

Abstract
It has long been known that fatty acids can either adversely or positively affect insulin signaling in skeletal muscle, depending on chain length or saturation, and can therefore be primary drivers of systemic insulin sensitivity. However, the detailed mechanisms linking fatty acids to insulin signaling in skeletal muscle have been elusive. In this issue of the JCI, Ferrara et al. suggest a model whereby membrane lipid remodeling mediates skeletal muscle insulin sensitivity. The authors demonstrate that membrane glycerophospholipid fatty acid remodeling by lysophosphatidylcholine acyltransferase 3 (LPCAT3) in skeletal muscle from subjects with obesity was induced, suppressing insulin signaling and glucose tolerance. Loss or gain of LPCAT3 function in mouse models showed that Lpcat3 was both required and sufficient for high-fat diet–induced muscle insulin resistance. These results suggest that the physiochemical properties of muscle cell membranes may drive insulin sensitivity and, therefore, systemic glucose intolerance.
Glycerophospholipid remodeling in obesity and diabetes
At first pass, de novo membrane glycerophospholipid biosynthesis via the canonical Kennedy pathway seems to make an imperfect lipid bilayer. In order to generate suitable membrane properties, including an asymmetric phospholipid biolayer, membrane curvature, and appropriate fluidity, membranes require further remodeling. Membrane glycerophospholipid fatty acid chain remodeling occurs via the Land’s cycle (1), whereby a phospholipase hydrolyzes a glycerophospholipid fatty acid that is then reesterified by a lysophospholipid acyltransferase (2). This process enables the cell to fine-tune its acyl chain diversity and adapt to environmental conditions to affect the biophysical properties of the cell, for example, incorporating important polyunsaturated fatty acids (PUFAs) into the membrane (Figure 1). One class of acyltransferases are the lysophosphatidylcholine acyltransferases, which influence the membrane lipid composition associated with a variety of pathologies in diverse tissues, such as the intestine, liver, and brain (3, 4). Lysophosphatidylcholine acyltransferase 3 (LPCAT3) is a relatively well-studied LPCAT family member that preferentially inserts arachidonic acid into glycerophospholipids in membranes. The loss of Lpcat3 results in perinatal lethality and has been shown to maintain systemic lipid homeostasis by regulating lipid absorption in intestine as well as lipoprotein secretion and de novo lipogenesis in liver (5, 6).
Figure 1. Model of diet-induced lipid bilayer changes in skeletal muscle that result in insulin resistance.
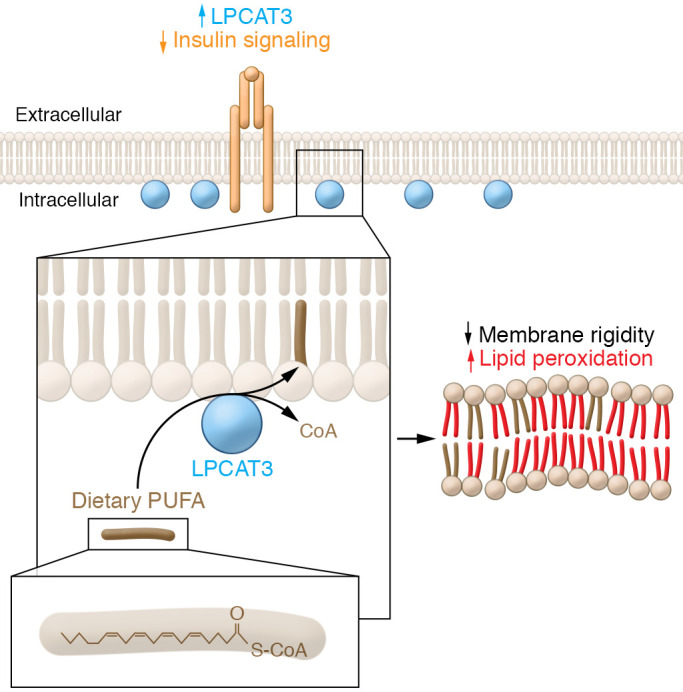
When incorporating lipids into the plasma membrane, LPCAT3, an enzyme involved with phospholipid transacylation, esterifies a fatty acid onto a lysophospholipid from an acyl-CoA donor. Ferrara et al. (7) showed that an imbalance of glycerophospholipid remodeling in skeletal muscle occurs with obesity. In people, skeletal muscle remodeling was associated with an increase in LPCAT3. Loss or gain of Lpcat3 in mouse models showed that Lpcat3 is required and sufficient for inducing muscle insulin resistance following a high-fat diet. Muscle cell membrane composition may drive insulin sensitivity and glucose intolerance by influencing oxidation and plasma membrane rigidity. LPCAT3-dependent changes in plasma membrane may increase susceptibility to lipid oxidation by increasing the quantity of oxidation-sensitive fatty acids. Moreover, membrane fluidity may directly affect insulin signaling efficacy by changing accessibility to insulin receptors or altering glucose transporter trafficking.
