

Abstract
Tandem mass tag (TMT)-based mass spectrometry (MS) enables deep proteomic profiling of more than 10,000 proteins in complex biological samples, but requires up to 100 micrograms protein in starting materials during a standard analysis. Here we present a streamlined protocol to quantify more than 9,000 proteins with 0.5 microgram protein per sample by 16-plex TMT coupled with two-dimensional liquid chromatography and tandem mass spectrometry (LC/LC-MS/MS). In this protocol, we optimized multiple conditions to reduce sample loss, including processing each sample in a single tube to minimize surface adsorption, increasing digestion enzymes to shorten proteolysis and function as carriers, eliminating a desalting step between digestion and TMT labeling, and developing miniaturized basic pH LC for prefractionation. By profiling 16 identical human brain tissue samples of Alzheimer’s disease (AD), vascular dementia (VaD) and non-dementia controls, we directly compared this new microgram-scale protocol to the standard-scale protocol, quantifying 9,116 and 10,869 proteins, respectively. Importantly, bioinformatics analysis indicated the microgram-scale protocol had adequate sensitivity and reproducibility to detect differentially expressed proteins in disease-related pathways. Thus, this newly developed protocol is of general application for deep proteomics analysis of biological and clinical samples at sub-microgram levels.
Keywords: proteomics, proteome, mass spectrometry, isobaric labeling, TMT, liquid chromatography, nanoscale, single cell proteomics, Alzheimer’s disease, vascular dementia
INTRODUCTION
Recent advances in mass spectrometry have allowed for near-global proteomic profiling (more than 10,000 proteins) of a variety of biological samples.1,2 Shotgun/bottom-up proteomics, based on the analysis of enzymatic peptides instead of intact proteins, takes advantage of the fact that peptides are much more uniform and compatible with liquid chromatography and MS analysis than proteins.3,4 Untargeted quantitative MS methods include DDA/DIA-based label-free quantification5,6 and stable isotope labeling, such as SILAC,7 dimethyl labeling,8 and isobaric labeling strategies (e.g. iTRAQ,9 TMT,10 and DiLeu11). Because of its high multiplexing capabilities (up to 16-plex12) and deep proteome coverage, TMT has become one of the most commonly used quantitative MS strategies.13,14 However, the accuracy of TMT measurement is often affected by ratio compression caused by co-eluted and co-fragmented ions.15 This problem has been mostly resolved by recent workflow improvements using the combination of numerous approaches including extensive LC fractionation,16,17 ion gas-phase purification,18 narrow-window MS2 isolation,19 multistage MS3,20,21 MS2 complement TMT ions,22 and computational correction during data analysis.23
Routine TMT proteomic analysis requires micrograms of protein in starting materials (e.g. 100 μg protein per sample in the manufacturer’s standard protocol). This standard-scale input prevents deep proteomic profiling of samples of minute quantity, such as proteomes of post-translational modifications at low stoichiometric levels,24 limited cells isolated by fluorescence activated cell sorting, and specimens captured by laser capture microdissection.25 These samples yield total protein at low microgram or even nanogram levels. More recently, the idea of single-cell proteomics has been proposed to quantify complex proteomes at sub-nanogram levels in individual cells.26 To minimize sample loss and enhance sensitivity when analyzing these sparse samples, some nanoproteomic strategies have been developed to reduce adsorptive loss by carrier proteins: single tube processing, online (on-column) processing, and automation (e.g. nanoPOTS).27–35 Upon these improvements, ~1,000 to 7,000 proteins can be quantified from hundreds to thousands of cells. For example, a stagetip-based on-column TMT labeling method has been reported to quantify about 7,000 proteins from ~2 μg protein samples.29 Thus, it is important to further develop these methods to achieve proteomic coverage similar to routine standard-scale studies.
Here we present a newly designed microgram-scale pipeline of TMT-LC/LC-MS/MS for deep protein identification and quantification of over 9,000 proteins. This method can process 0.5 μg protein per sample using surplus enzymes to expedite digestion and work as a carrier. The desalting step before TMT labeling is skipped to reduce sample loss. The TMT-labeled peptides are pooled and further resolved into 30 concatenated fractions to increase the depth of proteome coverage. This microgram-scale pipeline shows comparable performance to the routine standard-scale pipeline (100 μg protein) when analyzing identical clinical brain samples.
METHODS
Sample extraction and protein quantification
The analysis of human frontal cortex samples from the Brain and Body Donation Program of the Banner Sun Health Research Institute was approved by institutional review board. Three AD cases, seven VaD cases, four non-dementia controls, and two internal reference samples (16 total) were included in this study (Supporting Table S1). The internal reference samples were mixed brain samples to function as universal standards for batch normalization, although only one batch result was reported here. Brain tissue samples were harvested and lysed with a denaturing buffer (50 mM HEPES, pH 8.5, 8 M urea, 0.5% sodium deoxycholate (NaDOC), and 10 μl buffer per mg tissue) with a Bullet Blender (Next Advance).36 The addition of ionic detergents (e.g. NaDOC and CHAPS) in the urea denaturing buffer may enhance the solubilization of hydrophobic proteins, and a recent study supports better performance of NaDOC than CHAPS.37 Protein amount was quantified by a BCA assay (Thermo Fisher Scientific) and confirmed by a short SDS-PAGE gel followed by Coomassie staining assay.38,39 The lysates were diluted and aliquoted for microgram-scale and standard-scale TMT studies.
Optimization of TMT labeling with TMT10 and TMTpro zero reagents
The TMT reagents (Thermo Fisher Scientific) were dissolved in anhydrous acetonitrile (ACN). We used TMT10 to test the reactivity of buffer components (see Supporting methods), and TMTpro zero to examine TMT/tissue ratios. TMTpro zero was added to multiple equal aliquots of a digested human brain sample (~0.5 μg protein from ~5 μg tissue, assuming 10% protein content) at different TMT/tissue ratios (w/w) for 30 min at 21 °C. The reactions were quenched by hydroxylamine (Thermo Fisher Scientific) for 15 min. The labeled and non-labeled samples were then analyzed by LC-MS/MS. TMT labeling efficiency was calculated by comparing the intensities of unlabeled precursor ions in each labeled sample with those in the unlabeled sample.
