

Abstract
To define the contribution of CD8+ T cell responses to control of SIV reactivation during and following antiretroviral therapy (ART), we determined the effect of long-term CD8+ T cell depletion using a rhesusized anti-CD8β monoclonal antibody on barcoded SIVmac239 dynamics on stable ART and after ART cessation in rhesus macaques (RMs). Among the RMs with full CD8+ T cell depletion in both blood and tissue, there were no significant differences in the frequency of viral blips in plasma, the number of SIV RNA+ cells and the average number of RNA copies/infected cell in tissue, and levels of cell-associated SIV RNA and DNA in blood and tissue relative to control-treated RMs during ART. Upon ART cessation, both CD8+ T cell–depleted and control RMs rebounded in fewer than 12 days, with no difference in the time to viral rebound or in either the number or growth rate of rebounding SIVmac239M barcode clonotypes. However, effectively CD8+ T cell–depleted RMs showed a stable, approximately 2-log increase in post-ART plasma viremia relative to controls. These results indicate that while potent antiviral CD8+ T cell responses can develop during ART-suppressed SIV infection, these responses effectively intercept post-ART SIV rebound only after systemic viral replication, too late to limit reactivation frequency or the early spread of reactivating SIV reservoirs.
Keywords: AIDS/HIV
Keywords: Adaptive immunity, T cells
Introduction
CD8+ T cells are crucial components of the adaptive immune response against HIV and SIV, and most examples of stringent, long-term, spontaneous and vaccine-associated immune control of these infections are either known, or strongly suspected to involve effective, antiviral CD8+ T cell responses (1–7). Given these well-established precedents, an optimized CD8+ T cell response has been invoked as a key immune mechanism to be exploited in HIV cure and remission strategies, either by direct CD8+ T cell–mediated elimination of rebound-competent virus that persists despite sustained ART, including latent viral reservoirs on ART (typically after induction of viral gene expression by latency reversing agents), or by CD8+ T cell–mediated control of viral reactivation and spread following ART discontinuation (ideally prior to systemic viral rebound) (8–10). Underlying these proposed therapeutic approaches is a concept of immune surveillance in which virus-specific, effector-differentiated CD8+ memory T cells continuously patrol lymphoid tissues in sufficient numbers to provide immediate killing of viral antigen–expressing cells before they return to full latency, or in the absence of ART, before they spread infection to neighboring susceptible cells.
Intrinsic to this concept and its application to HIV cure/remission therapy are several assumptions, including that CD8+ T cells with antiviral activity (a) can be generated in the setting of ART-suppressed HIV/SIV infection and maintained in sufficient numbers to ensure early intercept of antigen-expressing infected cells, (b) can manifest appropriate effector differentiation to provide for prompt virus-infected cell elimination or suppression, and (c) have in their regulated homing/migration behavior access to all sites of infected cell persistence. Most of these assumptions for CD8+ T cell effectiveness have not been rigorously tested, and their applicability to HIV cure/remission are potentially undermined by the findings that (a) immune escape can obviate the effectiveness of HIV/SIV reservoir recognition (11); (b) ART suppression (and consequent reduction in viral antigen exposure) leads to contraction of virus-specific CD8+ T cell responses and loss of effector differentiation (12, 13); (c) CD8+ T cells are largely excluded from some key known sites of HIV/SIV-infected-cell persistence, in particular the reservoir maintained in CD4+ follicular helper T cells within B cell follicles (14); and (d) cells harboring reactivating virus may be intrinsically resistant to CD8+ T cell–mediated cytolysis (15). Together, these observations raise the question whether HIV/SIV-specific CD8+ T cells can perform or can be therapeutically modulated to perform the role they have been envisaged to perform in HIV cure/remission therapies. To address this critical issue, we turned to SIV infection in rhesus macaques (RMs), a model that closely recapitulates the virology and immunology of HIV infection in people, while allowing for both experimental control of infection parameters and immune manipulation (16, 17). Using barcoded, highly pathogenic SIVmac239 and ART initiation at peak plasma viremia in primary infection, we demonstrate that long-term CD8+ T cell depletion had no significant effect on the frequencies of viral blips in plasma or SIV-infected cells in blood and tissue during ART, nor on the rate and magnitude of viral recrudescence following ART withdrawal. However, effective CD8+ T cell depletion resulted in a stable, approximately 2-log increase in post-ART plasma viremia relative to IgG-control treatment. Collectively, these studies indicate that while CD8+ T cell responses can effectively intercept post-ART SIV rebound, this antiviral activity is too slow to limit either reactivation frequency or the systemic spread of reactivating SIV reservoirs.
Results
Study design.
To evaluate the role of SIV-specific CD8+ T cells in ART-suppressed SIV infection and post-ART viral rebound, we used an RM model in which highly pathogenic SIVmac239 is suppressed by ART at or about peak viral replication in primary infection (day 12), with the goal of achieving full reservoirs, rapid and complete viral suppression, and limited or no immune system damage or immune escape (18). In addition, we hypothesized that day 12 ART initiation would provide sufficient infection to prime CD8+ T cells with antiviral activity. To ensure that this hypothesis was correct, we compared post-ART viral replication dynamics in RMs infected with SIVmac239 that were placed on ART starting on day 12 and maintained for 1 year to that of SIV-naive RMs given the same viral challenge and followed through primary infection (Figure 1). As shown in the figure, the ART-treated RM rebounded rapidly after ART cessation, but controlled viral replication to levels that were approximately 2-logs reduced from the peak replication of the same RMs during primary infection, and from the peak and plateau replication of different RMs given the same viral challenge without ART. These data strongly suggest that in this early-ART model, the immune sensitization inherent in transient high-level viral exposure with subsequent stringent ART-mediated viral control generates and maintains anti-SIV immune responses that are substantially more effective in controlling viral replication and spread following ART cessation than primary immune responses — a vaccine-like effect similar to the efficacy of the best T cell–targeted prime-boost prophylactic vaccines (19).
Figure 1. Substantial control of post-ART rebound in RMs with day 12 ART–suppressed SIV infection.

Mean (+SEM) SIVmac239X pvl profiles of RMs 0–12 days pi (maroon; n = 6) or rebound pvl following 1 year of ART (teal; n = 6) in comparison with RMs with primary SIVmac239X infection with no ART (black; n = 6). The 2-sample Wilcoxon rank-sum test was used to determine significance of differences in the area under curve (AUC) of pvl between days 28 and 70 after SIV infection or after ART cessation (unadjusted P values shown).
