

Abstract
Objective:
Intraductal papillary mucinous neoplasms (IPMNs) are non-invasive precursor lesions that can progress to invasive pancreatic cancer and are classified as low-grade or high-grade based on the morphology of the neoplastic epithelium. We aimed to compare genetic alterations in low-grade and high-grade regions of the same IPMN in order to identify molecular alterations underlying neoplastic progression.
Design:
We performed multi-region whole exome sequencing on tissue samples from 17 IPMNs with both low-grade and high-grade dysplasia (76 IPMN regions, including 49 from low-grade dysplasia and 27 from high-grade dysplasia). We reconstructed the phylogeny for each case, and we assessed mutations in a novel driver gene in an independent cohort of 63 IPMN cyst fluid samples.
Results:
Our multi-region whole exome sequencing identified KLF4, a previously unreported genetic driver of IPMN tumorigenesis, with hotspot mutations in one of two codons identified in >50% of the analyzed IPMNs. Mutations in KLF4 were significantly more prevalent in low-grade regions in our sequenced cases. Phylogenetic analyses of whole exome sequencing data demonstrated diverse patterns of IPMN initiation and progression. Hotspot mutations in KLF4 were also identified in an independent cohort of IPMN cyst fluid samples, again with a significantly higher prevalence in low-grade IPMNs.
Conclusion:
Hotspot mutations in KLF4 occur at high prevalence in IPMNs. Unique among pancreatic driver genes, KLF4 mutations are enriched in low-grade IPMNs. These data highlight distinct molecular features of low-grade and high-grade dysplasia and suggest diverse pathways to high-grade dysplasia via the IPMN pathway.
Keywords: intraductal papillary mucinous neoplasm, pancreatic precursor lesion, whole exome sequencing, KLF4, phylogenetic analysis, cyst fluid, somatic mutation
INTRODUCTION
Intraductal papillary mucinous neoplasms (IPMNs) are non-invasive cyst-forming pancreatic neoplasms that can progress to aggressive invasive pancreatic ductal adenocarcinoma (PDAC). IPMNs are classified based on the morphological dysplasia of their neoplastic epithelium – low-grade IPMNs have minimal atypia and low risk of malignant transformation, while high-grade IPMNs have severe atypia and are at higher risk for progression to invasive cancer.1 IPMNs are frequently diagnosed incidentally on abdominal imaging, providing an important opportunity to prevent pancreatic cancer.2,3 Guidelines for surveillance or surgical intervention in IPMN patients must balance the opportunity for cancer prevention with the morbidity and even mortality associated with overtreatment of low-risk lesions.4–6 Current decision making relies largely on clinical and radiological features, but these approaches are not adequately sensitive nor are they specific for high-risk IPMNs.5,6 Thus, there is a critical need to better understand the molecular alterations that drive the progression of low-risk IPMNs to those at high-risk for progression to invasive carcinoma, as these represent potential biomarkers for cysts requiring clinical intervention.
In contrast to the trove of genomic data describing invasive pancreatic cancers, the genomes of relatively few IPMNs have been analyzed. Previous comprehensive sequencing of small cohorts of IPMNs mostly focused on advanced lesions and revealed characteristic driver genes,7–9 while targeted analyses in larger, more diverse cohorts have confirmed the prevalence of specific driver gene mutations that correlate with grade of dysplasia or histological subtype.10 These initial studies relied on analysis of a single region from each IPMN, followed by comparison of clinical, pathological, and molecular characteristics across different patients. More recently, multi-region targeted next generation sequencing of IPMNs has revealed a surprising degree of intratumoral genetic heterogeneity, even with respect to well-characterized driver gene mutations in IPMNs, highlighting previously unappreciated genetic complexity in precancerous pancreatic lesions.11–13 However, comprehensive multi-region sequencing has not yet been performed on IPMNs. Such analyses can define the genomic alterations associated with progression in individual lesions, uncover new driver genes, and define unique evolutionary patterns in precancerous lesions.
In this study, we report multi-region whole exome sequencing of distinct low-grade and high-grade regions of 17 human IPMNs without associated invasive carcinoma. The resulting data define evolutionary trajectories in IPMN progression and highlight genetic heterogeneity throughout these lesions. In addition, we identify a novel driver of early IPMN tumorigenesis with a unique evolutionary pattern and validate patterns of mutations in an independent cohort of IPMN cyst fluid samples. Taken together, our results provide several key insights not possible through analysis of advanced cancers, highlighting the importance of direct analysis of precancerous lesions.
METHODS
Clinical Data
Electronic medical records were reviewed to document clinical information such as age, sex, family history, clinical presentation, imaging diagnosis, and outcome. These clinical data are summarized in online supplementary table 1.
Case selection and specimen acquisition
We retrospectively reviewed surgical pancreatectomy specimens from patients diagnosed with IPMN without associated invasive carcinoma between 2007 and 2016. Diagnostic hematoxylin-and-eosin stained slides were reviewed by pancreatic pathologists (WH, LDW) to identify IPMNs with distinct components of both low-grade and high-grade dysplasia and to select 1–3 blocks per case with regions of both grades of dysplasia. Because of recent previous reports of polyclonal origin in IPMNs,12 we carefully selected IPMN cases in which morphological features suggested that the high-grade component arose in association with the co-existing low-grade component. We set the following histologic criteria for the case selection: IPMN with adequate quantities of both low-grade and high-grade components for genomic analysis in which (1) a high-grade component was in direct contact with a low-grade component, OR (2) if the low-grade component was close to but not directly attached to the high-grade component (in most instances growing in a different cystic space), then both components were located within the same formalin-fixed paraffin-embedded (FFPE) block. Of 118 resected specimens of high-grade IPMN retrieved from the database, we found 24 high-grade IPMN cases that met the aforementioned criteria, and morphologically distinct regions of each grade were identified in 32 blocks from these cases and selected for subsequent laser capture microdissection. The histological subtype of each sequenced region was determined by consensus of four pathologists (KF, EDT, RHH, and LDW).