Microgram-scale digestion, TMT labeling and prefractionation
Each protein sample was processed in one protein LoBind microcentrifuge tube (Eppendorf), containing 0.5 μg protein in 0.5 μl lysis buffer. The protein was first digested with Lys-C (50 ng in 0.5 μl lysis buffer, Wako Chemicals) for 10 min at 21 °C, followed by dilution with a trypsin-containing buffer (2.5 μg in 3 μl of 50 mM HEPES, pH 8.5, Promega) and incubation for 30 min at 21 °C. The digested tissue lysates were directly labeled with 16-plex TMT reagents (15 μg in 1 μl of ACN, at a TMT/tissue ratio of 3:1, w/w) for 30 min at 21 °C, and quenched with 0.5 μl of 5% hydroxylamine for 15 min. The quenched samples (~5.5 μl) were equally pooled (~88 μl containing 18% ACN), diluted with 1% TFA to acidify and reduce ACN to ~5% (final 320 μl). The final pH was confirmed by pH paper. The acidified mix was centrifuged at 21,000 × g for 5 min, and the supernatant was desalted using a reported protocol.40 The peptides were washed three times with 10 x bed volumes of 5% ACN plus 0.1% TFA, eluted with 5 x bed volumes of 60% ACN plus 0.1% TFA, and finally dried by SpeedVac.
Prefractionation was performed by basic pH RPLC using a microscale HPLC system (Agilent 1220) coupled with a 3-way flow splitter (IDEX Health & Science). The desalted TMT-labeled peptides were resuspended in solvent A (10 mM ammonium formate, pH 8.0) and centrifuged at 21,000 × g for 5 min. With the flow splitter closed, the supernatant was loaded onto an XBridge C18 column (1 mm × 50 mm, 3.5 μm beads, Waters) at a flow rate of 20 μl/min by 95% of solvent A. After loading, the flow splitter was opened and the peptides were eluted with a 45 min gradient starting from 15-35% of solvent B (90% acetonitrile, 10 mM ammonium formate, pH 8.0). The HPLC was operated at 500 μl/min and the column was eluted at 20 μl/min. 90 fractions were manually collected by microvolume inserts (J.G Finneran Associates) in every 30 sec and concatenated back into 30 fractions. The peptide levels in fractions were measured by OD205 (absorption wavelength for a peptide bond). All fractions were dried by SpeedVac.
Standard-scale TMT labeling and prefractionation
Standard-scale protein samples were processed by the previously reported method.40 Each protein sample (100 μg protein) was digested, desalted, TMT-labeled and pooled. The pooled sample was desalted again, dried, and dissolved in solvent A for basic pH RPLC. The sample was fractionated by an offline XBridge C18 column (4.6 mm × 250 mm, 3.5 μm beads, Waters) with a 160 min gradient of 15-50% solvent B. A total of 40 concatenated fractions were collected and dried by SpeedVac.
Acidic pH RPLC-MS/MS analysis and data processing
The acidic pH LC-MS/MS analysis was performed on an UltiMate ™ 3000 RSLCnano system and Q Exactive HF Orbitrap MS (Thermo Fisher Scientific). While the microgram-scale analysis used a 50 μm i.d. x 50 cm LC column (1.7 μm C18 resin, CoAnn Technologies, ~400 bar under 125 nl/min of solvent A), the standard-scale analysis used a self-packed 75 μm i.d. x 18 cm LC column (1.9 μm C18 resin, Dr. Maisch GmbH, ~160 bar under 250 nl/min of solvent A). All fractions were solubilized in 5% formic acid, loaded sequentially on the column (heated at 65 °C to reduce backpressure), and eluted by a 20-50% gradient (solvent A: 0.2% formic acid, 5% DMSO; solvent B: solvent A plus 65% ACN) in 90 min. The MS spectra were collected with m/z range of 410–1,600, resolution of 60,000, AGC of 1 × 106, and maximum injection time of 50 ms. Selected precursor ions were fragmented by HCD with 32% normalized collision energy. The resulting product ions were acquired at resolution of 60,000, AGC of 1 × 105 and maximum injection time of ~110 ms. Dynamic exclusion was set at 15 s to reduce repeat sequencing.
The JUMP software suite41,42 was used for protein identification and quantification. Search parameters included precursor ion and product ion mass tolerance (10 ppm), maximal modification sites (n = 3), full trypticity, maximal missed cleavage (n = 2), static modification of TMT tag (+304.20715), methionine oxidation dynamic modification (+15.99491), and cysteine carbamidomethyl static modification (+57.02146) if the residue was alkylated with iodoacetamide. To achieve protein FDR <1%, the peptide assignments were filtered by mass accuracy and matching scores (Jscore and ΔJn). If one peptide was shared by multiple homologous proteins, the peptide was assigned to the protein with highest PSM number.
The quantification of accepted peptides/proteins was based on TMT reporter ion intensities using the reported method and software.23 Microgram-scale and standard-scale datasets were first normalized to remove batch bias (see Supporting methods). Principal component analysis (PCA) was performed by an R statistical analysis package to illustrate the differences among AD, VaD, and ND groups, after removing two outliers from the control and VaD samples. Differential expression (DE) analysis was performed in the comparison between ND and VaD groups by following steps: (i) p values were calculated using the moderated t-test and a threshold of 0.01 was applied; (ii) fold change based on the standard deviation (z-score > 2) was used to filter the results to generate the final list of DE proteins; (iii) a permutation analysis (n = 1,000 permutations) was performed to estimate the final FDR of the accepted DE proteins (less than 10%).43
RESULTS AND DISCUSSION
Design of a microgram-scale TMT-LC/LC-MS/MS pipeline
The standard TMT-based pipeline (Fig. 1A) can be used to perform deep proteomic profiling of complex biological samples at a standard-scale level (~100 μg protein per sample in the protocol of the manufacturer). When the initial sample amount is limited to a microgram-scale level (around 1 μg protein per sample), the standard pipeline cannot be directly applied, largely because of possible sample loss during processing and limited sensitivity of routine LC and MS settings. Therefore, we designed a protocol for handling these microgram-scale samples (Fig. 1B).
Figure 1.
