

Abstract
Nonsense-mediated messenger RNA decay (NMD) controls mRNA quality and degrades physiologic mRNAs to fine-tune gene expression in changing developmental or environmental milieus. NMD requires that its targets are removed from the translating pool of mRNAs. Since the decay steps of mammalian NMD remain unknown, we developed assays to isolate and sequence direct NMD decay intermediates transcriptome-wide based on their co-immunoprecipitation with phosphorylted UPF1, which is the active form of this essential NMD factor. We show that, unlike steady-state UPF1, phosphorylated UPF1 binds predominantly deadenylated mRNA decay intermediates and activates NMD cooperatively from 5′- and 3′-ends. We leverage method modifications to characterize the 3′-ends of NMD decay intermediates, show that they are ribosome-bound, and reveal that some are subject to the addition of non-templated nucleotide. Uridines are added by TUT4 and TUT7 terminal uridylyl transferases and removed by the Perlman syndrome-associated exonuclease DIS3L2. The addition of other non-templated nucleotides appears to inhibit decay.
Nonsense-mediated mRNA decay (NMD) is a key safeguard against routine mistakes made during mammalian-cell gene expression that would otherwise result in the accumulation of aberrant mRNAs and their encoded proteins; it also plays essential roles in maintaining cellular homeostasis during developmental or other environmental changes1–5.
Given the importance of NMD to mammalian cells, it is remarkable that NMD decay intermediate sequences and translational status remain undefined. Considering the numerous types of RNAs in cells and their various modes of constitutive and regulated elimination, comprehensive profiling of NMD decay intermediates necessitates targeted approaches. For all mammals that have been examined, NMD requires translation and the ATP-dependent RNA helicase UPF1. It is known that translation termination of the type that triggers NMD, for example upstream of an exon-junction complex (EJC), modulates UPF1 binding to the 3′ -untranslated region (3′ UTR) of NMD targets1–5. Unlike promiscuous UPF1 binding to physically accessible nucleotides, including those of transcripts that are not NMD targets, regulated UPF1 binding to NMD targets leads to UPF1 phosphorylation by the kinase SMG11–5. UPF1 phosphorylation inhibits further translation initiation events on the NMD target1,3 and recruits ribonucleolytic activities that degrade the NMD target1–5. It follows that phosphorylated UPF1 (p-UPF1), which is enriched on the 3′UTR and additionally binds to the coding region of NMD targets, provides a direct and reliable marker for cellular transcripts undergoing NMD6–8. While the RNA decay steps of NMD in mammalian cells are known to involve the endonuclease SMG6, which targets capped and polyadenylated mRNAs9–11, decapping and the 5′ -to-3′ exonuclease XRN1, and deadenylases and the 3′ -to-5′ exonucleolytic exosome12–15, no methodology has been developed for the comprehensive analysis of direct NMD decay intermediates. Identification of NMD decay intermediates in human cells is warranted given that mammalian-cell NMD differs from the better-characterized generic mRNA decay pathway and NMD in yeast, which do not involve SMG616,17.
Here, we describe a transcriptome-wide method that captures and sequences decay intermediates derived from direct NMD targets that we call NMD-degradome sequencing (NMD-DegSeq). Unlike methods that identify NMD targets by their upregulation on the downregulation of one or more NMD factors, which cannot distinguish between direct and indirect NMD targets, our method identifies direct NMD targets based on their co-immunoprecipitation with p-UPF1. We find that, unlike steady-state UPF1, which is hypophosphorylated, p-UPF1 binds predominantly deadenylated NMD decay intermediates. By ligating different adapters to each end of the p-UPF1-bound transcripts, we capture both ends of the NMD decay intermediates, and we focus here on 3′ -ends.
We also describe additional methodologies that not only verify results obtained using NMD-DegSeq but offer utility in defining new NMD nucleolytic activities (NMD-DegNAs), new NMD tailing activities (NMD-DegTAs), or NMD-decay intermediates in immunoprecipitations of ribosomal protein L5 (NMD-DegRPL5) or in polysome profiles (NMD-DegRibo). Among our findings, 3′ -to-5′ mRNA decay is mediated by the exosome-free nuclease DIS3L2, which is mutated in the Perlman syndrome of fetal overgrowth and causes susceptibility to Wilms and bilateral tumors18–20. Results indicate that DIS3L2 functions in a mechanism that is accompanied by the addition of non-templated uridines, by the terminal transferases TUT4 and TUT7, and also non-templated cytidines, adenosines, guanines, or some combination thereof. While widespread RNA 3′ -end oligouridylation has been shown to be a molecular mark for the decay of cellular mRNAs, pre-microRNAs, and long noncoding RNAs21–29, our data provide a demonstration that 3′ -end oligouridylation also occurs during NMD. We find that NMD decay intermediates are associated with one or more ribosomes along with p-UPF1, and that the decay steps of mammalian-cell NMD occur co-translationally through repetitive cycles of 3′ -to-5′ exonucleolytic decay followed by the addition of non-templated nucleotides. While most non-templated nucleotides are uridines, which are known to be destabilizing, those that are not uridines appear to interfere with further decay.
Results
Transcriptome-wide NMD-DegSeq reveals non-templated nucleotide heterogeneity at the 3′-ends of NMD decay intermediates.
Since the binding of p-UPF1 can be used to differentiate transcripts that are direct NMD targets from transcripts that are either indirect NMD targets or not NMD targets6,8,30, and given the role of decapping in NMD10,12,31–33, we rationalized that cellular NMD decay intermediates could be analyzed based on their simultaneous co-immunoprecipitation with p-UPF1 and loss of the 5′ -cap. The p-UPF1 immunoprecipitation step proved to be critical: without this step, transfer RNAs, small nuclear RNAs, small nucleolar RNAs, and miRNAs were five orders of magnitude more abundant than uncapped RNA species deriving from NMD targets (data not shown). Thus, we performed anti-p-UPF1 immunoprecipitation and, in parallel as a negative control, rabbit IgG (rIgG) immunoprecipitation of lysates of HEK293T cells that had been cultured in the presence of okadaic acid (Fig. 1a and Supplementary Fig. 1a,b). Okadaic acid inhibits the serine-threonine protein phosphatases PP1 and PP2A so as to increase cellular p-UPF1 abundance34–36 by inhibiting p-UPF1 recycling to a hypophosphorylated state after p-UPF1 is removed from an NMD target6. Thus, p-UPF1 immunoprecipitation after okadaic acid treatment enriches for p-UPF1-bound decay intermediates actively undergoing degradation.
Fig. 1 |. Transcriptome-wide NMD-DegSeq maps decay intermediate 3′-ends, revealing heterogenous non-templated nucleotide additions.
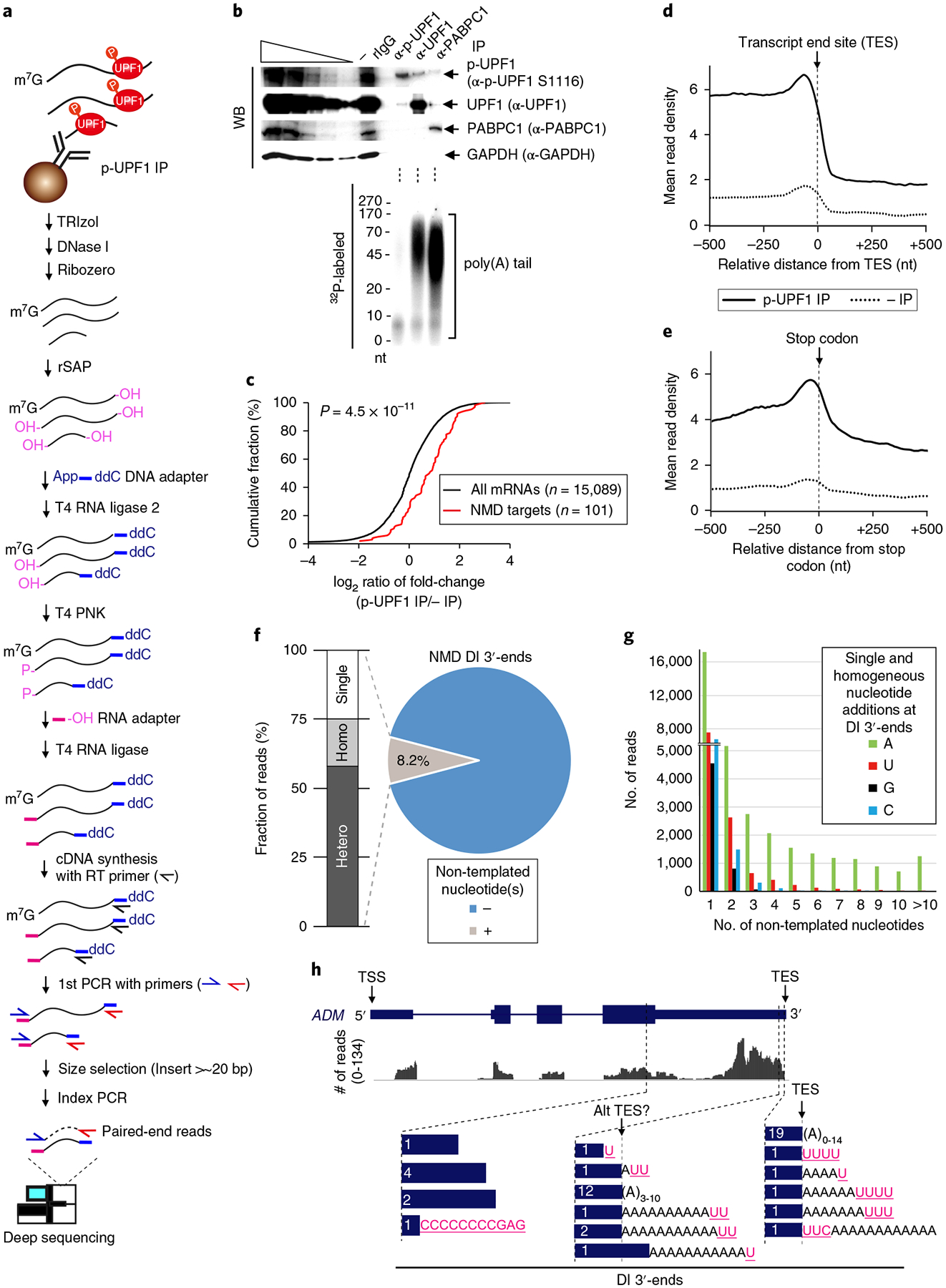
a, Schematic workflow used to identify decay intermediates (DI) of direct NMD targets using lysates of okadaic acid-treated HEK293T cells. Control immunoprecipitations (IPs) performed in parallel with anti-p-UPF1 immunoprecipitations utilized rabbit (r)IgG. b, Top, western blot (WB) of cell lysates prior to (−) or after immunoprecipitation. Leftmost five lanes are 3-fold serial dilutions of lysate. Bottom, primer extension assays analyze poly(A)-tail length of immunoprecipitated RNAs. (Schematic protocol depicted in Supplementary Fig. 1d.) Results are representative of two independently performed experiments. Uncropped images are shown in Supplementary Data Set 1. c, Cumulative fraction of log2 ratio of RNA sequence reads after anti-p-UPF1 immunoprecipitation relative to before immunoprecipitation. P value derived from the Wilcoxon rank-sum test. d,e, Exonic paired-end sequence reads ± 500 nucleotides (nt) to either side of the transcript end site (TES) (d) or stop codon (e) in 10-nucleotide bins before (dotted lines) or after (solid lines) p-UPF1 immunoprecipitation. f, Pie chart of genome-aligned paired-end reads with (grey = 146,018 reads) or without (blue = 1,770,844 reads) non-templated nucleotide additions. Genome-aligned sequence reads with ≥ 1 non-templated nucleotides are shown as percentages for single (Single), heterogeneous (Hetero), or homogeneous (Homo) non-templated nucleotides. Homogeneous A stretches probably reflect poly(A) tail remnants. g, Number of sequencing reads for each 3′ -decay intermediate addition that consists of either single or homogeneous stretches of non-templated nucleotides, shown according to length. h, Graphic view of NMD-DegSeq paired-end reads for ADM mRNA, aligned to the coding strand of the ADM gene, plus the number (in white) and location (blue bars) of the corresponding 3′ -decay intermediate ends. Pink, non-templated nucleotide additions; black, poly(A) tail remnants. Alt TES, alternative transcript end site.
