GFAP expression, was evident in the neocortex and hippocampus CA1 of 4 weeks old cKO mice. In contrast, thalamus in the cKO (where Emx1-Cre is not active) showed normal expression of GFAP at both age groups. Scale bar 50 µm. (B) GFAP expression increases with age being statistically significantly increased at four weeks of age. The expression levels were quantified by immunoblotting of hippocampi from wild-type (WT) and Glul cortical knockout (cKO) mice: five pairs of WT and cKO littermates in each age group: 2 weeks of age n = 5 pairs: 3 pairs of males, 2 pairs of females; 4 weeks of age n = 5 pairs: 2 pairs of males, 3 pairs of females; >12 weeks of age n = 5 pairs: 4 pairs of males, 1 pair of females; *p<0.05, **p<0.01.
3.5. Loss of Glul impacts the molecular anatomy and physiology of cerebral blood vessels
Interestingly, the expression of GFAP was increased in astroglial processes along blood vessels in the cKO mice as early as 3 weeks of age (Fig. 6AB). Further, at later stages, the vessels became markedly dilated in the areas of the neocortex and of the hippocampus where neurodegeneration was occurring. (Fig. 6C). We therefore asked whether the astroglial reactivity was associated with changes in the physiology of cerebral blood vessels. MRI cerebrovascular reactivity (CVR), defined as the relative increase in Blood Oxygen Level Dependent (BOLD) signal during a CO2 challenge, revealed significant differences between WT and cKO mice at 4 weeks and 12–15 weeks of age (Fig. 6D). In both age groups, CVR was reduced in neocortical areas, such as cingulate and sensorimotor cortices in the cKO mice (4 weeks: WT, 5.44 ± 0.65%; cKO, 2.8 ± 0.58%; 12–15 weeks: WT, 5.65 ± 1.17%; cKO, 1.67 ± 0.44%). In the dorsal hippocampus, a reduction of CVR was noted at 4 weeks of age in the cKO mice, but not at 12–15 weeks of age (4 weeks: WT, 4.37 ± 0.75%; cKO, 2.24 ± 0.35%; 12–15 weeks: WT, 3.35 ± 0.37%; cKO, 3.03 ± 0.33%). In subcortical areas spared from Glul deletion like the basal forebrain, no significant group effect was found at either age (4 weeks: WT, 3.22 ± 0.59%; cKO, 2.81 ± 0.41%; 12–15 weeks: WT, 2.39 ± 0.37%; cKO, 2.88 ± 0.46%).
Fig. 6.
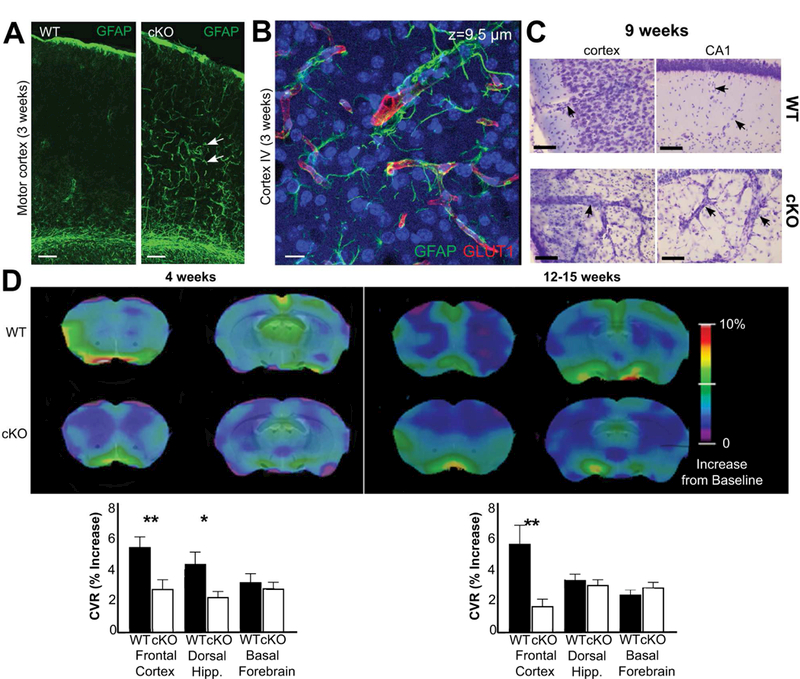
Glul knockout mice (cKO) had altered cerebral blood vessels with reduced ability to react to increases in CO2 levels. (A) There was increased GFAP labeling (green) in astroglial processes in 3 weeks old Glul cKO mice. The images from wildtype (WT) and cKO mice represent montages of three overlapping images acquired from the motor cortex. Scale bar 100 µm. (B) Z-stack image (total thickness 9.5 µm) acquired from motor cortex (layer VI) of the same cKO section as in A. Note increased GFAP labeling in astroglial processes surrounding blood vessels (red, GLUT1). Scale bar 100 µm. (C) Nissl staining shows changed vascular structures in the motor cortex and hippocampus CA1 of 9 weeks old cKO mice compared to WT littermates (processed in parallel). Scale bar 50 µm. (D) Impaired cerebrovascular reactivity in (as indicated) 4 and 12 – 15 weeks old cKO mice. Voxel-level maps of cerebrovascular reactivity (CVR), as defined by the relative increase of signal during CO2 challenge, averaged