Laser capture microdissection
Twenty to thirty 10um serial tissue sections from selected FFPE tissue blocks were cut onto membrane slides (Carl Zeiss MembranSlide 1.0 PEN; Carl Zeiss, Oberkochen, Germany). Deparaffinization and staining were performed as previously described.14 Two to six morphologically distinct regions were microdissected from each case using laser capture microdissection (LMD7000, Leica, Wetzlar, Germany), resulting in DNA samples of adequate quantity and quality for whole exome sequencing from 76 IPMN regions from 17 cases, as well as a matched normal sample from each case. We did not obtain adequate DNA from the other 7 microdissected cases, which were excluded from further analyses.
Whole exome sequencing and data analysis
Genomic DNA libraries were prepared from the 93 FFPE DNA samples (76 IPMN samples, 17 normal samples) following Illumina’s (Illumina, San Diego, CA) suggested protocol. Human exome capture was performed following a protocol from Agilent SureSelect Human All Exon 50Mb Kit 5.0 (Agilent, Santa Clara, CA). The captured libraries were sequenced with XTEN sequencer (Illumina) with 150bp paired-end reads. Nonsynonymous mutations were called using a well-validated pipeline based on Mutect.15 Details of the pipeline are presented in the online supplementary methods.
Phylogenetic analysis
To extract per-site features (mismatch frequency, insertion frequency, and deletion frequency), BAM alignment files were converted to tab delimited format using jvarkit (https://github.com/lindenb/jvarkit). We inferred phylogenies with Treeomics v1.7.12 based on all mutations that passed the filtering described above. Each phylogeny is rooted at the subject’s normal sample and the leaves represent the distinct regions of the IPMN. Treeomics uses a Bayesian inference model to account for error-prone sequencing and varying neoplastic cell content to infer globally optimal trees using Mixed Integer Linear programming.16 Gene names along lineages indicate an acquired non-synonymous mutation in the corresponding gene – genes are listed twice in the same phylogeny only if multiple distinct somatic mutations in that gene were identified. We display mutations in previously identified PDAC driver genes (significantly mutated genes in TCGA and ICGC PDAC studies),17,18 as well as mutations in genes that were mutated in at least three separate IPMNs and had a nonsynonymous mutation frequency >0.5 mutations per kb gene size in our cohort.
Analysis of IPMN cyst fluid
Pancreatic cyst fluid was collected from resected specimens in the surgical pathology laboratory (n=55) or at the time of endoscopic ultrasound (n=8). Genomic DNA was purified from pancreatic cyst fluid as described previously.19 The hotspot loci in the KLF4 gene were analyzed in IPMN cyst fluid using the Safe Sequencing System (Safe-SeqS),20 which has been described in detail previously.21,22 Details are provided in the online supplementary methods.
RESULTS
Overall approach
In order to dissect the molecular events in low-grade and high-grade components of precancerous pancreatic lesions, we performed multi-region whole exome sequencing of 17 IPMNs containing regions of both low-grade and high-grade dysplasia (online supplementary table 2). We analyzed a total of 76 IPMN exomes, including 49 from low-grade regions and 27 from high-grade regions – the number of exomes analyzed per IPMN ranged from 2 to 6 (online supplementary table 2). In the majority of IPMNs in our cohort (14/17), the low-grade regions showed gastric differentiation, while the high-grade regions in the same IPMN were pancreatobiliary (online supplementary table 2). In addition, our cohort contained two mixed gastric-intestinal type IPMNs, as well as one that showed gastric differentiation in all regions analyzed (online supplementary table 2). In each case, matched normal samples were also analyzed by whole exome sequencing to exclude germline variants and to identify somatic mutations. The average distinct coverage for our IPMN whole exome sequencing was 170x (range 38x–367x) (online supplementary table 3).
From the multi-region whole exome sequencing data, we identified a total of 3,090 nonsynonymous somatic mutations, with a mean of 41 nonsynonymous somatic mutations per analyzed IPMN region, corresponding to a mutation burden of 1.11 nonsynonymous mutations per megabase (Mb) (online supplementary table 4, online supplementary figure 1). Surprisingly, the mean number of somatic mutations did not differ between low-grade and high-grade regions – we identified a mean of 41 nonsynonymous somatic mutations (range 20–103) in low-grade regions compared to a mean of 41 (range 22–76) in high-grade regions (p=0.55, two-tailed Mann-Whitney U test) (figure 1A). These correspond to a mutation burden of 1.10 nonsynonymous mutations per Mb in low-grade regions and 1.12 non-synonymous mutations per Mb in high-grade regions. A mean of 10 nonsynonymous somatic mutations were shared among all samples analyzed from a given IPMN, while a mean of 26 were unique/private to low-grade regions and a mean of 24 were unique/private to high-grade regions. We then compared the neoplastic cell fraction (NCF) of mutations in low-grade and high-grade regions (figure 1B). There was no significant difference between low-grade and high-grade regions in the NCF of mutations shared in all samples of a given IPMN (p=0.51, two-tailed Mann-Whitney U test). In contrast, we identified a significant difference between low-grade and high-grade regions in the NCF of unshared mutations, with mutations in low-grade regions having a significantly lower NCF than those in high-grade regions (p<0.0001, two-tailed Mann-Whitney U test). Regions of high-grade dysplasia had a larger proportion of unshared mutations with a NCF approaching 1, suggestive of clonal mutations shared in all analyzed cells (figure 1B). Still, mutation signatures were similar among all samples analyzed, with an enrichment for C-to-T transitions (figure 1C).
Figure 1: Whole exome sequencing of multi-region low-grade (LG) and high-grade (HG) IPMN samples.