To ensure that decay intermediates were not generated non-specifically after cell lysis and that immunoprecipitated p-UPF1 was not promiscuously bound to RNA, in vitro-synthesized, uniformly 32P-labeled mouse β-actin mRNA was added to lysates prior to p-UPF1 immunoprecipitation. We confirmed that no decay intermediates deriving from this mRNA were detected nor was this mRNA detected after p-UPF1 immunoprecipitation (Supplementary Fig. 1c). Using primer extension assays (Fig. 1b and Supplementary Fig. 1d), we found that the 3′-ends of p-UPF1-bound RNAs were characterized by short (<~10-nucleotide) poly(A) stretches, whereas RNAs bound by the bulk of cellular UPF1, which is largely hypophosphorylated6,34,36, or by poly(A)-binding protein 1 (PABPC1) contained longer (30–170-nucleotide) poly(A) stretches (Fig. 1b). Consistent with a role for deadenylation in the decay of premature termination codon (PTC)-containing mRNAs12,13, these data demonstrate that the activation of UPF1 by phosphorylation triggers NMD at least in part by promoting poly(A) shortening.
To capture and sequence decay intermediates derived from direct NMD targets, we performed NMD-DegSeq (Fig. 1a and Supplementary Fig. 1a,b). After computationally removing adapter sequences, we obtained ~5–10 × 106 HEK293T-cell sequence reads that uniquely mapped to the hg19 human genome database. We excluded RefSeq annotated transcripts for which we could not detect sequence reads in either input, that is RNA prior to immunoprecipitation, or immunoprecipitated samples, thereby defining 15,089 transcripts as HEK293T-expressed mRNAs (“All mRNAs” in Fig. 1c). As expected, RNA fragments that derived from each of the 101 previously validated NMD targets6,10,37–40 were significantly enriched in libraries from p-UPF1-bound samples relative to all mRNAs (Fig. 1c and Supplementary Table 1). Since p-UPF1-bound RNAs are predominantly deadenylated (Fig. 1b) and > 90% are < 120 nucleotides (Supplementary Fig. 1e), which is much shorter than the average > 1,200-nucleotide length of human mRNAs41,42, we conclude that we are detecting NMD decay intermediates that lack a 5′ -cap and have either a short or no poly(A) tail.
To exclude the possibility that nucleotides identified at decay intermediate 5′ - and 3′ -ends are sequencing artifacts, we analyzed paired-end sequencing reads that spanned the full length of NMD decay intermediates and included the adapters: this ensured that the 3′ -end of reads beginning at the 5′-end of an NMD decay intermediate were consistent with the 5′-end of reads beginning at the 3′ -end of that same NMD decay intermediate (Fig. 1a and Supplementary Fig. 1a). This was possible since ~80% of insert reads were ≤ 86 nucleotides (Supplementary Fig. 1e).
Paired-end sequencing revealed that NMD decay intermediate sequences mapped with comparable abundance throughout the transcribed regions of p-UPF1-bound transcripts except for (i) a relative deficiency of reads that extended from the first transcribed nucleotide, that is the transcript start site (TSS), to nucleotide ~500 (Supplementary Fig. 1f), and (ii) a relative enrichment of reads from the 3′ -ends of transcripts, in particular from ~200 nucleotides upstream of the poly(A) tails that extended to the poly(A) tail, that is the transcript end site (TES) (Fig. 1d). The relative lack of sequence reads from transcript 5′ -ends might be attributable to the efficiency with which decay intermediates lacking 5′ -cap structures are subjected to cellular 5′ -to-3′ exonucleolytic decay. The enrichment of reads from the 3′ -ends of transcripts peaked with an additional increase in decay intermediate abundance immediately upstream of the TES (Fig. 1d), at least partially reflecting p-UPF1 binding, which is known to predominate on NMD target 3′ UTRs6,8. When reads were aligned relative to the termination codon, we also observed an enrichment of NMD decay intermediates in coding regions that peaked ~40 nucleotides upstream of the stop codon (Fig. 1e). In contrast, there was no enrichment when reads were aligned relative to start codons (Supplementary Fig. 1g). This peak upstream of stop codons may reflect the position of the terminating ribosome, which could serve as a barrier to 5′ -to-3′ and 3′ -to-5′ exonucleolytic decay, and to SMG6-initiated endonucleolytic decay, which is known to occur adjacent to the termination codon that triggers NMD9–11. Bound ribosomes may also explain why decay intermediates identified in paired-end reads are enriched in the coding region relative to the 3′ UTR (Fig. 1e). These findings were confirmed using data from NMD decay intermediate single-end reads, which were found to exceed 86 nucleotides (data not shown).
We next focused on the 3′ -end of decay intermediates at a single-nucleotide level. Surprisingly, NMD-DegSeq revealed that ~8.2% of decay intermediate 3′ -ends harbored ≥ 1 non-templated nucleotide (Fig. 1f, g). These non-templated nucleotides included remnants of the poly(A) tail that were < 20 nucleotides, consistent with results from primer extension assays (Fig. 1b). Notably, we found that ~32% of these poly(A) remnants harbored one or more non-templated nucleotide(s) at their 3′ -end. For comparison, analyses of total HeLa-cell and NIH3T3-cell mRNA 3′ -ends demonstrated that ~1–2% were characterized by a poly(A) tail that had been shortened to 8–25 nucleotides and terminated in one or more non-templated uridines26,43. All told, ~58% of NMD decay intermediate 3′ -end non-templated nucleotides were heterogeneous stretches, ~25% were single-nucleotide additions, and ~17% were homogeneous stretches (Fig. 1f). In each population, when remnants of the poly(A) tail are discounted, one or more non-templated U additions were the most frequent 3′ -end modification, occupying ~4% of total paired-end reads (Fig. 1f, g). As examples of heterogeneity at decay intermediate 3′ -ends, decay intermediate termini within the transcribed body or poly(A) tail are detailed for NMD targets encoding adrenomedullin (ADM) (Fig. 1h) and DNA-damage inducible protein 4 (DDIT4) (Supplementary Fig. 1h).
A streamlined variation of NMD-DegSeq (Supplementary Fig. 1i, j; see Supplementary Note 1) was performed in which only the 3′ -ends of p-UPF1-bound transcripts were ligated to an adapter, there was no size-selection step, and PCR-amplified sequences extending from a transcript-specific primer through to the downstream 3′ -end adapter were individually cloned and sequenced. Results confirmed the existence of non-templated nucleotides at the 3′ -ends of some NMD decay intermediates, for example some that derived from the dRLUC-Gl PTC NMD reporter mRNA44 (Supplementary Fig. 1k,l), but not others, for example those that derived from the endogenous NMD target6,10,37,39,40 encoding growth arrest and DNA damage inducible A (GADD45A) (Supplementary Fig. 1m,n).
NMD-DegNAs demonstrates that NMD involves non-templated nucleotide removal by DIS3L2 and the exosome.
Our results from NMD-DegSeq indicate that nucleotide transferase and nucleolytic activities that, respectively, add and remove non-templated nucleotides at decay intermediate 3′ -ends are integral to the process of NMD. To begin to define these activities, obtain information on their relative order and efficiency of function, and determine if different NMD targets manifest differences in their decay pathways, HEK293T cells were transiently co-transfected with (i) two reporter plasmids that were either nonsense-free (Norm) or harbored an NMD triggering PTC, and (ii) a reference plasmid that harbored the mouse major urinary protein (MUP) gene, whose product mRNA is not an NMD target6,44. One reporter expressed a deletion-bearing (d) Renilla luciferase (RLUC) gene fused to a β -globin (Gl) gene (dRLUC-Gl) (Fig. 2a), and the other expressed the glutathione peroxidase 1 (GPx1) gene (Fig. 2b). Co-transfections were executed after using short interfering RNA(s) (siRNA(s)) to downregulate one of the 3′ -to-5′ exonucleases that are active in nuclear, cytoplasmic or both fractions12,27,45–47, either PMScl100, DIS3, DIS3L1, or DIS3L2, alone or in combination with downregulating the 5′ -to-3′ exonuclease XRN1, with or without downregulating the SMG6 endonuclease (Supplementary Fig. 2a,b).
Fig. 2 |. NMD-DegNas identifies NMD degradative activities.
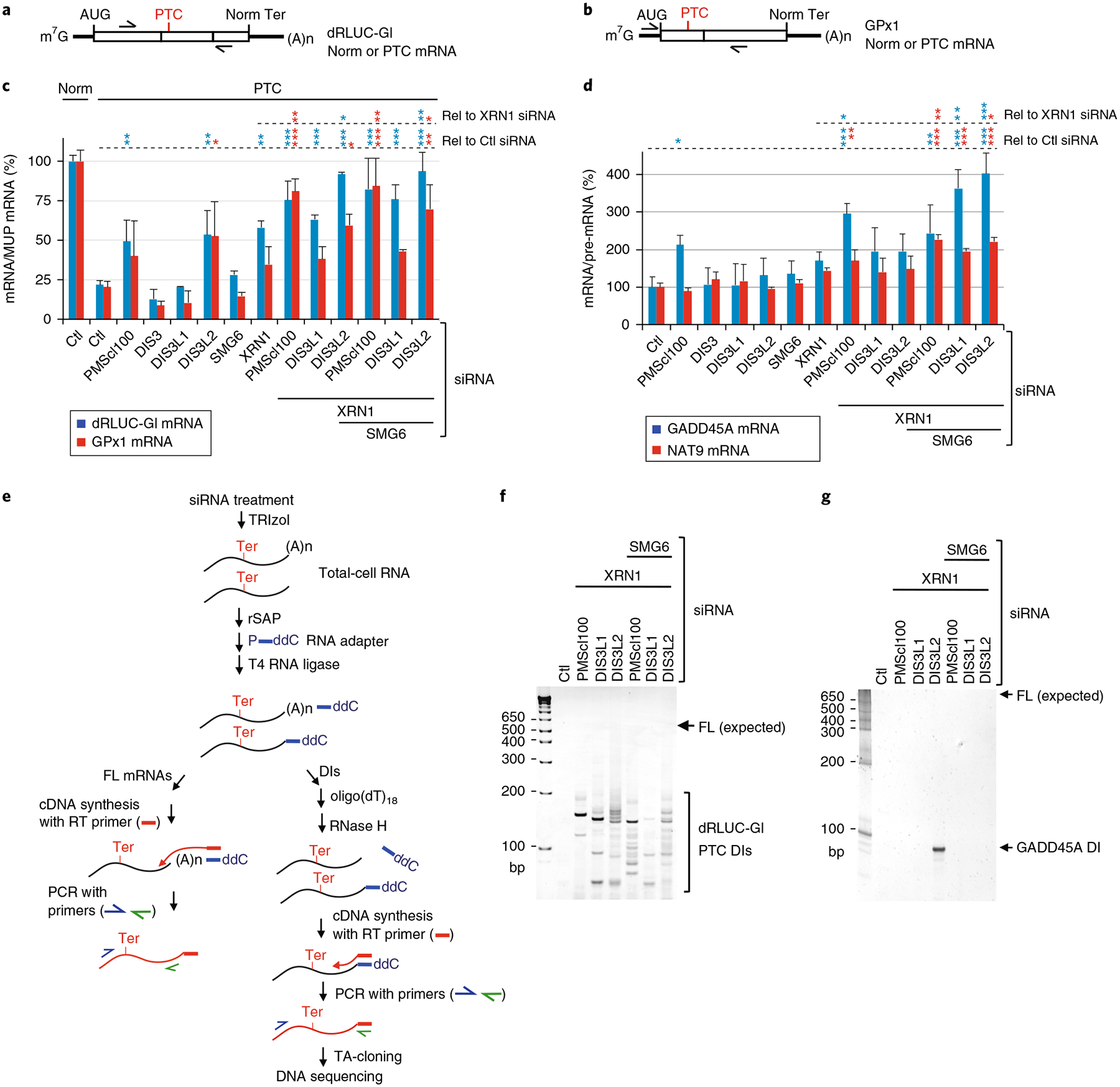
a,b, Diagram of hybrid dRLUC-Gl (a) or glutathione peroxidase 1 (GPx1) (b) Norm or PTC reporter mRNAs; arrows position PCR primers used to amplify cDNA. c, RT–qPCR using lysates of HEK293T cells expressing dRLUC-Gl and GPx1 reporter mRNAs, both Norm or both PTC, and the MUP reference mRNA after transfection with the denoted siRNAs. The level of each Norm or PTC mRNA was normalized to the level of MUP mRNA, and the normalized level of Norm mRNA in the presence of Ctl siRNA is defined as 100%. d, As in c, but for the cellular NMD targets GADD45A mRNA and NAT9 mRNA. The level of each mRNA was normalized to the level of its pre-mRNA, and the normalized level in the presence of Ctl siRNA is defined as 100%. For RT–qPCR results, *P < 0.05, **P < 0.01, and ***P < 0.001 pertain to comparisons to Ctl siRNA samples (Rel to Ctl siRNA) or XRN1 siRNA samples (Rel to XRN1 siRNA) using one-way ANOVA and Dunnett’s multiple comparison test; n = 2–4 independent experiments, showing means with s.d. See source data for details. e, Schematic workflow for NMD-DegNAs to define cellular NMD nuclease activities. Ter signifies the termination codon triggering NMD. FL, full-length; ddC, dideoxy C. f, SYBR-Gold-stained dRLUC-Gl PTC decay intermediates analyzed following the experimental scheme shown in e. FL (expected) denotes the position where FL dRLUC-Gl mRNA would migrate. The sequences of dRLUC-Gl PTC decay intermediates are detailed in Supplementary Fig. 2e. g, As in f, but analyzing GADD45A NMD target decay intermediates; sequences are detailed in Supplementary Fig. 2f. Results are representative of two independently performed experiments. Uncropped images are shown in Supplementary Data Set 1. Source data for panels c and d are available online.
