

Abstract
BACKGROUND
Uterine fibroids are a common cause of heavy menstrual bleeding and pain. Treatment with the combination of relugolix (an oral gonadotropin-releasing hormone-receptor antagonist), estradiol, and norethindrone acetate, administered once daily, may have efficacy in women with uterine fibroids and heavy bleeding while avoiding hypoestrogenic effects.
METHODS
We conducted two replicate international, double-blind, 24-week, phase 3 trials involving women with fibroid-associated heavy menstrual bleeding. Participants were randomly assigned in a 1:1:1 ratio to receive once-daily placebo, relugolix combination therapy (40 mg of relugolix, 1 mg of estradiol, and 0.5 mg of norethindrone acetate), or delayed relugolix combination therapy (40 mg of relugolix monotherapy, followed by relugolix combination therapy, each for 12 weeks). The primary efficacy end point in each trial was the percentage of participants with a response (volume of menstrual blood loss <80 ml and a ≥50% reduction in volume from baseline) in the relugolix combination therapy group, as compared with the placebo group. Key secondary end points were amenorrhea, volume of menstrual blood loss, distress from bleeding and pelvic discomfort, anemia, pain, fibroid volume, and uterine volume. Safety and bone mineral density were assessed.
RESULTS
A total of 388 women in trial L1 and 382 in trial L2 underwent randomization. A total of 73% of the participants in the relugolix combination therapy group in trial L1 and 71% of those in trial L2 had a response (primary end point), as compared with 19% and 15%, respectively, of those in the placebo groups (P<0.001 for both comparisons). Both relugolix combination therapy groups had significant improvements, as compared with the placebo groups, in six of seven key secondary end points, including measures of menstrual blood loss (including amenorrhea), pain, distress from bleeding and pelvic discomfort, anemia, and uterine volume, but not fibroid volume. The incidence of adverse events was similar with relugolix combination therapy and placebo. Bone mineral density was similar with relugolix combination therapy and placebo but decreased with relugolix monotherapy.
CONCLUSIONS
Once-daily relugolix combination therapy resulted in a significant reduction in menstrual bleeding, as compared with placebo, and preserved bone mineral density in women with uterine fibroids. (Funded by Myovant Sciences; LIBERTY 1 [L1] and LIBERTY 2 [L2] Clinicaltrials.gov numbers, NCT03049735 and NCT03103087, respectively.)
Uterine fibroids are common; the cumulative incidence by 50 years of age is approximately 70% among White women and 80% among Black women.1,2 Approximately 25% of women with uterine fibroids have symptoms,3,4 most often heavy menstrual bleeding, which is often associated with anemia.5–7 Uterine fibroid–associated pain is the second most debilitating problem.8–10
Although contraceptives are first-line medical treatments for uterine fibroid symptoms,11 the quality of evidence for their use is low.7,12 Injectable long-acting gonadotropin-releasing hormone (GnRH) agonists (e.g., leuprolide acetate) are effective; however, hypoestrogenic sequelae limit their duration of use or lead to the administration of additional hormonal therapy to mitigate side effects.5,13 The GnRH antagonist elagolix, administered with estradiol and norethindrone acetate, reduces heavy menstrual bleeding in women with uterine fibroids14 and is approved for the treatment of uterine fibroids for 24 months.15 However, elagolix involves twice-daily administration because of its short half-life,16 and its use has been associated with a loss of bone mineral density at 1 year and with adverse effects on blood pressure and levels of lipids and liver enzymes.15
The selective progesterone-receptor modulator ulipristal acetate has been approved to treat uterine fibroids in some countries12,17; however, it has been linked to rare cases of serious liver injury, and the European Commission, on the basis of guidance from the European Medicines Agency, has recommended that ulipristal acetate for the treatment of uterine fibroids be used only in premenopausal women in whom surgical procedures (including uterine fibroid embolization) are not appropriate or have not worked.18 Surgery remains a common treatment option, although uterine-sparing procedures are associated with a substantial incidence of reoperation,19 and hysterectomy (both with and without ovarian conservation) has long-term sequelae, including increased mortality and risk of cardiovascular disease.20–22 With an estimated $34 billion in health care costs associated with fibroids in the United States alone,22 there is a major need for a non-surgical long-term treatment option,7 particularly one that addresses women’s usual preference for uterine-sparing alternatives regardless of their reproductive plans.4
Relugolix is an orally active nonpeptide GnRH-receptor antagonist that is suitable for daily use. It competitively binds to pituitary GnRH receptors, blocking the binding and signaling of endogenous GnRH23 and thus leading to reversible, dose-dependent decreases in gonadotropin concentrations and subsequent suppression of ovarian estradiol and progesterone production. In previous phase 3 trials involving Japanese women with symptomatic fibroids, relugolix at a dose of 40 mg led to improvements similar to those observed with leuprolide acetate with regard to heavy menstrual bleeding, anemia, and pain24 and to a significant reduction in pain as compared with placebo.25 To achieve efficacy, minimize hypoestrogenic side effects, and preserve bone mineral density, relugolix combination therapy (consisting of 40 mg of relugolix, 1 mg of estradiol, and 0.5 mg of norethindrone acetate) was developed as a once-daily treatment for maintaining estradiol levels within the physiologic range of the early follicular phase of the menstrual cycle,26,27 with the addition of a progestin to mitigate the unopposed estrogen action that could lead to endometrial hyperplasia.28
We report the results of two replicate international, double-blind, randomized, placebo-controlled, phase 3 trials (LIBERTY 1 and LIBERTY 2) in which we assessed the efficacy and safety of once-daily relugolix combination therapy in women with fibroid-associated heavy menstrual bleeding. We also evaluated a delayed relugolix combination therapy regimen, which consisted of relugolix monotherapy for 12 weeks followed by 12 weeks of relugolix combination therapy, in an additional group of participants to assess the benefit and safety of the addition of estradiol and norethindrone acetate.
METHODS
PARTICIPANTS
In these two trials, we enrolled women in Africa, Europe, North America, and South America. LIBERTY 1 (trial L1) was conducted at 80 sites, and LIBERTY 2 (trial L2) at 99 sites. Premenopausal women 18 to 50 years of age who had a diagnosis of fibroids as confirmed on ultrasonography and who had heavy menstrual bleeding, as assessed by the alkaline hematin method,29 were eligible. The alkaline hematin method directly measures the volume of menstrual blood loss by comparing hematin from menstrual products against calibration curves created from a simultaneous venous blood sample.29 Heavy menstrual bleeding was defined as a volume of menstrual blood loss of 80 ml or more per cycle for two cycles or a volume of 160 ml or more during one cycle.
