

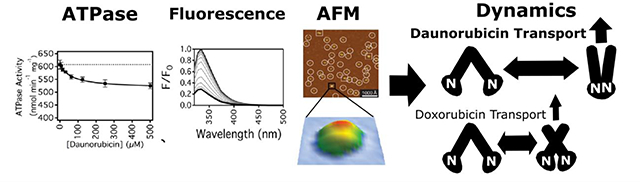
Abstract
P-glycoprotein (Pgp) is an ATP-dependent efflux transporter and plays a major role in anti-cancer drug resistance by pumping a chemically diverse range of cytotoxic drugs from cancerous tumors. Despite numerous studies with the transporter, the molecular features that drive anti-cancer drug efflux are not well understood. Even subtle differences in the anti-cancer drug molecular structure can lead to dramatic differences in their transport rates. To unmask these structural differences, this study focused on two closely-related anthracycline drugs, daunorubicin (DNR), and doxorubicin (DOX), with mouse Pgp. While only differing by a single hydroxyl functional group, DNR has a 4 to 5-fold higher transport rate than DOX. They both non-competitively inhibited Pgp-mediated ATP hydrolysis below basal levels. The Km of Pgp-mediated ATP hydrolysis extracted from the kinetics curves was lower for DOX than DNR. However, the dissociation constants (KDs) for these drugs determined by fluorescence quenching were virtually identical. Acrylamide quenching of Pgp tryptophan fluorescence to probe the tertiary structure of Pgp suggested that DNR shifts Pgp to a “closed” conformation, while DOX shifts Pgp to an “intermediate” conformation. The effects of these drugs on the Pgp conformational distributions in a lipid bilayer were also examined by atomic force microscopy (AFM). Analysis of AFM images revealed that DNR and DOX cause distinct and significant shifts in the conformational distribution of Pgp. The results were combined to build a conformational distribution model for anthracycline transport by Pgp.
Keywords: atomic force microscopy (AFM), multidrug resistance transporters, protein conformation, anti-cancer drug resistance
Graphical Abstract
1. Introduction
P-glycoprotein (Pgp) is a member of the ATP binding cassette (ABC) transporter superfamily and can efflux a chemically and structurally diverse range of drugs from the cytosol (C) to the extracellular (EC) space of cells [1–3]. Although the exact molecular transport mechanism remains to be elucidated, Pgp-mediated efflux is believed to involve the coordinated interplay of drug binding, ATP hydrolysis, and conformational changes [3,4]. Pgp expression is significant in about 50% of cancers [5–7]. Therefore, the effluxing of cytotoxic drugs by the transporter is a major driver of anti-cancer drug resistance in cancerous tumors [8–10]. Complicating matters, the cancer treatment itself can upregulate the transporter expression in these tumors leading to reduced efficacy of the chemotherapeutics and higher cancer mortality [e.g. 5,11]. Even silent synonymous genetic polymorphisms of the transporter can significantly affect the pharmacokinetics and the efficacy of cancer treatment [12]. To combat Pgp-mediated anti-cancer drug resistance, attempts have been made to design specific transporter inhibitors to improve the sensitivity of resistant tumors to anti-cancer drugs [13–16]. Unfortunately, these drugs have had limited success in the clinic due to their lack of specificity and because they often have unacceptable toxicity [13,17]. Another strategy to combat anti-cancer drug resistance is to design anti-cancer drugs that are poor Pgp substrates. Unfortunately, the functional groups of drugs that make good transporter substrates are not well defined. For these reasons, there is considerable interest in identifying the molecular factors that drive anti-cancer drug transport by Pgp.
The anthracycline antibiotics are a class of anti-cancer drug that is widely used in human chemotherapy [18]. Statistics showed that anthracyclines are used to treat a substantial portion of the cancer population, including about 32% of breast cancer patients, 70% of elderly lymphoma patients, and 60% of childhood cancer survivors [19]. The drugs have a tetracyclic aglycone structure of four cyclohexane chains with a daunosamine sugar moiety [18]. The primary anti-cancer drug function is to intercalate into double-stranded DNA within cancer cells [20,21]. The interaction of the DNA intercalated drugs with topoisomerase II leads to double-stranded DNA breaks and cell death [19,20,22,23]. Unfortunately, cancer cells become resistant to anthracycline drugs because of the overexpression of Pgp [6,7,24]. The Pgp-mediated transport rate also varies widely between therapeutics of this drug class [25].
Fig. 1 shows the molecular structures of the anthracyclines, daunorubicin (DNR), and doxorubicin (DOX). Despite differing by a single hydroxyl functional group, these drugs show significant differences in their Pgp-mediated transport rates. The transport rate for DNR in the K562/DNR leukemia cell line expressing Pgp was significantly higher than DOX [26]. In human Pgp expressing K562/ADR leukemia cells, the active efflux of the anti-cancer drug was 5-fold higher for DNR than DOX [27]. In an S1(1.1)400 cell line stably transfected with Pgp, there was almost a 4-fold difference in their transport rates between DNR and DOX [25]. Unfortunately, these studies were unable to resolve exactly why there were differences in their Pgp-mediated transport rates.
Fig. 1.

The molecular structures of DNR and DOX. The figure legend shows the R group of DNR and DOX.
This study bridges gaps in knowledge of anthracycline transport by Pgp. In this study, we examined their effects on Pgp-mediated ATP hydrolysis and their affinity to the transporter. Anthracycline-induced changes in the Pgp conformation were investigated by fluorescence spectroscopy and atomic force microscopy (AFM). The AFM experiments were particularly informative because it allowed us to monitor the extracellular (EC) and cytosolic (C) domains of the transporter in a lipid bilayer [28]. These experiments represent the first time that drug-induced conformational changes have been monitored directly in a lipid bilayer for any mammalian transporter. These results, combined with previous transport studies [25–27], allowed us to define a mechanism of anthracycline transport.
2. Materials and Methods
2.1. Reagents
Daunorubicin hydrochloride and doxorubicin hydrochloride were purchased from BIOTANG Inc. (Lexington, MA). Cholesterol, Tris-HCl, and disodium ATP were purchased from Amresco (Solon, OH). Dithiothreitol (DTT) was purchased from Gold Biotechnology (Olivette, MO). Ethylene glycol tetraacetic acid (EGTA), and imidazole were purchased from Alfa Aesar (Tewksbury, MA). Escherichia (E.) coli total extract lipid and E. coli polar extract lipid were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). The detergent n-dodecyl-β-D-maltoside (DDM) was purchased from MilliporeSigma (Formerly, EMD Millipore Corporation) (Burlington, MA). 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and acrylamide was purchased from MilliporeSigma (Formerly, Calbiochem) (Burlington, MA). Sodium orthovanadate (Na3VO4) was purchased from Enzo Life Sciences (Farmingdale, NY). All remaining chemicals were purchased from ThermoFisher Scientific (Waltham, MA).