across each age/group. Maps are projected on two coronal slices of a structural mouse atlas. Region of interest-averaged CVR amplitudes are shown on bar graphs (bottom); cKO mice have impaired CVR response in the dorsal hippocampus at 12 weeks, and in the cortex at both time-points. Subcortical areas like the basal forebrain are unaffected in cKO animals. The most cranial slices (the left images on each sub-figure) are +1.2 mm from bregma, while the caudal slices (the right images on each sub-figure) are −0.6 mm from bregma. *p < 0.05; ** p < 0.01.
3.6. Young mice lacking cortical Glul have abnormal glutamate signaling
As Glul is critical for the homeostasis of glutamate, glutamine and GABA, and as cerebral blood flow is regulated through glutamate-mediated astrocyte responses (Attwell et al., 2010), we quantified the levels of these and other amino acids in the cKO and WT brains (Table 1). As expected, the concentrations of glutamine, glutamate, aspartate and GABA in the tissue were significantly lower in the cKO cortex than in the WT cortex, whereas no changes were observed in the cerebellum, which expressed normal levels of Glul (Fig. 1BC).
We also assessed several molecules involved in glutamate signaling by Taqman RT-PCR (Fig. 7A) and found reduced levels of mRNA encoding the major glial glutamate transporter EAAT2 and of mRNA encoding the glutamate receptor subunits GRIA1 (GluR1) and GRIN2A (GluN2A) in the hippocampi of 5 weeks old cKO mice. Immunoblotting of EAAT2 showed that the reduction in EAAT2 protein levels was present already at 2 weeks of age (Fig. 7B). Another glial glutamate transporter, EAAT1, displayed a similar expression pattern albeit less pronounced, while the expression of EAAT3 (which is the major neuronal glutamate transporter at the plasma membrane: Holmseth et al., 2012b) and neuronal vesicular glutamate transporter VGluT1 were not changed significantly.
Fig. 7.
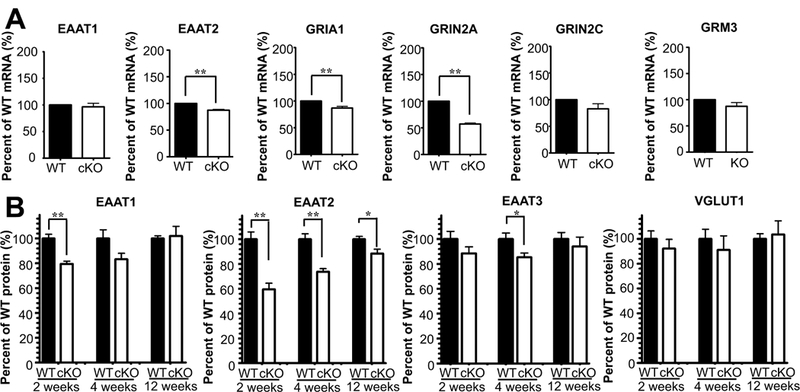
Several of the components of the glutamatergic signaling system were altered in Glul cortical knockout (cKO) mice. (A) Taqman RT-PCR revealed statistically significant reductions in the expression of glutamate transporter EAAT2 (**p<0.01) and of the glutamate receptor subunits GRIA1 (**p < 0.01) and GRIN2A (**p < 0.01) in the hippocampi of 5 weeks old cKO mice compared to their wild-type (WT) littermates (n = 4 pairs of male mice). In contrast, the expression of GRIN2C (p = 0.119), GRM3 (p = 0.128) and EAAT1 (p = 0.606) were not changed. The expression levels were normalized with that of GAPDH (control). (B) Immunoblots showed statistically significant (*p < 0.05; ** p < 0.01) reduction in expression of glutamate transporter proteins in the hippocampus of young cKO mice compared to their WT littermates (2 weeks of age n = 5 pairs: 3 pairs of males, 2 pairs of females; 4 weeks of age n =5 pairs: 2 pairs of males, 3 pairs of females; >12 weeks of age n = 5 pairs: 4 pairs of males, 1 pair of females). Note that EAAT2 was the most affected subtype and that the strongest reduction was in the youngest mice. In contrast, the vesicular glutamate transporter 1, VGLUT1, was hardly reduced (n = 3 pairs of WT and cKO mice).