To evaluate the contribution of CD8+ T cells to this post-rebound control and to determine the ability of such antiviral CD8+ T cells to modulate reservoirs and reactivation dynamics, we set up an experiment to analyze reservoir and viral dynamics in SIV-infected RMs, on and subsequently off stable ART, that were systemically and specifically depleted of CD8+ T cells, compared to nondepleted controls. Previous studies exploring this question in the RM model (20, 21) used cell-depleting monoclonal antibodies (mAbs) targeting the CD8α molecule, which is expressed on both classical MHC-Ia–restricted, TCR-αβ+, CD8αβ+ T cells and other distinct CD8αα+ cell types, including NK cells, nonclassically restricted TCR-αβ+ T cells, TCR-γδ+ T cells, and some CD4+ memory T cells (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI141677DS1). Here, we used a depleting, rhesusized anti-CD8β mAb that specifically induces and maintains depletion of classical (MHC-Ia–restricted, TCR-αβ–expressing) CD8αβ+ T cells, preserving the various CD8αα+ cell types (22). In addition, we have previously shown that CD8α-targeted cell depletion results in a massive increase in IL-15 bioactivity, which strongly drives activation, differentiation, and proliferation of residual cells, in particular CCR5-expressing CD4+ memory T cells (23), which would potentially increase both CD4+ memory T cell viral susceptibility, and in latently infected cells, viral reactivation (21, 24). These off-target effects are substantially mitigated by CD8β targeting (see below), facilitating experimental interpretation. Other important design elements of our study include (a) intravenous SIVmac239 challenge (200 infectious units [IU]) with ART initiation at peak acute-phase viral replication (day 12 postinfection [pi]), designed to provide for a full, disseminated reservoir and rapid viral suppression, prior to both CD8+ T cell immune escape and any significant loss of viral genomic integrity (e.g., generation of defective proviruses) (2, 18, 25); (b) use of RMs with defined MHC-Ia allomorphs associated with moderately (Mamu-A*01; n = 10) and highly (Mamu-B*08; n = 10) effective SIV-specific CD8+ T cell responses (6, 26); (c) use of barcoded SIVmac239M virus, which contains approximately 10,000 different barcodes and therefore allows discrimination and quantification of individual viral clonotypes (27, 28); and (d) stratification of RMs within the Mamu-A*01 and Mamu-B*08 groups to anti-CD8β– versus isotype control mAb–treated cohorts at week 31 pi (week 29 on ART) to ensure pretreatment equivalence between anti-CD8β–treated and control groups (Figure 2A and Supplemental Table 1). With regard to the last point, prior to treatment initiation, the RMs in the anti-CD8β–treated versus control groups were indistinguishable with respect to (a) plasma viral load (pvl) dynamics (including pre-ART peak viral loads and rate and extent of post-ART suppression) (Figure 2B), (b) the number and distribution of detected individual barcodes contributing to the infection (Figure 2C), and (c) cell-associated SIV DNA and RNA levels in peripheral blood mononuclear cells (PBMCs) and lymph nodes (LNs) at both 3 days and 28 weeks after ART initiation (Figure 2, D and E). In addition, there were no differences between the anti-CD8β–treated and control RM groups with respect to the overall magnitude of the SIV-specific, CD4+ and CD8+ T cell response in blood and lung airspace (Figure 3, A and B), and within the Mamu-A*01 and Mamu-B*08 groups, the magnitude of the CD8+ T cell responses to the relevant immunodominant epitopes presented by these allomorphs (i.e., A*01: Gag CM9, Tat SL8, and B*08: Nef RL10, Vif RL9) in blood (Figure 3, C and D).
Figure 2. Equivalence of plasma- and cell-associated viral loads between study groups prior to mAb treatment.

(A) Schematic representation of the study protocol showing SIVmac239M infection, ART initiation 12 days pi, and anti-CD8β or IgG isotype control mAb administration, which occurred every other week from weeks 31–47 pi. Both the anti-CD8β and isotype control groups included n = 5 Mamu-A*01+ and n = 5 Mamu-B*08+ RMs. (B) Mean (+SEM) pvl profiles of anti-CD8β (red) and IgG controls (blue) (n = 10 each) prior to treatment initiation. (C) Comparison of SIVmac239M barcode clonotypes identified by high-throughput sequencing in plasma, PBMCs, and LNs between anti-CD8β (red) or IgG controls (blue) at 3 days after ART. Each data point represents the number of detectable barcodes in an individual RM. (D and E) Comparison of SIV RNA and DNA levels in PBMCs and LNs (copies per 1 × 106 cell equivalents) between anti-CD8β (red) and IgG controls (blue) at 3 days (15 days pi) and 28 weeks after ART. Each data point represents a single determination from an individual RM. Plots show jittered points with a box from first to third quartiles (IQR) and a line as the median, with whiskers extending to the farthest data point within 1.5 × IQR above and below the box, respectively. In C–E, closed circles indicate Mamu-A*01+ RMs and open circles indicate Mamu-B*08+ RMs. In B–E, Wilcoxon’s rank-sum test was used to determine the significance of differences between the anti-CD8β and IgG control treatment groups (unadjusted P values shown).
Figure 3. Equivalence of SIV-specific T cell response development between study groups prior to mAb treatment.

(A and B) Mean (+SEM) frequencies of CD4+ and CD8+ memory T cells (Tm) in peripheral blood and lung airspace (obtained by bronchoalveolar lavage) specific for SIV Gag, Rev/Tat/Nef, Env, and Pol following SIVmac239M infection and ART initiation at 12 days pi in RMs selected for the anti-CD8β treatment group (red; n = 10) versus the IgG isotype control group (blue; n = 10). These response frequencies were determined by intracellular expression of TNF-α and/or IFN-γ after stimulation with mixes of consecutive, overlapping 15-mer peptides for each SIV protein (Gag, Env, Pol) or protein combination (Rev/Tat/Nef) with the total response to these SIV proteins, reflecting the sum of the Gag + Rev/Tat/Nef + Env + Pol responses, shown. (C and D) Mean (+SEM) frequencies of peripheral blood CD8+ Tm specific for SIV Gag CM9, Tat SL8, Nef RL10, and Vif RL9 determined by tetramer staining for each SIV epitope as described in the Methods section. In A–D, Wilcoxon’s rank-sum test was used to determine the significance of differences between the treatment groups (unadjusted P values shown).
Anti-CD8β selectively depletes classical CD8+ T cells in blood and tissues.
Treatment with the control versus anti-CD8 mAbs was initiated at week 31 pi, while the RMs were on stable ART and continued every 2 weeks until week 47 pi, with ART release occurring at week 41 pi. Thus, anti-CD8β versus control treatment was maintained for 10 weeks during continued ART and for 6 weeks after ART release. Anti-CD8β treatment resulted in massive depletion of CD8+ T cells in blood, including nearly complete (99%) depletion of the naive subset (Tn) and high-level (80%) depletion of the memory (Tm) subset (Figure 4A and Supplemental Figure 2). As expected, NK cells, TCR-γδ+ T cells, and CD4+ Tm (each subset expressing CD8α in whole or in part; Supplemental Figure 1) were not depleted by anti-CD8β mAb. Indeed, both NK cells and TCR-γδ+ T cells manifested transient increases in numbers in CD8β cell–depleted versus control RM at 5–7 weeks after mAb treatment (Figure 4, B–D). The profound depletion of CD8+ Tn, which was maintained throughout and subsequent to the treatment period, demonstrated the depletion potency of the anti-CD8β treatment against a population that is entirely CD8αβ+ (Supplemental Figure 1). The negligible rebound of CD8+ Tn after cessation of anti-CD8β treatment is consistent with diminished thymopoiesis in HIV/SIV infection (29). In contrast with homogeneous CD8αβ+ Tn, circulating (TCR-αβ+) CD8+ Tm include a mixture of CD8αβ+ conventional T cells and CD8αα+ (nonclassical) innate-type T cells that are not susceptible to CD8β-targeted cell depletion (30, 31), likely explaining the small residual posttreatment CD8+ Tm population in blood (Figure 4A and Supplemental Figure 2). Although anti-CD8β mAb treatment had no effect on the absolute counts of any CD4+ T cell subset, there was a modest boost in CD4+ effector memory T cell (Tem) proliferation (%Ki-67) that peaked 2 weeks after the first dose and was maintained for 6 weeks (4 weeks prior to ART cessation) before returning to control levels (Figure 5). Of note, this CD4+ Tem proliferation is substantially less than that previously observed with anti-CD8α depletion, which was shown to be due to IL-15 (23).
Figure 4. Profound selective depletion of CD8+ T cells by anti-CD8β treatment.

Mean (+SEM) of the percentage change from baseline in the absolute counts of total, memory, and naive CD8+ T cells (A), NK cells (B), TCR-γδ+ T cells (C), and total CD4+ T cells (D) in blood following anti-CD8β (red; n = 10) versus IgG isotype control (blue; n = 10) mAb treatment. Arrows indicate anti-CD8β or IgG control mAb administration and time of ART cessation. Wilcoxon’s rank-sum test was used to determine significance of differences in the AUC between weeks 0 and 16 after mAb treatment (unadjusted P values shown).
Figure 5. Effect of anti-CD8β mAb treatment on CD4+ T cell subset dynamics in blood.