2.2. Expression, Purification, and Reconstitution of Pgp
The his-tagged wild type mouse Pgp (Abcb1a, MDR3) was purified from Pichia (P.) pastoris using nickel-nitrilotriacetic acid (Ni-NTA) (Thermo Fisher Scientific) followed by diethylaminoethyl cellulose (DEAE) resin (Thermo Fisher Scientific) [29,30]. Pgp solubilized by DDM was reconstituted into 250 nm liposomes using the previous procedure so that the nucleotide-binding domains (NBDs) projected outside [31]. Liposomes were prepared with lipid-to-protein ratio of 6.25 μM Pgp (mg ml−1 lipid)−1 using 80% wt/vol Avanti Total E. coli Lipid Extract (Avanti Polar Lipids) and 20% wt/vol cholesterol [31–33]. A thin lipid film was created by evaporating the mixture of lipid extract and cholesterol dissolved in chloroform. Rehydration of the dried lipid film in 0.1 mM EGTA and 50 mM Tris-HCl buffer following by ten cycles of freeze-thaw in liquid nitrogen generated liposomes of various sizes. These liposomes were extruded through a 400 nm filter by LIPEX extruder to create normal distribution of liposomes with an average size of 250 nm. Detergent-solubilized Pgp was dialyzed in HEPES buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 2 mM DTT, pH 7.4) for 2 hours to remove excess detergent. Dialyzed-protein was incubated with the extruded liposome for an hour and dialyzed for another 2 hours to make proteoliposomes. The concentration of protein reconstituted in liposome was determined with the DC Protein Assay Kit II (Bio-Rad, Hercules, CA).
2.3. ATPase activity measurements of Pgp
The ATP hydrolysis mediated by Pgp was determined using the Chifflet method as previously described [31,34]. The assay was performed on a FlexStation 3 spectrometer (Molecular Devices, Sunnyvale, CA) in the presence and absence of DNR and DOX. The Chifflet assay detects the inorganic phosphate (Pi) following ATP hydrolysis by Pgp. In the Chifflet assay, inorganic phosphate forms a complex with molybdenum resulting in the absorbance signal at 850 nm. The ATPase activity of DNR and DOX was measured in the presence of 50 nM Pgp reconstituted in liposomes in Chifflet buffer (150 mM NH4Cl, 5 mM MgSO4, 0.02% wt/vol NaN3, 50 mM Tris-HCl, pH 7.4) [34–37].
The Pgp-mediated ATP hydrolysis kinetics was fit by nonlinear regression using Igor Pro 6.2 software (Wavemetrics, Tigard, OK), as done previously [e.g., 31,36]. Monophasic ATPase kinetics curves were fit with the modified Michaelis-Menten equation, where v is the ATP hydrolysis rate, Vsat is the maximum ATP hydrolysis rate at saturating drug, [L] is the ligand concentration, Km is the Michaelis-Menten constant, and vbasal is the basal ATPase activity in the absence of the drug [38].
| (1) |
2.4. Pgp affinity measurements by fluorescence quenching
Quenching of intrinsic protein fluorescence has been used to measure the dissociation constants of Pgp and various ligands [e.g., 31,39,40]. This technique was utilized to measure the binding affinity between Pgp and anthracycline drugs. Protein fluorescence emission was recorded between 300 and 500 nm with an emission maximum at around 330 nm after excitation at 295 nm using an Olis DM 45 spectrofluorimeter (Olis Corporation, Bogart, GA) [41]. Proteoliposome samples for the fluorescence experiments contained 1 μM Pgp and 100 mM potassium phosphate buffer with 2 mM DTT to prevent protein aggregation [31,36]. Fluorescence quenching of Pgp by the drugs was corrected for background, volume, and inner filter effects using the following equation [39,41].
| (2) |
where F is the measured protein fluorescence at 330 nm, B is the background, [Q] is the ligand concentration, εex, εem, bex and bem are the extinction coefficients and pathlengths for excitation and emission, respectively. The extinction coefficients at 295 nm and 330 nm for DNR were 7.8 mM−1cm−1 and 1.0 mM−1cm−1, respectively. The extinction coefficients for DOX at 295 nm and 330 nm were 6.9 mM−1cm−1 and 0.8 mM−1cm−1, respectively. The extinction coefficient of Pgp was 0.092 μM−1 cm−1 at 295 nm and was transparent at 330 nm. At 1 μM Pgp in a narrow 1 mm pathlength cuvette, the absorbance contribution will have a negligible effect on the Fcorrected, so this contribution was ignored.
Quenching of protein fluorescence can occur by static and dynamic mechanisms. A static quenching mechanism is the result of a complex between the drug and the protein receptor [41]. In contrast, the dynamic quenching of fluorescence occurs because of random collisions between the drug and the protein receptor [41]. The two fluorescence quenching mechanisms can be differentiated by looking at the effect of different temperatures on the titration curves [41]. The corrected fluorescence (Fcorrected) of a monophasic fluorescence quenching curve was fit to the modified Stern-Volmer equation:
| (3) |
where Fcorrected,0 is the fluorescence in the absence of a quencher, Funquenched is an offset related to unquenched fluorescence, KSV is the Stern-Volmer constant, and [Q] is the concentration of quencher.
Acrylamide quenching of protein fluorescence is a technique used to probe the conformational changes in protein by changes in solvent accessibility of fluorescent amino acids, such as tryptophans [41–43]. Acrylamide can specifically probe solvent accessible amino acids because it is a polar molecule that cannot diffuse through the lipid bilayer and is excluded from the hydrophobic core [41–43]. Typically, in these experiments, protein fluorescence is measured at 330 nm after excitation at 295 nm with a range of acrylamide concentrations [e.g., 31,44,45]. Unfortunately, the absorbance bands of DNR and DOX have significant overlap with the protein absorbance at 295 nm leading to significant interference from fluorescence by these drugs. To minimize fluorescence interference from these drugs, the protein was excited at 280 nm. Exciting at this wavelength will have fluorescence contributions from phenylalanine and tyrosine, as well as tryptophan. The values were corrected for background, volume, and inner filter effects using Eq. (2). The extinction coefficients for DNR and DOX at 280 nm was 8.55 Mm−1 cm−1 and 6.32 mM−1 cm−1, respectively. Because of the relatively low concentration of protein (i.e., 1 μM) with respect to the drugs’ concentrations, the protein’s contribution to the absorbance was considered negligible. To determine the maximum protein fluorescence, a mixture of the 11 μM tryptophan analog n-acetyl-l-tryptophanamide (NATA), 36 μM tyrosine, and 67 μM phenylalanine that correlates to their relative concentrations in 1 μM Pgp were used. These amino acid concentrations were based on the primary amino acid sequence of mouse Pgp, which has 11 tryptophans, 36 tyrosines, and 67 phenylalanines. The Stern-Volmer plot was generated by plotting Fcorrected,0/Fcorrected versus acrylamide concentration. The KSV value, estimated from the slope of the plot, reflects the degree of tryptophan accessibility to solvent as described previously [41].