Patients were excluded from the trials if they had a z score of less than −2.0 for bone mineral density at the lumbar spine, total hip, or femoral neck; had other causes of heavy menstrual bleeding30; or were using hormonal therapy. Additional exclusion criteria are summarized in Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org.
TRIAL DESIGN AND OVERSIGHT
Participants were randomly assigned, in a 1:1:1 ratio, by means of an interactive website to receive blinded placebo for 24 weeks, relugolix combination therapy for 24 weeks, or delayed relugolix combination therapy (relugolix monotherapy, followed by relugolix combination therapy, each for 12 weeks) (Fig. S3). A 40-mg relugolix tablet and a capsule containing estradiol and norethindrone acetate, or a placebo tablet and capsule, were packaged together in blister cards for once-daily coadministration. The delayed relugolix combination therapy group received the 40-mg relugolix tablet and a placebo capsule for 12 weeks, followed by the active-agent tablet and capsule for 12 weeks. The delayed relugolix combination therapy group was included to allow for the comparison of bone mineral density and vasomotor symptoms in the combination and monotherapy groups during the first 12 weeks of the trial. Trial visits occurred at baseline and every 4 weeks for 24 weeks.
The trials were conducted in accordance with the guidelines of the International Council for Harmonisation and the principles of the Declaration of Helsinki. All the participants provided written informed consent. The sponsor, Myovant Sciences, designed the trials and analyzed the data; the investigators and the sponsor jointly conducted the trials and collected the data. A steering committee of academic gynecologists provided strategic and scientific guidance. A data and safety monitoring committee that comprised practicing gynecologists and a statistician monitored trial progress and reviewed safety data. The seventh author was responsible for data analysis and vouches for the accuracy of the data. The sponsor held the data, and the authors had full access to the data analyses, reviewed the analyses, and vouch for fidelity of the trial to the protocol, available at NEJM.org. The first draft of the manuscript was written by the penultimate author. The other authors critically reviewed and provided feedback on the first draft and subsequent versions. A medical writer, who was funded by the sponsor, assisted with the preparation of the manuscript and its submission for publication.
EFFICACY END POINTS
In the primary efficacy analysis in each trial, a response was defined as both a volume of menstrual blood loss of less than 80 ml and a reduction of at least 50% from the baseline volume of menstrual blood loss, as measured by the alkaline hematin method, over the last 35 days of the treatment period. The primary comparison was the percentage of participants who had a response to relugolix combination therapy, as compared with placebo.
Key secondary end points at week 24 included the following: the percentage of women who reported amenorrhea; the mean percent reduction in the volume of menstrual blood loss; reduction in distress related to bleeding, the passing of blood clots, and tightness or pressure in the pelvic area, as measured by the Bleeding and Pelvic Discomfort scale31; the percentage of women with a baseline hemoglobin level of no more than 10.5 g per deciliter who had an increase of more than 2 g per deciliter; the percentage of women with moderate-to-severe pain at baseline (numerical rating scale score of ≥4, on a scale from 0 [no pain] to 10 [worst imaginable pain]) who had minimal-to-no fibroid-associated pain according to the numerical rating scale score in a daily electronic diary; the percent change in the volume of the largest fibroid; and the percent change in uterine volume. (The last two end points were assessed by means of transvaginal ultrasonography.) Details about each end point are provided in Table S2.
SAFETY ASSESSMENTS
Safety evaluations included the monitoring of vital signs, physical examination, adverse events, clinical laboratory variables, and 12-lead electrocardiography. Changes in bone mineral density were assessed by means of dual-energy x-ray absorptiometry at baseline and every 3 months during the trials. Endometrial biopsies were performed at baseline and at week 24 or the end of the treatment period (i.e., after the participant’s last dose of relugolix combination therapy or placebo).
STATISTICAL ANALYSIS
We calculated that the enrollment of approximately 390 participants in each trial would provide each trial with more than 90% power to detect a difference of at least 30 percentage points in the primary end point, at a two-sided alpha level of 0.05, between the relugolix combination therapy groups and placebo groups, assuming that 25% of the participants in the placebo group would have a response and that 20% of the participants would withdraw. Efficacy and safety analyses were performed in the modified intention-to-treat population, which included all the participants who underwent randomization and received at least one dose of relugolix (as combination therapy or monotherapy) or placebo.
The comparison of the primary end point between relugolix combination therapy and placebo was analyzed with the use of a Cochran– Mantel–Haenszel test for proportions, with stratification according to the baseline mean volume of menstrual blood loss (<225 ml vs. ≥225 ml) and geographic region (North America vs. rest of world). Rules for the handling of missing data were implemented for deriving the response status at week 24 or the end of the treatment period (last 35 days of the treatment period), with consideration for the duration of exposure to treatment or placebo and for adherence to the collection of menstrual products against entries in the electronic diary (i.e., the number of days with returned menstrual products, divided by the number of days with reported bleeding and product use, according to the data recorded in the electronic diary). In participants with 100% adherence, response status was based on the observed volume of menstrual blood loss. Participants who reported amenorrhea or “spotting or negligible bleeding,” as confirmed by data collected in the electronic diary, were considered to have had a response. Participants who received treatment or placebo for less than 4 weeks or who withdrew to undergo surgical intervention for uterine fibroids were considered not to have had a response. Detailed rules regarding the handling of missing data were prespecified (Table S3).
Analyses of the primary and key secondary efficacy end points were performed at an overall alpha level of 0.05 (two-sided) for the comparison of relugolix combination therapy with placebo. A gate-keeping, mixed-sequence testing procedure was used to maintain the familywise type I error. In each trial, the primary end point was tested first; if the P value was less than 0.05, the key secondary efficacy end points were tested as prespecified in the statistical analysis plans. In trial L1, the first four key secondary end points were tested sequentially in the order listed (see above), and the remaining three secondary end points were to be tested with the use of the Hochberg step-up procedure (Fig. S1). In trial L2, the first, second, third, and fifth secondary end points were tested sequentially, followed by testing of the other three key secondary end points (fourth, sixth, and seventh) with the use of the Hochberg procedure (Fig. S2). (This change in the order of hierarchical testing was made on the basis of the results of trial L1 before unblinding and the analysis of data in trial L2.)
Efficacy analyses comparing the delayed relugolix combination therapy groups with the placebo groups were not prespecified for any end points other than the percentage of participants with a response regarding menstrual blood loss (which was not included among the key secondary end points in the gatekeeping statistical testing procedure that was adjusted for multiplicity). Thus, data from the delayed relugolix combination therapy groups regarding the secondary end points are considered to be supportive.