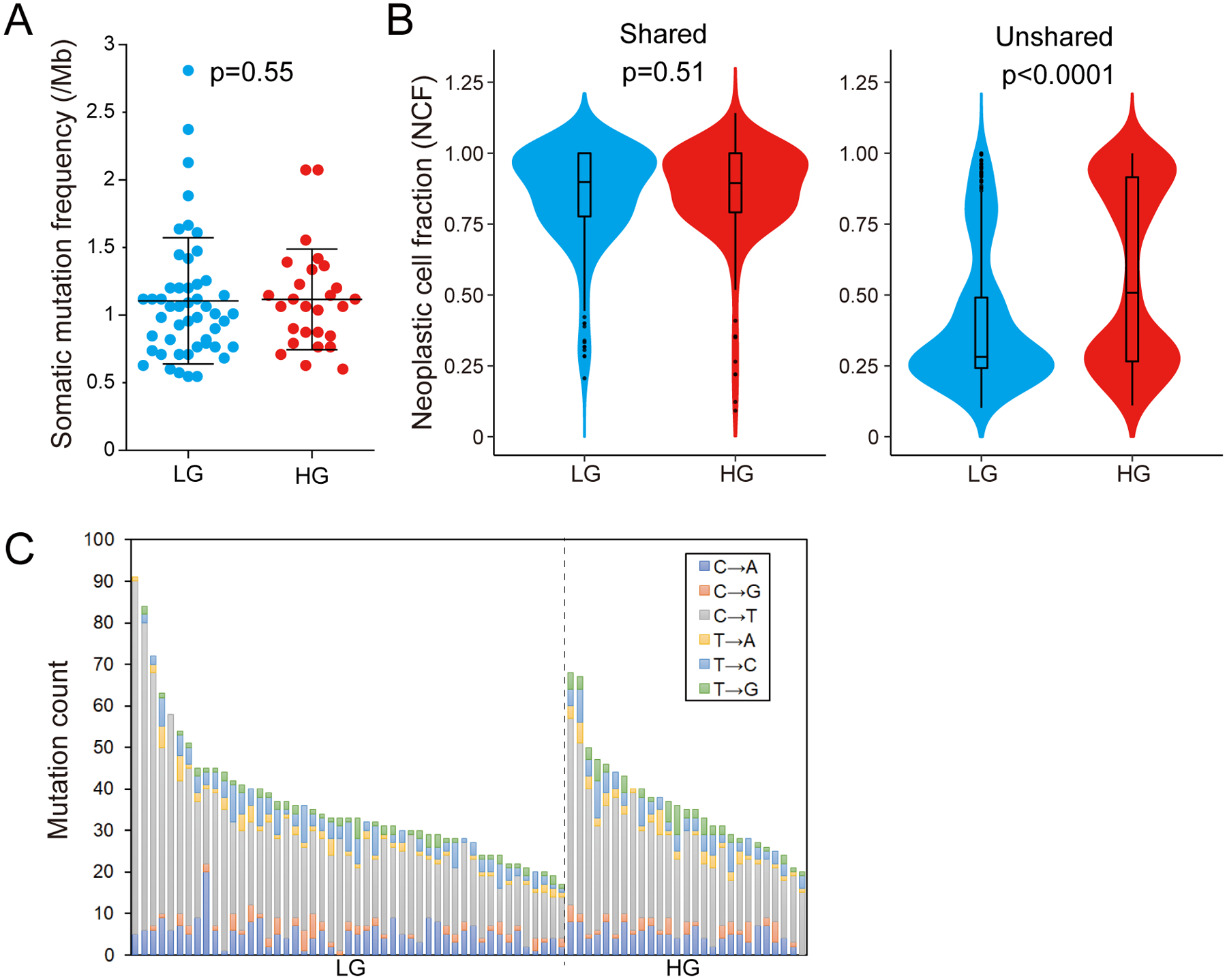
(A) Comparison of the tumor mutation burden per megabase (TMB/Mb) between LG (n=49) and HG (n=27) IPMN regions. The lines and error bars indicate mean ± 1 standard deviation. (B) Violin plots showing the neoplastic cell fraction (NCF) of all mutations detected in LG (n=49) and HG (n=27) regions. The NCFs were calculated separately for shared mutations among all regions (left panel) and unshared mutations (right panel). (C) Proportion of base changes observed in each IPMN sample. Samples are organized by grade of dysplasia and descending total number of alterations.
Copy number analyses utilizing the whole exome sequencing data revealed scattered alterations without striking differences between low-grade and high-grade components in most IPMNs (online supplementary figure 2, online supplementary table 5). However, a minor subset of cases showed increased copy number alterations in high-grade regions (for example, IP2, IP9, IP29), suggesting accumulation of copy number alterations during neoplastic progression in some IPMNs (online supplementary figure 2).
Driver genes of IPMN tumorigenesis
Through multi-region whole exome sequencing, we confirmed the high prevalence of mutations in some previously identified pancreatic driver genes in IPMN cases, including GNAS (15/17 cases; 88%), KRAS (14/17 cases; 82%), RNF43 (10/17 cases; 59%), and CDKN2A (5/17 cases; 29%) (figure 2A, online supplementary figure 3). Intriguingly, TP53 mutations were uncommon in our cohort (2/17 cases; 12%), and we identified no mutations in SMAD4 or TGFBR2. We also identified hotspot mutations in CTNNB1 (3/17 cases; 18%) and inactivating mutations in APC (3/17 cases; 18%), demonstrating WNT signaling alterations in a subset of IPMNs as has been previously reported.10 In addition, nonsynonymous mutations in RBM10 were identified in 24% (4/17) of IPMNs in our study – mutations in this gene have been previously reported in both IPMNs and PDACs.13,23
Figure 2: Driver mutations in low-grade (LG) and high-grade (HG) IPMN.