2.5. Atomic force microscopy imaging and analysis
Atomic force microscopy (AFM) is a technique initially developed to measure the curvature and other physical properties of solid-state sample surfaces [46]. Recently, it has been adapted for analysis of protein dynamics and protein structure, which allows investigation of protein conformational changes induced by drugs [28,46]. Pgp reconstituted in liposome was diluted to a concentration of 100 nM in imaging buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, pH 7.4). A volume of this solution was then deposited on a freshly cleaved mica surface (Grade v1; Ted Pella, Redding, CA) and incubated for 45 minutes at ~30°C, which allows the proteoliposomes to rupture and form a planar lipid bilayer on the top of the mica surface. After imaging ligand-free Pgp (apoPgp), 500 μM DNR or 100 μM DOX was added to the buffer. These drug concentrations were chosen to be at least five times larger than the KD or Km. AFM imaging of the EC-side and C-side domains of Pgp protruding from lipid bilayer were performed as previously described [28]. Only protrusions in the AFM images that could be positively identified as EC or C-side domain features of Pgp were used for the analysis.
To demonstrate that the drug-induced shifts in the Pgp conformation were significant, a statistical bootstrapping approach was applied to the height distributions [47]. In the bootstrapping method [47], data is deleted and randomly replaced by resampled data to produce a new distribution. The statistical approach was applied to all the datasets, and the mean and standard error of the mean (SEM) were calculated. This information is shown as insets in the corresponding figures.
All images were obtained in imaging buffer at 32°C in tapping mode using a commercial instrument (Asylum Research Cypher, Santa Barbara, CA) and tips (BL-AC40TS, Olympus, Tokyo, Japan) with an estimated tip-sample force < 100 pN, which minimizes the probability of protein distortion. The protrusions in AFM images of proteoliposome were analyzed using custom software written in Igor Pro 7 (Wavemetrics, Portland, OR), as described previously [28,48]. Smoothed histograms were generated in Igor Pro 7 using kernel density estimation with Epanechnikov kernels.
3. Results
3.1. Effects of DNR and DOX on Pgp-mediated ATP Hydrolysis
Fig. 2 shows the effect of DNR and DOX on Pgp-mediated ATPase activity in the presence of 3.2 mM ATP. In the absence of drugs, basal ATPase activity of Pgp was 586 ± 6 nmol min−1 mg−1, which is consistent with previous studies [e.g., 31,37]. In the presence of the non-competitive inhibitor Na3VO4 [31,49], the ATP hydrolysis activity was reduced to 119 ± 5 nmol min−1 mg−1, which is similar to previous measurements [e.g., 31]. The difference between the vbasal and the Pgp-mediated ATPase velocity in the presence of Na3VO4 gives a Na3VO4 sensitive vbasal for Pgp of 467 ± 8 nmol min−1 mg−1, which is similar to what we was obtained previously [31]. The kinetics for ATPase activity were monophasic for both two drugs, suggesting that each drug has a single binding site on the transporter. The curves in Figs. 2A and 2B were fit to Eq. (1) to determine the ATPase velocity with saturating drug and the Km values. For DNR in Fig. 2A, the Vsat and the Km were 515 ± 10 nmol min−1 mg−1 (12.2 ± 1.9 % inhibition) and 62.55 ± 9.20 μM, respectively. For DOX in Fig. 2B, the Vsat, and Km of DOX were 459 ± 18 nmol min−1 mg−1 (21.7 ± 3.2 % inhibition) and 7.80 ± 3.47 μM, respectively. DOX had stronger inhibitory effects on the ATPase activity at saturating drug concentration and had a significantly lower Km implying a higher affinity to Pgp. Considering that the transport rate of DNR is 4–5 fold higher [25,27], the lower Km value for DOX versus DNR for Pgp-mediated ATP hydrolysis might be surprising when one might expect the opposite. Another way to look at it is that DNR has less inhibitory effect on Pgp-mediated ATP hydrolysis than DOX. Overall, these results are consistent with previous studies that showed that DNR and DOX inhibited Pgp-induced ATPase activity [50–52].
Fig. 2.

The effects of DNR and DOX on Pgp mediated ATP hydrolysis. ATPase activity in response to varying concentrations of (A) DNR and (B) DOX. The points represent the average of at least three experiments, and the error bars reflect the standard deviation. Fits to the points with Eq. (1) are shown as solid lines.
3.2. DNR and DOX Affinity with Pgp Determined by Protein Fluorescence Quenching
The binding affinities of DNR and DOX to Pgp were estimated through drug-induced quenching of Pgp protein fluorescence and is shown in Fig. 3. Figs. 3A and 3C show the Pgp fluorescence with a range of DNR and DOX concentrations. Saturating DNR causes about an 80% decrease in Pgp fluorescence (Fig. 3A) while saturating DOX quenched ~65% of Pgp fluorescence (Fig. 3B). In Figs. 3B and 3D, the Pgp fluorescence in the presence of a range of DNR and DOX concentrations were monitored at 330 nm and adjusted for inner filter effects using Eq. (2). Figs. 3B and 3D show that quenching of Pgp fluorescence was monophasic by DNR and DOX, so they were fit to Eq. (3) to estimate KSV value. Fitting the protein fluorescence quenching curve in Fig. 3B gave a KSV value of 0.061 ± 0.003 μM−1. The KSV value decreased at a higher temperature (i.e., 37°C), indicating that DNR quenched Pgp protein fluorescence by a static quenching mechanism (data not shown), which correlates to DNR affinity to Pgp. The corresponding KD value was 16.3 ± 0.7 μM. Fitting the curve shown in Fig. 3D to Eq. (3) gave a KSV value of 0.059 ± 0.004 μM−1. Like DNR, the KSV value decreased at 37°C, indicating a static quenching mechanism (data not shown). The calculated KD to Pgp was similar to DNR at 16.9 ± 1.3 μM. The fact that there were no significant differences in the KDs suggests that binding is not driving the differences in the transport rates. These KDs are also similar to the low-affinity sites determined on hamster Pgp [40].
Fig. 3.

Protein fluorescence quenching of Pgp by DNR and DOX. Pgp fluorescence emission spectra after excitation at 295 nm in the presence of a range of (A) DNR and (C) DOX concentrations. Pgp fluorescence in the absence and the presence of saturating drug is shown as thin and thick black lines, respectively, while Pgp fluorescence with intermediate drug concentrations are shown as gray lines. The fluorescence at 330 nm after adjusting for inner filter effects with Eq. (2) with a range of (B) DNR and (D) DOX concentrations. The data points represent the average of at least three experiments with the error bars representing the standard deviation. Fits to the points by Eq. (3) are shown as solid lines.