RESULTS
PARTICIPANTS
From April 2017 through October 2018, a total of 388 women underwent randomization in trial L1; from June 2017 through December 2018, a total of 382 women underwent randomization in trial L2. A total of 308 women (79%) in trial L1 and 302 women (79%) in trial L2 completed the trial regimen. The percentages of participants who completed the trial were similar across all the trial groups (77 to 82%) (Fig. S4). Within each trial, the demographic and clinical characteristics of the participants at baseline were similar across the trial groups (Tables 1 and S4).
Table 1.
Demographic and Clinical Characteristics of the Participants at Baseline.*
| Characteristic | Trial L1 | Trial L2 | ||||
|---|---|---|---|---|---|---|
| Placebo (N = 127) | Relugolix Combination Therapy (N = 128) | Delayed Relugolix Combination Therapy (N = 132) | Placebo (N = 129) | Relugolix Combination Therapy (N = 125) | Delayed Relugolix Combination Therapy (N = 127) | |
| Age — yr | 42.2±5.7 | 42.5±5.0 | 41.3±5.4 | 41.8±5.3 | 42.4±5.4 | 42.1±5.3 |
| Race or ethnic group — no. (%)† | ||||||
| White | 56 (44) | 64 (50) | 53 (40) | 49 (38) | 58 (46) | 50 (39) |
| Black | 65 (51) | 59 (46) | 67 (51) | 74 (57) | 62 (50) | 66 (52) |
| Other | 6 (5) | 5 (4) | 12 (9) | 5 (4) | 2 (2) | 8 (6) |
| Hispanic ethnic group† | 23 (18) | 34 (27) | 33 (25) | 32 (25) | 18 (14) | 34 (27) |
| Body-mass index‡ | 32.3±7.5 | 31.4±7.6 | 31.4±7.3 | 32.1±7.6 | 31.0±6.6 | 30.8±5.7 |
| Bone mineral density — g/cm2 | ||||||
| Lumbar spine§ | 1.23±0.17 | 1.16±0.17 | 1.21±0.19 | 1.24±0.16 | 1.22±0.17 | 1.22±0.18 |
| Total hip | 1.07±0.15 | 1.03±0.15 | 1.06±0.15 | 1.07±0.13 | 1.06±0.14 | 1.06±0.15 |
| Menstrual blood loss | ||||||
| Volume — ml | 218.8±125.0 | 239.4±180.3 | 228.9±159.6 | 211.8±129.9 | 246.7±186.0 | 227.4±134.4 |
| Distribution — no. (%) | ||||||
| <225 ml | 85 (67) | 84 (66) | 86 (65) | 86 (67) | 80 (64) | 80 (63) |
| ≥225 ml | 42 (33) | 44 (34) | 46 (35) | 43 (33) | 45 (36) | 47 (37) |
| Hemoglobin concentration — g/dl | 11.4±1.4 | 11.2±1.6 | 11.1±1.7 | 11.1±1.6 | 11.3±1.5 | 11.1±1.6 |
| Uterine-fibroid volume — cm3 | 71.8±124.0 | 71.9±128.1 | 93.8±143.8 | 74.1±123.0 | 73.7±126.7 | 78.9±157.5 |
| Uterine volume — cm3 | 397.8±324.9 | 379.1±316.8 | 469.9±427.9 | 407.9±402.0 | 387.7±344.0 | 402.7±371.1 |
| Bleeding and Pelvic Discomfort scale score¶ | 71.4±21.3 | 66.8±22.1 | 68.5±22.9 | 70.0±20.3 | 70.7±20.8 | 72.0±22.9 |
| Maximum numerical rating scale score for uterine fibroid–associated pain ≥4 — no. (%)‖ | 95 (75) | 84 (66) | 89 (67) | 95 (74) | 93 (74) | 92 (72) |
Plus–minus values are means ±SD. L1 denotes LIBERTY 1, and L2 LIBERTY 2.
Race and ethnic group were reported by the participant. Other race or ethnic group included Asian, American Indian or Alaska Native, other race or ethnic group, and multiple races or ethnic groups. Percentages may not total 100 because of rounding or because of missing data: in trial L2, race or ethnic group was not reported by 1 participant in the placebo group, by 3 in the relugolix combination therapy group, and by 3 in the delayed relugolix combination therapy group. In trial L1, Hispanic ethnic group was not reported by 1 participant in the placebo group and by 2 in the delayed relugolix combination therapy group; in trial L2, Hispanic ethnic group was not reported by 1 participant in the placebo group, by 2 in the relugolix combination therapy group, and by 2 in the delayed relugolix combination therapy group.
The body-mass index is the weight in kilograms divided by the square of the height in meters.
Bone mineral density at the lumbar spine was assessed in the area from L1 through L4.
The Bleeding and Pelvic Discomfort scale score is calculated as the sum of scores for three symptoms (heavy bleeding during menstrual period, passing blood clots during menstrual period, and feeling tightness or pressure in pelvic area), each of which is scored on a scale from 1 to 5, with higher scores indicating greater symptom severity. Raw scores range from 3 to 15 and are normalized by dividing the difference between the reported score and the lowest possible raw score (i.e., 3) by the raw score range (i.e., 12) and then multiplying by 100.
Numerical rating scale scores for pain attributed to uterine fibroids range from 0 (no pain) to 10 (worst imaginable pain). Scores were recorded in a daily electronic diary. The numerical rating scale score at baseline was defined as the maximum score from the 35 days of data collected before the date of the first dose of relugolix or placebo. In the placebo groups and relugolix combination therapy groups, the baseline numerical rating scale scores were missing for 1 participant and 1 participant, respectively, in trial L1 and for 3 and 2 participants, respectively, in trial L2. No participants in the delayed relugolix therapy group in either trial had missing scores.
PRIMARY EFFICACY END POINT
In the relugolix combination therapy groups, 73% of the participants in trial L1 and 71% of those in trial L2 had a response, as compared with 19% and 15%, respectively, of the participants in the placebo groups (P<0.001 for both comparisons) (Fig. 1). In each trial, the observed treatment effects appeared to be similar, regardless of race or other characteristics of the participants at baseline (Fig. S5). In the delayed relugolix combination therapy groups, the percentages of participants with a response were similar in the two trials: 80% in trial L1 and 73% in trial L2.
Figure 1. Participants with Reduction in Heavy Menstrual Bleeding.