Change in the proliferative fraction (left panels) and absolute counts (right panels) of CD4+ T subsets, including naive (Tn) and total memory (Tm), central memory (Tcm), transitional memory (Ttrm), and effector memory (Tem) in blood following anti-CD8β mAb (n = 10) versus IgG control mAb (n = 10) treatment. Results are shown as mean (+SEM) change from baseline of %Ki-67 (left panels) and percentage of baseline absolute counts (right panels). Anti-CD8β or IgG control mAb administration is indicated by arrows. Wilcoxon’s rank-sum test was used to determine the significance of differences in AUC prior to ART cessation (weeks 0–10), value at time of ART cessation (day 74), and AUC after ART cessation (weeks 10.5–16) between the 2 treatment groups (unadjusted P values shown).
Anti-CD8β depletion also induced profound CD8+ T cell deficiency in the LNs of 7 of 10 treated RMs (sampled on ART, 9 weeks after mAb treatment initiation), with CD8+ Tn and Tm depleted to <1% and ≤3% of T cells, respectively (Figure 6A and Supplemental Figure 3), with a greater proportion of CD8+ T cell depletion occurring within the T cell zone of the LNs (Figure 6, C–F). Similarly, at the same time point, the fraction of CD8+ Tn and Tm were depleted to <1% and <10%, respectively, in the bone marrow (BM) in 8 of 10 anti-CD8β–treated RMs (Figure 6B). Taken together, these data indicate that whereas CD8+ Tn were depleted across the board in anti-CD8 mAb–treated RMs, only 7 of 10 of these RMs demonstrated similarly high levels of CD8+ Tm depletion across both tissues (operationally defined as full depletion), whereas 3 of 10 RMs manifested less CD8+ Tm depletion in LNs, BM, or both tissues (>4.5% in LNs and >15% in BM; operationally defined as incomplete depletion).
Figure 6. Differential depletion of CD8+ T cells in tissues after anti-CD8β.

(A and B) Comparison of the fraction of memory (Tm) and naive CD8+ T cells of total T cells in lymph nodes (LNs) and bone marrow (BM) after 9 weeks of treatment with anti-CD8β or IgG control mAb. RMs with full CD8+ Tm depletion are shown in red (n = 7; see main text), RMs with incomplete CD8+ Tm depletion are shown in green (n = 3) with individual RMs delineated by different symbols, and IgG isotype controls are shown in blue (n = 10). Each data point represents an individual RM. Plots show jittered points with a box from first to third quartiles (IQR) and a line as the median, with whiskers extending to the farthest data point within 1.5 × IQR above and below the box, respectively. Wilcoxon’s rank-sum test was used to determine the significance of differences between treatment groups (unadjusted P values shown). (C) Immunofluorescence microscopy of representative LN sections from an RM with maximal (“complete”) CD8 depletion (middle panel), an RM with incomplete CD8 depletion (right panel), and an IgG control RM (left panel). CD8+ cells are in red, while CD20+ cells are in blue. The white line is used to demarcate the T cell zone (TCZ) and the B cell zone (BCZ). Scale bars: 100 μm. (D–F) Quantification of the number of CD8+ cells per 1 × 105 cells in LNs in TCZ, BCZ, and medullary cords (MC) of LNs at 16 days after ART release in the treatment groups. Each data point represents the average number of CD8+ cells derived from quantitative measures from 2–3 LN sections from a single time point from an individual RM. Wilcoxon’s rank-sum test was used to determine the significance of differences between treatment groups, first excluding RMs with incomplete CD8+ Tm depletion, and then including all treated RMs versus IgG controls (unadjusted P values shown). In A, B, and D–F closed symbols indicate Mamu-A*01+ RMs and open symbols indicate Mamu-B*08+ RMs.
Selective CD8+ T cell depletion had no effect on SIV dynamics during ART.
Plasma SIV RNA was monitored by a high sensitivity assay at least weekly 7 days prior to and during anti-CD8β versus control mAb treatment to look for treatment-related on-ART plasma viral blips, defined as increases in plasma viremia above the standard threshold of 15 SIV RNA copies per milliliter. Overall, there was no significant difference in the number of such viral blips observed among control and fully CD8β-depleted RM groups over the 10 weeks of observation on ART (Figure 7A). We note, however, that whereas 10 of 10 control RMs showed 0–1 above-threshold viral blips, 2 of 7 RMs in the fully CD8β-depleted group manifested more than 5 such blips, suggesting that in these RMs, CD8+ T cell depletion might have induced a modest increase in on-ART viral replication. However, it should also be noted that the majority of these blips were observed between 1 and 6 weeks after treatment initiation, coinciding with the increased proliferation of CD4+ Tem (Figure 5), and suggesting that these increased blips might have resulted from increased latency reversal due to infected cell activation. Importantly, levels of cell-associated SIV RNA and DNA in blood and LNs were not significantly different 10 days prior to ART release (week 9 after anti-CD8β mAb treatment initiation; Figure 7B), and anti-CD8β mAb treatment did not significantly increase the frequency of SIV RNA+ cells in LNs, or the number of copies of SIV RNA per SIV RNA+ cell, relative to control treatment (Figure 7C). Taken together, these data suggest that during ART levels of persistently infected cells in blood and LNs were largely unchanged by prolonged CD8+ T cell depletion.
Figure 7. Effect of CD8+ T cell depletion on SIV infection dynamics prior to ART withdrawal.

(A) Individual pvl profiles monitored by a high-sensitivity assay (limit of detection, 1 RNA copy/mL) prior to and during anti-CD8β or IgG isotype control mAb treatment, prior to ART cessation. The area in gray denotes pvl values below threshold of the standard assay (15 RNA copies/mL). There was no significant difference between treatment groups in instance and duration of above–standard threshold pvl as determined by Wilcoxon’s rank-sum and Fisher’s exact tests. (B) Comparison of cell-associated SIV RNA (left panel) and DNA (right panel) levels in PBMCs, LNs, and BM (copies per 1 × 106 cell equivalents) after 9 weeks of treatment with anti-CD8β or IgG control mAb. Each data point represents a single determination from an individual RM. Plots show jittered points with a box from first to third quartiles (IQR) and a line as the median, with whiskers extending to the farthest data point within 1.5 × IQR above and below the box, respectively. (C) Quantification of the number of SIV RNA+ cells per 1 × 105 cells (left panel) and the average number of SIV RNA copies per cell measured in LN tissue sections by RNAscope. In B and C, Wilcoxon’s rank-sum test was used to determine the significance of differences between treatment groups, first excluding RMs with incomplete CD8+ Tm depletion, and then including all treated RMs versus IgG controls (unadjusted P values shown). In all panels, RMs with effective CD8+ T cell depletion (n = 7) are shown in red, RMs with incomplete CD8+ T cell depletion (n = 3) are shown in green, and IgG isotype controls (n = 10) are shown in blue. Closed symbols indicate Mamu-A*01+ RMs and open symbols indicate Mamu-B*08+ RMs.
Selective CD8+ T cell depletion had no effect on the rate of SIVmac239M reactivation following ART withdrawal but had a substantial impact on post-ART viremia.
After 40 weeks of ART administration and 10 weeks of anti-CD8β versus IgG control mAb treatment, ART was discontinued while maintaining mAb treatment for an additional 6 weeks, so as to study the effect of CD8+ T cell depletion on the dynamics of viral rebound. All study RMs manifested viral rebound within 12 days of ART release, with no significant differences in the time to measurable rebound viremia between RMs with or without CD8+ T cell depletion (Figure 8A). Next, we determined the growth rate of total plasma RNA and used the proportional representation of individual SIVmac239M barcode–defined clones in relation to total rebound viremia at each time point to estimate the average clonal viral reactivation rates in individual RMs (27, 32). Of note, SIVmac239M clones that contributed to post-ART viremia were generally observed at higher proportions early after ART initiation (Supplemental Figure 4). In RMs with complete CD8+ T cell depletion, neither the overall growth rate of plasma viremia nor the average rates of viral clone reactivation were significantly different from those of the control-treated RMs (Figure 8, B and C). There was also no correlation between either the growth rate or reactivation rate with cell-associated SIV DNA and SIV RNA in blood and LNs just prior to ART cessation (Supplemental Figure 5), indicating that in the conditions of this experiment, overall levels of residual SIV did not predict viral rebound dynamics.