3.3. Conformational Changes of P-glycoprotein in the Presence of DNR and DOX Deduced by Acrylamide Quenching of Protein Fluorescence
The acrylamide quenching technique exploits the fact that the solvent accessibility of fluorescence residues changes as a result of tertiary conformational changes [41–43]. Acrylamide quenching of protein fluorescence was used to probe the effect of DNR and DOX on the tertiary structure of Pgp. When Pgp assumes an “open” conformation, fluorescent residues within the binding cavity become more exposed to bulk solvent, and the degree of acrylamide quenching is relatively high [e.g., 31,36,40]. When Pgp assumes a “closed” conformation, fluorescent residues within the binding cavity become sequestered to bulk solvent, and the degree of acrylamide quenching is relatively low [e.g., 31,36,40]. The relative degree of acrylamide quenching and the solvent accessibility of Pgp was estimated by the slopes of Stern-Volmer plots [41].
Fig. 4 shows the acrylamide quenching experiments with Pgp and anthracycline drugs. In Fig. 4A, a control experiment was performed to determine the degree of quenching in the presence of 530 mM acrylamide for phenylalanine, tyrosine, and the tryptophan analog n-Acetyl-L-tryptophanamide (NATA) at their relative concentrations in Pgp, while exciting at 280 nm. The first two columns show the fluorescence of 36 μM tyrosine and 67 μM in the absence and presence of 530 mM acrylamide. The 40,000 relative fluorescence units of tyrosine and phenylalanine fluorescence were virtually abolished with acrylamide. The next two columns show the level of 11 μM NATA fluorescence in the absence and presence of 530 mM acrylamide. In the absence of acrylamide, the fluorescence for NATA was about 5-fold larger than the fluorescence of Phe and Tyr. Therefore, the contribution of Pgp tryptophan fluorescence will be about 80% of the total Pgp fluorescence. Again, acrylamide virtually abolished fluorescence in the next column. The last two columns show the degree of fluorescence from all three amino acids (Phe+Tyr+NATA) with and without acrylamide. The relative fluorescence amplitude of all three amino acids was about 240,000 in the absence of acrylamide and obliterated with 530 mM acrylamide.
Fig. 4.

Acrylamide quenching of Pgp in the presence of DNR and DOX. (A) Acrylamide quenching of 67 μM Phe and 36 μM Tyr (Phe+Tyr) (a), 11 μM NATA (NATA) (b), and a combination of (a) and (b), or NATA+Phe+Tyr (c) in the absence (closed columns) and presence of 530 mM acrylamide (open columns). The relative fluorescence units (RFUs) are shown on the y-axis. (B) The Stern-Volmer plot of NATA+Phe+Tyr (open circles), Pgp in the absence of ligands (apoPgp, open diamonds), Pgp with 3.2 mM AMPPNP (AMPPNP, open triangles) with a range of acrylamide concentrations. Stern-Volmer plots of Pgp in the presence of 16 μM (open circles) and 500 μM (open squares) for (C) DNR and (D) DOX. The data points represent the average of at least three experiments, and the error bars are the standard deviation. The Stern-Volmer curves for apoPgp and AMPPNP are shown as gray and black dashed lines, respectively, for comparison.
Fig. 4B shows the Stern-Volmer plot with Phe+Tyr+NATA and Pgp in the absence and presence of the non-hydrolyzable ATP analog adenylyl-imidodiphosphate (AMPPNP). The steepest slope in the Stern-Volmer plot was from quenching of Phe+Tyr+NATA, which had a KSV value of 35.79 ± 2.29 M−1. This would be the KSV value obtained if all the fluorescent residues of Pgp were accessible to bulk solvent. Pgp in the absence of ligands (apoPgp) has a significantly lower KSV value of 4.85 ± 0.15 M−1 (Fig. 4B, open diamonds), showing that most fluorescent residues remain buried away from the bulk solvent. Because the samples were excited at 280 nm instead of 295 nm, this value is considerably higher than previously reported KSV values [e.g., 31,36]. The binding cavity of several X-ray crystal structures solved of apoPgp had the binding cavity exposed to the bulk solvent with the nucleotide-binding domains (NBDs) separated in an “open” conformation [53–55]. Therefore, apoPgp in the acrylamide quenching experiments is assumed to be in a similar conformation. The slope of the Stern-Volmer plots of Pgp in the presence of 3.2 mM AMPPNP decreased to 1.84 ± 0.02 M−1 (Fig. 4B, open triangles), which means that fluorescent residues are inaccessible to bulk solvent and implies that the binding cavity has become occluded. Likewise, the binding cavity of a cryo-electron microscopy (cryo-em) structure of Pgp under similar conditions with AMPPNP was closed to the bulk solvent in a “closed” conformation [56].
In the presence of 16 μM DNR, which is close to the KD for DNR, the KSV value obtained from the Stern-Volmer decreased to 2.87 ± 0.06 M−1 from the KSV value determined for apoPgp (Fig. 4C). Increasing the DNR concentration to 500 μM decreased the KSV value further to 1.91 ± 0.02 M−1 (Fig. 4C). This KSV value is similar to the KSV value of Pgp with saturating AMPPNP, indicating that most fluorescent residues became inaccessible to acrylamide and imply that Pgp is in a “closed” conformation. The KSV value determined from the Stern-Volmer plot of Pgp in the presence of 16 μM DOX only decreased to 3.76 ± 0.06 M−1 (Fig. 4D). In the presence of saturating 500 μM DOX concentration, the KSV value was 2.94 ± 0.07 M−1 (Fig. 4D), which is between the value determined for Pgp with AMPPNP and the value of apoPgp. This result implies that Pgp in the presence of saturating 500 μM DOX is in an “intermediate” conformation.
3.4. Effect of DNR and DOX on the conformational distributions of Pgp analyzed by AFM
In a previous work [28], we were able to identify the extracellular (EC) and cytosolic (C) domains of Pgp from AFM images. In this study, the AFM approach also allowed us to examine the effect of DNR and DOX on the conformational distributions of Pgp. Fig. 5 shows AFM images of Pgp in the absence of drugs and the presence of DNR and DOX. The white spots in the AFM images are Pgp protrusions emanating from the bilayer. The EC and C domains are circled and squared in the AFM images, respectively, with a 1000 Å bar indicating a lateral scale and a contrast bar indicating the vertical scale. In the AFM images, there were significantly more EC than C domains, which is consistent with previous observations [28].
Fig. 5.

500 × 500 nm representative AFM images of apoPgp (A), with 500 μM DNR (B), and with 100 μM DOX (C). The extracellular side of Pgp (EC side) is circled and the cytosolic side (C side) of Pgp is squared.
Fig. 6 shows the height distributions of Pgp in the absence and presence of anthracycline drugs. Two separate height distributions of the EC and C-side domains were measured in the absence of DNR and DOX, and are shown as thick black lines in the panels. Qualitatively, the height distributions for the EC and C-side domains in the absence of drugs were essentially identical. In the absence of DNR (Figs. 6A and B), the heights of 1662 and 766 AFM features of the EC and C-sides were tabulated. The average for the EC and C-side domains was 30.1 ± 3.6 Å and 54.8 ± 14.0 Å, respectively. For Pgp in the absence of DOX, the heights of 1791 EC-side and 645 C-side AFM features were collected. The average height of the EC and C-sides were 25.9 ± 6.1 Å and 55.7 ± 14.7 Å, respectively, which are similar to the average height values obtained in the absence of DNR. The average of averages for the height of the EC and C-side domains was 28.0 ± 4.9 Å and 55.3 ± 15.0 Å, respectively. The large standard deviation of the C-side domain is consistent with the Pgp conformational dynamics observed in a previous study [28].