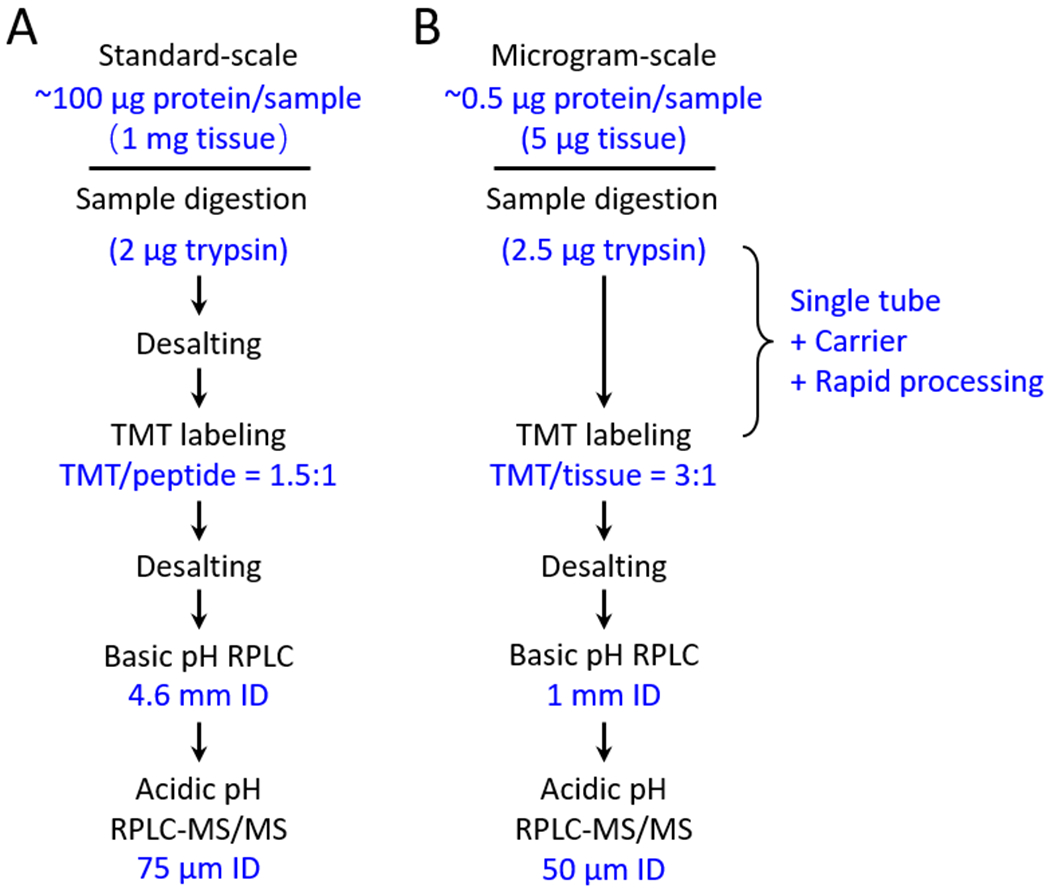
Workflow of standard-scale and microgram-scale of TMT-LC/LC-MS/MS. (A) Standard-scale pipeline. Digested proteins (peptides) are desalted and labeled by TMT (TMT/peptide ratio of 1.5:1). The pooled samples are then desalted and analyzed by LC/LC-MS/MS. (B) Microgram-scale pipeline. Proteins in the tissue lysate are digested into peptides with excess trypsin to shorten digestion time. Without desalting, the digested tissue lysate is directly labeled by TMT (TMT/tissue ratio of 3:1). The pooled sample is analyzed by LC/LC-MS/MS with highly sensitive LC columns.
First, the microgram-scale pipeline aims to limit sample contact with tubes, so each sample is processed in a single tube until TMT labeling and pooling. During the basic pH RPLC prefractionation, the pooled samples are directly collected in microvolume inserts that are loaded in the autosampler for acidic pH RPLC-MS/MS, without the need of sample transfer. In addition, we digest the protein samples with a high level of trypsin (2.5 μg per sample) that not only expedites the digestion to as short as 30 min, but also functions as a carrier (Fig. S1), while the standard TMT method requires at least 3 h for trypsin digestion.
Second, our microgram-scale pipeline removes the desalting step after enzymatic digestion, even though this step is commonly used for purifying peptides and removing non-peptide components that are often TMT-reactive. The desalting step may cause the loss of ~30% of peptides.38 When removing this desalting step, we expect to improve sample recovery but still retain TMT-reactive non-peptide components. Therefore, sufficient TMT reagents are required to fully label these samples. This strategy was reported in the SCoPE-MS method for single-cell proteomics.44
Third, to increase the sensitivity of the LC setting, a small-sized C18 column is used to match the reduced input during basic pH RPLC prefractionation. To run the small column in a standard microscale HPLC, we insert a flow splitter to modulate the flow rate (see METHODS). Moreover, a smaller, nanoscale column (i.e. 50 μm i.d.) instead of a regular 75 μm i.d. column is implemented for acidic pH RPLC-MS/MS. In summary, these adjustments in the microgram-scale pipeline are expected to increase detection sensitivity while maintaining the separation power of LC/LC-MS/MS for deep proteomic analysis.
Optimization of tissue lysis and TMT labeling in the microgram-scale pipeline
In the new pipeline, because the TMT reagents directly label undesalted lysate digest, the lysis buffer must be compatible with the TMT reaction. We tested if any components in the lysis buffer react with TMT reagents, which are claimed to form covalent bonds with an amine group (Fig. 2A). Although there is no amine group in the commonly used lysis buffer (e.g. fresh urea, HEPES, NaDOC, and DTT), we examined these components individually with a TMT reagent (Fig. S2A) and confirmed that the former three compounds indeed cannot react with TMT (Fig. S2B), but that DTT can be conjugated to TMT effectively by the thiol group (Fig. 2B). Therefore, we removed DTT from the lysis buffer and also skipped the alkylation step by iodoacetamide. As Cys-containing peptides occupy only 2% of total tryptic peptides, eliminating the reduction/alkylation step does not significantly affect the outcome of proteomic analysis.45,46
Figure 2.
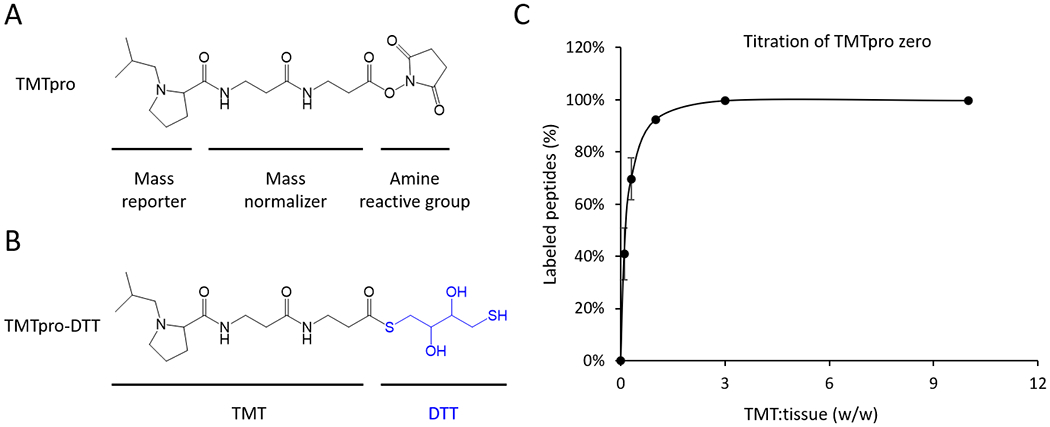
TMT reactivity of lysis buffer components and TMTpro titration. (A) Structure of TMTpro. (B) Hypothetical product of TMTpro-DTT, showing that TMT reacts with the thiol group in DTT. (C) Titration of the TMTpro zero reagent. Human brain tissue lysate (5 μg tissue containing ~0.5 μg protein) was digested and directly labeled by TMTpro zero with various TMT/tissue ratios (w/w in triplicates) for 30 min. The labeled peptides were analyzed by LC-MS/MS. The percentage of labeled peptides was defined by quantifying unlabeled peptides in each run, compared to the control run (i.e. unlabeled sample). The titration experiment was replicated (n = 3) with the standard deviation of the mean shown.