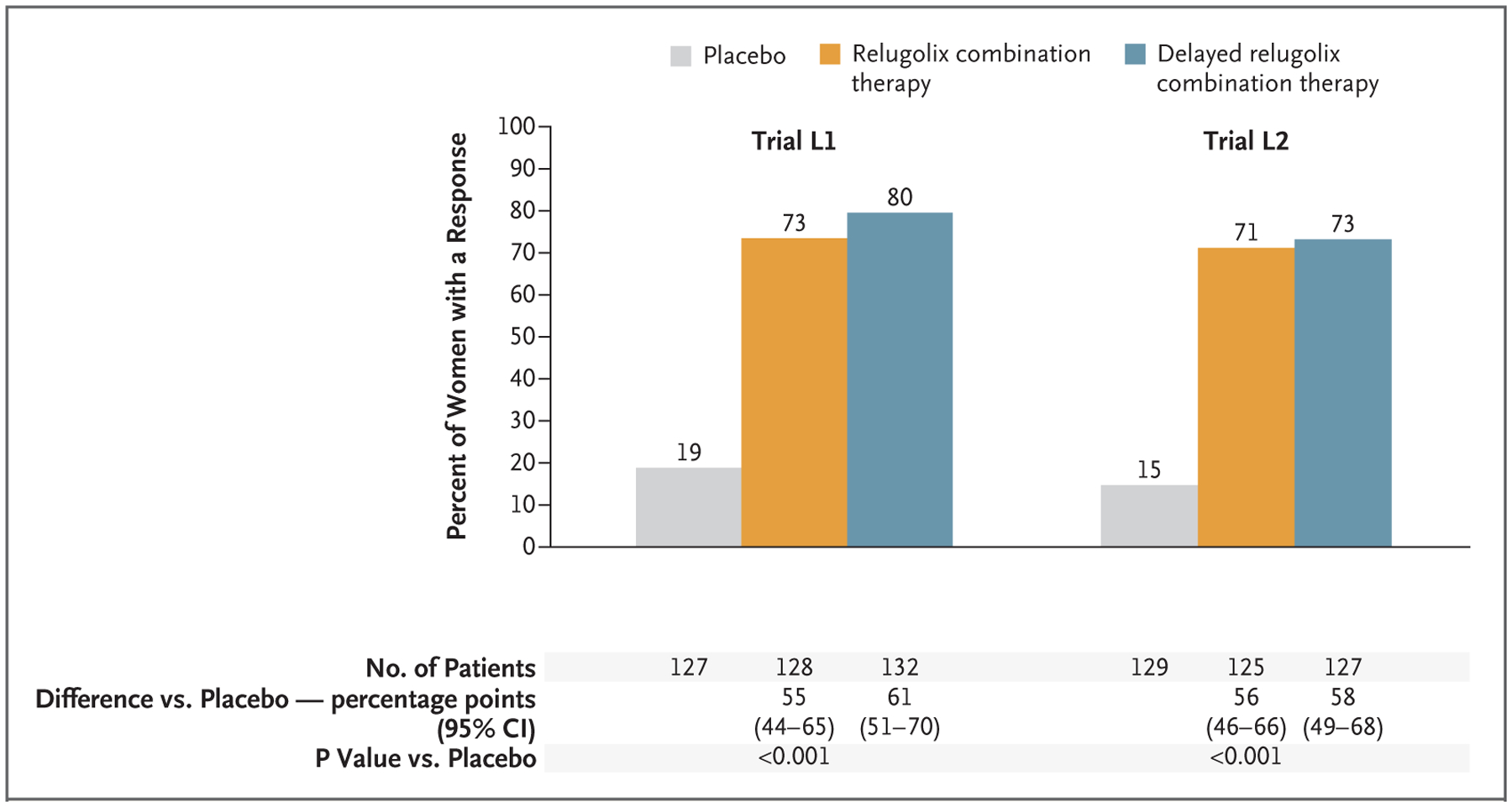
Shown are the percentages of women who had a response, which was defined as a volume of menstrual blood loss of less than 80 ml and a reduction of at least 50% from the baseline volume of menstrual blood loss, as measured by the alkaline hematin method, over the last 35 days of the treatment period. The primary end-point analysis in each trial was the comparison of relugolix combination therapy with placebo. CI denotes confidence interval, L1 LIBERTY 1, and L2 LIBERTY 2.
KEY SECONDARY EFFICACY END POINTS
Relugolix combination therapy was superior to placebo with regard to six of seven key secondary end points that were tested hierarchically in trial L1 and trial L2 (Table 2). Amenorrhea over the last 35 days of the treatment period occurred in 52% and 50% of the participants receiving relugolix combination therapy in trials L1 and L2, respectively, as compared with 6% and 3%, respectively, of those receiving placebo (P<0.001 for both comparisons).
Table 2.
Key Secondary Efficacy End Points, with Adjustment for Multiplicity.*
| End Point | Trial L1 | Trial L2 | ||||
|---|---|---|---|---|---|---|
| Placebo (N = 127) | Relugolix Combination Therapy (N = 128) | Delayed Relugolix Combination Therapy (N = 132) | Placebo (N = 129) | Relugolix Combination Therapy (N = 125) | Delayed Relugolix Combination Therapy (N = 127) | |
| Amenorrhea over last 35 days of treatment period — no. (%) | 7 (6) | 67 (52) | 76 (58) | 4 (3) | 63 (50) | 63 (50) |
| Difference vs. placebo (95% CI) — percentage points | 47 (37 to 56) | 47 (38 to 57) | ||||
| P value vs. placebo | <0.001 | <0.001 | ||||
| Percent change from baseline to wk 24 in menstrual blood-loss volume | −23.2±4.6 | −84.3±4.7 | −88.2±4.6 | −15.1±5.5 | −84.3±5.5 | −89.4±5.7 |
| Difference vs. placebo (95% CI) — percentage points | −61.1 (−73.5 to −48.6) | −69.2 (−84.1 to −54.3) | ||||
| P value vs. placebo | <0.001 | <0.001 | ||||
| Change from baseline to wk 24 in Bleeding and Pelvic Discomfort scale score | −16.1±2.8 | −45.0±2.9 | −51.3±2.9 | −18.3±2.9 | −51.7±2.9 | −48.9±3.0 |
| Difference vs. placebo (95% CI) | −28.9 (−36.3 to −21.5) | 33.4 (−41.2 to −25.5) | ||||
| P value vs. placebo | <0.001 | <0.001 | ||||
| Participants with anemia at baseline and an increase in hemoglobin level of >2 g/dl at wk 24 — no. (%)† | 5/23 (22) | 15/30 (50) | 18/32 (56) | 2/37 (5) | 19/31 (61) | 18/31 (58) |
| Difference vs. placebo (95% CI) — percentage points | 28 (4 to 53) | 56 (37 to 75) | ||||
| P value vs. placebo | 0.04 | <0.001 | ||||
| Maximum numerical rating scale score ≤1 over last 35 days of treatment period among participants in pain-evaluation subgroup — no. (%)‡ | 7/69 (10) | 25/58 (43) | 27/65 (42) | 14/82 (17) | 32/68 (47) | 24/58 (41) |
| Difference vs. placebo (95% CI) — percentage points | 33 (18 to 48) | 30 (16 to 44) | ||||
| P value vs. placebo | <0.001 | <0.001 | ||||
| Percent change from baseline to wk 24 in volume of primary uterine fibroid | −0.3±5.4 | −12.4±5.6 | −22.7±5.5 | −7.4±5.9 | −17.4±5.9 | −30.2±6.3 |
| Difference vs. placebo (95% CI) — percentage points | −12.1 (−26.3 to 2.0) | −10.0 (−25.8 to 5.8) | ||||
| P value vs. placebo§ | 0.09 | 0.22 | ||||
| Percent change from baseline to wk 24 in uterine volume | 2.2±3.0 | −12.9±3.1 | −17.9±3.0 | −1.5±3.4 | −13.8±3.4 | −17.7±3.5 |
| Difference vs. placebo (95% CI) — percentage points | −15.1 (−23.0 to −7.3) | −12.2 (−21.3 to −3.2) | ||||
| P value vs. placebo | <0.001 | 0.008 | ||||
Plus–minus values are least-squares means ±SD. In trial L1, the first four key secondary end points were tested sequentially in the order listed, and the remaining three secondary end points were to be tested with the use of the Hochberg step-up procedure. In trial L2, the first, second, third, and fifth secondary end points were tested sequentially, followed by testing of the other three key secondary end points (fourth, sixth, and seventh) with the use of the Hochberg procedure. CI denotes confidence interval.