4. Discussion
The exact role of Glul in the etiology of CNS disease, including epilepsy, is poorly understood due to the lack of viable and specific in vivo approaches. To overcome this hurdle, we selectively deleted Glul in the cerebral cortex of mice. This resulted in a viable Glul knockout model and proved that a selective loss of cortical Glul is, by itself, sufficient to cause epileptic seizures and progressive neurodegeneration resembling hippocampal sclerosis. The mutation had a high penetrance considering that the C57BL6 mouse strain used here is relatively resistant to epileptic seizures (McLin and Steward, 2006).
However, the first behavioral seizures or neurodegeneration were only evident in animals six weeks of age or older, even though changes in brain chemistry, glial cells and cortical blood vessels had been present for several weeks. Thus, seizures and neurodegeneration are likely not directly caused by the Glul deficiency, but rather occur as a consequence of a pathological process involving reactive astrocytes and impaired neurovascular coupling. These slowly developing effects of Glul deficiency are distinctly different from the rapid, excitotoxic syndrome with early seizures and increased mortality seen in EAAT2 knockout mice (Tanaka et al., 1997; Zhou et al., 2014).
It is well known that brain tissues from humans with epilepsy and from animal models of the disease contain reactive astrocytes and altered blood vessels (Eyo et al., 2017; Wolf et al., 2017). While these changes were originally regarded as secondary responses to injury, more recent studies have shown that reactive astrocytes can induce neuronal hyperexcitability and spontaneous seizures irrespective of the primary initiating event (Ortinski et al., 2010; Robel et al., 2015). In line with this gliopathy hypothesis of epilepsy (Verkhratsky et al., 2012; Coulter and Eid, 2012; Robel and Sontheimer, 2016), we observed reactive astrocytes weeks before the first seizure and neuron loss were detected.
Further, astrocytes, particularly their cytoplasmic processes, are integral elements of the neurovascular unit and can control vessel diameter (Attwell et al., 2010). Numerous changes in the molecular anatomy of microvessels in Glul-deficient brain regions in patients with TLE and animal models of the disorder have been demonstrated (Eid et al., 2016). However, whether these changes are a cause or consequence of seizures has remained unclear. Here we show, for the first time, that deletion of astrocytic Glul leads to increased vascular caliber and lack of vasodilation in response to CO2. These changes were present before the occurrence of seizures and significant neuron loss, and were probably induced by dysfunctional glutamate-ammonia handling as the only known reaction catalyzed by Glul is the production of glutamine; a process which also results in inactivation of glutamate and detoxification of ammonia (Eid et al., 2016). Here we demonstrated that deletion of Glul causes a major reduction in brain tissue levels of glutamine, glutamate and GABA. The reductions in glutamate and GABA probably reflect decreases in the neuronal transmitter pool as most tissue glutamate and GABA is in neurons (Danbolt, 2001). When glutamate cannot be amidated to glutamine, due to lack of Glul, glutamate is likely degraded via the glial tricarboxylic acid cycle rather than being transferred back to neurons as glutamine. Because glutamine is a precursor for the synthesis of neuronal glutamate and GABA, the lack of glutamine in the cKO cortex probably explains the low levels of the neurotransmitters. However, it is important to note that neurons may be able to synthesize glutamate de novo (Hassel and Bråthe, 2000) and that glutamate transporters in axon-terminals represent a mechanism for direct recycling independent of Glul (Danbolt et al., 2016b; Zhou et al., 2018). This may explain why in vitro electrophysiological recordings show that axon-terminals can maintain basal glutamatergic neurotransmission during low extracellular glutamine conditions, whereas glutamine was only required for sustained high-frequency firing (Tani et al., 2014). A reduction in total tissue glutamate does not necessarily indicate decreased extracellular glutamate. The episodes of wild-running and the occurrence of seizures in the Glul cKO mice are, however, suggestive of hyperexcitability, which may indicate increased extracellular glutamate. In support, these cKO mice have reductions in expression of glutamate transporters (EAAT1 and EAAT2), but these were moderate. Other factors that may contribute to increased excitability comprise reduced GABA levels and reductions in the GRIN2A glutamate receptor subunit. In addition, a conceptually distinct pathway of the control of cerebral blood flow mediated by glutamate transporters has been suggested, though it is unclear at the moment whether it is a direct or indirect effect (Petzold et al., 2008; Schummers et al., 2008). Further, the brain has no urea cycle, and detoxification of ammonia by Glul is thereby the only significant mechanism apart from removal via blood. Consequently, Glul deficiency probably raises ammonia levels. The similarity between the ammonium ion and the potassium ion implies that ammonium ions can impair potassium buffering by interfering with several transporter proteins and subsequently weaken neuronal inhibition (Rangroo Thrane et al., 2013). A raised [K+] could affect the function of smooth muscle via inward rectifier K+ channels and dilates the vessels (Knot et al., 1996). In conclusion, there are several factors that are likely to increase excitability, but we do not at this stage know their relative importance.