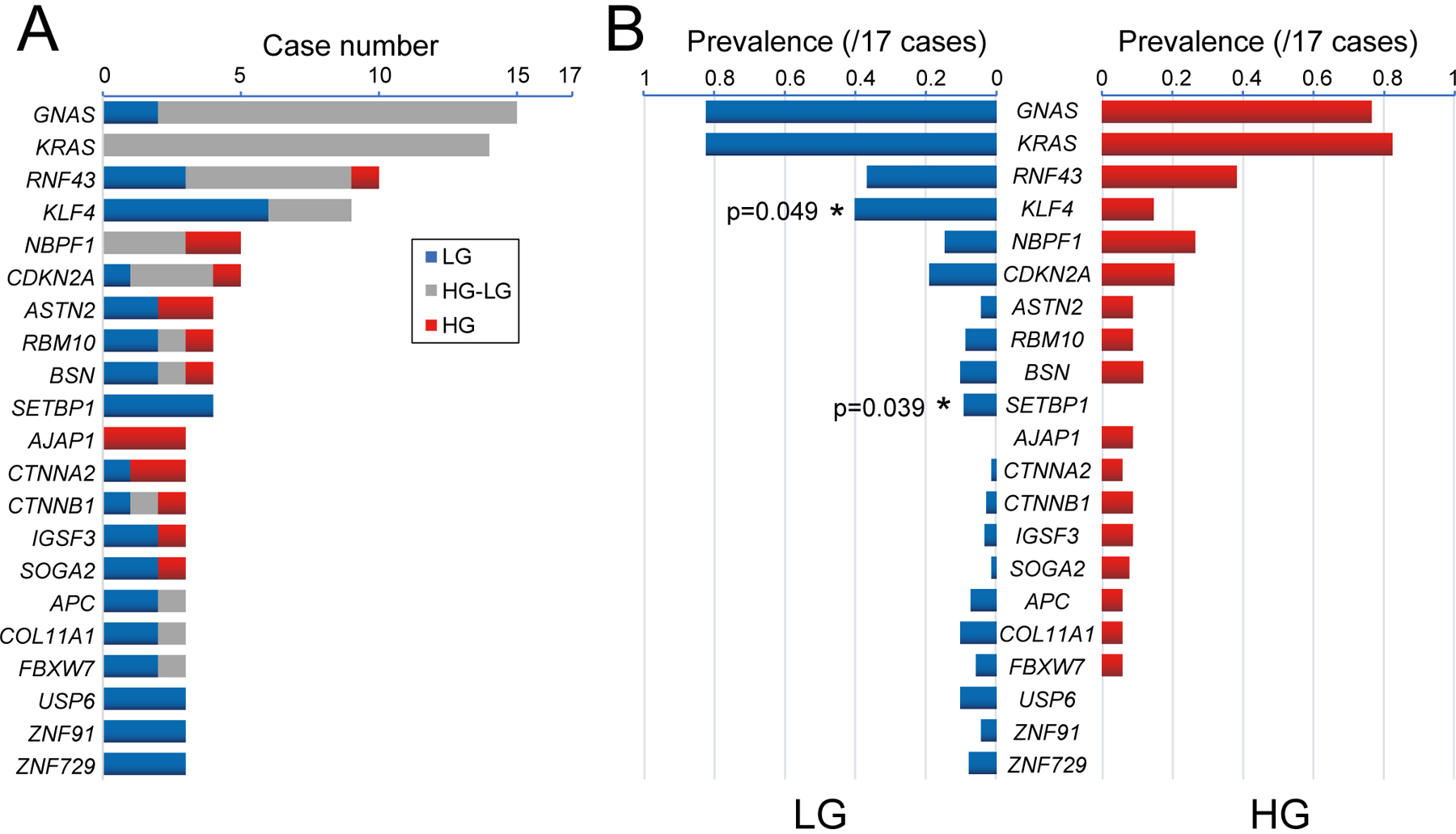
(A) Somatic nonsynonymous mutations in the most frequently mutated genes are categorized as shared between LG and HG IPMN (gray), limited to LG (blue), or limited to HG (red). Genes with mutations in >3 IPMNs and >0.5 mutations per kb gene size are included. (B) Comparison of prevalence nonsynonymous mutations between LG and HG samples. Genes showing significantly different mutation frequencies between LG and HG are indicated by asterisks (two-tailed Mann-Whitney U test).
In addition to confirming the prevalence of mutations in previously characterized driver genes, our study also identified novel drivers of pancreatic tumorigenesis these IPMNs. The most striking of these new drivers was KLF4¸ which encodes a member of the Kruppel family of transcription factors. We identified somatic mutations in one of two hotspot codons (amino acids K409 and S411) of KLF4 in 53% (9/17) of IPMNs in our study (figure 2A–2B). Both hotspots are located in the highly conserved C2H2 zinc finger domains in KLF4 (figure 3A–3B). A total four different amino acid substitutions (K409Q, K409E, S411Y and S411F) were detected in these two hotspots, and two IPMNs in our cohort had two different KLF4 mutations (K409E and S411F in IP5; K409Q and S411Y in IP7) (2/17 cases; 12%). We analyzed the four different KLF4 hotspot mutations using Cancer-specific High-throughput Annotation of Somatic Mutations plus (CHASMplus).24 All four different KLF4 hotspot mutations had high CHASMplus PDAC scores (K409Q, p=0.042; K409E, p=0.041; S411Y, p=0.038; S411F, p=0.038), suggesting that these mutations are likely to be drivers of IPMN tumorigenesis. None of these amino acid substitutions were detected in the Genome Aggregation Database (gnomAD) of germline alterations (>250,000 alleles). Only KLF4 K409Q has been previously reported in meningiomas and in three cases in previous IPMN analyses, but our study represents the first report of four different KLF4 hotspot mutations at this high prevalence in epithelial neoplasms.8,13,25,26
Figure 3: Characterization of recurrent hotspot mutations in KLF4.