Fig. 6.

Height histogram showing the distribution of the EC and C-sides of Pgp in the presence of 500 μM DNR and 100 μM DOX. The height distribution of the (A) EC and (B) C sides of Pgp in the absence (black) and presence of DNR (gray), taken from 1662 and 746 features respectively for the EC side, and 766 and 444 features for the C side. The height distribution of the (C) EC and (D) C sides of Pgp in the absence (black) and presence of DOX (gray), taken from 1791 and 1459 features respectively for the EC side, and 645 and 375 features for the C side. The insets show the mean values ±SEM of the protrusion heights for each observed dataset without using (closed shapes) and with using a statistical bootstrapping approach (open shapes). The horizontal axis of all the insets is the height (Å).
After adding 500 μM DNR to apoPgp (Fig. 6B), the average height of the EC-side decreased to 29.2 ± 3.8 Å. The average height of the C-side domain increased almost 5 Å to 59.9 Å, and the standard deviation of the conformational distribution increased about 3 Å to 16.9 Å. The large standard deviation suggests that Pgp samples a wider range of conformations in the presence of DNR. These shifts are also consistent with a shift to the “closed” Pgp conformation, where the EC-side height decreases, while the C-side height increases [28].
Figs. 6C and 6D show the effect of adding DOX on the height distributions of the EC-side and C-side domains of Pgp. The average height of the EC-side Pgp domain in the presence of saturating 100 μM DOX decreased 2 Å to 23.9 ± 6.3 Å. The C-side domain increased by about the same amount (2 Å) to 57.4 ± 15.6 Å, which is modest compared to the C-side shifts observed with DNR.
To determine the robustness and significance of the Pgp height distributions in the presence of the anthracylines, a statistical bootstrapping method was applied to the height distributions shown in Fig. 6. The mean (± SEM) with and without using the statistical bootstrapping method are shown as insets in the figure panels. In all cases, there was a significant separation in the mean values ± drug showing that the drug-induced conformational changes were significant.
Fig. 7 shows representative 3D AFM images at oblique angles of Pgp roughly in their ensemble average conformation to provide a sense of drug-induced conformational changes. The leftmost images of the EC and C-side domains show the AFM image of an individual Pgp transporter molecule in the absence of ligands. The protrusion height of the EC and C-side domain of this AFM image was approximately 30 Å and 60 Å, respectively. The addition of saturating 500 μM DNR induced significant changes in the Pgp conformation. DNR reduced the heights of the EC-side domain several angstroms and increased the C-side domain almost 10 Å (Fig. 7, middle column). The conformational changes induced by saturating 100 μM DOX on Pgp were more modest (Fig. 7, right column). The protrusion heights of the EC and C-side domains of Pgp decreased and increased, respectively, by several angstroms.
Fig. 7.

Representative 3D oblique AFM images of the (top row) EC and (bottom row) C-sides of Pgp (left column) in the absence of drugs, (middle column) in the presence of 500 μM DNR, and (right column) in the presence of 100 μM DOX.
4. Discussion
The mechanism of Pgp-mediated drug transport remains unresolved. Transport models have been proposed where substrates and ATP are cooperative during transport [31,57]. Other models have been posited where substrates occupy different sites during efflux [4,58,59]. Transport models have also been put forward where conformational changes drive efflux [36,60].
In previous studies with the transporter, the Pgp conformations were simplified into a three-state model with “closed,” “intermediate,” and “open” conformations [e.g., 35,37]. In contrast, a cryo-electron microscopic (cryo-em) study showed that detergent-solubilized apoPgp had a range of distinct conformations [61]. Also, a recent AFM study with the transporter showed that apoPgp embedded in a lipid bilayer is very conformationally dynamic [28]. In Fig. 8, we propose that the Pgp-mediated transport rate differences between DNR and DOX are primarily driven by shifts in the conformational distribution of the transporter. In [28], changes in the protrusion height of the C-side domain were significantly larger than changes in the heights of the EC-side domain making them more sensitive to changes in Pgp conformation. Also, the C-side domain height was higher for the “closed” Pgp conformation than the “open” Pgp conformation [28]. Therefore, the C-side protrusion height was used to indicate a shift toward the “closed,” the “open,” or the “wide open” Pgp conformations. The dynamic range of Pgp conformations was calculated using the average C-side protrusion height ± one standard deviation.
Fig. 8.

The conformational distribution model of Pgp in a lipid bilayer in the presence of DNR and DOX. The top of the figure shows a height scale in Å for the C-side Pgp domain going from a “wide open,” “open,” and “closed” Pgp conformations. The range of Pgp conformations A) for AFM simulations for the Pgp X-ray crystal structure in the absence of ligands (PDB ID: 4M1M, [53]) and the cryo-em structure in the presence of AMPPNP (PDB ID: 6C0V, [56]), B) for Pgp, C) for Pgp with saturating DNR, and D) for Pgp with saturating DOX. The parallel lines represent the lipid bilayer. The solid black figure is a cartoon of Pgp with the linear parts and the circular parts reflecting the transmembrane (TM) and nucleotide-binding domains (NBDs), respectively. The NBDs are also labeled with the letter “N.” Next to one of the NBDs is the protrusion height of the C-side Pgp domain in Å.
The first row of Fig. 8A shows the protrusion heights of Pgp in the “open” and “closed” conformations deduced from AFM simulations using the Pgp X-ray crystal structure and the Pgp cryo-em structure with AMPPNP [28,53,56]. The “open” Pgp conformation has a minimum C-side protrusion height of 67.5 Å, while the “closed” Pgp conformation has a maximum C-side protrusion height of 78.4 Å (Fig. 8A and [28]).
The next row (Fig. 8B) shows the minimum and maximum C-side protrusion heights of apoPgp calculated from the average of averages and the standard deviation. The minimum protrusion height was 41.3 Å. We propose that Pgp is in a conformation where the NBDs are highly separated, and Pgp is in a “wide open” conformation such as the inhibitor and epitope-bound Pgp X-ray crystal structures [62,63]. The maximum C-side protrusion height of 69.3 Å is close to the simulated height of the X-ray crystal structure of the Pgp in the “open” conformation [28]. We propose that this protrusion height corresponds to Pgp in the “open” conformation. In the absence of ligands, Pgp goes between a “wide open” and “open” conformations.