In addition to the buffer, the digested tissue lysate also includes TMT-reactive metabolites47, peptides, and added enzymes (i.e. 50 ng Lys-C and 2.5 μg trypsin). We determined the optimal amount of TMTpro zero reagent to fully label peptides in a titration experiment for TMT/tissue (w/w) ratios of 0.1:1, 0.3:1, 1:1, 3:1, and 10:1. Because of the sample complexity before TMT labeling, TMT/tissue ratio was used in the microgram-scale pipeline instead of the TMT/peptide ratio in the standard-scale pipeline. Given that protein contributes about 10% of tissue weight,48 the 0.1:1 TMT/tissue ratio is equivalent to a 1:1 TMT/peptide ratio, close to the 1.5:1 TMT/peptide ratio in the standard standard-scale pipeline.40 As anticipated, the 0.1:1 TMT/tissue ratio was insufficient for labeling (i.e. only 40% of peptides were labeled), the 1:1 ratio led to ~90% labeling efficiency, and the 3:1 ratio achieved full labeling (Fig. 2C). Thus, the TMT/tissue ratio of 3:1 (e.g. 15 μg TMT reagent for 5 μg tissue) is recommended in this microgram-scale pipeline. Still, labeling efficiency should be always monitored in different experiments.
Implementation of modified LC/LC-MS/MS for microgram-scale profiling
Basic pH RPLC prefractionation of TMT-labeled samples not only enhances peptide separation to deepen the proteome coverage,49,50 but also reduces the complexity in each fraction to alleviate ratio compression.23 In the microgram-scale pipeline, we adjusted LC/LC settings to fit the low loading amount of the sample. During basic pH RPLC, a 1 mm × 50 mm C18 column (bed volume of ~39 μl) was used for collecting concatenated fractions. A low-cost flow splitter was used to run the column on a regular microscale HPLC (Fig. 3). With the flow splitter, the HPLC pump was operated at 530 μl/min, while the column had at a steady flow rate of 20 μl/min. The flow splitting system greatly reduces delay due to HPLC dead volume, and improves the gradient accuracy under a regular high flow rate.51,52 For the acidic pH LC-MS/MS, we selected the 50 μm i.d. column to balance the sensitivity and robustness. Although narrow bore columns (e.g. 20-30 μm i.d.) provide a higher sensitivity for LC-MS/MS analysis, their performance is often compromised by technical challenges.53–55
Figure 3.
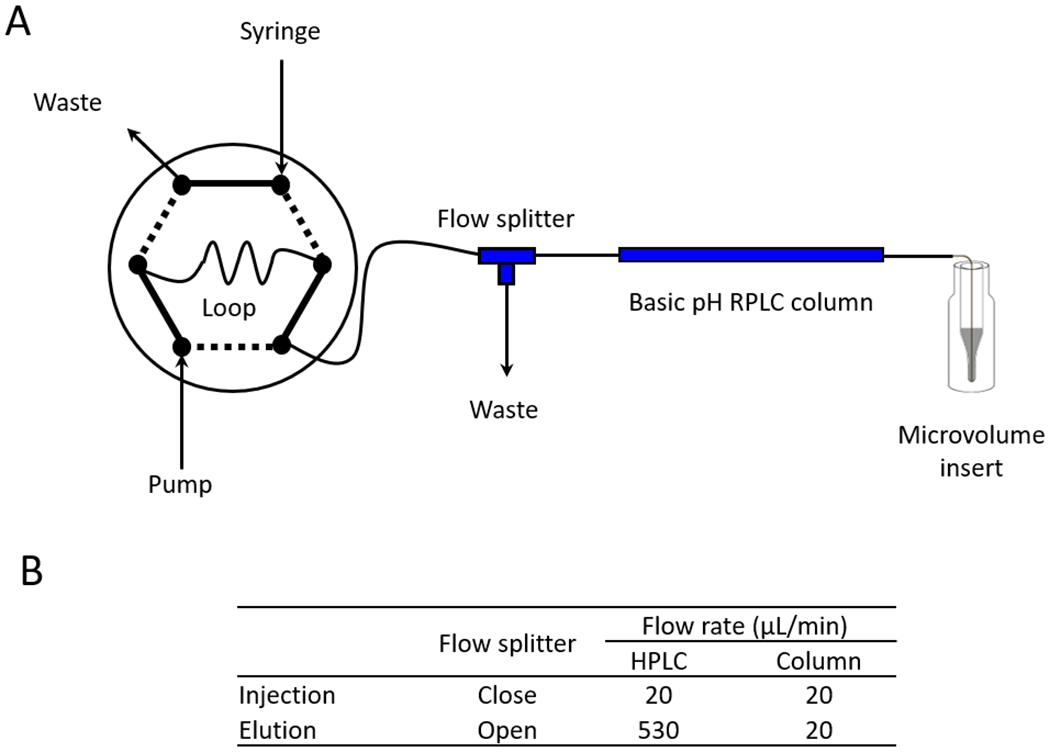
A flow splitter implemented in microscale HPLC. (A) Integrated scheme including a sample loading loop, HPLC pump, a reverse-phase LC column, and one flow splitter with a waste line (63.5 μm i.d. x 60 cm tubing). The pooled TMT sample was first loaded onto a 20 μl sample loop, and injected to the column at flow rate of 20 μl/min for 20 min with the splitter closed. After injection, the sample was eluted with the splitter open, which allowed for 20 μl/min column flow rate using 530 μl/min HPLC flow rate. (B) The flow rates with the flow splitter at different positions.
After setting up the basic steps for the microgram-scale protocol, it is not straightforward to determine the minimal amount of input for a deep proteome analysis. We performed a pilot 11-plex TMT experiment with 0.1 μg protein per sample and surplus trypsin, and collected 10 fractions during basic pH RPLC for the LC-MS/MS analysis, identifying 28,302 unique peptides from 4,928 proteins. We also performed another analysis in parallel using regular amount of trypsin, identifying 22,591 unique peptides from 3,988 proteins. The data confirmed that the use of a high level of trypsin improves proteome coverage, possibly due to its speedy digestion and carrier effect. To pursue deep proteome profiling, we increased the amount of starting sample from 0.1 to 0.5 μg, and collected 30 fractions during basic pH RPLC in a 16-plex TMT pipeline.