Figure 8. Effect of CD8+ T cell depletion on SIV infection dynamics after ART withdrawal.

(A) Kaplan-Meier analysis of SIV rebound kinetics in RMs with effective CD8+ T cell depletion (red; n = 7) and RMs with incomplete CD8+ T cell depletion (green; n = 3) versus IgG isotype controls (blue; n = 10). (B and C) Quantification of overall viral growth rates and SIVmac239M clonal reactivation rates in plasma by high-throughput sequencing after ART cessation. (D) Individual pvl profiles of RMs in each treatment group. Left panel shows anti-CD8β–treated RMs with effective CD8+ T cell depletion versus RMs with incomplete CD8+ T cell depletion (green), while the middle panel shows IgG isotype controls (blue). Right panel shows mean (+SEM) pvl profiles of RMs stratified by treatment group (limit of detection, 15 RNA copies/mL). Wilcoxon’s rank-sum test was used to determine significance of differences in the AUC of pvl between 14 and 20 weeks after mAb treatment (unadjusted P values shown). (E) Quantification of cell-associated SIV RNA in PBMCs, LNs, and BM (copies per 1 × 106 cell equivalents) at 8 and 16 days after ART cessation. Plots show jittered points with a box from first to third quartiles (IQR) and a line as the median, with whiskers extending to the farthest data point within 1.5 × IQR above and below the box, respectively. In B, C, and E, Wilcoxon’s rank-sum test was used to determine the significance of differences between treatment groups, first excluding RMs with incomplete CD8+ Tm depletion, and then including all treated RMs versus IgG controls (unadjusted P values shown). Closed symbols indicate Mamu-A*01+ RMs and open symbols indicate Mamu-B*08+ RMs.
In contrast, complete CD8+ T cell depletion did result in a striking, approximately 2-log increase in post-ART viremia both during ongoing anti-CD8β versus control mAb treatment and for 30 weeks after treatment was discontinued (Figure 8D). Cell-associated SIV RNA levels in PBMCs significantly increased in fully CD8+ T cell–depleted RMs by day 8 in PBMCs, but not in LNs or BM; these tissues only showed significant differences on day 16 after ART (Figure 8E). This antiviral activity is likely due to antigen-driven expansion of SIV-specific CD8+ T cells in response to post-ART viremia (Supplemental Figure 6). Of note, the late-arriving antiviral activity in control-treated RMs was similar in both Mamu-A*01+ and -B*08+ RMs (Supplemental Figure 7). This indicates that the CD8+ T cell responses that developed in response to pre-ART primary infection and maintained during ART can provide efficient intercept of rebound SIV infection (Supplemental Figure 8), but this intercept occurs only after viral amplification and spread. Also, the 3 anti-CD8β mAb–treated RMs with incomplete depletion manifested post-ART release viral dynamics similar to control-treated RMs (Figure 8D), suggesting that the residual SIV-specific CD8+ T cell populations in these RMs were sufficient to mount an effective (and similarly late arriving) response.
Discussion
Although these data were developed in an optimized model system, they illustrate several fundamental aspects of the primate immune response to AIDS-causing lentivirus infections that are highly germane to development of HIV cure/remission strategies. First, primary SIV infection can prime SIV-specific immune responses that, while largely ineffective in mediating efficient control of primary infection, can, when protected from viral pathogenesis by early ART initiation, mediate substantial, durable viral control upon ART cessation. Second, this effect is completely abrogated by anti-CD8β mAb treatment, indicating that classical CD8αβ+ T cells are required for (and almost certainly mediate) this enhanced viral control. Third, CD8αβ+ T cell responses that are able to achieve an approximately 2-log reduction in long-term chronic post-ART SIV replication rates have no discernable effect on early parameters of post-ART viral rebound, including the timing, number, and growth rates of reactivating viral clonotypes, suggesting that this antiviral activity requires coordination of the CD8+ T cell response to an expanding infection, not immediate effector activity acting preemptively to effectively abort individual viral reactivation foci. Fourth, restoration of CD8αβ+ Tm after post-ART viral replication set points are established does not result in subsequent reduction in viremia, suggesting that a coordinated CD8+ T cell response during initial viral rebound is required for this enhanced viral control. These conclusions are consistent with the effects of effective therapeutic vaccination of ART-suppressed SIV, which, upon ART release, resulted in durable post-ART viral control with little to no effect on rebound dynamics (33), and suggest that CD8+ T cell–based immunotherapeutic strategies designed to induce off-ART remission should focus on enhancing the effectiveness of the response to rebound, enhancing the effector potency of the CD8+ T cell responses at the time of ART release to provide an earlier, stronger immune intercept of the nascent viral rebound.
These results have important implications for efforts to exploit CD8+ T cell responses for piecemeal destruction of infected cells supporting spontaneous or induced viral reactivation on ART, so-called shock and kill cure strategies (8–10). The lack of difference between RMs with and without CD8αβ+ T cells in the kinetics and extent of early SIV rebound in this study suggests that CD8αβ+ T cells are not effective against early reactivation foci, possibly due to insufficient numbers of cells relative to the number of these sites, an inability of these cells to access these sites (e.g., reactivation within B cell follicles), or inappropriate functional differentiation (3, 14, 34–36). In keeping with this conclusion, we also found no evidence that measures of persistent virus parameters during ART were affected by the presence or absence of CD8αβ+ T cells. Two RMs in our study showed an increased frequency of viral blips during anti-CD8β mAb treatment, but these blips coincided with increased CD4+ Tem proliferation/activation that could account for increased viral reactivation from latently infected cells. Indeed, a more dramatic increase in plasma viral blip frequency following administration of an anti-CD8α depleting mAb in ART-suppressed RMs has also been reported, which was further enhanced by exogenous IL-15 administration (20, 21).
In conclusion, our results call into question the widely held assumption that de novo virus-specific CD8+ T cells will mediate the kill part of shock and kill under most circumstances, or without specific intervention, will mediate immediate, complete control of viral outgrowth after ART cessation. We would suggest that rigorous analysis of SIV reservoir modulation in the nonhuman primate model, specifically enhancing CD8+ T cell numbers and/or functionality (e.g., with therapeutic vaccines, adjuvants, immune checkpoint blockade, cytokines) and/or removing other barriers to CD8+ T cell–mediated antiviral activity (e.g., B cell follicles), will be required to delineate whether and under what circumstances CD8+ T cell responses can contribute to reduction of the rebound-competent HIV/SIV reservoir on ART, or mediate sufficiently effective viral control after ART discontinuation to qualify as a virologic remission.
Methods
Animals.
A total of 32 purpose-bred male and female RMs (Macaca mulatta) of Indian genetic background were used for these experiments. These RMs were specific pathogen free as defined by being free of cercopithecine herpesvirus 1, D-type simian retrovirus, simian T-lymphotropic virus type 1, rhesus rhadinovirus, and Mycobacterium tuberculosis. MHC-1 genotyping for common Mamu alleles such as Mamu-A*01/-A*02 and Mamu-B*08/-B*17 was performed by sequence-specific priming PCR, essentially as described previously (6). The 20 RMs (3–6 years of age) used for the main study were intravenously inoculated with 200 IU of SIVmac239M (27) and placed on ART starting at 12 days pi. The 200 IU inoculum challenge dose was chosen as it represents a compromise between a lower (more physiologic) challenge dose and a dose that also allows for seeding of a sufficient number of barcodes for quantification of reactivation rates. As previously described, the average number of detectable barcodes at peak viremia at the 200 IU dose is 29 in comparison to 698 at the 10,000 IU dose. Using a low-dose intravenous challenge with day 12 ART provides for a full, disseminated reservoir prior to both CD8+ T cell immune escape and any significant loss of viral genomic integrity, while still ensuring rapid viral suppression within a reasonable experimental timeframe. ART was maintained for up to 290 days pi. In addition, RMs received intravenous infusions of the CD8β-depleting mAb CD8β255R1 (n = 10) or IgG isotype control mAb (n = 10) at 50 mg/kg on days 217, 231, 245, 259, 273, 286, 301, 315, and 329 pi. An additional 12 male RMs (3–5 years of age) were intravenously inoculated with 2 IU of SIVmac239X (37) and received either no treatment (n = 6) or started on ART 12 days pi and maintained on ART for 1 year (n = 6). ART consisted of subcutaneous injection of 5.1 mg/kg/d tenofovir disoproxil, 40 mg/kg/d emtricitabine, and 2.5 mg/kg/d dolutegravir in a solution containing 15% (v/v) kleptose at pH 4.2, as previously described (38).