The next row (Fig. 8C) shows the range of Pgp conformations in the presence of saturating DNR. DNR increases the range of C-side protrusion heights sampled by Pgp from 28 Å to 33 Å. At the low end (43.0 Å), Pgp is in a “wide open” conformation. At the other end (76.8 Å), the height of the C-side domain is close to the C-side height determined from the AFM simulation of the “closed” Pgp conformation. In addition, the KSV value for Pgp in the presence of saturating DNR that was determined from acrylamide quenching suggests that Pgp is in a “closed” conformation (Fig. 4C). Here, we propose that Pgp is in a “closed” conformation.
The last row (Fig. 8D) shows that Pgp samples a narrower range of conformations in the presence of saturating DOX than saturating DNR. At the lowest C-side heights, the NBDs of Pgp are proposed to be highly separated in the “wide open” conformation like the Pgp X-ray crystal structures [62,63]. The highest C-side height in the presence of DOX lies between the C-side heights of the “open” and “closed” Pgp conformations that are shown in the first row. Also, the acrylamide quenching experiments with Pgp and saturating DOX produced a KSV value between the KSV values for apoPgp and Pgp with AMPPNP, which are purportedly in the “open” and “closed” Pgp conformation, respectively (Fig. 4D). Therefore, we propose that Pgp conformation under these conditions is “intermediate” between the “open” and the “closed” Pgp conformations.
The conformational distribution model shown in Fig. 8 correlates well with the biochemical data in this study and the observed transport rates [25–27]. In the conformational distribution model, the higher Pgp-mediated transport rates of DNR are driven by the drug shifting the conformational distribution of Pgp toward the “closed” conformation where there is a higher probability of transport. This shift also explains the lower inhibition of Pgp-mediated ATP hydrolysis rates in the presence of saturating DNR than saturating DOX (Fig. 2). Shifting the Pgp conformation toward the “closed” conformation brings the NBDs closer together, where the ATP hydrolysis rate will likely increase. Interestingly, both DNR and DOX non-competitively inhibited Pgp-mediated ATP hydrolysis (Figure 2). Previous molecular dynamics study showed that the drug verapamil, which activates ATP hydrolysis, aligns the NBDs promoting ATP hydrolysis [64]. In-kind, we propose that DNR and DOX, which partially inhibits ATP hydrolysis (Figure 2), shifts the NBDs in non-optimal trajectories for ATP hydrolysis. DOX, which has a lower Km and is more inhibitory than DNR (Figure 2), more strongly restricts the NBDs in this non-optimal position. Furthermore, the higher ATP hydrolysis rates of Pgp in the absence of ligands (i.e., 600 nmol min−1 mg−1) can be attributed to less restricted degrees of freedom with respect to the trajectories of the NBDs.
In this study, AFM images revealed that DNR and DOX cause distinct effects on the conformational distribution of Pgp. Drug-induced effects on the conformational distribution of Pgp by DNR and DOX are consistent with previous fluorescence/luminescence studies [58,65,66]. In a fluorescence resonance energy transfer study of Pgp with fluorescently labeled NBDs, the drugs, verapamil, and cyclosporine, were found to alter the separation distances of NBDs during transport [58]. A luminescence resonance energy transfer study with Pgp found that the drug verapamil shifted the distance distribution of the NBDs into two distinct populations that were sensitive to nucleotides [65]. A single-molecule Förster resonance energy transfer study using hidden Markov models found that the “closed” conformation with the NBDs together was more populated than the other conformations in the presence of verapamil [66]. This observation aligns well with our results showing a drug-induced shift to the “closed” and “intermediate” Pgp conformations by DNR and DOX, respectively.
Despite differing by a single hydroxyl group, DNR and DOX caused distinct and significant effects on the conformational distribution of Pgp that correlates to their transport rate differences. Since these drugs have similar effects on Pgp-mediated ATP hydrolysis implying similar binding sites, a single hydrogen bond from the hydroxyl group of DOX may be driving these conformational and transport rate differences. This study focused on only two anthracycline drugs, but drug-induced shifts in the conformational distribution are likely to be a common mechanism of Pgp-mediated transport. This study is also the first to directly measure the effects of drugs on the conformational distribution of any mammalian transporter.
Acknowledgments
We give thanks to Dr. Ina L. Urbatsch of Texas Tech University Health Sciences Center for her generous gift of P. pastoris with the wild-type mouse Pgp transporter gene. We would also like to thank Dr. Laura A. Wilt of St. Jude Children’s Research Hospital and Morgan E. Gibbs of the University of North Carolina at Chapel Hill for their help with the techniques that were used in this study. Without their contribution and generosity, this research would not be possible. This work was supported by the National Institutes of Health (1R01CA204846-01A1, A.G.R.), the National Science Foundation (1054832, G.M.K.), and the Burroughs Wellcome Fund (1016386, K.P.S.).
Footnotes
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References
- [1].George AM, ed., ABC Transporters - 40 Years on, 1st ed. 2016 edition, Springer, New York, NY, 2015. [Google Scholar]
- [2].Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM, Biochemical, cellular, and pharmacological aspects of the multidrug transporter, Annu. Rev. Pharmacol. Toxicol 39 (1999) 361–398. 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- [3].Chufan EE, Sim H-M, Ambudkar SV, Molecular basis of the polyspecificity of P-Glycoprotein (ABCB1): Recent biochemical and structural studies, in: Schuetz JD, Ishikawa T (Eds.), Adv. Cancer Res, Academic Press, 2015: pp. 71–96. 10.1016/bs.acr.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Al-Shawi MK, Omote H, The remarkable transport mechanism of P-glycoprotein; a multidrug transporter, J. Bioenerg. Biomembr 37 (2005) 489–496. 10.1007/s10863-005-9497-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Clarke R, Leonessa F, Trock B, Multidrug resistance/P-glycoprotein and breast cancer: review and meta-analysis, Semin. Oncol 32 (2005) S9–15. 10.1053/j.seminoncol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- [6].Leonard GD, Fojo T, Bates SE, The role of ABC transporters in clinical practice, Oncologist. 8 (2003) 411–24. 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- [7].Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I, Genetic analysis of the multidrug transporter, Annu. Rev. Genet 29 (1995) 607–649. 10.1146/annurev.ge.29.120195.003135. [DOI] [PubMed] [Google Scholar]