Even though the Glul cKO mice replicate several features of human TLE, there are notable differences. First, the mice are deficient in Glul in large portions of the cerebral cortex from early embryonic life, whereas patients with MTLE exhibit focal losses of Glul in the hippocampus and amygdala (Eid et al., 2004; van der Hel et al., 2005). Moreover, the Glul loss in human MTLE is probably not present during embryogenesis, but likely occurs secondary to another insult, later in life. Despite these differences, we have clearly demonstrated that loss of Glul in the cerebral cortex leads to epilepsy and progressive neurodegeneration, which are key features of human MTLE. We have also shown that loss of astrocytic Glul leads to significant changes in cerebrovascular physiology, suggesting new and important roles of Glul in neurovascular biology.
5. Conclusions
In summary, Glul deficiency does not immediately lead to seizures via a classical excitotoxic syndrome, but rather triggers a prolonged pathological process involving early glial and cerebrovascular changes before progressive neuron loss become apparent.
Highlights:
# Most mice lacking cortical Glul survive despite abnormal glutamate signaling
# Lack of cortical Glul causes late-onset epilepsy and progressive neurodegeneration
# Progressive astrogliosis and vascular abnormalities preceded neurodegeneration
Acknowledgements
We are grateful to the staff at the animal facility at The Norwegian Governmental Institute of Public Health for assisting with animal husbandry. This study was supported by the Norwegian Research Council (Grant no. 240844 to NCD), Novo Nordisk Fonden (Grant no. NNF14OC0010959 and NNF15OC0016528 to NCD), University of Oslo (SERTA to NCD; UNIFOR-FRIMED to NCD; PhD fellowship to HQX), and the National Institutes of Health (NIH, Grants number NS070824 to TE, RD and SEG; and MH067528 and NS052519 to FH). This work was also made possible by a grant from the National Center for Advancing Translational Sciences (NCATS; UL1 TR000142), a component of the NIH and the NIH Roadmap for Medical Research.
Abbreviations:
- CAS
Chemical Abstracts Service Registry Number
- CVR
cerebrovascular reactivity
- Glul
glutamate-ammonia ligase (a.k.a. glutamine synthetase, GS)
- cKO
conditional knockout
- Cre
Cyclization recombinase
- EAATs
excitatory amino acid transporters
- EEG
electroencephalogram
- EAAC1
excitatory amino acid carrier (EAAT3; slc1a1)
- GLAST
glutamate aspartate transporter (EAAT1; slc1a3)
- EAAT2
glutamate transporter 2 (EAAT2; slc1a2)
- GFAP
glial fibrillary acidic protein
- fMRI
functional MRI
- MRI
magnetic resonance imaging
- MTLE
mesial temporal lobe epilepsy
- NaPi
sodium phosphate buffer at pH 7.4
- NCS
newborn calf serum
- SDS
sodium dodecyl sulfate
- WT
wild-type in the sense that the Glul gene is intact
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no competing financial interests.