The percentages of participants with a response were calculated in the subgroup of participants with anemia (hemoglobin level ≤10.5 g per deciliter) at baseline and who had hemoglobin data reported at week 24. This end point was tested fourth in trial L1 and fifth in trial L2.
The percentages of participants with a response were calculated in the subgroup of participants who had pain ratings that could be evaluated (maximum numerical rating scale score of ≥4 at baseline, with ≥28 days [80% of the last 35 days of the treatment period] of pain scores recorded in an electronic diary). This end point was tested fifth in trial L1 and fourth in trial L2.
The P value for the comparison in the percentage change from baseline to week 24 in the volume of the primary uterine fibroid did not meet the cutoff for statistical significance according to the Hochberg procedure.
The mean reduction in menstrual blood loss from baseline to week 24 in the relugolix combination therapy groups was 84.3% in both trial L1 and trial L2, as compared with 23.2% and 15.1%, respectively, in the placebo groups (P<0.001 for both comparisons). Reduction in blood loss occurred by week 4 and was sustained through week 24 (Fig. S6). In both trials, scores on the Bleeding and Pelvic Discomfort scale improved significantly from baseline in the relugolix combination groups, as compared with those in the placebo groups (Fig. S6), and more than 50% of the participants who had anemia at baseline had an increase of more than 2 g per deciliter in hemoglobin levels with relugolix combination therapy, as compared with placebo.
In addition, among the approximately 50% of the participants with moderate-to-severe pain at baseline who met the trial pain-evaluation requirements, the percentages of participants who had reductions to minimal or no pain (maximum numerical rating scale score, ≤1) over the last 35 days of the treatment period were significantly greater in the relugolix combination therapy groups than in the placebo groups (43% in trial L1 and 47% in trial L2 vs. 10% in trial L1 and 17% in trial L2; P<0.001 for both comparisons) (Fig. S6).
The overall uterine volume was decreased to a greater extent with relugolix combination therapy than with placebo (P<0.001). However, changes in the volume of the largest fibroid with relugolix combination therapy did not differ significantly from those with placebo (Table 2). Additional secondary end points, which were not tested hierarchically, are reported in Table S5.
SAFETY
In trial L1, the overall incidence of adverse events was 66% in the placebo group, 62% in the relugolix combination therapy group, and 73% in the delayed relugolix combination therapy group; in trial L2, the incidence was 59%, 60%, and 71%, respectively (Table 3). Serious adverse events were reported infrequently; each serious adverse event that was reported occurred in one participant in a given trial group (Table S6). No deaths were reported.
Table 3.
Adverse Events.*
| Event | Trial L1 | Trial L2 | ||||
|---|---|---|---|---|---|---|
| Placebo (N = 127) | Relugolix Combination Therapy (N = 128) | Delayed Relugolix Combination Therapy (N = 132) | Placebo (N = 129) | Relugolix Combination Therapy (N = 126) | Delayed Relugolix Combination Therapy (N = 126) | |
| number of participants with event (percent) | ||||||
| Any adverse event | 84 (66) | 79 (62) | 96 (73) | 76 (59) | 76 (60) | 90 (71) |
| Adverse event leading to discontinuation | 5 (4) | 7 (5) | 16 (12) | 6 (5) | 3 (2) | 14 (11) |
| Serious adverse event | 2 (2) | 7 (5) | 3 (2) | 4 (3) | 1 (1) | 2 (2) |
| Adverse event reported in >5% of participants in any group | ||||||
| Hot flash | 10 (8) | 14 (11) | 47 (36) | 5 (4) | 7 (6) | 44 (35) |
| Headache | 19 (15) | 14 (11) | 14 (11) | 15 (12) | 11 (9) | 28 (22) |
| Hypertension | 0 | 7 (5) | 3 (2) | 4 (3) | 5 (4) | 7 (6) |
| Arthralgia | 4 (3) | 4 (3) | 7 (5) | 4 (3) | 1 (1) | 8 (6) |
| Cough | 7 (6) | 1 (1) | 0 | 4 (3) | 0 | 1 (1) |
| Nausea | 6 (5) | 4 (3) | 5 (4) | 10 (8) | 6 (5) | 4 (3) |
| Upper respiratory tract infection | 3 (2) | 1 (1) | 7 (5) | 7 (5) | 6 (5) | 3 (2) |
| Anemia | 6 (5) | 4 (3) | 0 | 8 (6) | 2 (2) | 2 (2) |
| Fatigue | 5 (4) | 4 (3) | 6 (5) | 2 (2) | 1 (1) | 7 (6) |
Adverse events were coded with the use of the Medical Dictionary for Regulatory Activities, version 22.0. The severity of adverse events was evaluated by the investigator according to the National Cancer Institute Common Terminology for Adverse Events, version 5.0. No group in either trial had an incidence of hyperhidrosis or night sweats of more than 3%. There were no deaths in either trial.