NMD was assayed by normalizing the level of each reporter mRNA to the level of the MUP reference mRNA, or by normalizing the level of an endogenous NMD target encoding either GADD45A or N-acetyltransferase 9 (NAT9)6,10,37–40 to the level of the corresponding pre-mRNA, which controlled for any siRNA-mediated transcriptional changes.
In summary of these results, different NMD targets can manifest distinct sensitivities when individual or combinations of nucleases are downregulated (Fig. 2c,d and Supplementary Fig. 2a,b). In general, NMD is largely mediated by exonucleases acting at mRNA 5′ - and 3′ -ends, rather than by endonucleolytic cleavage. Moreover, nucleases can function cooperatively as evidenced from the finding, for all NMD targets tested, that downregulating either PMScl100 or DIS3L2 and, in the case of cellular NMD targets, SMG6 tended to increase XRN1 siRNA-mediated effects (Fig. 2c,d; see Supplementary Note 2). This increase would promote NMD15 so as to minimize the effects of SMG6 depletion, presumably most notably for those NMD targets that are more susceptible to the SMG5–SMG7 pathway. It is worth noting that the modest increase in abundance of three of the four NMD targets in the presence of SMG6 siRNA alone (Fig. 2c, d) may be attributable to an increase in the interaction of p-UPF1 with the SMG5–SMG7 heterodimer7.
In control experiments, no single siRNA or combination of siRNAs that was assayed significantly upregulated the level of dRLUC-Gl Norm mRNA, GPx1 Norm mRNA, or cellular GAPDH mRNA (data not shown).
To define the 3′-ends of NMD decay intermediates that result from the downregulation of nuclease activities shown above to function in NMD, including the nature and frequency of the various modifications to decay intermediate 3′-ends, we developed ‘NMD-Deg to Define Nuclease Activities’ or NMD-DegNAs, which assays total cell RNA, instead of p-UPF1-bound RNA, after the downregulation of one or more nucleolytic activities (Fig. 2e; see Supplementary Note 2).
While the number of clones obtained for dRLUC-Gl PTC mRNA was comparable regardless of which siRNA(s) were used, different siRNAs resulted in different decay intermediate 3′ -ends (Fig. 2f,g and Supplementary Fig. 2c,d). In the presence of Control (Ctl) siRNA, no decay intermediates were detected for dRLUC-Gl PTC by PCR with reverse transcription (RT–PCR) (Fig. 2f), indicating that they were efficiently degraded. Sequencing the multiple decay intermediates observed on downregulating PMScl100, DIS3L1, or DIS3L2 concomitantly with downregulating either XRN1 or XRN1 and SMG6 (Fig. 2f and Supplementary Fig. 2c,e) revealed that the 3′ -end of most decay intermediates (170/172) mapped to upstream of the PTC, and some (17/172) 3′ -ends were modified with non-templated (U)1–4, UA, A, or C (Supplementary Fig. 2e). Thus, during NMD, DIS3L2, the exosome, or some combination thereof can degrade non-templated 3′-end additions. Using Quadruplex forming G-Rich Sequence (QGRS) Mapper48, we identified sequences capable of forming G-quadruplex structures at the 3′-ends of the dRLUC-Gl PTC decay intermediates (Supplementary Fig. 2e), suggesting that these structures may stall DIS3L2 and the exosome, and that the addition of unstructured non-templated nucleotides may facilitate progressive 3′ -to-5′ decay through such structures (see Discussion).
Analysis of the endogenous NMD target encoding GADD45A using NMD-DegNAs after downregulating XRN1 and DIS3L2 (Fig. 2g and Supplementary Fig. 2d) revealed that, remarkably, all 29 decay intermediates obtained had a 3′-end situated 123 nucleotides upstream of the termination codon, 3% of which ended in a non-templated U (Supplementary Fig. 2f). As for dRLUC-Gl mRNA, QGRS Mapper predicted that a G-quadruplex structure situated as part of a very (~78%) G- and C-rich region spanning the 3′ -end of the decay intermediate might also form a stable RNA structure that blocks further decay intermediate degradation. Given the importance of SMG6 function in the NMD of GADD45A mRNA when an exonuclease is concomitantly downregulated (Fig. 2d), our finding that GADD45A decay intermediates were not detected on the concomitant downregulation of SMG6 with XRN1 and DIS3L2 (Fig. 2g) probably reflects a dominant role of SMG6 in GADD45A NMD. These results further illustrate that different NMD targets can manifest differential nuclease sensitivities depending on the balance of cellular nucleases.
NMD-DegTAs reveal that terminal U transferases promote NMD.
While this is the first report of non-templated nucleotides at RNA 3′ -ends during NMD, 3′ -end oligouridylation is widespread in mammalian cells26,43,49–52, and the length of added Us negatively correlates with RNA stability43. Oligouridylation is often mediated by terminal uridylyl transferases (TUTases) TUT4 and TUT7, also known as ZCCHC11 and ZCCHC626,28,53. Moreover, TUT4 and TUT7 have been recently reported to uridylate the 3′ -ends of maternal mRNAs in a mechanism that is essential for oocyte maturation and fertility in the mouse29. Consistent with our findings that stretches of ≤ 20 Us are enriched at NMD decay intermediate 3′ -ends (Fig. 1f,g) and that DIS3L2-and exosomal-mediated 3′ -to-5′ decays contribute to NMD (Fig. 2 and Supplementary Fig. 2), the individual or combined downregulation of TUT4 and TUT7 (Fig. 3a,b and Supplementary Fig. 3a) inhibited the decay of reporter and cellular NMD targets when XRN1 was concomitantly downregulated, and inhibition was beyond that observed when XRN1 alone was downregulated (Fig. 3a,b and Supplementary Fig. 3a). In contrast, the corresponding Norm and ß-actin mRNAs were not significantly upregulated (data not shown).
Fig. 3 |. NMD-DegTas demonstrate that terminal uridylyl transferases function in NMD.
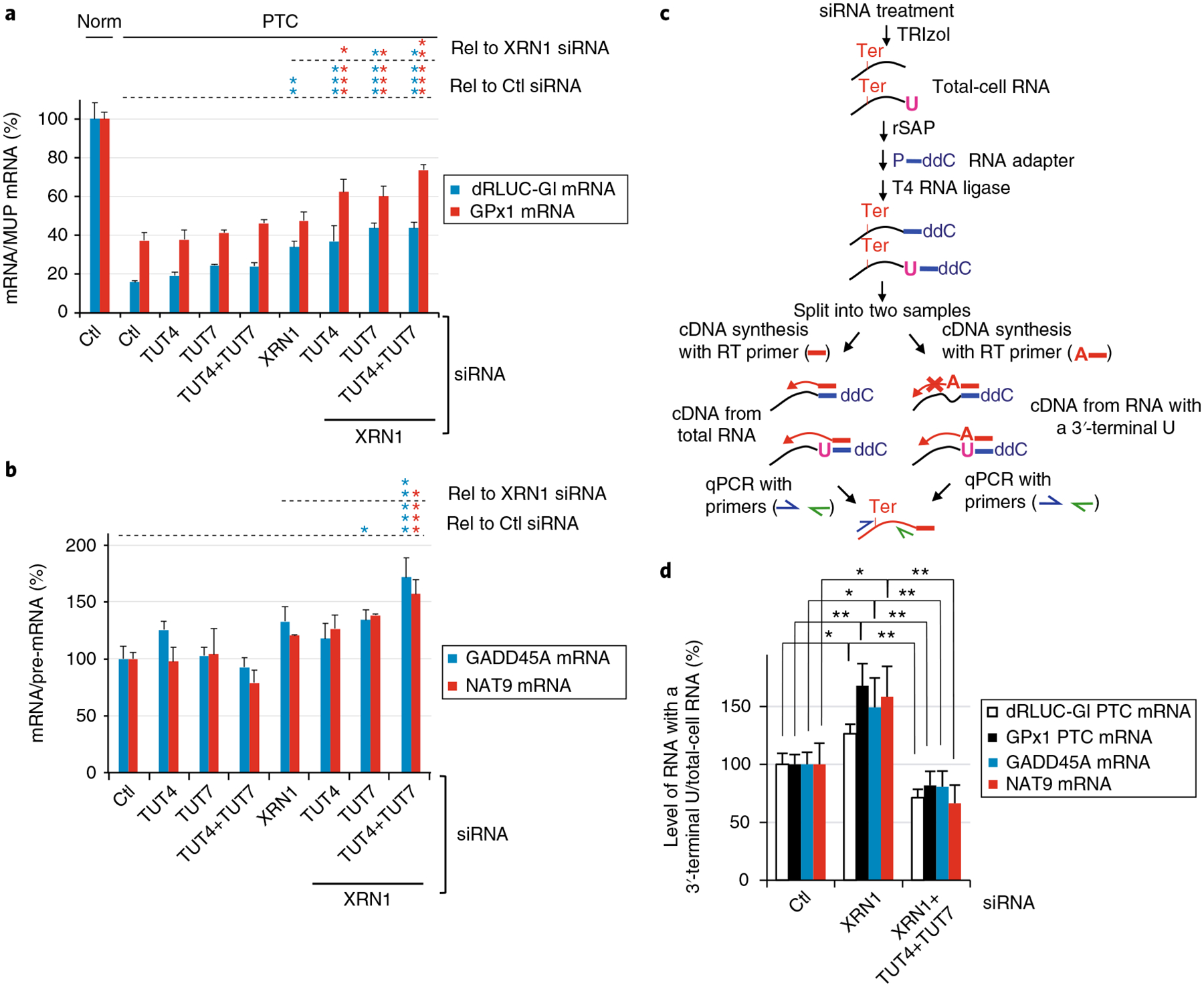
a, Relative levels of dRLUC-Gl and GPx1 mRNAs, either Norm or PTC, were measured after normalization to MUP mRNA. Normalized level of Norm mRNA in the presence of Ctl siRNA is defined as 100%. *P < 0.05, **P < 0.01, and ***P < 0.001 pertain to comparisons to Ctl siRNA samples (Rel to Ctl siRNA) or XRN1 siRNA samples (Rel to XRN1 siRNA) using one-way ANOVA and Dunnett’s multiple comparison test; n = 3 independent experiments, showing means with s.d. See source data for details. b, As in a, but for GADD45A or NAT9 mRNA, which are cellular NMD targets, each normalized to the level of their pre-mRNA. c, Schematic workflow for NMD-DegTAs to define cellular NMD tailing activities. NMD-DegTAs measure the level of an NMD target containing a 3′-most uridine (pink U) (right workflow), relative to the level of the NMD target without selecting for the terminal nucleotide (left workflow). See Methods for experimental procedures. d, For the specified RNAs, relative levels of RNAs ending with U after normalization to levels without selection for a 3′-terminal U, each as defined following the workflow in c; normalized levels in the presence of Ctl siRNA are defined as 100%. For RT–qPCR results, *P < 0.05 and **P < 0.01 pertain to comparisons to Ctl siRNA or XRN1 siRNA samples (unpaired two-tailed t-test); n = 3–4 independent experiments, showing means with s.d. See source data for details. Source data for panels a, b, and d are available online.