Finally, the optimized pipeline was applied to whole proteome analysis of 16 human brain tissue specimens (0.5 μg protein each sample), including Alzheimer’s disease (AD, 3 cases), vascular dementia (VaD, 7 cases), and non-dementia control (ND, 4 cases) (shown as samples 3-16, Supporting Table S1). Two internal reference samples (samples 1-2) were also included for normalizing batch-to-batch variations, which were not further investigated as only one batch was analyzed here (see METHODS). A total of 1,080,421 high-resolution MS/MS scans were collected and 196,498 peptide-spectrum matches (PSMs) were identified, corresponding to 82,981 unique peptides and 9,116 unique proteins (Supporting Table S2), with a protein false discovery rate (FDR) less than 1%. To our knowledge, this is one of the deepest proteomic studies from microgram-scale starting materials. The throughput of this analysis may be further increased by combining the TMT 11-plex and 16-plex reagents to analyze up to 27 samples in a single experiment.40
Comparison of microgram-scale and standard-scale TMT datasets
To further evaluate the performance of the microgram-scale protocol, we analyzed the same set of 16 samples using regular standard-scale protocol (Supporting Table S3), and compared the identified PSMs peptides, and proteins between the two datasets (Fig. 4A). The PSMs, peptides and proteins identified by the microgram-scale protocol were 57%, 63%, and 84% of those identified by the standard-scale protocol, respectively. As expected, the majority of peptides (66%) and proteins (90%) analyzed in the microgram-scale experiment were re-identified by the standard-scale experiment (Fig. 4B, 4C). The difference in peptide/protein identification may result from different protocols and the known undersampling issue of data-dependent acquisition in shotgun mass spectrometry.5
Figure 4.
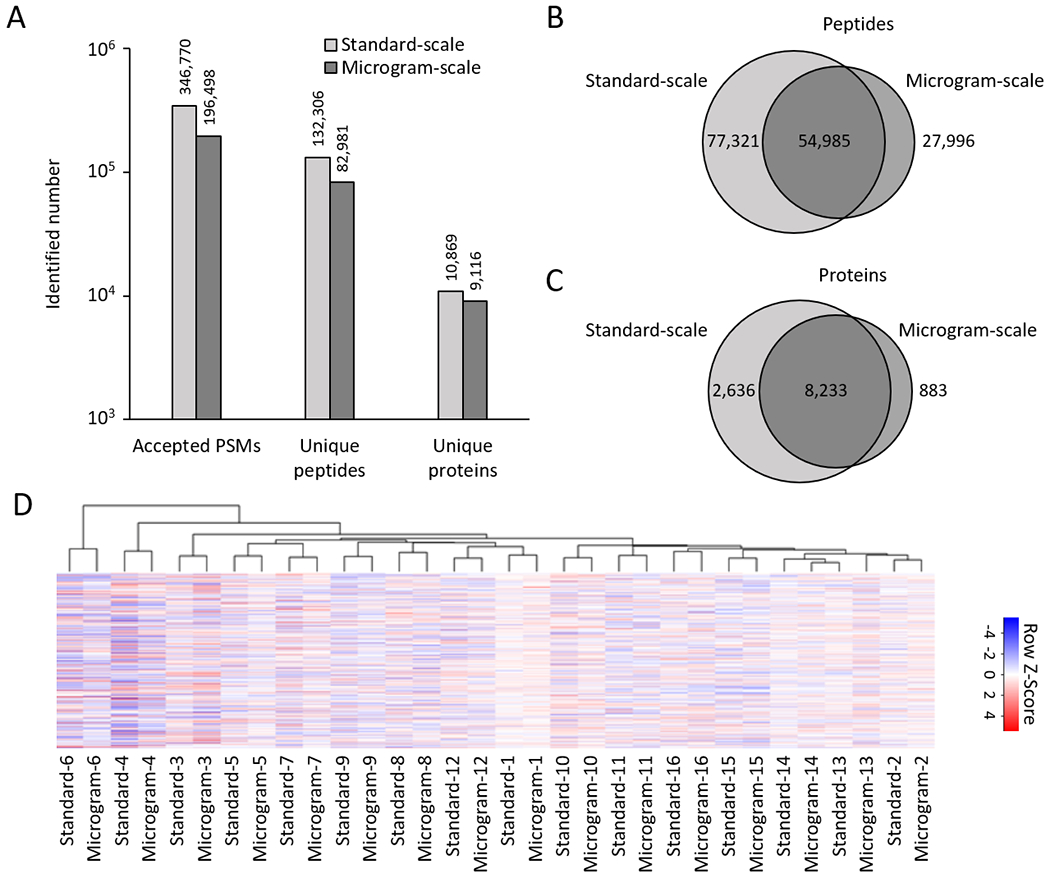
Microgram-scale protocol achieved comparable proteome profiling depth compared to the standard-scale method. (A) Comparison of accepted peptide-spectrum-match (PSM), peptide, and protein numbers. (B-C) Overlaps of identified proteins and peptides from the microgram- and standard-scale datasets. (D) Cluster heatmap of 32 samples analyzed by the microgram- and standard-scale methods using the top 20% most variable proteins.
We also analyzed the variations of protein quantification in the two datasets by evaluating standard deviation of null comparisons, such as intragroup pairwise comparisons (e.g. AD versus AD, and ND versus ND). As the outcome of protein quantification may be affected by PSM number assigned to each protein, we plotted PSM distribution of all proteins. In the microgram-scale dataset, 13.1% and 8.5% of proteins were matched by one and two PSMs, respectively. In the standard-scale dataset, the one- and two-PSM proteins dropped to 4.3% and 4.5%, respectively (Fig. S3A). As expected, one- and two-PSM proteins in the microgram-scale dataset had higher standard deviations than their overlapped proteins in the standard-scale dataset (Fig. S3B). Once PSM number increased to 3 or more, standard deviations of overlapped proteins in both datasets became comparable. The results implicate that the quality of protein quantification of the microgram-scale analysis is slightly impacted by an increased percentage of one- and two-PSM proteins.
We then evaluated the reproducibility of the microgram- and standard-scale datasets. After merging the two datasets (Fig. S4), hierarchical clustering analysis clearly indicated that all identical samples (i.e. technical replicates) were essentially grouped together, regardless of the pipeline used (Fig. 4D). These comparisons strongly support that the microgram-scale pipeline can generate reproducible results with high proteome coverage, similar to that of the standard-scale pipeline.