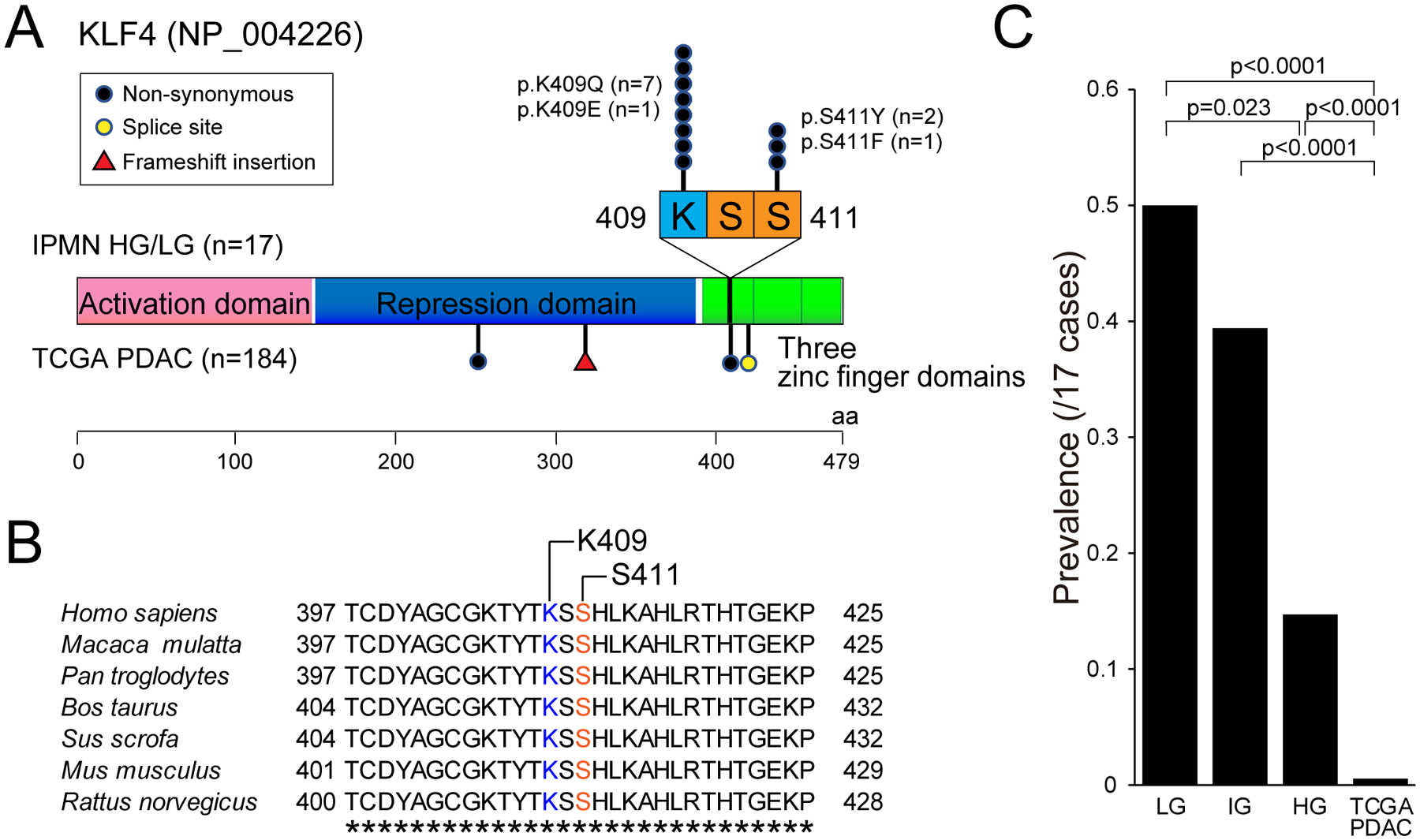
(A) Gene schematic showing mutations identified in KLF4 in our IPMN cohort (n=17 cases, 76 regions) and TCGA PDAC cohort (n=184 cases). All mutations in our IPMN cohort are located in two hotspots (K409 and S411) in first C2H2 zinc finger domain. (B) Amino acid sequences of different species around the hotspot mutations of KLF4. Completely conserved amino acids across all species are indicated by asterisks. (C) Comparison of KLF4 mutation prevalence among “low-grade”, “intermediate-grade” and “high-grade” IPMN in our cohort and TCGA PDAC. The histologic grade in this panel is based on the previous three-tiered grading system for IPMN. p-values were calculated based on two-tailed Mann-Whitney U test.
In addition to these frequent mutations in KLF4, we report nonsynonymous mutations in FBXW7 and HUWE1, each in 18% (3/17) of the IPMNs studied – these genes encode the components of an E3 ubiquitin ligase and are previously characterized tumor suppressor gene in other tumor types (figure 2A–2B, online supplementary figure 3).27,28 We also report mutations in SETBP1 in 24% (4/17) of IPMNs; mutations in this gene have been reported in several other tumor types but have not been previously highlighted in pancreatic neoplasms.29,30 Other genes with prevalent somatic mutations in our cohort include MUC16 (6/17 cases; 35%), TTN (5/17 cases; 29%), and NBPF1 (5/17 cases; 29%). However, the large size of MUC16 and TTN suggest that many (if not all) of these mutations may be passengers.
The comprehensive genomic analysis of multiple regions per IPMN, including regions of both low-grade and high-grade dysplasia, provides a unique opportunity to assess the timing of mutations in specific driver genes. In IPMNs with KRAS mutations, at least one mutation in this gene was shared in both low-grade and high-grade components (figure 2A–2B). Similarly, mutations in GNAS were almost always shared between low-grade and high-grade components. Most other mutated genes had a mixture of mutational patterns, including mutations limited to both low-grade and high-grade components. Of note, TP53 mutations were consistently limited to high-grade components, as were mutations in the adherens junction protein AJAP1 (figure 2A–2B).