- [8].Eckford PD, Sharom FJ, ABC efflux pump-based resistance to chemotherapy drugs, Chem. Rev 109 (2009) 2989–3011. 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- [9].Gottesman MM, Pastan IH, The role of multidrug resistance efflux pumps in cancer: Revisiting a JNCI publication exploring expression of the MDR1 (P-glycoprotein) gene, J Natl Cancer Inst. 107 (2015). 10.1093/jnci/djv222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S, Drug Resistance in Cancer: An Overview, Cancers. 6 (2014) 1769–1792. 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tada Y, Wada M, Kuroiwa K, Kinugawa N, Harada T, Nagayama J, Nakagawa M, Naito S, Kuwano M, MDR1 gene overexpression and altered degree of methylation at the promoter region in bladder cancer during chemotherapeutic treatment, Clin. Cancer Res 6 (2000) 4618–4627. [PubMed] [Google Scholar]
- [12].Sauna ZE, Kimchi-Sarfaty C, Ambudkar SV, Gottesman MM, Silent Polymorphisms Speak: How They Affect Pharmacogenomics and the Treatment of Cancer, Cancer Res. 67 (2007) 9609–9612. 10.1158/0008-5472.CAN-07-2377. [DOI] [PubMed] [Google Scholar]
- [13].Callaghan R, Luk F, Bebawy M, Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy?, Drug Metab. Dispos 42 (2014) 623–631. 10.1124/dmd.113.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG, Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells, Sci. Rep 8 (2018) 967. 10.1038/s41598-018-19325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wu C-P, Calcagno AM, Ambudkar SV, Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: Evaluation of current strategies, Curr. Mol. Pharmacol 1 (2008) 93–105. 10.2174/1874-470210801020093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Binkhathlan Z, Lavasanifar A, P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives, Curr. Cancer Drug Targets 13 (2013) 326–346. 10.2174/15680096113139990076. [DOI] [PubMed] [Google Scholar]
- [17].Chung FS, Santiago JS, Jesus MFMD, Trinidad CV, See MFE, Disrupting P-glycoprotein function in clinical settings: what can we learn from the fundamental aspects of this transporter?, Am. J. Cancer Res 6 (2016) 1583–1598. [PMC free article] [PubMed] [Google Scholar]
- [18].Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L, Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity, Pharmacol. Rev 56 (2004) 185–229. 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- [19].McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM, Anthracycline chemotherapy and cardiotoxicity, Cardiovasc. Drugs Ther 31 (2017) 63–75. 10.1007/s10557-016-6711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yang F, Kemp CJ, Henikoff S, Anthracyclines induce double-strand DNA breaks at active gene promoters, Mutat. Res 773 (2015) 9–15. 10.1016/j.mrfmmm.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Leonard GA, Brown T, Hunter WN, Anthracycline binding to DNA, Eur. J. Biochem 204 (1992) 69–74. 10.1111/j.1432-1033.1992.tb16606.x. [DOI] [PubMed] [Google Scholar]
- [22].Dal Ben D, Palumbo M, Zagotto G, Capranico G, Moro S, DNA topoisomerase II structures and anthracycline activity: insights into ternary complex formation, Curr. Pharm. Des 13 (2007) 2766–2780. 10.2174/138161207781757105. [DOI] [PubMed] [Google Scholar]
- [23].Nitiss JL, Targeting DNA topoisomerase II in cancer chemotherapy, Nat. Rev. Cancer 9 (2009) 338–350. 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gottesman MM, Fojo T, Bates SE, Multidrug resistance in cancer: role of ATP-dependent transporters, Nat Rev Cancer. 2 (2002) 48–58. 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- [25].Wielinga PR, Westerhoff HV, Lankelma J, The relative importance of passive and P-glycoprotein mediated anthracycline efflux from multidrug-resistant cells, Eur J Biochem. 267 (2000) 649–57. 10.1046/j.1432-1327.2000.01030.x. [DOI] [PubMed] [Google Scholar]
- [26].Praet M, Stryckmans P, Ruysschaert JM, Cellular uptake, cytotoxicity, and transport kinetics of anthracyclines in human sensitive and multidrug-resistant K562 cells, Biochem Pharmacol. 51 (1996) 1341–8. 10.1016/0006-2952(96)00042-1. [DOI] [PubMed] [Google Scholar]
- [27].Marbeuf-Gueye C, Ettori D, Priebe W, Kozlowski H, Garnier-Suillerot A, Correlation between the kinetics of anthracycline uptake and the resistance factor in cancer cells expressing the multidrug resistance protein or the P-glycoprotein, Biochim. Biophys. Acta BBA - Mol. Cell Res 1450 (1999) 374–384. 10.1016/S0167-4889(99)00060-9. [DOI] [PubMed] [Google Scholar]
- [28].Sigdel KP, Wilt LA, Marsh BP, Roberts AG, King GM, The conformation and dynamics of P-glycoprotein in a lipid bilayer investigated by atomic force microscopy, Biochem. Pharmacol 156 (2018) 302–311. 10.1016/j.bcp.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bai J, Swartz DJ, Protasevich II, Brouillette CG, Harrell PM, Hildebrandt E, Gasser B, Mattanovich D, Ward A, Chang G, Urbatsch IL, A Gene Optimization Strategy that Enhances Production of Fully Functional P-Glycoprotein in Pichia pastoris, PLoS One. 6 (2011) e22577. 10.1371/journal.pone.0022577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lerner-Marmarosh N, Gimi K, Urbatsch IL, Gros P, Senior AE, Large scale purification of detergent-soluble P-glycoprotein from Pichia pastoris cells and characterization of nucleotide binding properties of wild-type, walker A, and walker B mutant proteins, J. Biol. Chem 274 (1999) 34711–34718. 10.1074/jbc.274.49.34711. [DOI] [PubMed] [Google Scholar]
- [31].Ledwitch KV, Gibbs ME, Barnes RW, Roberts AG, Cooperativity between verapamil and ATP bound to the efflux transporter P-glycoprotein, Biochem. Pharmacol 118 (2016) 96–108. 10.1016/j.bcp.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Eckford PDW, Sharom FJ, Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: effects on ATPase activity, drug binding and transport, Biochemistry. 47 (2008) 13686–13698. 10.1021/bi801409r. [DOI] [PubMed] [Google Scholar]
- [33].Rothnie A, Theron D, Soceneantu L, Martin C, Traikia M, Berridge G, Higgins CF, Devaux PF, Callaghan R, The importance of cholesterol in maintenance of P-glycoprotein activity and its membrane perturbing influence, Eur Biophys J. 30 (2001) 430–42. [DOI] [PubMed] [Google Scholar]
- [34].Chifflet S, Torriglia A, Chiesa R, Tolosa S, A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases, Anal. Biochem 168 (1988) 1–4. 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- [35].Ledwitch KV, Barnes RW, Roberts AG, Unravelling the complex drug–drug interactions of the cardiovascular drugs, verapamil and digoxin, with P-glycoprotein, Biosci. Rep 36 (2016). 10.1042/BSR20150317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wilt LA, Nguyen D, Roberts AG, Insights into the molecular mechanism of triptan transport by P-glycoprotein, J. Pharm. Sci 106 (2017) 1670–1679. 10.1016/j.xphs.2017.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gibbs ME, Wilt LA, Ledwitch KV, Roberts AG, A conformationally-gated model of methadone and loperamide transport by P-glycoprotein, J. Pharm. Sci (2018). 10.1016/j.xphs.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Segel IH, Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, John Wiley & Sons, Inc., New York, 1975. [Google Scholar]