References
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA, 2010. Glial and neuronal control of brain blood flow. Nature 468, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard-Helary K, Ardourel M-Y, Hevor T, Cloix J-F, 2002. In vivo and in vitro glycogenic effects of methionine sulfoximine are different in two inbred strains of mice. Brain Res 929, 147–155. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, Varghese GI, Purcaro MJ, Motelow JE, Enev M, McNally KA, Levin AR, Hirsch LJ, Tikofsky R, Zubal IG, Paige AL, Spencer SS, 2009. Cortical and subcortical networks in human secondarily generalized tonic-clonic seizures. Brain 132, 999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Eid T, 2012. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 60, 1215–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg D, Ivanovic J, Hassel B, 2014. High extracellular concentration of excitatory amino acids glutamate and aspartate in human brain abscess. Neurochem Int 69, 41–47. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, 2001. Glutamate uptake. Prog Neurobiol 65, 1–105. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Furness DN, Zhou Y, 2016b. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem Int 98, 29–45. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Lehre KP, Dehnes Y, Chaudhry FA, Levy LM, 1998. Localization of transporters using transporter-specific antibodies. Methods Enzymol 296, 388–407. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Zhou Y, Furness DN, Holmseth S, 2016a. Strategies for immunohistochemical protein localization using antibodies: What did we learn from neurotransmitter transporters in glial cells and neurons. Glia 64, 2045–2064. [DOI] [PubMed] [Google Scholar]
- Eid T, Ghosh A, Wang Y, Beckstrom H, Zaveri HP, Lee T-SW, Lai JC, Malthankar-Phatak GH, de Lanerolle NC, 2008. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 131, 2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Gruenbaum SE, Dhaher R, Lee T-SW, Zhou Y, Danbolt NC, 2016. The Glutamate-Glutamine Cycle in Epilepsy. In: The Glutamate/GABA-Glutamine Cycle - Amino Acid Neurotransmitter Homeostasis Schousboe A and Sonnewald U, eds. Springer International Publishing. Switzerland. [Google Scholar]
- Eid T, Thomas MJ, Spencer DD, Rundenpran E, Lai JCK, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, Delanerolle NC, 2004. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37. [DOI] [PubMed] [Google Scholar]
- Eyo UB, Murugan M, Wu L-J, 2017. Microglia-Neuron Communication in Epilepsy. Glia 65, 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson RE, Carroll HP, Harris A, Maher ER, Selby PJ, Banks RE, 2005. Housekeeping proteins: a preliminary study illustrating some limitations as useful references in protein expression studies. Proteomics 5, 566–571. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR, 2002. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle J, Shahbeck N, Ibrahim K, Schmitt B, Scheer I, O’Gorman R, Chaudhry FA, Ben-Omran T, 2012. Glutamine supplementation in a child with inherited GS deficiency improves the clinical status and partially corrects the peripheral and central amino acid imbalance. Orphanet J Rare Dis 7, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel B, Bråthe A, 2000. Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J Neurosci 20, 1342–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Hakvoort TB, Vermeulen JL, Labruyere WT, De Waart DR, Van Der Hel WS, Ruijter JM, Uylings HB, Lamers WH, 2010. Glutamine synthetase deficiency in murine astrocytes results in neonatal death. Glia 58, 741–754. [DOI] [PubMed] [Google Scholar]
- He Y, Hakvoort TB, Vermeulen JL, Lamers WH, Van Roon MA, 2007. Glutamine synthetase is essential in early mouse embryogenesis. Dev Dyn 236, 1865–1875. [DOI] [PubMed] [Google Scholar]
- Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, Plachez C, Zhou Y, Furness DN, Bergles DE, Lehre KP, Danbolt NC, 2012b. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J Neurosci 32, 6000–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Scott HA, Real K, Lehre KP, Leergaard TB, Bjaalie JG, Danbolt NC, 2009. The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neuroscience 162, 1055–1071. [DOI] [PubMed] [Google Scholar]