Hot flash was the most frequently reported adverse event in both trials. In trial L1, hot flash occurred in 8% of the participants in the placebo group, in 11% of those in the relugolix combination therapy group, and in 36% of those in the delayed relugolix combination therapy group; in trial L2, the incidence was 4%, 6%, and 35%, respectively.
In trial L1, hypertension as an adverse event was reported in no participants in the placebo group, in 5% of the participants in the relugolix combination therapy group, and in 2% of those in the delayed relugolix combination therapy group. In trial L2, the incidence was 3%, 4%, and 6%, respectively. Data regarding participants with a history of hypertension are reported in Table S4.
The percent changes from baseline to weeks 12 and 24 in bone mineral density at the lumbar spine (L1 through L4) and the total hip were similar in the relugolix combination therapy group and the placebo group in both trials (Fig. 2A and 2B and Table S7). As expected, in the delayed relugolix combination therapy group, the bone mineral density at the lumbar spine and the total hip decreased from baseline at week 12 with relugolix monotherapy, which was followed by a plateau after the initiation of relugolix combination therapy (Fig. 2A and 2B).
Figure 2. Change in Bone Mineral Density.
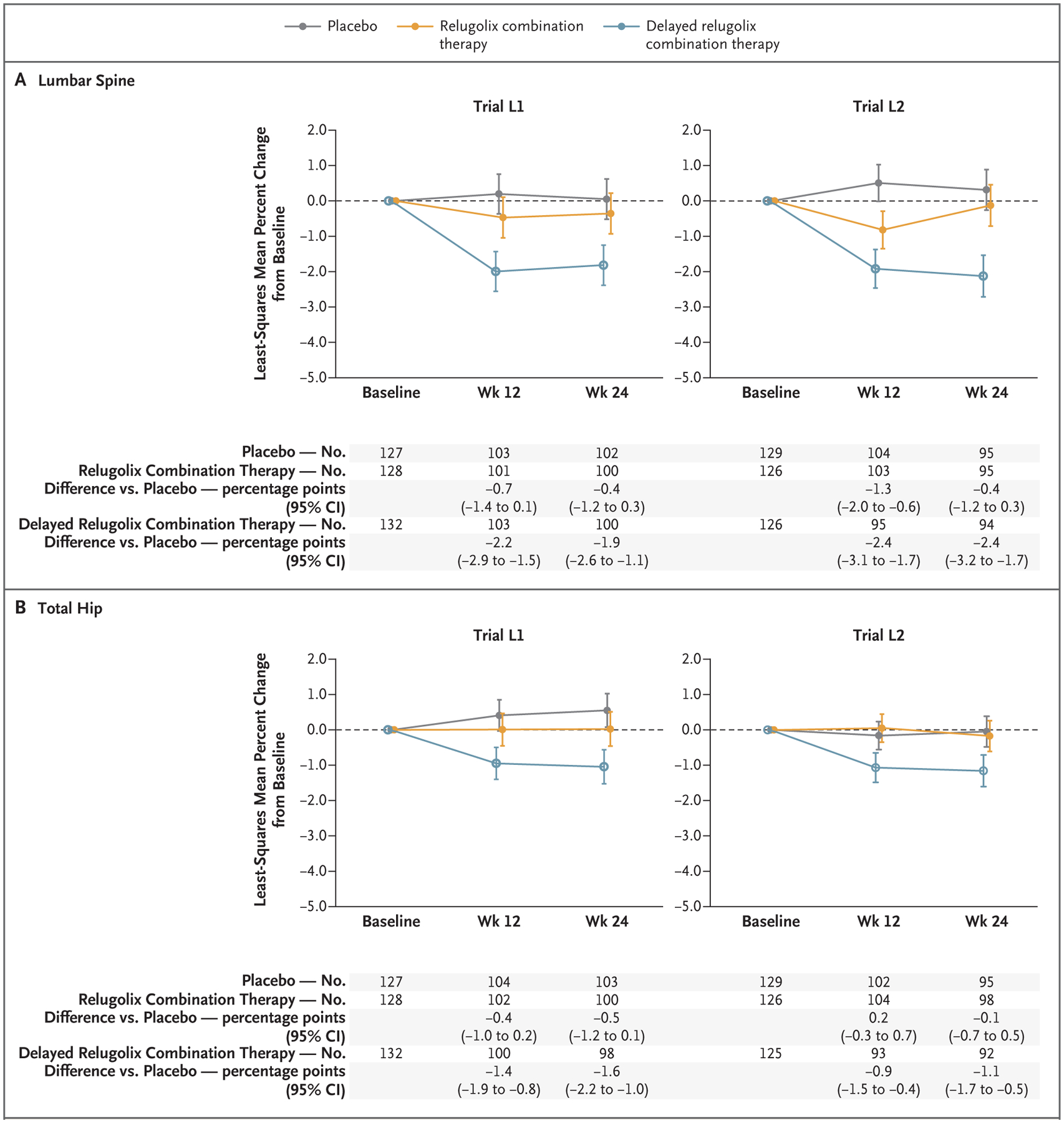
Least-squares means were based on mixed-effects models with baseline volume of menstrual blood loss, geographic region, age at baseline, body-mass index at baseline, bone mineral density at baseline, race, visit, and trial group–by–visit interaction as fixed effects. The dashed line indicates baseline, and the I bars 95% confidence intervals.
Laboratory tests and vital signs, including systolic and diastolic blood pressures, were similar among the groups. There were no meaningful differences in the mean changes from baseline or in the percentages of participants who met prespecified limits of change for any analysis, including liver-function tests and lipid levels (Tables S8 and S9).
At week 24, no cases of endometrial hyperplasia or endometrial cancer had occurred in the relugolix groups (i.e., the relugolix combination therapy group and the delayed relugolix combination therapy group). Endometrial hyperplasia without atypia was observed in two participants in the placebo group in trial L1. No pregnancies were reported in the relugolix groups in either trial.
DISCUSSION
In these two randomized, placebo-controlled, phase 3 trials involving women with symptomatic uterine fibroids, the percentage of participants with a response (i.e., volume of menstrual blood loss of <80 ml and a ≥50% reduction in volume from baseline) was significantly higher with relugolix combination therapy than with placebo. In addition, benefits were observed with relugolix combination therapy for six of seven key secondary end points, including amenorrhea in half the participants; an average (least-squares mean) 84.3% reduction in menstrual blood loss, with the decrease first observed at 4 weeks and persisting through 24 weeks; an improvement in hemoglobin levels in participants with anemia; and pain reduction in those with moderate-to-severe pain at baseline.