To further verify TUTase function in NMD, we used total-cell RNA and modified NMD-DegNAs to selectively reverse-transcribe decay intermediates having at least one U, A, G, or C abutting their 3′ -most transcribed nucleotide. We call this method “NMD-Deg to Define Tailing Activities” or NMD-DegTAs. The levels of each for the four NMD targets assayed were normalized to the levels when RT did not select for a specific 3′ -terminal nucleotide (Fig. 3c). Results for all four NMD targets revealed that, unlike Ctl siRNA, XRN1 siRNA upregulated the cellular abundance of NMD targets that harbored a 3′ -terminal U (Fig. 3d) but not a 3′ -terminal A, G, or C (Supplementary Fig. 3b–d). Similar results were observed when NMD targets having UUUU, instead of a single U, at their 3′ -ends were selectively reverse-transcribed (Supplementary Fig. 3e). This suggests that NMD target decapping, XRN1 activity, or both are promoted by ≥ 1 3′ -terminal U, consistent with evidence that the presence of uridines at RNA 3′ -ends promote 5′ -decapping and 5′ -to-3′ exonucleolytic decay21,22. In contrast, the combined addition of XRN1, TUT4, and TUT7 siRNAs significantly and specifically reduced the levels of NMD targets harboring a 3′ -terminal U (Fig. 3d and Supplementary Fig. 3e), but was of no consequence to the abundance of decay intermediates harboring a 3′ -terminal A, G, or C (Supplementary Fig. 3b–d). Thus, TUT4 and TUT7 are not likely to add 3′ -terminal A, G, or C nucleotides.
We conclude that XRN1-mediated exonucleolytic decay during NMD is facilitated by TUT4- and TUT7-mediated uridylation at decay intermediate 3′ -ends, which possibly also promotes DIS3L2- and exosome-mediated 3′ -to-5′ exonucleolytic decay. Since 12 non-templated Us is the minimal length required for DIS3L2 function25, and NMD-DegSeq demonstrated that only a small percentage (~0.1%) of non-templated U stretches at decay intermediate 3′ -ends are ≥ 12 (Fig. 1g), our steady-state analyses probably detect the most stable decay intermediates during cyclical additions of non-templated nucleotides followed by their complete or partial removal. Moreover, a 3′ -terminal nucleotide that is not U may inhibit further decay (see Discussion).
Evidence that p-UPF1 resides at translationally active cellular sites.
Although NMD in yeast has been shown to occur on polysomes17, whether the steps of mRNA degradation during mammalian-cell NMD also occur on polysomes or in cytoplasmic bodies continues to be debated (see Supplementary Note 3).
To resolve this debate, we used multiple experimental approaches. First, using two different antibodies that are specific to p-UPF1, which is functionally critical for NMD6, as assayed using western blotting (Supplementary Fig. 4a), immunofluorescence microscopy (Supplementary Fig. 4b,c) revealed that human-cell UPF1 as well as p-UPF1 exist in the cytoplasm (Supplementary Fig. 4d), diffusely throughout the cytosol (Fig. 4a and Supplementary Fig. 4b,e), and they are barely detectable in P-bodies as determined by co-staining with DCP1a (Fig. 4a and Supplementary Fig. 4e). These results contrast reports that UPF1 and p-UPF1 accumulate together with NMD reporter transcripts in P-bodies when exogenously expressed from plasmid DNAs31,32,54,55, possibly because the conditions used for exogenous expression may result in mis-localization. The diffuse cytosolic localization of cellular p-UPF1 also typified ribosomal proteins RPS6 and RPL10a (Fig. 4a and Supplementary Fig. 4e). Moreover, immunofluorescence of HeLa cells exposed to okadaic acid so as to upregulate cellular p-UPF1 abundance (Supplementary Fig. 4f) demonstrated that p-UPF1, like UPF1, resides diffusely in the cytoplasm and is not detectably enriched with the P-body marker DCP1a (Supplementary Fig. 4g). Second, using lysates of cells exposed to an inhibitor of translation initiation or translation elongation (Supplementary Fig. 4h), the co-immunoprecipitation of both NMD target mRNAs and RPL5 protein with p-UPF1 was blocked when translation was inhibited (Fig. 4b and Supplementary Fig. 4h,i). Third, p-UPF1 is present on polysomes as are other proteins that bind to NMD targets primarily during the pioneer round of translation, on which 3′ UTR EJC-dependent NMD relies, including the cap-binding protein CBP80, NMD factors UPF3X, SMG5, and SMG6, the EJC anchor eIF4A3, NMD degradative activities XRN1, PMScl100, and DIS3L2, constituents of the translational apparatus such as the translation termination factor eRF1, and ribosomal proteins RPL5 and RPS6 (Supplementary Fig. 4j). Taken together these data are consistent with NMD occurring on polysomes, and not in P-bodies.
Fig. 4 |. p-uPF1 localizes diffusely throughout the cytoplasm and fails to co-immunoprecipitate with NMD targets when translation is inhibited.
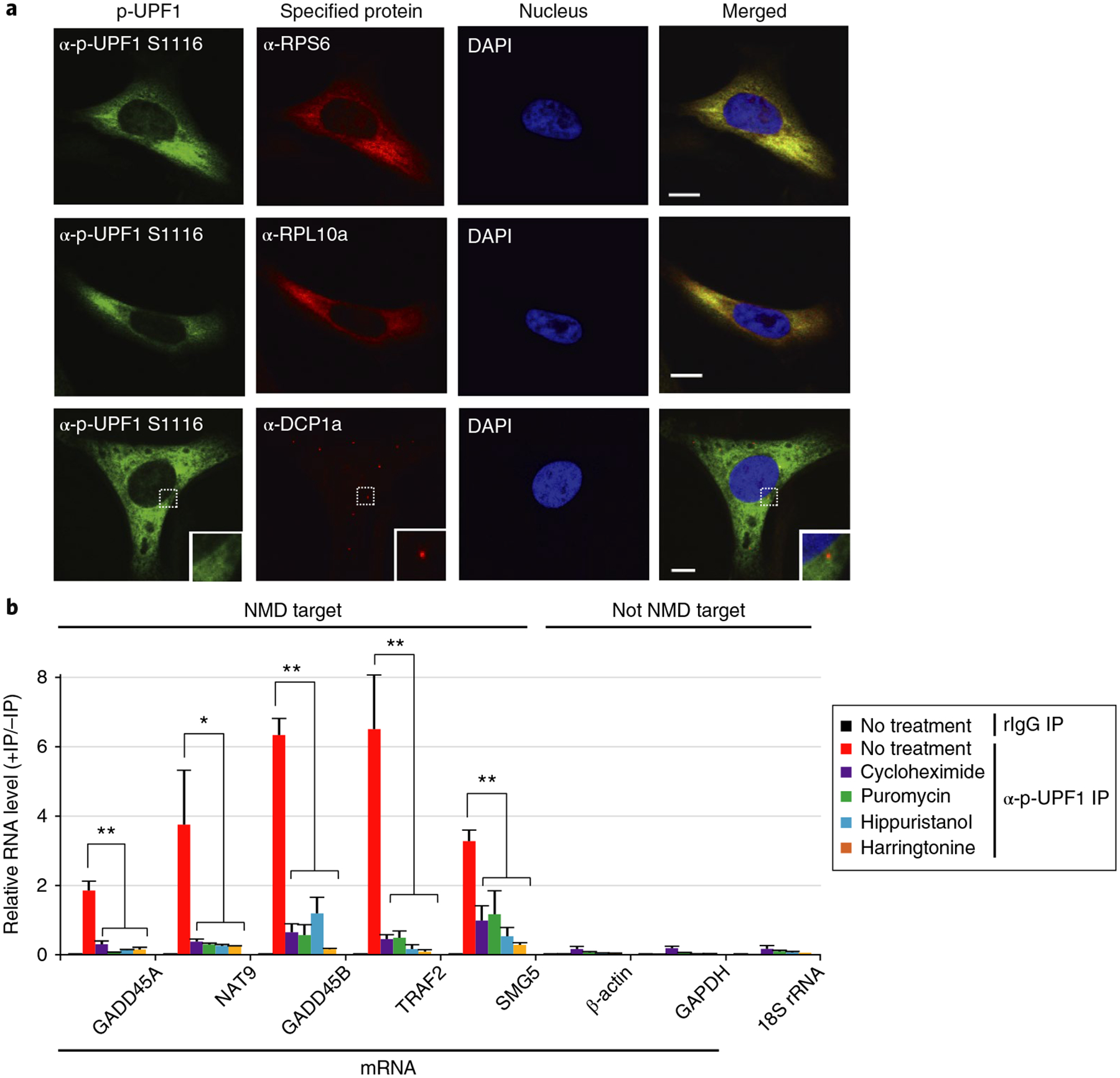
a, Immunofluorescence of HeLa cells using the specified antibody or DAPI to stain nuclei. Scale bar, 10 μm. Solid boxed regions are 3-fold magnifications of the dotted boxed regions. Uncropped images are shown in Supplementary Data Set 2. b, RT–qPCR analyses of RNAs from HEK293T cells that were cultured in the absence (no treatment) or for 3 h in the presence of the specified translational inhibitor before (−) or after (+) immunoprecipitation using rIgG or anti-p-UPF1 S1116. For RT–qPCR results, *P < 0.05 and **P < 0.01 pertain to comparisons to untreated samples (unpaired two-tailed t-test). n = 3 independent experiments, showing means with s.d. See source data for details. Source data for panel b are available online.
NMD is co-translational, generating ribosome-associated decay intermediates with 3′-end non-templated nucleotide additions.
We developed two additional approaches to determine whether NMD is co-translational. The first approach, which we call NMD-DegRPL5, analyzed NMD decay intermediates that lack a poly(A) and co-immunoprecipitate with RPL5 (Fig. 5a, leftmost steps). As the fourth indication that NMD is co-translational, decay intermediates deriving from dRLUC-Gl PTC and GADD45A NMD targets were associated with RPL5 when XRN1 and DIS3L2, but not XRN1 and PMScl100, were concomitantly downregulated (Fig. 5b–d). We found that RPL5-bound dRLUC-Gl PTC decay intermediates had 3′ -ends residing either 9 nucleotides downstream or 48 nucleotides upstream of the PTC that were respectively appended with the non-templated sequence AAAAG or AC (Supplementary Fig. 5a). Remarkably, the non-templated sequence AAAAG residing 9 nucleotides downstream of the PTC was also observed in the immunoprecipitation of p-UPF1 (Supplementary Fig. 1l). We also found that RPL5-bound GADD45A transcripts had a 3′ -end (Supplementary Fig. 5b) situated exactly as observed in the immunoprecipitation of p-UPF1 (Supplementary Fig. 1n). While both DIS3L2 and the exosome have been reported to remove non-templated U nucleotides from decay intermediate 3′ -ends25,51, our finding that NMD decay intermediates were detectable on the simultaneous downregulation of XRN1 and DIS3L2 but not XRN1 and PMScl100 suggests a more prominent role for DIS3L2 relative to PMScl100 in the NMD of these targets in HEK293T cells (Fig. 5c,d).