Ferrara et al. used lipidomics to demonstrate increased lysophospholipid species in human-cultured myotubes from subjects with obesity as compared with lean controls (7). The myotubes in culture conditions retained their in vivo phenotype while avoiding exposure to confounding contaminating intramuscular adipocytes. The increase in lysophospholipid species suggests that obesogenic conditions influenced membrane remodeling and generated imbalanced transacylation in skeletal muscle. The authors found that Lpcat3 was transcriptionally induced in myotubes and muscle biopsies from subjects with obesity. Indeed, the genetic or pharmacological inhibition of LPCAT3 increased muscle insulin sensitivity. Conversely, increasing LPCAT3 genetically in mouse models suppressed insulin sensitivity. These data align with the notion that an obesogenic increase in skeletal muscle LPCAT3 suppresses glucose uptake in humans. Consistent with the present study (7), membranes enriched in arachidonyl phosphatidylcholine were previously shown to inhibit AKT signaling (8), although Ferrara et al. argue that increased LPCAT3 specifically impairs insulin-mediated AKT signaling in muscle. These results link membrane glycerophospholipid dynamics to the development of diet-induced insulin resistance (Figure 1).
PUFA, ROS, and insulin signaling
Dietary intake of PUFA has been implicated in insulin sensitivity. Dietary sourced PUFA is particularly relevant to skeletal muscle, which synthesizes very little de novo fatty acid and relies largely on exogenous fatty acid uptake, not only for the generation of contractile energy, but also glycerolipid synthesis. In Ferrara et al., Lpcat3 expression clearly altered the membrane PUFA composition of cells, suggesting a possible mechanistic link among dietary PUFA intake, LPCAT3-dependent membrane remodeling, and insulin resistance (7).
Ferroptosis is a nonapoptotic cell death associated with iron accumulation and lipid peroxidation in which hyperoxidation ultimately causes cell death. The loss of the arachidonic acid–preferring acyl–coenzyme A (acyl-CoA) synthetase-4 determines ferroptosis sensitivity. Similarly, the loss of LPCAT3 makes cells resistant to ferroptosis (9, 10). LPCAT3 deficiency likely limits oxidation-sensitive fatty acids at the cell membrane. It seems reasonable to suggest that an LPCAT3-dependent increase in membrane arachidonic acid content would make the muscle membranes more susceptible to lipid oxidation, particularly in obesogenic and/or inflammatory conditions. This raises the question of whether antioxidants might improve glucose intolerance associated with obesogenic Lpcat3 expression.
Can membrane fluidity directly affect insulin signaling?
While it is clear that excess fatty acids can affect insulin signaling in muscle in vitro and in vivo, the detailed mechanisms remain elusive. The loss of Lpcat3 ultimately changes many structural and signaling properties of cells, making it difficult to define a strict cause-and-effect relationship. While disrupting membrane PUFA is likely the root of the issue, is it possible that membrane fluidity could directly affect insulin signaling efficacy? In cell culture models, the addition of saturated fatty acids, such as palmitate, can cause lipotoxicity and generate insulin resistance, which can generally be alleviated by also including unsaturated fatty acids. It is tempting to speculate that membrane physiochemical properties alone could predominately drive defective insulin signaling by changing signaling effectors’ affinity for membrane or altering glucose transporter trafficking (11).
One oft-cited insulin-sensitizing agent is the adipocyte-secreted hormone adiponectin. The putative receptors for adiponectin have been identified as AdipR1 and AdipR2, which have become attractive drug targets in type II diabetes (12). Interestingly, in the evolutionary record, AdipoRs predate the appearance of adiponectin or its paralogous C1QTNF family members. Emerging evidence suggests that AdipoRs and their orthologues act as membrane fluidity sensors (13–15). In line with the idea that AdipoRs sense and alter membrane lipid composition, adiponectin and C1QTNF family members have been shown to bind membrane lipids (16). These data suggest that some of the pleotropic observations of adiponectin and AdipoRs may be due to membrane lipid composition rather than strictly canonical hormone receptor interactions.