The trial population was representative of women with symptomatic fibroids in the general population.5,8,13,32 Approximately half the women were Black. Overall, the mean body-mass index (the weight in kilograms divided by the square of the height in meters) of the participants was in the obese range (≥30.0), and the mean volume of menstrual blood loss (218.8 to 239.4 ml across groups in trial L1 and 211.8 to 246.7 ml across groups in trial L2) was almost 3 times the upper limit of what has historically been considered the normal range. The majority of women had moderate-to-severe uterine fibroid–associated pain, and most experienced marked distress because of their symptoms and substantially impaired quality of life. The treatment effect of relugolix combination therapy on reducing menstrual bleeding was consistent, regardless of race or other characteristics of the participants or fibroids.
Pain is common in women with uterine fibroids, and the distress that women experience from bleeding and pain is underappreciated.33 Yet the effects of treatment on fibroid-associated pain are infrequently assessed in clinical trials. The LIBERTY phase 3 trials showed a reduction in rigorously assessed fibroid-associated pain with the use of a daily electronic diary and a validated pain-outcome measure, a finding that is supported by a 12-week, phase 3 trial of relugolix monotherapy.25 Participants in our two trials described considerable discomfort and distress associated with their fibroids at baseline, with those in the relugolix combination therapy group reporting significantly less distress from bleeding, passing of blood clots, and tightness or pressure in the pelvic area at the end of the treatment period than those who received placebo. There was no significant reduction in the volume of the largest fibroid, but the reduction in uterine volume, as measured by means of ultrasonography, suggests a reduced fibroid burden. The incidence of serious adverse events and non-serious adverse events was similar overall in the relugolix combination therapy groups and the placebo groups.
Fibroid growth and extracellular matrix production are stimulated by estrogen and progesterone by means of paracrine mechanisms.34 Although a potential direct antiproliferation effect of GnRH antagonists against human fibroid cells has been reported,35 a recent review of medical management of uterine fibroids suggests that the direct effect on fibroids of GnRH-receptor agonists and antagonists is probably modest at best.36 The estrogen-threshold hypothesis proposes that maintenance of the estradiol concentration between 20 and 50 pg per milliliter (70 and 180 pmol per liter) can decrease fibroid growth while minimizing hypoestrogenic adverse effects.26 In a phase 1 study, the median trough concentrations of estradiol were consistently less than 10 pg per milliliter (40 pmol per liter) with the administration of 40 mg of relugolix alone for 6 weeks but remained above 20 pg per milliliter when relugolix was coadministered with 1 mg of estradiol and 0.5 mg of norethindrone acetate.37
The inclusion of the delayed relugolix combination therapy group in the LIBERTY trials allowed for the comparison of the effects of combination therapy with monotherapy. We found that 12 weeks of monotherapy resulted in a loss of bone mineral density and a higher incidence of vasomotor adverse events, as compared with relugolix combination therapy, and although the transition to relugolix combination therapy prevented further loss of bone mineral density, it did not reverse the changes in bone mass. The initiation of treatment with relugolix combination therapy, as compared with relugolix monotherapy (followed later by relugolix combination therapy), did not meaningfully affect efficacy with respect to the volume of menstrual blood loss. The present trials showed that relugolix combination therapy reduced menstrual bleeding and fibroid-associated pain to a greater extent than placebo, without substantive hypoestrogenic effects, over a period of 6 months.
These trials had limitations. Many women with self-reported heavy menstrual bleeding and uterine fibroids did not pass screening owing to strict assessment criteria, which is a situation that could limit generalizability, and the duration of the trial regimen was only 6 months. Participants who completed these trials were offered enrollment in a 28-week extension study with open-label relugolix combination therapy and a subsequent 52-week randomized-withdrawal trial; these studies may provide more information regarding the long-term benefits and risks of relugolix therapy.
In these trials, once-daily relugolix combination therapy resulted in a substantial reduction in heavy menstrual bleeding in women with uterine fibroids, with resolution of anemia, a reduction in pain, and reduced distress related to bleeding and pelvic discomfort, while preserving bone density and minimizing the incidence of hot flashes associated with relugolix monotherapy.
Supplementary Material
Acknowledgments
Supported by Myovant Sciences.
We thank W. Mark Roberts, Ph.D., for medical writing and editing assistance with an earlier draft of the manuscript, funded by Myovant Sciences.
Footnotes
REFERENCES
- 1.Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol 2003; 188: 100–7. [DOI] [PubMed] [Google Scholar]
- 2.Wise LA, Laughlin-Tommaso SK. Epidemiology of uterine fibroids: from menarche to menopause. Clin Obstet Gynecol 2016; 59: 2–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laughlin-Tommaso SK, Stewart EA. Moving toward individualized medicine for uterine leiomyomas. Obstet Gynecol 2018; 132: 961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borah BJ, Nicholson WK, Bradley L, Stewart EA. The impact of uterine leiomyomas: a national survey of affected women. Am J Obstet Gynecol 2013; 209(4): 319.e1–319.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart EA. Uterine fibroids. N Engl J Med 2015; 372: 1646–55. [DOI] [PubMed] [Google Scholar]
- 6.Nelson AL, Ritchie JJ. Severe anemia from heavy menstrual bleeding requires heightened attention. Am J Obstet Gynecol 2015; 213(1): 97.e1–97.e6. [DOI] [PubMed] [Google Scholar]
- 7.Al-Hendy A, Myers ER, Stewart E. Uterine fibroids: burden and unmet medical need. Semin Reprod Med 2017; 35: 473–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.David M, Pitz CM, Mihaylova A, Siedentopf F. Myoma-associated pain frequency and intensity: a retrospective evaluation of 1548 myoma patients. Eur J Obstet Gynecol Reprod Biol 2016; 199: 137–40. [DOI] [PubMed] [Google Scholar]