Fig. 5 |. NMD-DegRPL5 and NMD-DegRibo show NMD decay intermediates are bound by ribosomes.
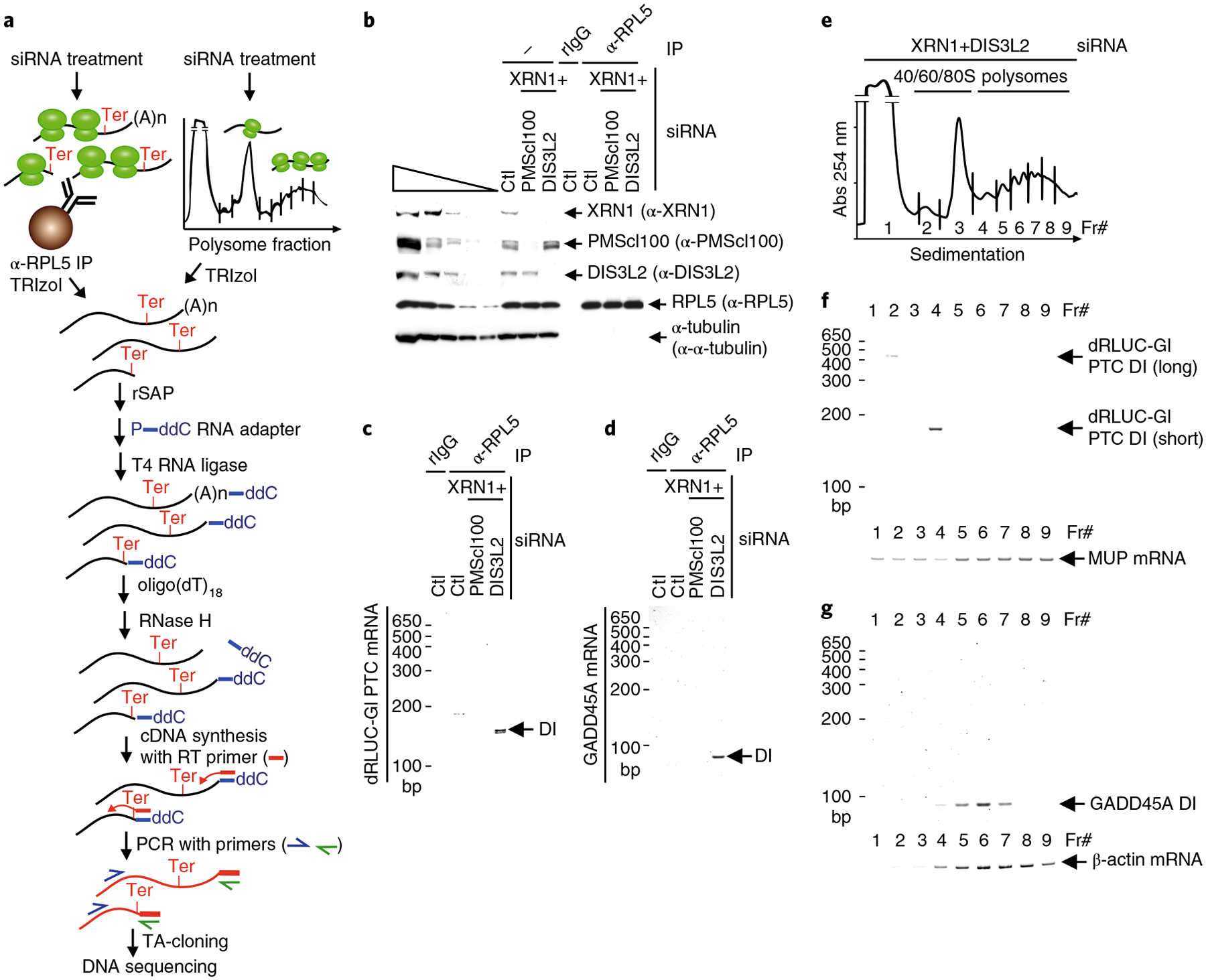
a, Schematic workflow used to identify the ribosome binding status of decay intermediates using anti(α)-RPL5 (NMD-DegRPL5, left steps) or polysome fractionation (NMD-DegRibo, right steps). b, Western blot of lysates of HEK293T cells, transfected with the specified siRNA and expressing the dRLUC-Gl PTC reporter and MUP reference mRNAs, before (−) or after (+) immunoprecipitation using anti(α) RPL5 or, as a negative control, rIgG. The five leftmost lanes represent 3-fold serial dilutions of lysate before immunoprecipitation. c,d, SYBR-Gold staining of dRLUC-Gl PTC decay intermediates (DI) (c) or decay intermediates of the GADD45A NMD target (d). d,e, Polysome profiles as derived using the scheme shown in a. f, SYBR-Gold staining of dRLUC-Gl PTC decay intermediates and, as a loading control, MUP mRNA isolated from polysome fractions represented in e. g, As in f, but for the GADD45A NMD target and β-actin mRNA. All results are representative of two independently performed experiments. Uncropped images are shown in Supplementary Data Set 1.
The second approach, NMD-DegRibo, analyzed decay intermediates that co-purify with one or more ribosomes in polysome profiles (Fig. 5a, rightmost steps). As a fifth indication that NMD is co-translational, decay intermediates deriving from dRLUC-Gl PTC mRNA in the presence of XRN1 and DIS3L2 siRNAs co-sedimented in polysome profiles (Fig. 5e). The major decay intermediate co-sedimented with disomes (Fr#4 in Fig. 5e,f and Supplementary Fig. 5c), which sequencing revealed had 3′ -ends appended with the non-templated AAAAG sequence as observed in the immunoprecipitations of p-UPF1 (Supplementary Fig. 1l) and RPL5 (Supplementary Fig. 5a). The minor decay intermediate co-sedimented with the 40S ribosome subunit (Fr#2 in Fig. 5e,f). Sequencing revealed that the 3′ -end of this decay intermediate, which resided 248 nucleotides downstream of the PTC, that is 73 nucleotides upstream of the normal termination codon, ended in three U nucleotides that probably come from the body of the transcript (Supplementary Fig. 5c). Considering that only the shorter decay intermediate was present in the p-UPF1 immunoprecipitation, it is conceivable that the less abundant but longer decay intermediate may derive from the fraction of dRLUC-Gl PTC mRNA that escapes NMD and is degraded as if it were PTC-free. Decay intermediates deriving from the GADD45A NMD target likewise co-sedimented with polysomes, specifically with 2–7 ribosomes (Fr#s4–7 in Fig. 5e,g), with 3′ -ends mapping (Supplementary Fig. 5d) as they did in the p-UPF1 immunoprecipitation (Supplementary Fig. 1n), total-cell lysates (Supplementary Fig. 2f), and in the RPL5 immunoprecipitation (Supplementary Fig. 5b).
In view of all of these results, we conclude that the decay steps of human-cell NMD initiate on transcripts that are bound by translationally active ribosomes and, in some cases, are accompanied by non-templated nucleotide additions to decay intermediate 3′ -ends.
Discussion
Here we develop and report the results of transcriptome-wide NMD-DegSeq and a variation thereof to elucidate the decay intermediates of direct NMD targets. Our findings allow us to conclude that NMD in HEK293T cells is mediated largely by exonucleolytic activities, and that the generation of decay intermediates can involve the addition of one or more non-templated nucleotides to their 3′ -ends (Fig. 6). By showing that decay intermediates are associated with translationally active ribosomes (Fig. 5 and Supplementary Note 4), we resolve a long-standing controversy over where the decay steps of NMD initiate while also describing how the 3′ -ends of human NMD decay intermediates are processed (Fig. 6 and Supplementary Note 5). It is important to note that NMD-DegSeq captures the decay intermediates bound by p-UPF1 and, thus, possibly not all decay steps of NMD. Future experiments that combine NMD-DegSeq with other methodologies will be required to understand the complete degradative process during NMD.
Fig. 6 |. Model for the decay steps of NMD.
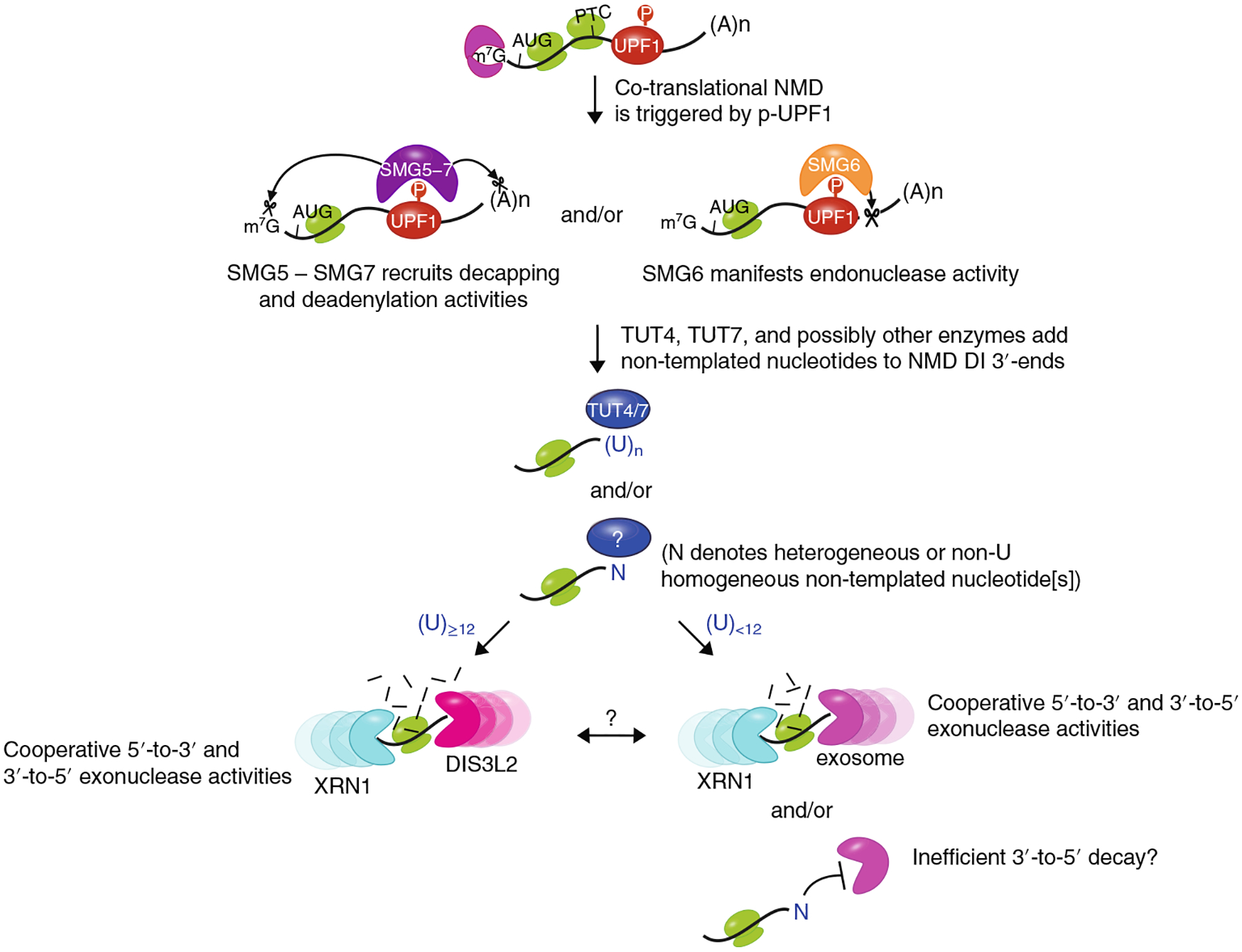
NMD decay intermediates, which can be identified by co-immunoprecipitation with p-UPF1, are generated co-translationally. p-UPF1 recruits either the SMG5–SMG7 complex, which in turn recruits decapping and deadenylation activities, the endonuclease SMG6, which cleaves near to the PTC, or some combination of both. Decay intermediates may be further processed by TUT4 and TUT7, which add non-templated U nucleotides, possibly at sites where 3′ -to-5′ DIS3L2 and exosomal exonucleolytic activities stall within the poly(A) tail or the transcribed body. These nucleotides promote further 3′ -to-5′ decay and also XRN1-mediated 5′ -to-3′ decay. Less frequently, undefined polymerases add non-U nucleotides that appear to inhibit further decay.