We further explored the biological implications of our large-scale proteomic analysis. First, the principal component analysis (PCA) was able to separate all samples into three distinct groups, consistent with the clinical diagnosis of AD, VaD, and ND (Fig. 5A). Considering the small number of AD cases (n = 3) and large heterogeneity of human clinical samples, it would be difficult to perform differential expression (DE) analysis with statistical rigor. We then selected the top 20 DE proteins from a recent AD proteomics study36 and found that 17 out of 20 proteins were identified and consistently altered in both microgram- and standard-scale datasets (Fig. 5B). For the VaD samples (n = 7), we performed a DE analysis to investigate protein dysregulation compared to the non-dementia controls. The microgram- and standard-scale datasets were combined together and filtered with a previously reported statistical method based on probability value and Z-score,36 identifying 96 DE proteins with false discovery rate (FDR) below 10% (Supporting Table S4). Among these DE proteins, IGKC, FGA, ATP5O, ATP5A1 and IDH2 were reported to be relevant to VaD.56 The DE proteins were divided into different functional groups by annotation including blood microparticles, immunity and mitochondrial function (Fig. 5C). Interestingly, some plasma microparticle components have been proposed as potential biomarkers of vascular injury/dysfunction and influenced by AD disease status, although the molecular mechanism has not been well studied.57 In our results, 15 upregulated proteins are related to the function of blood microparticles, providing new evidence to support the role of blood microparticles in vascular pathogenesis and cognitive decline. Importantly, the DE protein pattern from the microgram-scale dataset is highly similar to that from the standard-scale dataset, further highlighting the reproducibility of our new microgram-scale pipeline.
Figure 5.
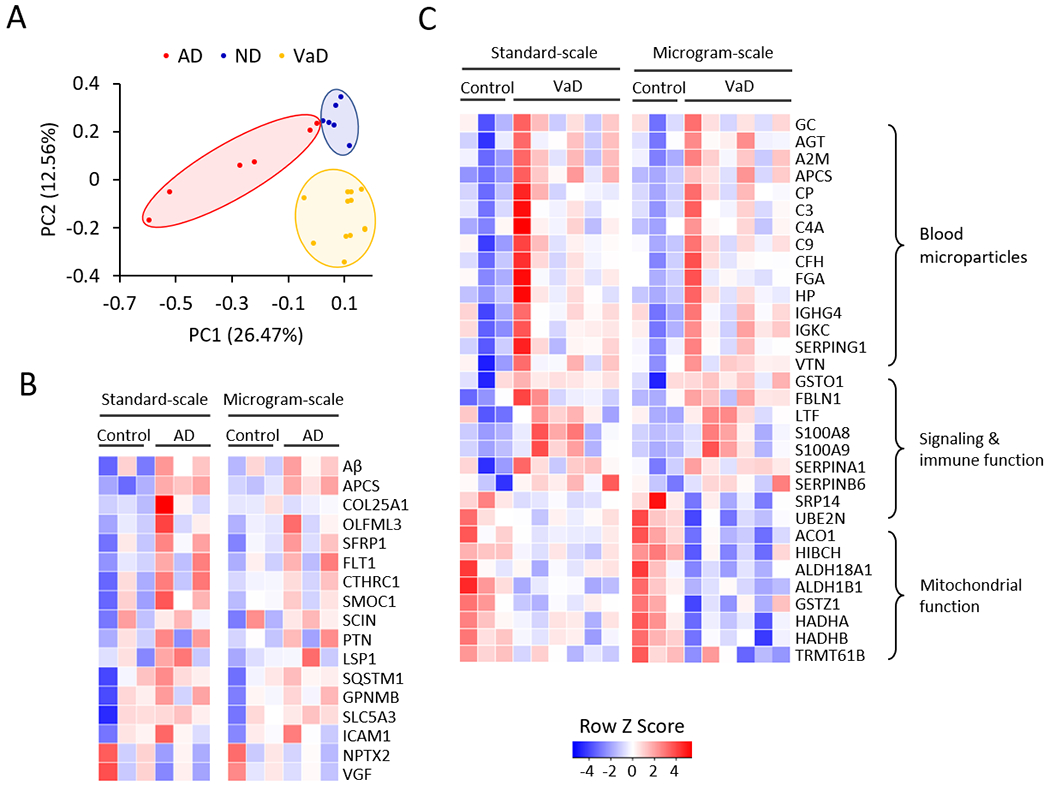
Bioinformatic analysis indicated the deregulated proteins in AD and VaD. (A) Principal component analysis (PCA) of identified proteins. Relative expression of all proteins was used as input of PCA, using an R statistical analysis package. (B) Heatmap of selected dysregulated AD proteins by standard- and microgram-scale TMT methods. (C) Heatmap of some DE proteins between the control and VaD samples, clustered in different pathways. Each protein was represented by a colored box after Z-score conversion.
CONCLUSIONS
We have designed and tested a new TMT-based LC/LC-MS/MS pipeline for deep proteomic analysis of protein samples at the sub-microgram scale, and compared the performance of this new pipeline with the routine standard-scale TMT pipeline. In this microgram-scale pipeline, we simplified the protocol and adjusted LC and MS settings to reduce sample loss and improve sensitivity, resulting in successful identification and quantification of 82,981 unique peptides and 9,116 unique proteins. The results obtained from the new pipeline were not only consistent with those from the conventional standard-scale pipeline, but also re-identified dysregulated proteins in AD from previous publications and revealed novel protein changes associated with VaD. Although the current experiment was performed as a proof-of-principle study, it is possible to harvest a small amount of cells/tissue by cell sorting or laser capture microdissection.58 Assuming 0.2 ng protein in one average mammalian cell (e.g. HeLa cell),59 2,500 cells can yield 0.5 μg protein, which is practical for collecting diverse cell types. Thus, we believe that this microgram-scale pipeline can be easily implemented in regular MS labs for deep global profiling of minute samples without additional instrumentation requirements.
Supplementary Material
Supporting methods; Figure S1,High amount of trypsin in the microgram-scale protocol is sufficient to digest proteins in 30 min; Figure S2, DTT is TMT-reactive in the reactivity assay; Figure S3, Analysis of PSM distribution and quantification variation of the standard- and microgram-scale datasets; Figure S4, Boxplot to show normalized intensities of the standard- and microgram-scale datasets; Supporting references.
Table S1, Summary of human cases used for microgram-scale and standard-scale proteomic studies; Table S2, Whole proteome profiling of AD, ND and VaD postmortem human brain tissues by TMT-LC/LC-MS/MS by the microgram-scale protocol; Table S3, Whole proteome profiling of AD, ND and VaD postmortem human brain tissues by TMT-LC/LC-MS/MS by the standard-scale protocol; Table S4; Differentially expressed (DE) proteins between normal control and Vascular Dementia (VaD) postmortem brain samples.
Acknowledgments
We thank Dr. Yuxin Li and other lab and center members for insightful suggestions and Ryan Z. Peng for editorial assistance. This work was partially supported by NIH grants R01GM114260 (J.P.), R01AG047928 (J.P.), R01AG053987 (J.P.), RF1AG064909 (J.P.), U54NS110435 (J.P.), U24NS072026 (T.G.B.), and P30AG19610 (T.G.B.), the Arizona Department of Health Services (contract 211002; T.G.B.), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901, and 1001; T.G.B.), and ALSAC (American Lebanese Syrian Associated Charities). MS analysis was performed in the Center of Proteomics and Metabolomics at St. Jude Children’s Research Hospital, partially supported by NIH Cancer Center Support Grant P30CA021765. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH.