- 9.Foth D, Röhl F-W, Friedrich C, et al. Symptoms of uterine myomas: data of an epidemiological study in Germany. Arch Gynecol Obstet 2017; 295: 415–26. [DOI] [PubMed] [Google Scholar]
- 10.Monleón J, Cañete ML, Caballero V, et al. Epidemiology of uterine myomas and clinical practice in Spain: an observational study. Eur J Obstet Gynecol Reprod Biol 2018; 226: 59–65. [DOI] [PubMed] [Google Scholar]
- 11.Yao X, Stewart EA, Laughlin-Tommaso SK, Heien HC, Borah BJ. Medical therapies for heavy menstrual bleeding in women with uterine fibroids: a retrospective analysis of a large commercially insured population in the USA. BJOG 2017; 124: 322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartmann KE, Fonnesbeck C, Surawicz T, et al. Management of uterine fibroids: comparative effectiveness review, no. 195. Rockville, MD: Agency for Health-care Research and Quality, 2017. [PubMed] [Google Scholar]
- 13.Pérez-López FR, Ornat L, Ceausu I, et al. EMAS position statement: management of uterine fibroids. Maturitas 2014; 79: 106–16. [DOI] [PubMed] [Google Scholar]
- 14.Schlaff WD, Ackerman RT, Al-Hendy A, et al. Elagolix for heavy menstrual bleeding in women with uterine fibroids. N Engl J Med 2020; 382: 328–40. [DOI] [PubMed] [Google Scholar]
- 15.Food and Drug Administration. Oriahnn (elagolix, estradiol, and norethindrone acetate capsules; elagolix capsules) prescribing information. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213388s000lbl.pdf). [Google Scholar]
- 16.Ng J, Chwalisz K, Carter DC, Klein CE. Dose-dependent suppression of gonadotropins and ovarian hormones by elagolix in healthy premenopausal women. J Clin Endocrinol Metab 2017; 102: 1683–91. [DOI] [PubMed] [Google Scholar]
- 17.Stewart EA, Laughlin-Tommaso SK, Catherino WH, Lalitkumar S, Gupta D, Vollenhoven B. Uterine fibroids. Nat Rev Dis Primers 2016; 2: 16043. [DOI] [PubMed] [Google Scholar]
- 18.Ulipristal acetate for uterine fibroids: EMA recommends restricting use. Amsterdam: European Medicines Agency, January 11, 2021. (https://www.ema.europa.eu/en/documents/referral/ulipristal-acetate-5mg-medicinal-products-article-31-referral-ulipristal-acetate-uterine-fibroids_en.pdf). [Google Scholar]
- 19.Donnez J, Dolmans M-M. Uterine fibroid management: from the present to the future. Hum Reprod Update 2016; 22: 665–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker WH, Feskanich D, Broder MS, et al. Long-term mortality associated with oophorectomy compared with ovarian conservation in the Nurses’ Health Study. Obstet Gynecol 2013; 121: 709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laughlin-Tommaso SK, Khan Z, Weaver AL, Smith CY, Rocca WA, Stewart EA. Cardiovascular and metabolic morbidity after hysterectomy with ovarian conservation: a cohort study. Menopause 2018; 25: 483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardozo ER, Clark AD, Banks NK, Henne MB, Stegmann BJ, Segars JH. The estimated annual cost of uterine leiomyomata in the United States. Am J Obstet Gynecol 2012; 206(3): 211.e1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miwa K, Hitaka T, Imada T, et al. Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}−3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor. J Med Chem 2011; 54: 4998–5012. [DOI] [PubMed] [Google Scholar]
- 24.Osuga Y, Enya K, Kudou K, Tanimoto M, Hoshiai H. Oral gonadotropin-releasing hormone antagonist relugolix compared with leuprorelin injections for uterine leiomyomas: a randomized controlled trial. Obstet Gynecol 2019; 133: 423–33. [DOI] [PubMed] [Google Scholar]
- 25.Osuga Y, Enya K, Kudou K, Hoshiai H. Relugolix, a novel oral gonadotropin-releasing hormone antagonist, in the treatment of pain symptoms associated with uterine fibroids: a randomized, placebo-controlled, phase 3 study in Japanese women. Fertil Steril 2019; 112(5): 922–929.e2. [DOI] [PubMed] [Google Scholar]
- 26.Friedman AJ, Lobel SM, Rein MS, Barbieri RL. Efficacy and safety considerations in women with uterine leiomyomas treated with gonadotropin-releasing hormone agonists: the estrogen threshold hypothesis. Am J Obstet Gynecol 1990; 163: 1114–9. [DOI] [PubMed] [Google Scholar]
- 27.Stricker R, Eberhart R, Chevailler M-C, Quinn FA, Bischof P, Stricker R. Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT analyzer. Clin Chem Lab Med 2006; 44: 883–7. [DOI] [PubMed] [Google Scholar]
- 28.Sabry M, Al-Hendy A. Medical treatment of uterine leiomyoma. Reprod Sci 2012; 19: 339–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hallberg L, Högdahl AM, Nilsson L, Rybo G. Menstrual blood loss — a population study: variation at different ages and attempts to define normality. Acta Obstet Gynecol Scand 1966; 45: 320–51. [DOI] [PubMed] [Google Scholar]
- 30.Munro MG, Critchley HOD, Fraser IS. The two FIGO systems for normal and abnormal uterine bleeding symptoms and classification of causes of abnormal uterine bleeding in the reproductive years: 2018 revisions. Int J Gynaecol Obstet 2018; 143: 393–408. [DOI] [PubMed] [Google Scholar]
- 31.Li J, Kang JB, Hunsche E, Hudgens S. Measuring patient-reported outcomes in women with heavy menstrual bleeding associated with uterine fibroids: the bleeding and pelvic discomfort scale. Fertil Steril 2019; 112: Suppl(3): e344. abstract. [DOI] [PubMed] [Google Scholar]
- 32.Higham JM, O’Brien PM, Shaw RW. Assessment of menstrual blood loss using a pictorial chart. Br J Obstet Gynaecol 1990; 97: 734–9. [DOI] [PubMed] [Google Scholar]
- 33.Soliman AM, Margolis MK, Castelli-Haley J, Fuldeore MJ, Owens CD, Coyne KS. Impact of uterine fibroid symptoms on health-related quality of life of US women: evidence from a cross-sectional survey. Curr Med Res Opin 2017; 33: 1971–8. [DOI] [PubMed] [Google Scholar]
- 34.Bulun SE. Uterine fibroids. N Engl J Med 2013; 369: 1344–55. [DOI] [PubMed] [Google Scholar]
- 35.Khan KN, Kitajima M, Hiraki K, et al. Cell proliferation effect of GnRH agonist on pathological lesions of women with endometriosis, adenomyosis and uterine myoma. Hum Reprod 2010; 25: 2878–90. [DOI] [PubMed] [Google Scholar]
- 36.Lewis TD, Malik M, Britten J, San Pablo AM, Catherino WH. A comprehensive review of the pharmacologic management of uterine leiomyoma. Biomed Res Int 2018; 2018: 2414609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukes A, Johnson B, Jones L, et al. Pharmacokinetics, pharmacodynamics, and safety of relugolix, a potent oral once-daily gonadotropin-releasing hormone (GnRH) receptor antagonist, as monotherapy and in combination with estradiol/norethindrone acetate add-back therapy. Hum Reprod 2017; 32: Suppl: i267–i268. abstract. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.