- [39].Sharom FJ, Russell PL, Qu Q, Lu P, Fluorescence techniques for studying membrane transport proteins: the P-glycoprotein multidrug transporter, Methods Mol. Biol. Clifton NJ 227 (2003) 109–128. 10.1385/1-59259-387-9:109. [DOI] [PubMed] [Google Scholar]
- [40].Liu R, Siemiarczuk A, Sharom FJ, Intrinsic fluorescence of the P-glycoprotein multidrug transporter: sensitivity of tryptophan residues to binding of drugs and nucleotides, Biochemistry. 39 (2000) 14927–14938. [DOI] [PubMed] [Google Scholar]
- [41].Lakowicz JR, Principles of Fluorescence Spectroscopy, 3rd edition, Springer, New York, NY, 2011. [Google Scholar]
- [42].Chang GG, Lee HJ, Monitoring protein conformational changes by quenching of intrinsic fluorescence, J Biochem Biophys Methods. 9 (1984) 351–5. 10.1016/0165-022X(84)90019-8. [DOI] [PubMed] [Google Scholar]
- [43].Möller M, Denicola A, Protein tryptophan accessibility studied by fluorescence quenching, Biochem. Mol. Biol. Educ 30 (2002) 175–178. 10.1002/bmb.2002.494030030035. [DOI] [Google Scholar]
- [44].Russell PL, Sharom FJ, Conformational and functional characterization of trapped complexes of the P-glycoprotein multidrug transporter, Biochem. J 399 (2006) 315–323. 10.1042/BJ20060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sonveaux N, Vigano C, Shapiro AB, Ling V, Ruysschaert JM, Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. A tryptophan fluorescence quenching analysis, J. Biol. Chem 274 (1999) 17649–54. 10.1074/jbc.274.25.17649. [DOI] [PubMed] [Google Scholar]
- [46].Jalili N, Laxminarayana K, A review of atomic force microscopy imaging systems: application to molecular metrology and biological sciences, Mechatronics. 14 (2004) 907–945. 10.1016/j.mechatronics.2004.04.005. [DOI] [Google Scholar]
- [47].Efron B, Hastie T, Computer Age Statistical Inference: Algorithms, Evidence, and Data Science, 1 edition, Cambridge University Press, New York, NY, 2016. [Google Scholar]
- [48].Marsh BP, Chada N, Gari RRS, Sigdel KP, King GM, The hessian blob algorithm: Precise particle detection in atomic force microscopy imagery, Sci. Rep 8 (2018) 978. 10.1038/s41598-018-19379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Litman T, Zeuthen T, Skovsgaard T, Stein WD, Competitive, non-competitive and cooperative interactions between substrates of P-glycoprotein as measured by its ATPase activity., BBA-Mol. Basis Dis 1361 (1997) 169–176. 10.1016/S0925-4439(97)00027-6. [DOI] [PubMed] [Google Scholar]
- [50].Borgnia MJ, Eytan GD, Assaraf YG, Competition of Hydrophobic Peptides, Cytotoxic Drugs, and Chemosensitizers on a Common P-glycoprotein Pharmacophore as Revealed by Its ATPase Activity, J. Biol. Chem 271 (1996) 3163–3171. 10.1074/jbc.271.6.3163. [DOI] [PubMed] [Google Scholar]
- [51].Doige CA, Yu X, Sharom FJ, ATPase activity of partially purified P-glycoprotein from multidrug-resistant Chinese hamster ovary cells, Biochim. Biophys. Acta BBA - Biomembr 1109 (1992) 149–160. 10.1016/0005-2736(92)90078-Z. [DOI] [PubMed] [Google Scholar]
- [52].Sharom FJ, Yu X, Chu JW, Doige CA, Characterization of the ATPase activity of P-glycoprotein from multidrug-resistant Chinese hamster ovary cells, Biochem J. 308 ( Pt 2) (1995) 381–90. 10.1042/bj3080381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li J, Jaimes KF, Aller SG, Refined structures of mouse P-glycoprotein, Protein Sci. 23 (2014) 34–46. 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Esser L, Zhou F, Pluchino KM, Shiloach J, Ma J, Tang W-K, Gutierrez C, Zhang A, Shukla S, Madigan JP, Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity, J. Biol. Chem 292 (2016) 446–461. 10.1074/jbc.M116.755884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jin MS, Oldham ML, Zhang Q, Chen J, Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans, Nature. 490 (2012) 566–9. 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kim Y, Chen J, Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation, Science. 359 (2018) 915–919. 10.1126/science.aar7389. [DOI] [PubMed] [Google Scholar]
- [57].Shapiro AB, Ling V, Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities, Eur. J. Biochem 250 (1997) 130–137. 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- [58].Verhalen B, Ernst S, Börsch M, Wilkens S, Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy, J. Biol. Chem. 287 (2012) 1112–1127. 10.1074/jbc.M111.301192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chufan EE, Kapoor K, Sim H-M, Singh S, Talele TT, Durell SR, Ambudkar SV, Multiple Transport-Active Binding Sites Are Available for a Single Substrate on Human P-Glycoprotein (ABCB1), PLoS One. 8 (2013) e82463. 10.1371/journal.pone.0082463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ritchie TK, Kwon H, Atkins WM, Conformational analysis of human ATP-binding cassette transporter ABCB1 in lipid Nanodiscs and inhibition by the antibodies MRK16 and UIC2, J. Biol. Chem 286 (2011) 39489–39496. 10.1074/jbc.M111.284554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Frank GA, Shukla S, Rao P, Borgnia MJ, Bartesaghi A, Merk A, Mobin A, Esser L, Earl LA, Gottesman MM, Xia D, Ambudkar SV, Subramaniam S, Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle, Mol. Pharmacol 90 (2016) 35–41. 10.1124/mol.116.104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ward AB, Szewczyk P, Grimard V, Lee C-W, Martinez L, Doshi R, Caya A, Villaluz M, Pardon E, Cregger C, Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain, Proc. Natl. Acad. Sci 110 (2013) 13386–13391. 10.1073/pnas.1309275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Szewczyk P, Tao H, McGrath AP, Villaluz M, Rees SD, Lee SC, Doshi R, Urbatsch IL, Zhang Q, Chang G, Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein, Acta Crystallogr. D 71 (2015) 732–741. 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Pan L, Aller SG, Allosteric Role of Substrate Occupancy Toward the Alignment of P-glycoprotein Nucleotide Binding Domains, Sci. Rep 8 (2018) 1–14. 10.1038/s41598-018-32815-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zoghbi ME, Mok L, Swartz DJ, Singh A, Fendley GA, Urbatsch IL, Altenberg GA, Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer–associated P-glycoprotein during ATP hydrolysis, J. Biol. Chem 292 (2017) 20412–20424. 10.1074/jbc.M117.814186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zarrabi N, Ernst S, Verhalen B, Wilkens S, Börsch M, Analyzing conformational dynamics of single P-glycoprotein transporters by Förster resonance energy transfer using hidden Markov models, Methods San Diego Calif. 66 (2014) 168–179. 10.1016/j.ymeth.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]