Our results demonstrating that the decay steps of NMD occur in association with translationally active ribosomes are consistent with our previous finding that the decay of Gl PTC mRNA occurs with a half-life of < 1 min once the mRNA reaches the cytoplasmic side of the nuclear envelope56. This half-life of < 1 min is consistent with a translation rate of approximately five codons per second57, or 8 s to reach the PTC56, which allows time not only for the association of multiple 80S ribosomes (Fig. 5) but also for 43S binding at the AUG36. While 3′ -end addition of non-templated nucleotides has been shown to recruit decapping enzymes31–33, setting in motion decay at the 5′ -end by XRN121,22, our data show that downregulating TUTases together with XRN1 efficiently stabilizes NMD targets (Fig. 3a,b). That downregulating XRN1 could stabilize decay intermediates harboring one or more terminal Us (Fig. 3c,d and Supplementary Fig. 3e) is consistent with the proposal that XRN1 activity could be promoted by 3′ -end uridylation21,22. Thus, it is reasonable to conclude that both XRN1-mediated 5′ -to-3′ decay and 3′ -to-5′ exonucleolytic decay followed by the addition of one or more non-templated Us cooperatively mediate NMD.
In keeping with results indicating that non-templated G additions protect decay intermediates from further decay43, our findings that non-U additions are not efficient targets for XRN1-mediated mRNA decay (Fig. 3d and Supplementary Fig. 3b–d) and are readily detected in the steady state (Figs. 1–3 and Supplementary Fig. 1–3) without an apparent barrier to 3′ -to-5′ exonucleolytic activity situated upstream argue that these additions do not efficiently promote the decay of NMD decay intermediates in the 3′ -to-5′ direction. In fact, NMD-DegSeq revealed that the relative abundance (mean read density) of decay intermediates mapping to near the transcript start sites was ~4-fold less abundant than decay intermediates mapping to near the transcript end sites (Fig. 1d and Supplementary Fig. 1f), suggesting that the efficiency of 5′ -to-3′ exonucleolytic decay exceeds the efficiency of 3′ -to-5′ exonucleolytic decay once the poly(A) tail has been removed (Fig. 6).
SMG6 has been shown to catalyze the endonucleolytic decay of capped and polyadenylated mRNAs and it has been proposed that it dominates NMD in HEK293 and HeLa cells9–11. Nevertheless, our data using HEK293T cells indicate that p-UPF1 predominantly facilitates deadenylation (Fig. 1b). Our finding is consistent with the report that poly(A) shortening initiates the decay of NMD targets in NIH3T3 cells13. Our data also demonstrate that the relative contributions of nucleolytic activities to NMD in HEK293T cells vary depending on the NMD target (Fig. 2 and Supplementary Fig. 2). It is reasonable to believe that perturbing one exonucleolytic activity may alter the balance of the remaining activities. For example, SMG6 is recruited to NMD targets by binding the amino terminus of p-UPF1 and sequences that overlap with the carboxy terminus of p-UPF1 that recruits SMG5− SMG758–60. Thus, downregulating SMG6 may upregulate SMG5–SMG7 binding to p-UPF17. If so, this would in turn promote the recruitment of the CCR4−NOT deadenylase complex to catalyze poly(A) tail shortening13–15, and 3′ -to-5′ decay by DIS3L2 and the PMScl100 exosome component.
Methods
Cell lines and cell culture.
Mycoplasma-free HEK293T (ATCC; CRL-11268) and HeLa (ATCC; CCL2) cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific). When specified, cells were treated with 50 μg ml−1 cycloheximide (Sigma) for 3 h, 50 μg ml−1 puromycin (Gibco) for 3 h, 0.5 μM hippuristinol (gift from J. Pelletier) for 3 h, 2 μg ml−1 harringtonine (Abcam) for 3 h, or 200 nM okadaic acid (Sigma) for 3 h; or transiently transfected with 22.5 nM siRNA (GE Healthcare Dharmacon; Supplementary Table 2) using Lipofectamine RNAiMAX (Life Technologies), and when specified, plasmid DNAs using Lipofectamine 2000 (Life Technologies). Cell lysates were prepared using Hypotonic Gentle Lysis Buffer (10 mM Tris (pH 7.4), 10 mM NaCl, 10 mM EDTA, 0.5% w/w Triton X-100) and cOmplete Protease Inhibitor Cocktail (Roche) or Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Protein was analyzed after the addition of NaCl to 150 mM, and RNA was extracted and purified using TRIzol reagent (Thermo Fisher Scientific).
Immunoprecipitations.
Samples were generated before and after immunoprecipitation as previously described6,30,44 using the specified antibodies (see below).
Western blotting.
Proteins were electrophoresed in 6–14% polyacrylamide and transferred to either a nitrocellulose membrane (GE Healthcare) or a polyvinylidene difluoride membrane (Millipore). Blots were probed using antibody to PMScl100 (1:1,000; Bethyl Laboratories, A300–988A), DIS3 (1:2,000; Bethyl Laboratories, A303–764A), DIS3L1 (1:1,000; ATLAS, HPA041805), DIS3L2 (1:2,000; ATLAS, HPA035796), XRN1 (1:2,000; Bethyl Laboratories, A300–443A), SMG6 (1:1,000; Abcam, ab87539), β-actin (1:1,000; Sigma, A2228), UPF1 (1:2,000)61, p-UPF1(S1089) (1:1,000; Millipore 07–1015), p-UPF1(S1116) (1:1,000; Millipore, 07–1016), p-UPF1(S1078, S1096) (1:1,000)62, calnexin (1:2,000; Enzo Life Sciences, ADI-SPA-865), UPF3X (1:1,000)63, eIF4A3 (1:1,000; Bethyl Laboratories, A302–981A), eRF1 (1:1,000; Sigma, E8156), SMG5 (1:1,000; Abcam, ab33033), CBP80 (1:1,000; Bethyl Laboratories, A301–793A), PABPN1 (1:5,000; Abcam, ab75855), PABPC1 (1:2,000; Abcam, ab21060), RPL5 (1:2,000; Bethyl Laboratories, A303–933A), RPS6 (1:2,000, Cell Signaling, no. 2317), EDC4 (1:2,000; Bethyl Laboratories, A300–745A), TUT4 (1:2,000; Bethyl Laboratories, A302–636A), TUT7(1:2,000; Bethyl Laboratories, A305–088A), CDC6 (1:500; Santa Cruz Biotechnology, sc-56273), p54nrb (1:2,000; Bethyl Laboratories, A300–582A), and α-tubulin (1:2,000, Cell Signaling, no. 3873). Blots were visualized using SuperSignal West Pico or Femto Chemiluminescent Substrate (Thermo Fisher Scientific).
RT–PCR and RT–qPCR.
RT–PCR or RT-coupled to real-time quantitative PCR (qPCR) was undertaken essentially as previously outlined6,30,44 using random hexamers (Thermo Fisher Scientific Invitrogen) for first-strand complementary DNA synthesis, gene-specific primers for PCR (Supplementary Table 3), and either 1 × TAQ DNA polymerase for RT–PCR for quantitations using SYBR-Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific) or Fast SYBR-Green Master Mix (Applied Biosystems) for quantitations using the 7500 Fast Real-Time PCR System (Applied Biosystems).
SYBR-Gold and ethidium-bromide staining.
RT–PCR products were detected after electrophoresis in 5% or 6% polyacrylamide using SYBR-Gold Nucleic Acid Gel Stain, and ribosomal RNAs were detected after electrophoresis in 1% agarose using ethidium bromide (Teknova). Gel images was visualized by ChemiDoc XRS + System (BioRad).
NMD-DegSeq: library preparations for transcriptome-wide NMD degradome sequencing.
HEK293T cells (6 × 108 in a total of eight 150-mm dishes) were treated with okadaic acid (200 nM) for 3 h. After immunoprecipitation using anti-p-UPF1 (EMD Millipore) and, in parallel as a negative control, rabbit IgG (rIgG; Sigma)6,30, RNA was purified using TRIzol Reagent, incubated following the manufacturer’s instruction first with DNase I (Promega) and, subsequently, with Ribozero Gold rRNA Removal Kit (Illumina). Samples were then incubated with recombinant shrimp alkaline phosphatase (rSAP; New England Biolabs) to remove 5′ -phosphates from any physiologically uncapped NMD targets. The 3′ -ends were ligated using T4 RNA ligase 2 (T4 RNA Ligase 2, truncated KQ; New England Biolabs) to a 34-nucleotide DNA adapter, referred to as the “3′ -DNA adapter for NMD-DegSeq” in Supplementary Table 3, having App appended at its 5′ -end and dideoxy-C (ddC; Integrated DNA Technologies) appended at its 3′ -end. Then 5′ -ends were phosphorylated using T4 polynucleotide kinase (T4 PNK; New England Biolabs) and ligated using T4 RNA ligase (Ambion T4 RNA Ligase; Thermo Fisher Scientific) to a 33-nucleotide RNA adapter, referred to as “5′ -RNA adapter for NMD-DegSeq” in Supplementary Table 3, having a hydroxyl moiety at both its 5′ - and 3′ -ends (Integrated DNA Technologies). cDNA was synthesized using SuperScript III reverse transcriptase (Thermo Fisher Scientific Invitrogen) and a DNA primer, referred to as “1st strand RT for NMD-DegSeq” in Supplementary Table 3, that is antisense to the 3′ -end DNA adapter, and subsequently PCR-amplified (20 cycles) using primers, referred to as “1st PCR sense and antisense primers for NMD-DegSeq” in Supplementary Table 3, complementary to the extreme ends of each adapter and Q5 High-Fidelity DNA Polymerase (New England Biolabs). Products with inserts of more than ~20 base-pairs (bp) were size-selected from 6% polyacrylamide using SYBR-Gold and Prestain Marker for Small RNA Plus (Diagnocine) to eliminate smaller inserts that could not be uniquely aligned to the genome, including products without inserts, PCR-amplified using index PCR primers, and submitted for deep sequencing (Illumina HiSeq2500v4 system).
NMD-DegSeq: construction of indexed cDNA library for multiplex PCR and Illumina sequencing.
The Illumina Nextera Indexing Kit (Illumina) was used to generate the final i5 and i7 indexed cDNA libraries for Illumina sequencing following the manufacturers’ protocols with modification to the PCR-thermocycler conditions (98 °C for 5 min, 20 cycles at 98 °C for 10 s, 66 °C for 30 s, 72 °C for 1 min, 72 °C for 2 min for the final extension). Indexed libraries were purified using Agencourt AMPURE SPRI beads (Beckman Coulter) followed by quality assessment and quantification using an Agilent Bioanalyzer 2100 and picogreen staining (Thermo Fisher), respectively. Libraries were diluted to 2 nM and sequenced using an Illumina HiSeq2500v4 system with paired-end 2 × 125-bp configuration and single-end 125-bp configuration (where the other end remained unmapped since it was more than 125-bp away). Only read pairs containing identifiable Illumina sequencing adapters were considered for downstream analysis.
NMD-DegSeq: read pre-processing and alignment.