Conclusions
Ferrara et al. demonstrate that human obesity results in an imbalance of glycerophospholipid remodeling in skeletal muscle associated with an increase in LPCAT3 (7). Given that LPCAT3 drives membrane reesterification, it remains unclear what the driver of membrane remodeling is. In order for LPCAT3 to affect glycerophospholipid fatty acid content, phospholipases must hydrolyze fatty acids to generate the LPCAT3 lysolipid substrate. How is this hydrolysis mediated? By what enzyme? And by what signal? Answering these questions will elucidate important mechanistic details. Could membrane rigidity, mediated by a Western high-fat diet high in saturated fatty acids activate the AdipoRs independently of their putative endocrine ligand in skeletal muscle, which uniquely depend on exogenous fatty acids? Prolonged dietary changes in membrane structure may have a primary physiochemical effect, signaling fatty acid hydrolysis and reesterification. Perhaps this process is why some diets high in PUFA effectively control glucose intolerance. LPCAT3 presents an intriguing tractable experimental handle for elucidating the role of tissue-specific membrane dynamics on systemic physiology.
Acknowledgments
MJW is supported by NIH grants R01DK120530 and R01DK116746.
Version 1. 04/15/2021
Electronic publication
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(8):e148176. https://doi.org/10.1172/JCI148176.
References
- 1.Hill EE, Lands WE. Incorporation of long-chain and polyunsaturated acids into phosphatidate and phosphatidylcholine. Biochim Biophys Acta. 1968;152(3):645–648. doi: 10.1016/0005-2760(68)90109-4. [DOI] [PubMed] [Google Scholar]
- 2.Hishikawa D, et al. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci U S A. 2008;105(8):2830–2835. doi: 10.1073/pnas.0712245105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang B, Tontonoz P. Phospholipid remodeling in physiology and disease. Annu Rev Physiol. 2019;81:165–188. doi: 10.1146/annurev-physiol-020518-114444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ichu TA, et al. ABHD12 and LPCAT3 interplay regulates a Lyso-phosphatidylserine-C20:4 phosphatidylserine lipid network implicated in neurological disease. Biochemistry. 2020;59(19):1793–1799. doi: 10.1021/acs.biochem.0c00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hashidate-Yoshida T, et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. Elife. 2015;4:e06328. doi: 10.7554/eLife.06328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rong X, et al. Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. Elife. 2015;4:e06557. doi: 10.7554/eLife.06557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrara PJ, et al. Lysophospholipid acylation modulates plasma membrane lipid organization and insulin sensitivity in skeletal muscle. J Clin Invest. 2021;131(8):e135963. doi: 10.1172/JCI135963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koeberle A, et al. Arachidonoyl-phosphatidylcholine oscillates during the cell cycle and counteracts proliferation by suppressing Akt membrane binding. Proc Natl Acad Sci U S A. 2013;110(7):2546–2551. doi: 10.1073/pnas.1216182110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kagan VE, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon SJ, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10(7):1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pilon M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis. 2016;15(1):167. doi: 10.1186/s12944-016-0342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada-Iwabu M, et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 2013;503(7477):493–499. doi: 10.1038/nature12656. [DOI] [PubMed] [Google Scholar]
- 13.Svensson E, et al. The adiponectin receptor homologs in C. elegans promote energy utilization and homeostasis. PLoS One. 2011;6(6):e21343. doi: 10.1371/journal.pone.0021343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruiz M, et al. AdipoR1 and AdipoR2 maintain membrane fluidity in most human cell types and independently of adiponectin. J Lipid Res. 2019;60(5):995–1004. doi: 10.1194/jlr.M092494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Svensk E, et al. PAQR-2 regulates fatty acid desaturation during cold adaptation in C. elegans. PLoS Genet. 2013;9(9):e1003801. doi: 10.1371/journal.pgen.1003801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye JJ, et al. Adiponectin and related C1q/TNF-related proteins bind selectively to anionic phospholipids and sphingolipids. Proc Natl Acad Sci U S A. 2020;117(29):17381–17388. doi: 10.1073/pnas.1922270117. [DOI] [PMC free article] [PubMed] [Google Scholar]