Viruses.
The SIVmac239M challenge stock used in this experiment was produced in transfected HEK-239T cells and the stock infectivity titer was determined using TZM-bl cells as previously described (27). The SIVmac239X challenge stocks were generated by expansion in RM PBMCs and titered by using the CMMT-CD4-LTR-β-Gal (sMAGI) cell assay, as previously described (37).
Viral detection assays.
Plasma SIV RNA levels were determined using a gag-targeted quantitative real-time/digital RT-PCR format assay, essentially as previously described, with 6 replicate reactions analyzed per extracted sample for an assay threshold of 15 SIV RNA copies/mL (35). Ultrasensitive determinations of plasma SIV RNA were obtained by concentrating virus from larger volumes of plasma by centrifugation. For ultrasensitive measurements, typically 1.7 mL of plasma was centrifuged in a refrigerated microfuge (21,000g, 1 hour, 4°C) and nucleic acid was extracted from pellets as described previously (39), and quantitative RT-PCR was performed with 12 reactions per extracted sample. Samples that did not yield any positive results across the replicate reactions are reported as a value of “less than” the value that would apply for 1 positive reaction out of 12 (35). As performed, the ultrasensitive assay provided a threshold sensitivity of 1 copy/mL plasma for a 1.7 mL sample. Quantitative assessment of SIV DNA and RNA in cells and tissues was performed using gag-targeted nested quantitative hybrid real-time/digital RT-PCR and PCR assays, as previously described (3, 35). SIV RNA or DNA copy numbers were normalized based on quantitation of a single-copy RM genomic DNA sequence from the CCR5 locus from the same specimen to allow normalization of SIV RNA or DNA copy numbers per 1 × 106 diploid genome cell equivalents, as described previously (40). Ten replicate reactions were performed with aliquots of extracted DNA or RNA from each sample, with 2 additional spiked internal control reactions performed with each sample to assess potential reaction inhibition. Samples that did not yield any positive results across the replicate reactions are reported as a value of “less than” the value that would apply for 1 positive reaction out of 10. Threshold sensitivities for individual specimens varied as a function of the number of cells or amount of tissue available and analyzed.
T cell response assays.
SIV-specific CD4+ and CD8+ T cell responses were measured in mononuclear cell preparations from blood and lung airspace (bronchoalveolar lavage samples) by flow cytometric intracellular cytokine analysis as previously described (3, 35). Briefly, mixes of sequential (11 amino acid overlapping) 15-mer peptides (AnaSpec) spanning the SIVmac239 Gag, Env, Pol, and Rev/Tat/Nef open reading frames were used as antigens in conjunction with anti-CD28 (CD28.2, purified 500 ng/test; eBioscience, custom bulk 7014-0289-M050) and anti-CD49d stimulatory mAb (9F10, purified 500 ng/test; eBioscience, custom bulk 7014-0499-M050). Mononuclear cells were incubated at 37°C with peptide mixes and antibodies for 1 hour, followed by an additional 8-hour incubation in the presence of brefeldin A (5 μg/mL; Sigma-Aldrich). Stimulation in the absence of peptides served as background control. After incubation, stimulated cells were stored at 4°C until staining with combinations of fluorochrome-conjugated monoclonal antibodies, including anti-CD3 (SP34-2, Pacific Blue; BD Biosciences, custom bulk 624034 and PerCP-Cy5.5; BD Biosciences, custom bulk 624060); anti-CD4 (L200, FITC; BD Biosciences, custom bulk 624044 and AmCyan; BD Biosciences, custom bulk 658025); anti-CD8α (SK1, APC-Cy7; eBioscience, custom bulk 7047-0087-M002); anti–TNF-α (MAB11, APC; BD Biosciences, custom bulk 624076 and FITC; BD Biosciences, custom bulk 624046 and PE; BD Biosciences, custom bulk 624049); anti–IFN-γ (B27, APC; BD Biosciences, custom bulk 624078 and FITC; BD Biosciences, 554700); and anti-CD69 (FN50, PE; eBioscience, custom bulk CUST01282 and PE–Texas Red; BD Biosciences, custom bulk 624005). Data were collected on an LSR II flow cytometer (BD Biosciences). Analysis was performed using FlowJo software (Tree Star). In all analyses, gating on the lymphocyte population was followed by the separation of the CD3+ T cell subset and progressive gating on CD4+ and CD8+ T cell subsets. Antigen-responding cells in both CD4+ and CD8+ T cell populations were determined by their intracellular expression of CD69 and either or both of the cytokines IFN-γ and TNF. After subtracting background, the raw response frequencies were memory corrected, as previously described (34, 36).
SIV-specific CD8+ T cells were also measured by tetramer staining. In brief, 100 μL whole blood or 0.5 × 106 to 2 × 106 mononuclear cells from the lung airspace were stained with 100 ng of tetramer. Mamu-A*01+ tetramers consisted of Gag CM9 and Tat SL8 biotinylated monomers conjugated with streptavidin BV421 and BV605, respectively. Mamu-B*08+ tetramers consisted of Nef RL10 and Vif RL9 biotinylated monomers conjugated with streptavidin BV421 and BV605, respectively. Cells were stained for 30 minutes, followed by washing and staining with combinations of fluorochrome-conjugated surface antibodies, including anti-CD3 (SP34-2, BUV395; BD Biosciences, custom bulk 624310); anti-CD4 (L200, BUV786; BD Biosciences, custom bulk 624159); anti-CD8α (RPA-T8, BV510; BD Biosciences, custom bulk 624144); anti-CD28 (CD28.2, PE-DAZZ; BioLegend, custom bulk 93364); anti-CD95 (DX2, BUV737; BD Biosciences, custom bulk 624231); anti-CCR7 (BV711; BD Biosciences, custom bulk 624386); anti-CXCR5 (MU5UBEE, PE; Life Technologies, custom bulk 7012-9185-M002); anti-CD20 (2H7, APC-Fire; BioLegend, custom bulk 93924); and anti-CCR5 (3A9, APC; BD Biosciences, custom bulk 624076). Cells were then washed, lysed/fixed, and permeabilized prior to intracellular staining with anti–Ki-67 (B56, FITC; BD Biosciences, custom bulk 624046) for 45 minutes. MHC-peptide complexes were provided as biotinylated monomers by the NIH Tetramer Core Facility and conjugated with streptavidin-BV421 (BD Biosciences, custom bulk 624337) or streptavidin-BV605 (BD Biosciences, custom bulk 624342) in a 4:1 molar ratio and prepared at a final concentration of 50 ng/μL in PBS.
Immunophenotyping.