Mutations in KLF4 had a strikingly different pattern, with mutations frequently limited to the low-grade component of the IPMNs – KLF4 hotspot mutations were limited to the low-grade component in 6 of 9 mutant cases and were shared between the low-grade and high-grade components of 3 of 9 (example in figure 4). In our cohort, the prevalence of KLF4 mutations in low-grade and high-grade components was significantly different (figure 2B; 40% vs. 15%, p=0.049, two-tailed Mann-Whitney U test). There were no KLF4 mutations that were limited to the high-grade components. When the previous three-tiered histologic grading system for IPMN was applied, with low-grade IPMN divided into “low-grade” and “intermediate-grade” dysplasia, the prevalence of KLF4 mutations was highest in “low-grade” (50%) compared to “intermediate-grade” (39%) or “high-grade” (15%; p=0.023, two-tailed Mann-Whitney U test vs. “low-grade”) (figure 3C). In addition, the prevalence of hotspot KLF4 mutations in The Cancer Genome Atlas (TCGA) analysis of invasive PDAC (0.5%, 1/184 cases) was much lower than the prevalence in our cohort of precancerous lesions.17
Figure 4: Distribution of somatic mutations in low-grade (LG) and high-grade (HG) regions of IP5.
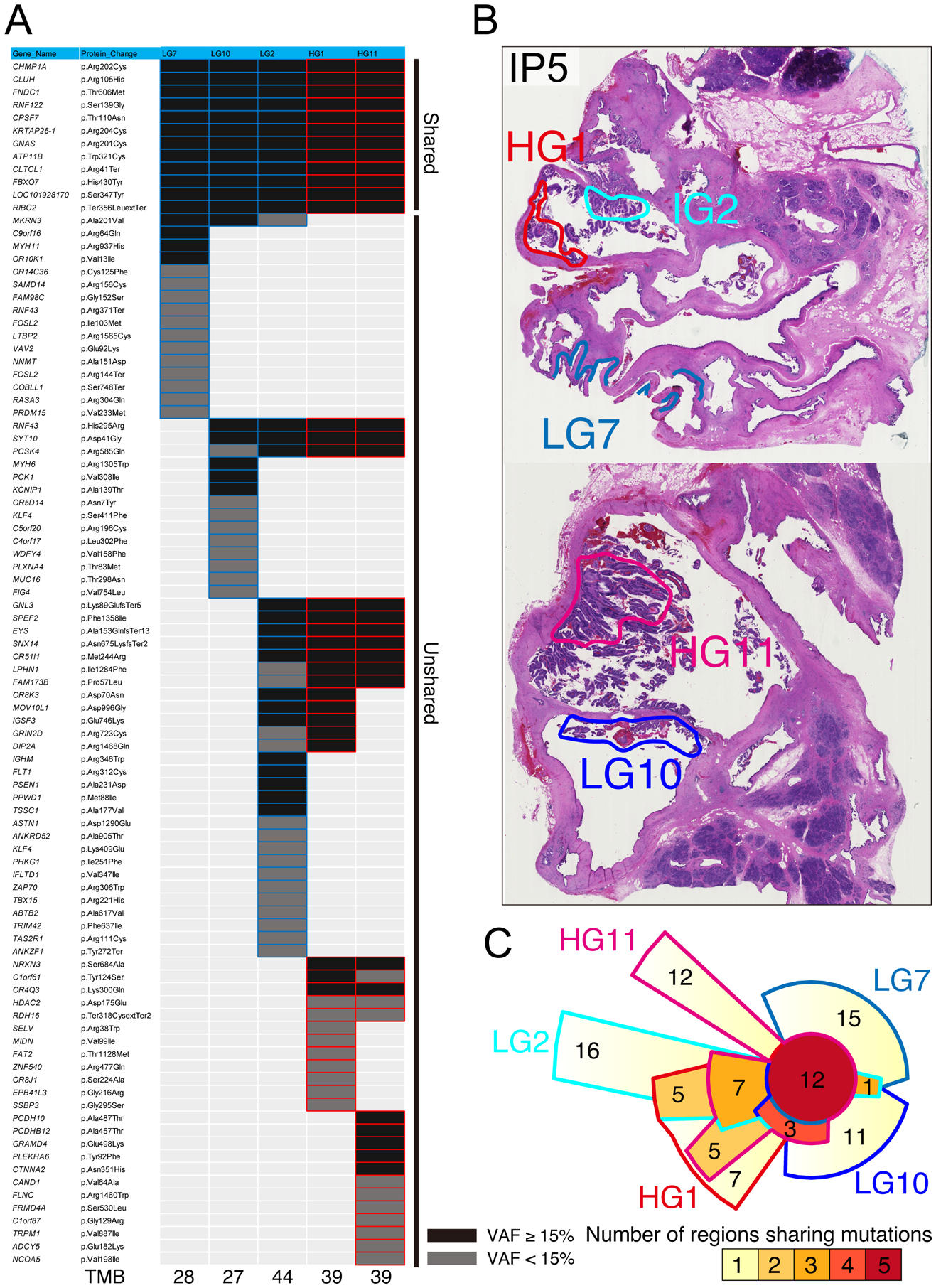
(A) Heatmap depicting the distribution of nonsynonymous somatic mutations among five different regions of IP5 (three LG and two HG). Black boxes indicate mutations with VAF >15%, and grey boxes indicate VAF of 5–15%. (B) Representative images of neoplastic tissue of IP5 stained by hematoxylin and eosin. The colored circles indicate the microdissected regions for sequencing analysis. (C) Chow–Ruskey plot of shared and unique somatic mutations among five different regions of IP5.