Raw reads generated from the Illumina HiSeq2500 sequencer were demultiplexed using configurebcl2fastq. pl version 2.19.0. CutAdapt 1.1564 was used to trim Nextera transposase sequences. An additional round of CutAdapt 1.15 removed reads with multiple transposase sequences or any other library preparation oligos. Processed and cleaned reads were then mapped to the human genome (hg19) using STAR_2.5.2b65 and the following parameters: “--twopassMode Basic --runMode alignReads --genomeDir ${GENOME} --readFilesIn ${SAMPLE} --outSAMtype BAM SortedByCoordinate --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonical”.
NMD-DegSeq: expression analyses.
Read counts per gene were derived in a strand-specific manner using featureCounts from the subread-1.5.0p3 package66 with the following parameters: “-s 1 -t exon -g gene_name”. Differential expression analyses and data normalizations were performed using DESeq2–1.16.167, R Bioconductor package68 with an adjusted P value (Benjamini-Hochberg) threshold of 0.05, and lfcshrink applied to the results within an R v3.4.1 environment (https://www.R-project.org). Heat maps were produced using the pHeatmap version 1.0.8 (https://CRAN.R-project.org/package=pheatmap) package with row scaling and hierarchical clustering of the rlog-transformed expression values.
NMD-DegSeq: non-templated nucleotide addition and coverage analyses.
Samtools 1.569 was used to isolate uniquely aligned reads: “view -q 255”. Duplicate reads were removed using picard-2.12.0 MarkDuplicates (http://broadinstitute.github.io/picard/). Coverage analysis was performed as described70 using deeptools-2.5.6 in “metagene” mode so that only exonic regions were considered. The number of reads mapping to each annotated meta-region for each gene was counted. Each gene region was binned into 800 bins, including 1 kilobase upstream or downstream of the annotated meta-region. To exclude any bias at the region boundaries during scaling, we specified that 500 nucleotides upstream and 500 nucleotides downstream of the annotated meta-region were to be unscaled. The mean of reads in each bin was calculated and plotted. We modified the removeclipping.py Python script from ngsutils-0.5.971 to output only soft-clipped bases from either the 3′ - or 5′ -end of the inserts. Fastx-toolkit-0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit/) was used to determine the nucleotide distribution of soft-clipped bases.
Control experiments for NMD-DegSeq: neither decay intermediate formation nor non-specific p-UPF1 binding occurs after cell lysis.
HEK293T cells (6 × 108 in a total of eight 150-mm dishes) were treated with okadaic acid (200 nM) for 3 h, and lysed using Hypotonic Gentle Lysis Buffer. Prior to immunoprecipitation, cell lysates were mixed with mouse β -actin mRNA, which was in vitro-transcribed in the presence of α-[32P]-ATP (PerkinElmer) using the MEGAscript T7 Transcription Kit and pTRI-β -actin-Mouse as a template (Thermo Fisher Scientific). After immunoprecipitation using anti-p-UPF1 or rIgG, RNA was purified using TRIzol Reagent. Radio-labeled RNAs were electrophoresed in 6 M urea-6% acrylamide and analyzed using the Typhoon FLA 9500 Phosphorimager (GE Healthcare).
Streamlined NMD-DegSeq: library preparations and analyses using p-UPF1 immunoprecipitation.
HEK293T cells (8 × 107 in one 150-mm dish) were transiently transfected with a reporter plasmid encoding dRLUC-Gl PTC mRNA.
Cells were treated with 200 nM of okadaic acid for 3 h, and lysed using Hypotonic Gentle Lysis Buffer (10 mM Tris [pH 7.4], 10 mM NaCl, 10 mM EDTA, 0.5% w/w Triton X-100, 1 × Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific)) prior to immunoprecipitation using anti-p-UPF1 or rIgG.
RNAs were purified using TRIzol Reagent, incubated following the manufacturer’s instruction with DNase I (Promega) and, to remove 5′ -phosphates, subsequently incubated with rSAP. RNA 3′ -ends were ligated using T4 RNA ligase to a 5′ -phosphorylated RNA adapter (3′ -RNA adapter in Supplementary Table 3). cDNA was synthesized using SuperScript III reverse transcriptase and adapter antisense DNA. Decay intermediates for dRLUC-Gl PTC were PCR-amplified using the sense primer (dRLUC-Gl FL mRNA sense in Supplementary Table 3) and an antisense primer that is complementary to the adapter (adapter antisense in Supplementary Table 3), and subsequently re-amplified using a sense primer of dRLUC-Gl exon 1 (2nd PCR dRLUC-Gl decay intermediate sense in Supplementary Table 3) and the same antisense primer (adapter antisense in Supplementary Table 3). Decay intermediates for GADD45A were PCR-amplified using the same sense primer (GADD45A mRNA sense in Supplementary Table 3) and an antisense primer that is complementary to the adapter (adapter antisense in Supplementary Table 3), and subsequently re-amplified using a sense primer of GADD45A exon 3 (2nd PCR GADD45A decay intermediate sense in Supplementary Table 3) and the same antisense primer (adapter antisense in Supplementary Table 3). PCR products were electrophoresed in 5% polyacrylamide, stained with SYBR-Gold, and subjected to TOPO TA-cloning (Thermo Fisher Scientific) followed by DNA sequencing (ACGT). All DNA or RNA oligonucleotides (Integrated DNA Technologies) are listed in Supplementary Table 1.
NMD-DegNAs: library preparations and analyses for defining NMD nucleolytic activities.
HEK293T cells (8 × 107 in a total of one 150-mm dish) were transiently transfected with the specified siRNA(s) and, subsequently, a reference plasmid encoding MUP mRNA and either (i) reporter plasmids encoding dRLUC-Gl PTC mRNA or GPx1 PTC mRNA, which harbors a PTC, respectively, at position 39 (where it was called dRLUC-Gl Ter mRNA)44 or position 46 (where it was called GPx1 Ter mRNA)6, or (ii) reporter plasmids encoding dRLUC-Gl Norm mRNA or GPx1 Norm mRNA. RNA was harvested, dephosphorylated using rSAP and ligated using T4 RNA ligase to an RNA adapter (3′ -RNA adapter). cDNA was synthesized using SuperScript III reverse transcriptase and adapter antisense DNA before (for full-length mRNA analyses) or after (for decay intermediate analyses) incubation with oligo(dT)18 and RNase H (Thermo Fisher Scientific) to remove poly(A). Full-length cDNA was PCR-amplified using a sense primer to dRLUC-Gl 5′ -UTR sequences in exon 1(dRLUC-Gl FL mRNA sense in Supplementary Table 3) and an antisense primer to dRLUC-Gl exon 3 (dRLUC-Gl FL mRNA antisense in Supplementary Table 3). Decay intermediates were PCR-amplified using the same sense primer (dRLUC-Gl FL mRNA sense in Supplementary Table 3) and an antisense primer that is complementary to the adapter (adapter antisense in Supplementary Table 3), and subsequently re-amplified using a sense primer of dRLUC-Gl exon 1(2nd PCR dRLUC-Gl decay intermediate sense in Supplementary Table 3) and the same antisense primer (adapter antisense in Supplementary Table 3). Full-length GADD45A cDNA was PCR-amplified using a sense primer to GADD45A exon 1 (GADD45A mRNA sense in Supplementary Table 3) and an antisense primer to GADD45A exon 2–3 junction (GADD45A mRNA antisense in Supplementary Table 3). decay intermediates were PCR-amplified using the same sense primer (GADD45A mRNA sense) and an antisense primer that is complementary to the adapter (adapter antisense in Supplementary Table 3), and subsequently re-amplified using a sense primer to GADD45A exon 3 (2nd PCR GADD45A decay intermediate sense in Supplementary Table 3) and the same antisense primer (Adapter antisense). In control PCRs, for MUP mRNA or β -actin mRNA was amplified using mRNA-specific primers (Supplementary Table 3). RT-PCR products were electrophoresed in 5% polyacrylamide, stained with SYBR-Gold, inserted into plasmid DNA using TOPO TA-cloning, and sequenced (ACGT). All DNA and RNA oligonucleotides (Integrated DNA Technologies) are listed in Supplementary Table 3.
NMD-DegTAs: Library preparations and analyses to define 3′-end tailing activities.
RNA from HEK293T cells (8 × 107 in one 150-mm dish) that had been transiently transfected with the specified siRNA(s) and, subsequently, a reference plasmid encoding MUP mRNA and either reporter plasmids encoding (i) dRLUC-Gl PTC mRNA or GPx1 PTC mRNA or (ii) dRLUC-Gl mRNA or GPx1 Norm mRNA was dephosphorylated using rSAP and ligated using T4 RNA ligase to an RNA adapter (3′ -RNA adapter). cDNA was synthesized using SuperScript III reverse transcriptase (i) adapter antisense for cDNA from total-cell RNA (Fig. 3c, d and Supplementary Fig. 3e), (ii) adapter antisense for a 3′ -terminal U (Fig. 3d) or a 3′ -terminal UUUU (Supplementary Fig. 3e), (iii) adapter antisense for a 3′ -terminal A (Supplementary Fig. 3b), (iv) adapter antisense for a 3′ -terminal G (Supplementary Fig. 3c) or (v) adapter antisense for a 3′ -terminal C (Supplementary Fig. 3d). The resulting cDNA libraries were analyzed using real-time quantitative PCR and RNA-specific primers.
Polysome analyses.
Prior to cell lysis, HEK293T cells (8 × 107/150-mm dish) were treated with 100 μg/ml cycloheximide (Sigma). Cells were collected in ice-cold Phosphate-Buffered Saline (PBS) containing 100 μg/ml cycloheximide. Cells were lysed with Hypotonic Gentle Lysis Buffer for Polysome Analyses (5 mM Tris-HCl [pH 7.4], 2.5 mM MgCl2, 1.5 mM KCl, 100 μg/ml of cycloheximide, 2 mM DTT, 200 U/ml RNaseOUT (Invitrogen), 0.5% TRITON X-100 (Sigma), 0.5% sodium deoxycholate). After adjusting the NaCl concentration to 150 mM, cell lysates were collected by centrifugation at 18,000 × g for 5 min at 4 °C. Supernatants were transferred to 10–50% sucrose gradients and centrifuged at 35,000 rpm for 2 h at 4 °C using an Optima LE-80K Ultracentrifuge (Beckman Coulter). Polysome fractions were isolated using a Biocomp Gradient Station (Biocomp).
NMD-DegRPL5 and NMD-DegRibo: library preparations and analyses using RPL5 immunoprecipitation or polysome fractionation.
HEK293T cells (8 × 107 in one 150-mm dish) were transiently transfected with specified siRNAs and one day later with a reporter plasmid encoding dRLUC-Gl PTC mRNA and a reference plasmid encoding MUP mRNA.
Cells were lysed on day 3 using Hypotonic Gentle Lysis Buffer CHX-Plus and EDTA-Minus (10 mM Tris [pH 7.4], 10 mM NaCl, 0.5% w/w Triton X-100, 1 × cOmplete Protease Inhibitor Cocktail [Roche], 100 μg/ml cycloheximide [Sigma]) prior to anti-RPL5 or rIgG immunoprecipitation or prior to polysome fractionation (see above).
RNAs were purified using TRIzol Reagent, incubated with DNase I and then rSAP before RNA 3′ -ends were ligated using T4 RNA ligase to a 5′ -phosphorylated RNA adapter (3′ -RNA adapter in Supplementary Table 3) as described in the section immediately above. Poly(A) tails were removed oligo(dT)18 and RNase H, and cDNA was synthesized using SuperScript III reverse transcriptase and adapter antisense DNA as described above. dRLUC-Gl PTC and GADD45A decay intermediates were detected as described for the NMD-DegNAs protocol.
Immunofluorescence microscopy.