To determine the phenotype of lymphocyte populations, whole blood or mononuclear cell preparations from LNs and BM were stained for flow cytometric analysis as previously described (23, 41, 42). Polychromatic (8–14 parameter) flow cytometric analysis was performed on a BD LSR II instrument using Pacific Blue, BUV395, BUV737, BV421, BV510, BV605, BV711, BV786, FITC, PE, PE–Texas Red (PE-CF594), PE-Cy7, PerCP-Cy5.5, APC, APC-Cy7, and Alexa Fluor 700 as the available fluorescence parameters. Instrument setup and data acquisition procedures were performed as previously described (23, 41, 42). List-mode multiparameter data files were analyzed using FlowJo software. Criteria for delineating Tn and Tm subsets and for setting positive versus negative markers for CCR5 and Ki-67 expression have been previously described in detail (23, 41, 42). In brief, Tn constitute a uniform cluster of cells with a CD28modCCR7+CCR5−CD95lo phenotype, which is clearly distinguishable from the phenotypically diverse memory population that is CD95hi or displays one or more of the following nonnaive phenotypic features: CD28−, CCR7−, and CCR5+. The Tcm, transitional memory T cell (Ttrm), and Tem components of the memory subset in the blood were further delineated based on the following phenotypic criteria: Tcm (CD28+CCR7+CCR5−), Ttrm (CD28+CCR7+/−CCR5+), and Tem (CD28−CCR7−CCR5dim). For each subset to be quantified, the percentages of the subset within the overall small lymphocyte and/or small T cell (CD3+ small lymphocyte) populations were determined. For quantification of peripheral blood subsets, absolute small lymphocyte counts were obtained using an AcT5diff cell counter (Beckman Coulter) and, from these values, absolute counts for the relevant subset were calculated based on the subset percentages within the light scatter–defined small lymphocyte population on the flow cytometer. Baseline values were determined as the average of values on days −14, −7, and 0. Absolute counts are indicated as percentage change from baseline, with baseline shown as 100%. Changes in proliferative fraction are indicated as the difference in the %Ki-67+ (Δ%Ki-67+) measured at the designated time points from baseline (0% = no change). Following anti-CD8β mAb administration, CD8+ T cell depletion was determined by gating on small lymphocytes that were CD3+, CD4–, TCR-γδ–, and CD8α+ (see Supplemental Figure 1). Combinations of fluorochrome-conjugated mAbs used for staining included anti-CD3 (SP34-2, BUV395; BD Biosciences, custom bulk 624310 and PerCP-Cy5.5; BD Biosciences, custom bulk 624060); anti-CD4 (L200, BV786; BD Biosciences, custom bulk 624159 and BUV395; BD Biosciences, custom bulk 624163); anti-CD8α (DK25, Pacific Blue; DAKO, PB98401-1 and SK1, BUV737; BD Biosciences, custom bulk 624235); anti-CD95 (DX2, PE; BioLegend, custom bulk 94203 and BV605; BioLegend, custom bulk 93384); anti-CD28 (CD28.2, PE-DAZZ; BioLegend, custom bulk 93364 and BV510; BD Biosciences, custom bulk 624339); anti-CCR5 (3A9, APC; BD Biosciences, custom bulk 624076); anti–Ki-67 (B56, FITC; BD Biosciences, custom bulk 624046); anti-CD14 (M5E2, PE-Cy7; BioLegend, custom bulk 93704); anti-CD16 (3G8, BV650; BD Biosciences, custom bulk 93384); anti–HLA-DR (L243, PE-DAZZ; BioLegend, custom bulk 93957); anti-CD20 (2H7, APC-Cy7; BioLegend, custom bulk 93924); anti-CCR7 (150503, biotin; R&D Systems, MAB197 and BV711; BD Biosciences, custom bulk 624386); anti-NKG2A (REA110, APC; Miltenyi Biotec, 130-095-212 and PE; Miltenyi Biotec, 130-095-212); anti-CD8β (2ST8.5H7, PE–Texas Red; Beckman Coulter, 6607123 and PE; Beckman Coulter, IM2217U); anti-CD56 (MEM-188, PerCP-Cy5.5, BioLegend, MHCD5618CS3); anti–TCR-γδ (B1, PE, BioLegend, custom bulk 95922 and FITC; BioLegend, 331208); anti-CD69 (CH/4, PerCP-Cy5.5; Life Technologies, MHCD6918); anti-CD27 (M-T271, BV421; BioLegend, custom bulk 86409); anti-CD21 (B-ly4, BV711; BD Biosciences, custom bulk 624148); anti-IgD (PE; Southern Biotech, 2030-09); and anti-streptavidin (BV421; BD Biosciences, custom bulk 624337, BV605, BD Biosciences custom bulk 624342 and BV786; BD Biosciences, custom bulk 624164).
SIV RNA in situ hybridization, immunofluorescence, and image analysis.
RNAscope and quantitative image analysis were performed as previously published (43, 44). RNAscope images, scanned at ×40 magnification on an Aperio AT2 (Leica Biosystems), were analyzed for the total number of SIV RNA+ cells/105 total cells (quantitative) and categorized according to the relative amount of SIV RNA present (semiquantitative) using the ISH module (v2.2) within HALO software (v3.0.311.405; Indica Labs). The relative amount of SIV RNA within a single infected cell was first estimated by quantifying the total area of the SIV RNA signal spot size (μm2). As the signal spot size is a function of several steps in the experimental procedures, module settings were established on concomitantly assayed, acutely infected SIV+ control slides. To estimate the signal spot size of a single SIV RNA molecule, we measured the signal area (minimum, mean and maximum) of more than 10 identifiable individual virions within B cell follicles, which corresponds to 2 copies of SIV RNA, and multiplied this by 0.5. We set the SIV RNA minimal signal spot size within the analysis module to only measure SIV RNA+ cells with 3 or more SIV RNA copies in order to exclude detection of a single SIV RNA molecule, which is indistinguishable from the signal generated by the integrated viral DNA. Relative SIV RNA copy number within SIV RNA+ cells was calculated as (signal spot size within SIV RNA+ cells [μm2])/(0.5 × mean signal size for a virion).
Staining for CD8+ cells was performed on formaldehyde-fixed, paraffin-embedded tissue sections (5 μm) according to our previously published protocol using a Biocare intelliPATH autostainer (45). Slides were retrieved in ACD P2 retrieval buffer (ACD, 322000), treated with 3% H2O2, and then stained with rabbit anti-CD8α (Sigma-Aldrich, HPA037756) and detected with rabbit Polink-2 HRP (GBI Labs, D39-110). CD8+ cells were analyzed using the cytonuclear (v2.0.9) module within Halo software (version v3.0.311.405, Indica Labs). Anatomic regions within the LNs were assigned to B cell follicles (BCFs), medullary cords (MCs), and the paracortical T cell zone (TCZ) based on a sequential tissue section stained with CD20 (Biocare, clone L26) and analyzed using the Halo AI classifier within Halo software (v3.0.311.405).
Fluorescence multiplex SIV RNAscope in situ hybridization with CD8+ T cell responses was performed as previously described (46). In brief, following RNAscope development with Tyr647 (Invitrogen, B40958), slides were stripped by boiling in ACD P2 retrieval buffer for 5 minutes. Slides were then stained with rabbit anti-IRF4 (Cell Signaling, clone E8H3S), developed with a rabbit Polink-1 HRP (GBI Labs, D13-110) using Tyr488 (Invitrogen, B40953), stripped in citrate buffer pH 6.0 (GBI Labs, B05C-100B) for 5 minutes, and then concurrently stained with rabbit anti-CD8α (Sigma-Aldrich, HPA037756) and mouse anti–granzyme B (Thermo Fisher Scientific, clone GZB01) for 1 hour at room temperature. After washing, CD8 was detected with donkey anti-rabbit secondary antibody labeled with Alexa Fluor 568 (Invitrogen, A10042), and granzyme B was detected with donkey anti-mouse secondary antibody labeled with DyLight 755 (Thermo Fisher Scientific, SA5-10171). Slides were counterstained with DAPI, coverslipped using ProLong Gold Antifade Mountant (Thermo Fisher Scientific, P36930), and scanned on an Axio Scan.Z1 using a Plan Apochromat 20× objective (Zeiss; NA = 0.8, FWD = 0.55 mm).
Barcode sequencing.