Our multi-region sequencing data also allows examination of genetic heterogeneity in IPMNs, confirming previous observations about precancerous pancreatic neoplasia.12 In IP24, two of the analyzed regions shared no somatic mutations with the other regions, suggesting that the analyzed tissue block contained multiple independent precancerous neoplasms. In this IPMN, the independent neoplasms demonstrably involved the same ductal space (figure 5). These observations provide further support for the hypothesis that at least a subset of IPMNs have polyclonal origin and are comprised of multiple independently arising clones that share no somatic mutations.12 We also identified a distinct pattern of mutation in both RNF43 and KLF4 in which multiple mutations in the same driver gene were present in different regions of the same IPMN, suggesting convergent evolution with respect to mutations in some driver genes (figure 4). This pattern has been previously reported in RNF43 but not KLF4, highlighting unique selective pressures on mutations in these two key driver genes.12,31
Figure 5: Distribution of somatic mutations in low-grade (LG) and high-grade (HG) regions of IP24.
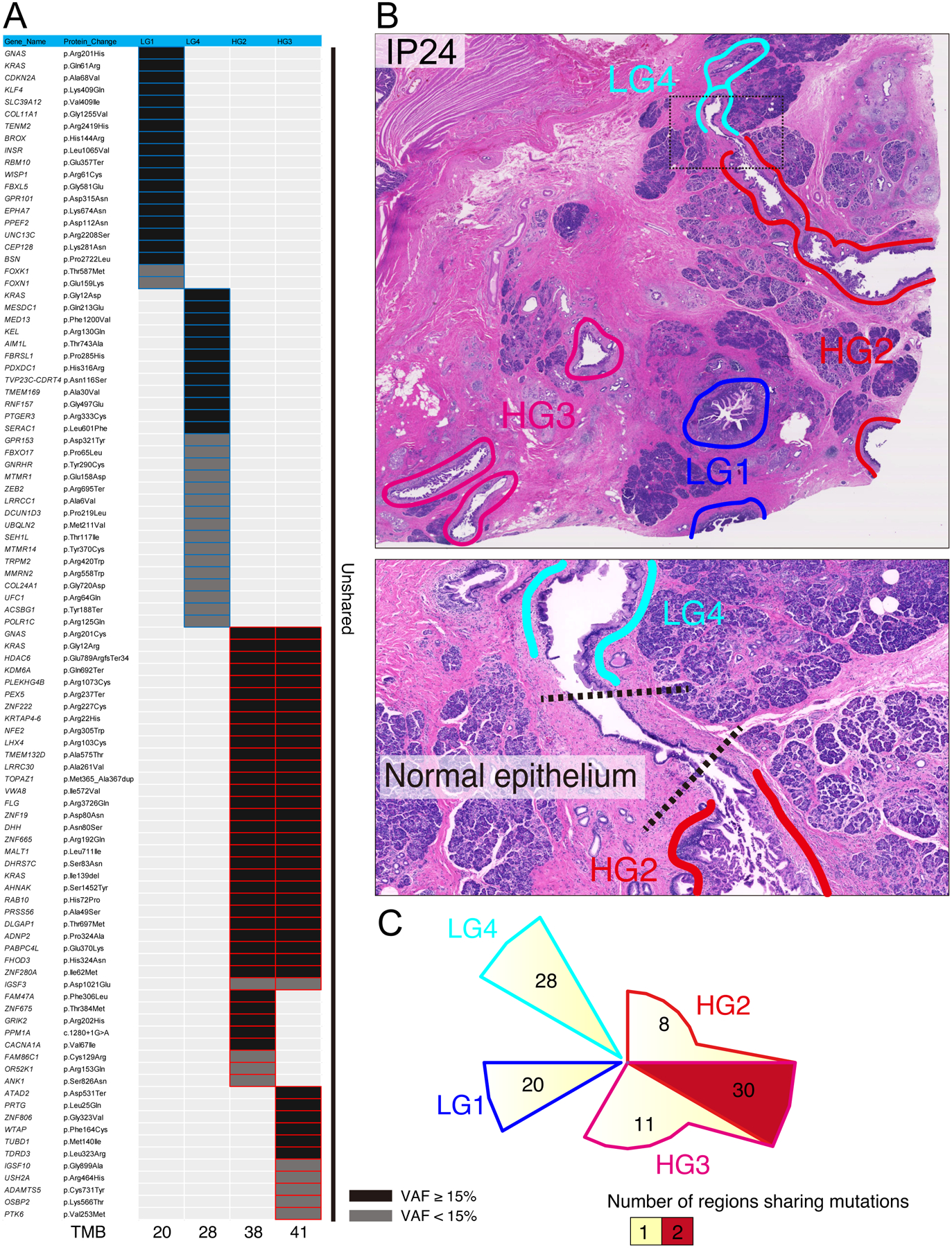
(A) Heatmap depicting the distribution of nonsynonymous somatic mutations among four different regions of IP24 (two LG and two HG). Black boxes indicate mutations with VAF >15%, and grey boxes indicate VAF of 5–15%. (B) Representative images of neoplastic tissue of IP24 stained by hematoxylin and eosin. The colored circles indicate the microdissected regions for sequencing analysis. The bottom image shows the enlarged view of the black dotted circle in upper image. (C) Chow–Ruskey plot of shared and unique somatic mutations among four different regions of IP24.
Evolutionary Analyses of Non-invasive IPMNs
In order to analyze the evolutionary relationships of IPMN samples in more detail, we reconstructed lesional phylogenies using Treeomics, a computational approach specifically designed for noisy next generation sequencing data from tumor samples.16 Treeomics calculates the probability that a mutation is present or absent in a particular sample based on the number of mutant and reference reads, with hyperparameters for the probability calculation depending on the estimated sample purity as well as other variables. Mutations are placed on the root of the tree by Treeomics only if that mutation was present with a high probability in all samples. Multiple important insights were evident from this analysis. First, our evolutionary analysis highlighted that in multiple samples (e.g. IP7, IP8, IP31), high-grade dysplasia arose without unique mutations in pancreatic driver genes, while in others (e.g. IP2, IP9) high-grade-specific mutations in driver genes such as TP53 were identified (online supplementary figure 4). Second, in some samples with multiple distinct high-grade regions (e.g. IP7, IP20), we demonstrate that such regions are more closely related to low-grade regions than to each other, suggesting that the transition to high-grade dysplasia can occur multiple times in the same IPMN (figure 6, online supplementary figure 4). However, this pattern is not universal, as in other IPMNs (e.g. IP2) the high-grade components have a recent common ancestor (online supplementary figure 4). Our Treeomics analysis also confirmed independent genetic origin of different regions of IP24 and highlighted the parallel evolution of multiple distinct RNF43 mutations in discrete subclones in IP20 (figure 6, online supplementary figure 4).