HeLa cells (5 × 104/24-well plate) were cultured on poly-L-lysine-coated coverslips, fixed using 4% paraformaldehyde in PBS (Affymetrix), and subsequently permeabilized using five successive 2-min incubations at room temperature in PBS containing 0.2% Triton X-100 (PBS-T). Coverslips were blocked using Image-iT signal enhancer (Invitrogen) or 3% bovine serum albumin (Rockland) in PBS for 30 min at room temperature, washed once with PBS, incubated overnight at 4 °C with primary antibody in PBS that had been diluted in PBS-T. Primary antibodies used were UPF1 (1:400)61, p-UPF1(S1089) (1:400; Millipore 07–1015), p-UPF1(S1116) (1:400; Millipore, 07–1016), Dcp1a (1:400; Abcam ab57654), RPS6 (1:100; Cell Signaling, no. 2317), RPL10a (1:100, Santa Cruz Biotechnology, sc-100827), and α-tubulin (1:500, Cell Signaling, no. 3873). Coverslips were then washed extensively using PBS-T, and subsequently incubated for 1 h at room temperature with 1:1,000 (v/v) Alexa Fluor 488-labeled goat anti-rabbit IgG (Invitrogen) or Alexa Fluor 568-labeled goat anti-mouse IgG (Invitrogen). After further extensive washing using PBS-T, coverslips were mounted using ProLong Gold Antifade Reagent with DAPI (Invitrogen). Images were captured with an Olympus FV-1,000 confocal laser-scanning microscope (Olympus) at the University of Rochester Medical Center Conventional and Confocal Microscopy Core.
Primer extension assay to estimate poly(A) length.
HEK293T cells (6 × 108 in a total of eight 150-mm dishes) were treated with okadaic acid (200 nM) for 3 h. After immunoprecipitation using anti-p-UPF1, anti-UPF1, anti-PABPC1 or rIgG, RNA was purified using TRIzol Reagent. Purified RNAs (100 ng) were incubated with DNase I and subsequently with rSAP, and RNA 3′ -ends were ligated to the 3′ -end adapter as in NMD-DegSeq. Samples were digested using RNase A and T1 (Thermo Fisher Scientific), and the resulting poly(A) tails were purified using phenol-chloroform-isoamyl alcohol that was saturated with 10 mM Tris (pH 8.0). cDNA was synthesized as in NMD-DegSeq but in the presence of α-[32P]-TTP (PerkinElmer) rather than all four dNTPs. Radio-labeled cDNAs were electrophoresed in 6 M urea-6% acrylamide and analyzed using the Typhoon FLA 9500 Phosphorimager.
Nuclear and cytoplasmic fractionations of HEK293T cells.
Fractionations were performed using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) following the manufacturer’s instructions.
Statistical analyses.
Statistical parameters are shown in the figures and listed in the figure legends. Statistical significance is claimed when P < 0.05 in either the unpaired two-tailed t-test or Wilcoxon rank-sum test, or one-way analysis of variance (ANOVA) is used with Dunnett’s multiple comparison test. In the figures, asterisks mark statistical significance as follows. *P < 0.05, **P < 0.01, or ***P < 0.001. See source data for details.
Software.
All scripts were written in Python or R and are available upon request.
Reporting Summary.
Further information on research design can be found in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank J. Pelletier for hippuristanol, S. Ohno and A. Yamashita for anti-p-UPF1(S1078, S1096), D. Ermolenko for Biocomp Gradient Fractionator usage, the University of Rochester Genomics Research Center for assistance in NGS library design, construction, and sequencing support, the University of Rochester Confocal Microscopy Core Facility for technical advice regarding confocal microscopy, and B. Lucas, M. Popp, and X. Rambout for comments on the manuscript. This work was supported by the National Institutes of Health (NIH) grant number R01 GM59614 to L.E.M. Salary support for T.K. was derived in part from a post-doctoral FRAXA Research Foundation Fellowship. The NIH through grant number S10 OD021489-01A1 supported purchase of the Typhoon FLA 9500 Phosphorimager.
Footnotes
online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability, and associated accession codes are available at https://doi.org/10.1038/s41594-018-0132-7.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41594-018-0132-7.
References
- 1.Kurosaki T & Maquat LE Nonsense-mediated mRNA decay in humans at a glance. J. Cell. Sci 129, 461–467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goetz AE & Wilkinson M Stress and the nonsense-mediated RNA decay pathway. Cell. Mol. Life Sci 74, 3509–3531 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lejeune F Nonsense-mediate mRNA decay at the crossroads of many cellular pathways. BMC Reports 50, 175–185 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nasif S, Contu L & Mühlemann O Beyond quality control: the role of nonsense-mediated mRNA decay (NMD) in regulating gene expression. Semin. Cell. Dev. Biol 75, 78–87 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Popp MW & Maquat LE Nonsense-mediated mRNA decay and cancer. Curr. Opin. Genet. Dev 48, 44–50 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurosaki T et al. A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev 28, 1900–1916 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durand S, Franks TM & Lykke-Andersen J Hyperphosphorylation amplifies UPF1 activity to resolve stalls in nonsense-mediated mRNA decay. Nat. Commun 7, 12434 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imamachi N, Salam KA, Suzuki Y & Akimitsu N A GC-rich sequence feature in the 3′ UTR directs UPF1-dependent mRNA decay in mammalian cells. Genome Res 27, 407–418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eberle AB, Lykke-Andersen S, Mühlemann O & Jensen TH SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol 16, 49–55 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Lykke-Andersen S et al. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev 28, 2498–2517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt SA et al. Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res 43, 309–323 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lejeune F, Li X & Maquat LE Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol. Cell 12, 675–687 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Yamashita A et al. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat. Struct. Mol. Biol 12, 1054–1063 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Jonas S, Weichenrieder O & Izaurralde E An unusual arrangement of two 14-3-3-like domains in the SMG5-SMG7 heterodimer is required for efficient nonsense-mediated mRNA decay. Genes Dev 27, 211–225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loh B, Jonas S & Izaurralde E The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev 27, 2125–2138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu W, Sweet TJ, Chamnongpol S, Baker KE & Coller J Co-translational mRNA decay in Saccharomyces cerevisiae. Nature 461, 225–229 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu W, Petzold C, Coller J & Baker KE Nonsense-mediated mRNA decapping occurs on polyribosomes in Saccharomyces cerevisiae. Nat. Struct. Mol. Biol 17, 244–247 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Astuti D et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat. Genet 44, 277–284 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Morris MR, Astuti D & Maher ER Perlman syndrome: overgrowth, Wilms tumor predisposition and DIS3L2. Am. J. Med. Genet. Part C Semin. Med. Genet 163, 106–113 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Wegert J et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27, 298–311 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Song M-G & Kiledjian M 3′ Terminal oligo U-tract-mediated stimulation of decapping. RNA 13, 2356–2365 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rissland OS & Norbury CJ Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat. Struct. Mol. Biol 16, 616–623 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi YS, Patena W, Leavitt AD & McManus MT Widespread RNA 3′ -end oligouridylation in mammals. RNA 18, 394–401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang HM, Triboulet R, Thornton JE & Gregory RI A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497, 244–248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faehnle CR, Walleshauser J & Joshua-Tor L Mechanism of Dis3l2 substrate recognition in the Lin28-let-7 pathway. Nature 514, 252–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim J et al. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159, 1365–1376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pirouz M, Du P, Munafò M & Gregory RI Dis3l2-mediated decay is a quality control pathway for noncoding RNAs. Cell Rep 16, 1861–1873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ustianenko D et al. TUT-DIS3L2 is a mammalian surveillance pathway for aberrant structured non-coding RNAs. EMBO J 35, 2179–2191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan M et al. mRNA 3′ uridylation and poly(A) tail length sculpt the mammalian maternal transcriptome. Nature 548, 347–351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurosaki T, Hoque M & Maquat LE Identifying cellular nonsense-mediated mRNA decay (NMD) targets: Immunoprecipitation of phosphorylated UPF1 followed by RNA sequencing (p-UPF1 RIP-Seq). Methods Mol. Biol 1720, 175–186 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Cho H, Kim KM & Kim YK Human proline-rich nuclear receptor coregulatory protein 2 mediates an interaction between mRNA surveillance machinery and decapping complex. Mol. Cell 33, 75–86 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Franks TM, Singh G & Lykke-Andersen J Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense-mediated mRNA decay. Cell 143, 938–950 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho H et al. SMG5-PNRC2 is functionally dominant compared with SMG5-SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res 41, 1319–1328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamashita A, Ohnishi T, Kashima I, Taya Y & Ohno S Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev 15, 2215–2228 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohnishi T et al. Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol. Cell 12, 1187–1200 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Isken O et al. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 133, 314–327 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F & Dietz HC Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet 36, 1073–1078 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Viegas MH, Gehring NH, Breit S, Hentze MW & Kulozik AE The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the nonsense mediated decay pathway. Nucleic Acids Res 35, 4542–4551 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tani H et al. Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA. Biol 9, 1370–1379 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lou C-H et al. Nonsense-mediated RNA decay influences human embryonic stem cell fate. Stem Cell Reports 6, 844–857 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Piovesan A, Caracausi M, Antonaros F, Pelleri MC & Vitale L GeneBase 1.1: a tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database 2016, baw153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang H, Lim J, Ha M & Kim VN TAIL-seq: genome-wide determination of poly(A) tail length and 3′ end modifications. Mol. Cell 53, 1044–1052 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Kurosaki T & Maquat LE Rules that govern UPF1 binding to mRNA 3′ UTRs. Proc. Natl Acad. Sci. USA 110, 3357–3362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brouwer R et al. Three novel components of the human exosome. J. Biol. Chem 276, 6177–6184 (2001). [DOI] [PubMed] [Google Scholar]
- 46.van Dijk EL, Schilders G & Pruijn GJM Human cell growth requires a functional cytoplasmic exosome, which is involved in various mRNA decay pathways. RNA 13, 1027–1035 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Staals RHJ et al. Dis3-like 1: a novel exoribonuclease associated with the human exosome. EMBO J 29, 2358–2367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kikin O, D’Antonio L & Bagga PS QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res 34, 676–682 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mullen TE & Marzluff WF Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev 22, 50–65 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi YS, Patena W, Leavitt AD & McManus MT Widespread RNA 3′ -end oligouridylation in mammals. RNA 18, 394–401 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slevin MK et al. Deep sequencing shows multiple oligouridylations are required for 3′ to 5′ degradation of histone mRNAs on polyribosomes. Mol. Cell 53, 1020–1030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Welch JD et al. EnD-Seq and AppEnD: sequencing 3′ ends to identify nontemplated tails and degradation intermediates. RNA 21, 1375–1389 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt M-J, West S & Norbury CJ The human cytoplasmic RNA terminal U-transferase ZCCHC11 targets histone mRNAs for degradation. RNA 17, 39–44 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Unterholzner L & Izaurralde E SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell 16, 587–596 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Fukuhara N et al. SMG7 is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol. Cell 17, 537–547 (2005). [DOI] [PubMed] [Google Scholar]
- 56.Trcek T, Sato H, Singer RH & Maquat LE Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev 27, 541–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ingolia NT, Lareau LF & Weissman JS Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okada-Katsuhata Y et al. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res 40, 1251–1266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chakrabarti S, Bonneau F, Schussler S, Eppinger E & Conti E Phospho-dependent and phospho-independent interactions of the helicase UPF1 with the NMD factors SMG5-SMG7 and SMG6. Nucleic Acids Res 42, 9447–9460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nicholson P, Josi C, Kurosawa H, Yamashita A & Mühlemann O A novel phosphorylation-independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res 42, 9217–9235 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gong C, Kim YK, Woeller CF, Tang Y & Maquat LE SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev 23, 54–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamashita A et al. SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev 23, 1091–1105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Serin G, Gersappe A, Black JD, Aronoff R & Maquat LE Identification and characterization of human orthologues to Saccharomyces cerevisiae Upf2 protein and Upf3 protein (Caenorhabditis elegans SMG-4). Mol. Cell. Biol 21, 209–223 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin M CutAdapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10–12 (2011). [Google Scholar]
- 65.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huber W et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H et al. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferrari F et al. ‘Jump Start and Gain’ model for dosage compensation in Drosophila based on direct sequencing of nascent transcripts. Cell Rep 5, 629–636 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Breese MR & Liu Y NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 29, 494–496 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.