Barcode sequencing was performed as previously described (27, 28). Briefly, RNA was isolated from plasma or viral stock using a Qiagen QIAamp Viral RNA Mini Kit per the manufacturer’s instructions. cDNA was then synthesized from the extracted DNA using Superscript III reverse transcriptase (Invitrogen) and a reverse primer (Vpr.cDNA3: 5′-CAGGTTGGCCGATTCTGGAGTGGATGC-3′). qRT-PCR was used to quantify the cDNA using the primers VpxF1 (5′-CTAGGGGAAGGACATGGGGCAGG-3′ at 6082–6101) and VprR1 (5′-CCAGAACCTCCACTACCCATTCATC-3′ at 6220–6199), and a fluorescently labeled probe (ACCTCCAGAAAATGAAGGACCACAAAGGG). Known quantities of the viral template were then PCR amplified with the same VpxF1 and VprR1 primers but with MiSeq adaptors directly synthesized onto the primers. Reactions were prepared using High Fidelity Platinum Taq per the manufacturer’s instructions, using primer VpxF1 and VprR1 with the following conditions: 94°C for 2 minutes followed by 40 cycles of 94°C, 15 seconds; 60°C, 90 seconds; 68°C, 30 seconds, with final extension of 68°C for 5 minutes. Following PCR cleanup, amplicons were pooled and sequenced directly on a MiSeq instrument (Illumina).
Estimation of the growth rate and reactivation rate.
We assumed that the growth of virus between 2 neighboring measurements follows the exponential law described by Equation 1:
(Equation 1)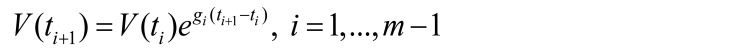
where m is the number of measurements of viral load and V(ti ) is the viral load at time ti. Thus, we can estimate the growth rate between 2 neighboring measurements (gi) as follows:
(Equation 2)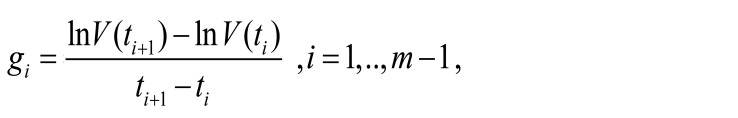
If the first measurement in the interval was below the detection threshold, then we estimated the growth rate assuming that the first measurement was equal to the detection threshold (15 copies/mL). Because the growth rate decreases toward the peak of viremia, we report the maximal growth rate as an estimate of the initial growth rate:
(Equation 3)
To estimate reactivation rate we assumed that all rebounders grow exponentially and have negligibly small difference of the growth rate (g); hence, the difference of natural logarithms of counts of barcode sequences will be proportional to delay between reactivations of these barcodes:
(Equation 4)
where τi is the time interval between i-th and i + 1 reactivations, and Si the number of sequences for i-th rebounder.
Knowing the time intervals between each rebounder and adopting the common assumption that waiting time between independent random events is exponentially distributed, we can find reactivation rate (r) as reciprocal to the arithmetic average of waiting times τi in each subject:
(Equation 5)
Statistics.
Statistical analyses were performed in R 3.6.0 with the package “survival” v3.1-8. We used Wilcoxon’s rank-sum test for all analyses comparing values across treatment groups. For analyses in Figure 6, B and C, Figure 7, B and C, and Figure 8, B–E, we first excluded RMs demonstrating incomplete CD8+ depletion. We also included analyses using the complete data set; however, these additional analyses were not considered to be independent sources of evidence and as such we did not adjust P values for multiple comparisons. Point values were transformed to the log10 scale where indicated. For analyses involving multiple time points, we calculated the area under the curve (AUC) for each RM and analyzed the resulting values in a similar fashion to that for single time point data. Time-to-event data are described with Kaplan-Meier estimates, and compared between groups using the Kruskal-Wallis test, with the difference being statistically significant at P < 0.05. Spearman’s test was used to conduct nonparametric correlation analysis of growth rate and reactivation rate with levels of cell-associated SIV DNA and RNA prior to ART release.
Sample size and treatment assignment.
Sample size was determined by logistical and resource considerations. Treatment assignments (CD8+ T cell depleted vs. IgG control treatment) were conducted after 70 days pi by rank ordering pvl AUC (28–70 days pi) and pvl 3 days after ART and assigning alternating treatments arbitrarily (but not randomly) and without revision. No RMs were excluded from any analysis in this study, apart from subanalyses by CD8 depletion status where noted. No blinding was possible due to the constraints of working with RMs.
Data and code availability.
The data sets generated and/or analyzed during the current study, as well as computer code used to perform statistical analysis, are available from the corresponding authors on reasonable request.
Study approval.
All animal procedures were approved by the Oregon Health & Science University, Oregon National Primate Research Center’s Animal Care and Use Committee, under the standards of the US NIH Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011).
Author contributions
LJP and AAO conceived and planned the study and wrote the manuscript. AAO supervised RM experiments, and analyzed immunological and virological data, assisted by DDD, SGH, BVM, HP, YF, MC, AM, ANS, RMG, and HB. SIV quantification was provide by JDL. BFK provided the SIVmac239M viral stock and performed SIVmac239M barcode sequencing and analysis. MP and MPD provided barcode modeling. RG provided a validated ART regimen. JDE provided in situ SIV RNA analysis and confocal images, assisted by KBS and MN. J Smedley and MKA managed the animal protocols. PTE and J Shao conducted statistical analyses.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH to LJP (grants UM1AI126611, UM1AI124377, 4R37AI054292) and to the Oregon National Primate Research Center (P51OD011092), by federal funds to JDL and BFK from the National Cancer Institute, NIH (contracts HHSN261200800001E and 75N91019D00024), and by an NIH grant to JDE for the Integrated Pathology Core at the Oregon National Primate Research Center (grant 1S10OD025002-01). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. The CD8β-depleting mAb CD8β255R1 and IgG isotype control mAbs were provided by the NIH Nonhuman Primate Reagent Resource Program. Tetramers were provided by the NIH Tetramer Core Facility at Emory University and ART drugs were provided by Gilead Sciences Inc. We thank the Quantitative Molecular Diagnostics Core of the AIDS and Cancer Virus Program of the Frederick National Laboratory for viral load analyses. We also thank A. Sylwester, S. Hagen, T. Swanson, M. Fischer, C. Shriver-Munsch, E. McDonald, K., W. Brantley, R. Lum, A. Maxwell, M. Lidell, D. Morrow, J. Ford, A. Townsend, and K. Rothstein for technical and/or administrative assistance.
Version 1. 02/25/2021
In-Press Preview
Version 2. 04/15/2021
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(8):e141677.https://doi.org/10.1172/JCI141677.
Contributor Information
Afam A. Okoye, Email: okoyea@ohsu.edu.
Derick D. Duell, Email: duell@ohsu.edu.
Yoshinori Fukazawa, Email: fukazawa@ohsu.edu.
Benjamin Varco-Merth, Email: varcomer@ohsu.edu.
Alejandra Marenco, Email: marenco@ohsu.edu.
Hannah Behrens, Email: behrensh@ohsu.edu.
Andrea N. Selseth, Email: selseth@ohsu.edu.
Roxanne M. Gilbride, Email: gilbride@ohsu.edu.
Jason Shao, Email: jshao@fredhutch.org.
Paul T. Edlefsen, Email: pedlefse@fhcrc.org.
Romas Geleziunas, Email: romas.geleziunas@gilead.com.
Mykola Pinkevych, Email: mpinkevych@kirby.unsw.edu.au.
Miles P. Davenport, Email: mdavenport@kirby.unsw.edu.au.
Kathleen Busman-Sahay, Email: busmansa@ohsu.edu.
Haesun Park, Email: parkh@ohsu.edu.
Michael K. Axthelm, Email: axthelmm@ohsu.edu.
Jacob D. Estes, Email: estesja@ohsu.edu.
Scott G. Hansen, Email: hansensc@ohsu.edu.
Brandon F. Keele, Email: keelebf@mail.nih.gov.
Jeffery D. Lifson, Email: lifson@ncifcrf.gov.
Louis J. Picker, Email: pickerl@ohsu.edu.