ABBREVIATIONS:
- RPLC
reverse-phase liquid chromatography
- MS
mass spectrometry
- TMT
tandem mass tag
- AD
Alzheimer’s disease
- VaD
vascular dementia
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Aebersold R; Mann M Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537 (7620), 347–55. [DOI] [PubMed] [Google Scholar]
- (2).Altelaar AM; Munoz J; Heck AJ Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2013, 14 (1), 35–48. [DOI] [PubMed] [Google Scholar]
- (3).Zhang Y; Fonslow BR; Shan B; Baek MC; Yates JR 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113 (4), 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gillet LC; Leitner A; Aebersold R Mass spectrometry applied to bottom-up proteomics: entering the high-throughput era for hypothesis testing. Annual Review of Analytical Chemistry 2016, 9 (1), 449–472. [DOI] [PubMed] [Google Scholar]
- (5).Liu H; Sadygov RG; Yates JR 3rd. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76 (14), 4193–201. [DOI] [PubMed] [Google Scholar]
- (6).Wang W; Zhou H; Lin H; Roy S; Shaler TA; Hill LR; Norton S; Kumar P; Anderle M; Becker CH Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal. Chem. 2003, 75 (18), 4818–26. [DOI] [PubMed] [Google Scholar]
- (7).Mann M Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7 (12), 952–8. [DOI] [PubMed] [Google Scholar]
- (8).Hsu J-L; Chen S-H Stable isotope dimethyl labelling for quantitative proteomics and beyond. Philos. Trans. R. Soc., A 2016, 374 (2079), 20150364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wiese S; Reidegeld KA; Meyer HE; Warscheid B Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7 (3), 340–350. [DOI] [PubMed] [Google Scholar]
- (10).Thompson A; Schafer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75 (8), 1895–1904. [DOI] [PubMed] [Google Scholar]
- (11).Frost DC; Greer T; Li L High-resolution enabled 12-plex DiLeu isobaric tags for quantitative proteomics. Anal. Chem. 2015, 87 (3), 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Li J; Van Vranken JG; Vaites LP; Schweppe DK; Huttlin EL; Etienne C; Nandhikonda P; Viner R; Robitaille AM; Thompson AH TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17 (4), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rauniyar N; Yates JR 3rd. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13 (12), 5293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hogrebe A; von Stechow L; Bekker-Jensen DB; Weinert BT; Kelstrup CD; Olsen JV Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9 (1), 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Savitski MM; Mathieson T; Zinn N; Sweetman G; Doce C; Becher I; Pachl F; Kuster B; Bantscheff M Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J. Proteome Res. 2013, 12 (8), 3586–98. [DOI] [PubMed] [Google Scholar]
- (16).Ow SY; Salim M; Noirel J; Evans C; Wright PC Minimising iTRAQ ratio compression through understanding LC–MS elution dependence and high–resolution HILIC fractionation. Proteomics 2011, 11 (11), 2341–2346. [DOI] [PubMed] [Google Scholar]
- (17).Zhou F; Lu Y; Ficarro SB; Adelmant G; Jiang W; Luckey CJ; Marto JA Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun. 2013, 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wenger CD; Lee MV; Hebert AS; McAlister GC; Phanstiel DH; Westphall MS; Coon JJ Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 2011, 8 (11), 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Savitski MM; Sweetman G; Askenazi M; Marto JA; Lang M; Zinn N; Bantscheff M Delayed fragmentation and optimized isolation width settings for improvement of protein identification and accuracy of isobaric mass tag quantification on Orbitrap-type mass spectrometers. Anal. Chem. 2011, 83 (23), 8959–8967. [DOI] [PubMed] [Google Scholar]
- (20).Ting L; Rad R; Gygi SP; Haas W MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 2011, 8 (11), 937–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).McAlister GC; Nusinow DP; Jedrychowski MP; Wühr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86 (14), 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wühr M; Haas W; McAlister GC; Peshkin L; Rad R; Kirschner MW; Gygi SP Accurate multiplexed proteomics at the MS2 level using the complement reporter ion cluster. Anal. Chem. 2012, 84 (21), 9214–9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Niu M; Cho JH; Kodali K; Pagala V; High AA; Wang H; Wu Z; Li Y; Bi W; Zhang H; Wang X; Zou W; Peng J Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal. Chem. 2017, 89 (5), 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Barber KW; Rinehart J The abcs of ptms. Nat. Chem. Biol. 2018, 14 (3), 188–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liao L; Cheng D; Wang J; Duong DM; Losik TG; Gearing M; Rees HD; Lah JJ; Levey AI; Peng J Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. Journal of Biological Chemistry 2004, 279 (35), 37061–37068. [DOI] [PubMed] [Google Scholar]
- (26).Specht H; Slavov N Transformative opportunities for single-cell proteomics. J. Proteome Res. 2018, 17 (8), 2565–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Huang EL; Piehowski PD; Orton DJ; Moore RJ; Qian W-J; Casey CP; Sun X; Dey SK; Burnum-Johnson KE; Smith RD SNaPP: simplified nanoproteomics platform for reproducible global proteomic analysis of nanogram protein quantities. Endocrinology 2016, 157 (3), 1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chen W; Wang S; Adhikari S; Deng Z; Wang L; Chen L; Ke M; Yang P; Tian R Simple and integrated spintip-based technology applied for deep proteome profiling. Anal. Chem. 2016, 88 (9), 4864–4871. [DOI] [PubMed] [Google Scholar]
- (29).Myers SA; Rhoads A; Cocco AR; Peckner R; Haber AL; Schweitzer LD; Krug K; Mani D; Clauser KR; Rozenblatt-Rosen O Streamlined protocol for deep proteomic profiling of FAC-sorted cells and its application to freshly isolated murine immune cells. Mol. Cell. Proteomics 2019, 18 (5), 995–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang P; Gaffrey MJ; Zhu Y; Chrisler WB; Fillmore TL; Yi L; Nicora CD; Zhang T; Wu H; Jacobs J Carrier-Assisted Single-Tube Processing Approach for Targeted Proteomics Analysis of Low Numbers of Mammalian Cells. Anal. Chem. 2018, 91 (2), 1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wu R; Pai A; Liu L; Xing S; Lu Y NanoTPOT: Enhanced Sample Preparation for Quantitative Nanoproteomic Analysis. Anal. Chem. 2020, 92 (2), 6235–6240. [DOI] [PubMed] [Google Scholar]
- (32).Zhu Y; Piehowski PD; Kelly RT; Qian W-J Nanoproteomics comes of age. Expert Rev. Proteomics 2018, 15 (11), 865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 2018, 9 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10 (10), 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wu R; Pai A; Liu L; Xing S; Lu Y NanoTPOT: Enhanced Sample Preparation for Quantitative Nanoproteomic Analysis. Anal. Chem. 2020, 92 (9), 6235–6240. [DOI] [PubMed] [Google Scholar]