- Holmseth S, Zhou Y, Follin-Arbelet VV, Lehre KP, Bergles DE, Danbolt NC, 2012a. Specificity controls for immunocytochemistry: the antigen pre-adsorption test can lead to inaccurate assessment of antibody specificity. J Histochem Cytochem 60, 174–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam K, Nicoll R, 2007. Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci 27, 9192–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ, Zimmermann PA, Nelson MT, 1996. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol 492 (Pt 2), 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK, 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG, 2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73, 56–65. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC, 1995. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15, 1835–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou Y, Danbolt NC, 2012. The rates of postmortem proteolysis of glutamate transporters differ dramatically between cells and between transporter subtypes. J Histochem Cytochem 60, 811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, 1953. The quantitative histochemistry of the brain. J Histochem Cytochem 1, 420–428. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Roberts NR, Leiner KY, Wu ML, Farr AL, Albers RW, 1954. The quantitative histochemistry of brain, III. ammon’s horn. J Biol Chem 207, 39–49. [PubMed] [Google Scholar]
- owry OH, Rosebrough NJ, Farr AL, Randall RJ, 1951. Protein measurement with the Folin phenol reagent. J Biol Chem 193, 265–275. [PubMed] [Google Scholar]
- Martinez-Hernandez A, Bell KP, Norenberg MD, 1977. Glutamine synthetase: glial localization in brain. Science 195, 1356–1358. [DOI] [PubMed] [Google Scholar]
- McLin JP, Steward O, 2006. Comparison of seizure phenotype and neurodegeneration induced by systemic kainic acid in inbred, outbred, and hybrid mouse strains. Eur J Neurosci 24, 2191–2202. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA, 2010. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 13, 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold GC, Albeanu DF, Sato TF, Murthy VN, 2008. Coupling of neural activity to blood flow in olfactory glomeruli is mediated by astrocytic pathways. Neuron 58, 897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ, Burnham WM, Gartner JG, Levitan D, 1973. Rates of motor seizure development in rats subjected to electrical brain stimulation: strain and inter-stimulation interval effects. Electroencephalogr Clin Neurophysiol 35, 553–556. [DOI] [PubMed] [Google Scholar]
- Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M, 2013. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med 19, 1643–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, Sutor B, Sontheimer H, 2015. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci 35, 3330–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Sontheimer H, 2016. Glia as drivers of abnormal neuronal activity. Nat Neurosci 19, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schummers J, Yu H, Sur M, 2008. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science 320, 1638–1643. [DOI] [PubMed] [Google Scholar]
- Shaw CA, Bains JS, 2002. Synergistic versus antagonistic actions of glutamate and glutathione: the role of excitotoxicity and oxidative stress in neuronal disease. Cell Mol Biol (Noisy-le-grand) 48, 127–136. [PubMed] [Google Scholar]
- Soriano P, 1997. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 124, 2691–2700. [DOI] [PubMed] [Google Scholar]
- Spodenkiewicz M, Diez-Fernandez C, Rufenacht V, Gemperle-Britschgi C, Haberle J, 2016. Minireview on Glutamine Synthetase Deficiency, an Ultra-Rare Inborn Error of Amino Acid Biosynthesis. Biology (Basel) 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K, 1997. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. [DOI] [PubMed] [Google Scholar]
- Tani H, Dulla CG, Farzampour Z, Taylor-Weiner A, Huguenard JR, Reimer RJ, 2014. A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron 81, 888–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hel WS, Notenboom RG, Bos IW, van Rijen PC, van Veelen CW, de Graan PN, 2005. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology 64, 326–333. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Sofroniew MV, Messing A, deLanerolle NC, Rempe D, Rodriguez JJ, Nedergaard M, 2012. Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro 44, art:e00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SA, Boddeke HW, Kettenmann H, 2017. Microglia in Physiology and Disease. Annu Rev Physiol 79, 619–643. [DOI] [PubMed] [Google Scholar]
- Wurst W, Joyner AL, 1999. Production of targeted embryonic stem cell clones. In: Gene Targeting: A Practical Approach Joyner AL, eds. Oxford University Press. New York. [Google Scholar]
- Zhou Y, Hassel B, Eid T, Danbolt NC, 2018. Axon-terminals expressing EAAT2 (GLT-1; Slc1a2) are common in the forebrain and not limited to the hippocampus. Neurochem Int, IN PRESS. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Holmseth S, Hua R, Lehre AC, Olofsson AM, Poblete-Naredo I, Kempson SA, Danbolt NC, 2012. The betaine-GABA transporter (BGT1, slc6a12) is predominantly expressed in the liver and at lower levels in the kidneys and at the brain surface. Am J Physiol Renal Physiol 302, F316–328. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Waanders LF, Holmseth S, Guo C, Berger UV, Li Y, Lehre A-C, Lehre KP, Danbolt NC, 2014. Proteome analysis and conditional deletion of the EAAT2 glutamate transporter provide evidence against a role of EAAT2 in pancreatic insulin secretion in mice. J Biol Chem 289, 1329–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]