References
- 1.Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27(3):406–416. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Goulder PJ, Watkins DI. HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol. 2004;4(8):630–640. doi: 10.1038/nri1417. [DOI] [PubMed] [Google Scholar]
- 3.Hansen SG, et al. Immune clearance of highly pathogenic SIV infection. Nature. 2013;502(7469):100–104. doi: 10.1038/nature12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walker BD, Yu XG. Unravelling the mechanisms of durable control of HIV-1. Nat Rev Immunol. 2013;13(7):487–498. doi: 10.1038/nri3478. [DOI] [PubMed] [Google Scholar]
- 5.International HIVCS, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330(6010):1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loffredo JT, et al. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J Virol. 2007;81(16):8827–8832. doi: 10.1128/JVI.00895-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitz JE, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283(5403):857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 8.Mylvaganam G, et al. Toward T cell-mediated control or elimination of HIV reservoirs: lessons from cancer immunology. Front Immunol. 2019;10:2109. doi: 10.3389/fimmu.2019.02109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones RB, Walker BD. HIV-specific CD8+ T cells and HIV eradication. J Clin Invest. 2016;126(2):455–463. doi: 10.1172/JCI80566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sengupta S, Siliciano RF. Targeting the latent reservoir for HIV-1. Immunity. 2018;48(5):872–895. doi: 10.1016/j.immuni.2018.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng K, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. 2015;517(7534):381–385. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casazza JP, et al. Decay kinetics of human immunodeficiency virus-specific CD8+ T cells in peripheral blood after initiation of highly active antiretroviral therapy. J Virol. 2001;75(14):6508–6516. doi: 10.1128/JVI.75.14.6508-6516.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Appay V, et al. Persistent HIV-1-specific cellular responses despite prolonged therapeutic viral suppression. AIDS. 2002;16(2):161–170. doi: 10.1097/00002030-200201250-00004. [DOI] [PubMed] [Google Scholar]
- 14.Fukazawa Y, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med. 2015;21(2):132–139. doi: 10.1038/nm.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang SH, et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest. 2018;128(2):876–889. doi: 10.1172/JCI97555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Rompay KKA. Tackling HIV and AIDS: contributions by non-human primate models. Lab Anim (NY) 2017;46(6):259–270. doi: 10.1038/laban.1279. [DOI] [PubMed] [Google Scholar]
- 17.Del Prete GQ, Lifson JD. Nonhuman primate models for studies of AIDS virus persistence during suppressive combination antiretroviral therapy. Curr Top Microbiol Immunol. 2018;417:69–109. doi: 10.1007/82_2017_73. [DOI] [PubMed] [Google Scholar]
- 18.Okoye AA, et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat Med. 2018;24(9):1430–1440. doi: 10.1038/s41591-018-0130-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Picker LJ, et al. New paradigms for HIV/AIDS vaccine development. Annu Rev Med. 2012;63:95–111. doi: 10.1146/annurev-med-042010-085643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao Y, et al. CD8+ lymphocyte control of SIV infection during antiretroviral therapy. PLoS Pathog. 2018;14(10):1007350. doi: 10.1371/journal.ppat.1007350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McBrien JB, et al. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8(+) cells. Nature. 2020;578(7793):154–159. doi: 10.1038/s41586-020-1946-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimura Y, et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature. 2017;543(7646):559–563. doi: 10.1038/nature21435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okoye A, et al. Profound CD4+/CCR5+ T cell expansion is induced by CD8+ lymphocyte depletion but does not account for accelerated SIV pathogenesis. J Exp Med. 2009;206(7):1575–1588. doi: 10.1084/jem.20090356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mueller YM, et al. IL-15 treatment during acute simian immunodeficiency virus (SIV) infection increases viral set point and accelerates disease progression despite the induction of stronger SIV-specific CD8+ T cell responses. J Immunol. 2008;180(1):350–360. doi: 10.4049/jimmunol.180.1.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bender AM, et al. The landscape of persistent viral genomes in ART-treated SIV, SHIV, and HIV-2 infections. Cell Host Microbe. 2019;26(1):73–85. doi: 10.1016/j.chom.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mothe BR, et al. Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol. 2003;77(4):2736–2740. doi: 10.1128/JVI.77.4.2736-2740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fennessey CM, et al. Genetically-barcoded SIV facilitates enumeration of rebound variants and estimation of reactivation rates in nonhuman primates following interruption of suppressive antiretroviral therapy. PLoS Pathog. 2017;13(5):1006359. doi: 10.1371/journal.ppat.1006359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khanal S, et al. In vivo validation of the viral barcoding of simian immunodeficiency virus SIVmac239 and the development of new barcoded SIV and subtype B and C simian-human immunodeficiency viruses. J Virol. 2019;94(1):e01420-19. doi: 10.1128/JVI.01420-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douek DC, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396(6712):690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 30.Van Kaer L, et al. CD8alphaalpha(+) innate-type lymphocytes in the intestinal epithelium mediate mucosal immunity. Immunity. 2014;41(3):451–464. doi: 10.1016/j.immuni.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambolez F, et al. Thymic differentiation of TCR alpha beta(+) CD8 alpha alpha(+) IELs. Immunol Rev. 2007;215:178–188. doi: 10.1111/j.1600-065X.2006.00488.x. [DOI] [PubMed] [Google Scholar]
- 32.Pinkevych M, et al. Predictors of SIV recrudescence following antiretroviral treatment interruption. Elife. 2019;8:e49022. doi: 10.7554/eLife.49022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borducchi EN, et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature. 2016;540(7632):284–287. doi: 10.1038/nature20583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansen SG, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011;473(7348):523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansen SG, et al. Addendum: Immune clearance of highly pathogenic SIV infection. Nature. 2017;547(7661):123–124. doi: 10.1038/nature22984. [DOI] [PubMed] [Google Scholar]
- 36.Hansen SG, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15(3):293–299. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del Prete GQ, et al. Molecularly tagged simian immunodeficiency virus SIVmac239 synthetic swarm for tracking independent infection events. J Virol. 2014;88(14):8077–8090. doi: 10.1128/JVI.01026-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Del Prete GQ, et al. Short communication: comparative evaluation of coformulated injectable combination antiretroviral therapy regimens in simian immunodeficiency virus-infected rhesus macaques. AIDS Res Hum Retroviruses. 2016;32(2):163–168. doi: 10.1089/aid.2015.0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, et al. Envelope residue 375 substitutions in simian-human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc Natl Acad Sci U S A. 2016;113(24):E3413–E3422. doi: 10.1073/pnas.1606636113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venneti S, et al. Longitudinal in vivo positron emission tomography imaging of infected and activated brain macrophages in a macaque model of human immunodeficiency virus encephalitis correlates with central and peripheral markers of encephalitis and areas of synaptic degeneration. Am J Pathol. 2008;172(6):1603–1616. doi: 10.2353/ajpath.2008.070967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okoye A, et al. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. J Exp Med. 2007;204(9):2171–2185. doi: 10.1084/jem.20070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okoye AA, et al. Naive T cells are dispensable for memory CD4+ T cell homeostasis in progressive simian immunodeficiency virus infection. J Exp Med. 2012;209(4):641–651. doi: 10.1084/jem.20112071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McBrien JB, et al. Author correction: robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature. 2020;578(7796):E21. doi: 10.1038/s41586-020-2002-9. [DOI] [PubMed] [Google Scholar]
- 44.Deleage C, et al. Defining HIV and SIV reservoirs in lymphoid tissues. Pathog Immun. 2016;1(1):68–106. doi: 10.20411/pai.v1i1.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chandrashekar A, et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science. 2020;369(6505):812–817. doi: 10.1126/science.abc4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harper J, et al. CTLA-4 and PD-1 dual blockade induces SIV reactivation without control of rebound after antiretroviral therapy interruption. Nat Med. 2020;26(4):519–528. doi: 10.1038/s41591-020-0782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated and/or analyzed during the current study, as well as computer code used to perform statistical analysis, are available from the corresponding authors on reasonable request.