- (36).Bai B; Wang X; Li Y; Chen P-C; Yu K; Dey KK; Yarbro JM; Han X; Lutz BM; Rao S Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105 (6), 975‐991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lau BYC; Othman A Evaluation of sodium deoxycholate as solubilization buffer for oil palm proteomics analysis. PLoS One 2019, 14 (8), e0221052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Xu P; Duong DM; Peng JM Systematical Optimization of Reverse-Phase Chromatography for Shotgun Proteomics. J. Proteome Res. 2009, 8 (8), 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bai B; Hales CM; Chen P-C; Gozal Y; Dammer EB; Fritz JJ; Wang X; Xia Q; Duong DM; Street C U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (41), 16562–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wang Z; Yu K; Tan H; Wu Z; Cho J-H; Han X; Sun H; Beach TG; Peng J 27-plex Tandem Mass Tag Mass Spectrometry for Profiling Brain Proteome in Alzheimer’s Disease. Anal. Chem. 2020, 92 (10), 7162–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Li Y; Wang X; Cho JH; Shaw TI; Wu Z; Bai B; Wang H; Zhou S; Beach TG; Wu G; Zhang J; Peng J JUMPg: An Integrative Proteogenomics Pipeline Identifying Unannotated Proteins in Human Brain and Cancer Cells. J. Proteome Res. 2016, 15 (7), 2309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wang X; Li Y; Wu Z; Wang H; Tan H; Peng J JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell. Proteomics 2014, 13 (12), 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Xie Y; Pan W; Khodursky AB A note on using permutation-based false discovery rate estimates to compare different analysis methods for microarray data. Bioinformatics 2005, 21 (23), 4280–4288. [DOI] [PubMed] [Google Scholar]
- (44).Budnik B; Levy E; Harmange G; Slavov N SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol. 2018, 19 (1), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wiśniewski JR; Zettl K; Pilch M; Rysiewicz B; Sadok I ‘Shotgun’proteomic analyses without alkylation of cysteine. Anal. Chim. Acta 2020, 1100, 131–137. [DOI] [PubMed] [Google Scholar]
- (46).Wiśniewski JR; Gaugaz FZ Fast and sensitive total protein and Peptide assays for proteomic analysis. Anal. Chem. 2015, 87 (8), 4110–4116. [DOI] [PubMed] [Google Scholar]
- (47).Murphy JP; Everley RA; Coloff JL; Gygi SP Combining amine metabolomics and quantitative proteomics of cancer cells using derivatization with isobaric tags. Anal. Chem. 2014, 86 (7), 3585–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Bai B; Tan H; Pagala VR; High AA; Ichhaporis VP; Hendershot L; Peng J Deep profiling of proteome and phosphoproteome by isobaric labeling, extensive liquid chromatography, and mass spectrometry. Methods Enzymol. 2017, 585, 377–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wang H; Yang Y; Li Y; Bai B; Wang X; Tan H; Liu T; Beach TG; Peng J; Wu Z Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J. Proteome Res. 2015, 14 (2), 829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dey KK; Wang H; Niu M; Bai B; Wang X; Li Y; Cho J-H; Tan H; Mishra A; High AA Deep undepleted human serum proteome profiling toward biomarker discovery for Alzheimer’s disease. Clin. Proteomics 2019, 16 (1), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Rogatsky E; Stein DT High dead volume pumps: Delay time reduction by short-term low-ratio split flow in LC/MS applications. J. Am. Soc. Mass Spectrom. 2007, 18 (2), 245–247. [DOI] [PubMed] [Google Scholar]
- (52).Licklider LJ; Thoreen CC; Peng J; Gygi SP Automation of nanoscale microcapillary liquid chromatography-tandem mass spectrometry with a vented column. Anal. Chem. 2002, 74 (13), 3076–83. [DOI] [PubMed] [Google Scholar]
- (53).Cong Y; Liang Y; Motamedchaboki K; Huguet R; Truong T; Zhao R; Shen Y; Lopez-Ferrer D; Zhu Y; Kelly RT Improved Single Cell Proteome Coverage Using Narrow-Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry. Anal. Chem. 2020, 92 (3), 2665–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhou F; Lu Y; Ficarro SB; Webber JT; Marto JA Nanoflow low pressure high peak capacity single dimension LC-MS/MS platform for high-throughput, in-depth analysis of mammalian proteomes. Anal. Chem. 2012, 84 (11), 5133–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhu Y; Zhao R; Piehowski PD; Moore RJ; Lim S; Orphan VJ; Paša-Tolić L; Qian W-J; Smith RD; Kelly RT Subnanogram proteomics: Impact of LC column selection, MS instrumentation and data analysis strategy on proteome coverage for trace samples. Int. J. Mass Spectrom. 2018, 427, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Datta A; Qian J; Chong R; Kalaria RN; Francis P; Lai MK; Chen CP; Sze SK Novel pathophysiological markers are revealed by iTRAQ-based quantitative clinical proteomics approach in vascular dementia. J. Proteomics 2014, 99, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Hosseinzadeh S; Noroozian M; Mortaz E; Mousavizadeh K Plasma microparticles in Alzheimer’s disease: the role of vascular dysfunction. Metab. Brain Dis. 2018, 33 (1), 293–299. [DOI] [PubMed] [Google Scholar]
- (58).Zhu Y; Dou M; Piehowski PD; Liang Y; Wang F; Chu RK; Chrisler WB; Smith JN; Schwarz KC; Shen Y Spatially resolved proteome mapping of laser capture microdissected tissue with automated sample transfer to nanodroplets. Mol. Cell. Proteomics 2018, 17 (9), 1864–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Dou M; Tsai C-F; Piehowski PD; Wang Y; Fillmore TL; Zhao R; Moore RJ; Zhang P; Qian W-J; Smith RD Automated nanoflow two-dimensional reversed-phase liquid chromatography system enables in-depth proteome and phosphoproteome profiling of nanoscale samples. Anal. Chem. 2019, 91 (15), 9707–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting methods; Figure S1,High amount of trypsin in the microgram-scale protocol is sufficient to digest proteins in 30 min; Figure S2, DTT is TMT-reactive in the reactivity assay; Figure S3, Analysis of PSM distribution and quantification variation of the standard- and microgram-scale datasets; Figure S4, Boxplot to show normalized intensities of the standard- and microgram-scale datasets; Supporting references.
Table S1, Summary of human cases used for microgram-scale and standard-scale proteomic studies; Table S2, Whole proteome profiling of AD, ND and VaD postmortem human brain tissues by TMT-LC/LC-MS/MS by the microgram-scale protocol; Table S3, Whole proteome profiling of AD, ND and VaD postmortem human brain tissues by TMT-LC/LC-MS/MS by the standard-scale protocol; Table S4; Differentially expressed (DE) proteins between normal control and Vascular Dementia (VaD) postmortem brain samples.