Figure 6: Representative IPMN phylogenies constructed using Treeomics.
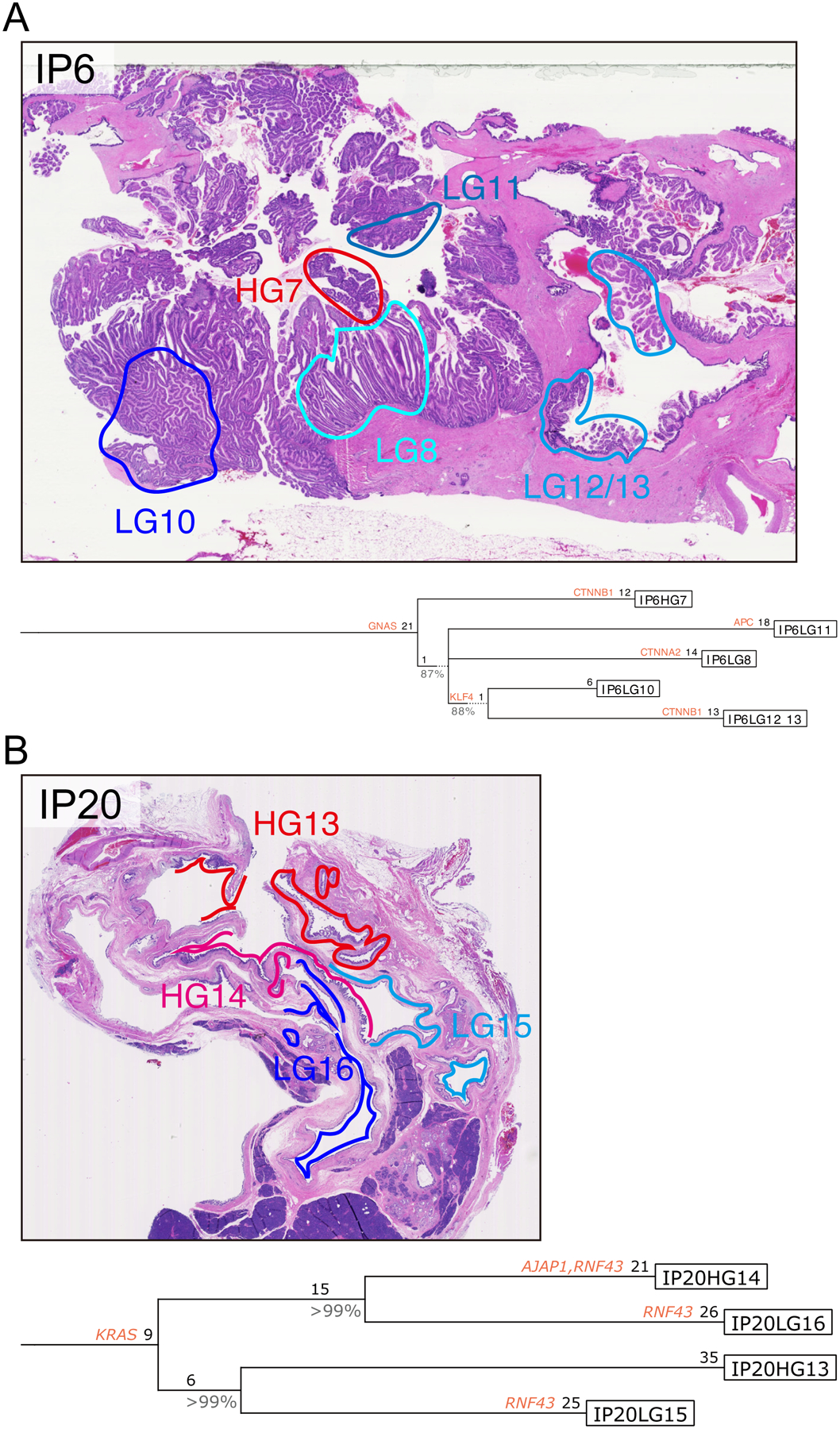
Treeomics generated phylogenetic trees from all nonsynonymous mutations identified in each IPMN region (A, IP6; B, IP20). Potential driver gene mutations (including those identified in previous pancreatic cancer genomics studies, as well as those mutated in >3 IPMNs and >0.5 mutations per kb gene size in the current study) are indicated by their gene name on the lineage in which they occur. Numbers indicate the number of nonsynonymous somatic mutations occurring in each trunk or branch. Representative images of neoplastic tissue stained by hematoxylin and eosin are presented for both cases, with colored circles indicating the microdissected regions for sequencing analysis.
We also assessed quantitative features of the Treeomics phylogenies in detail. We observed varying lengths of the “trunk” of the phylogenetic tree, ranging from 0 mutations shared in all samples to 27 (online supplementary figure 4). The mean was ~12 truncal mutations, but there was a broad range - seven IPMNs had <10 truncal mutation while two IPMNs had >20. This highlights distinct evolutionary features in different IPMNs, underscoring that the selective forces governing neoplastic evolution may be variable between patients. Next, we compared the genetic relatedness of low-grade and high-grade regions using “genetic distance”, defined as the total number of non-shared somatic mutations between two samples.32 High-grade/high-grade sample comparisons had a lower mean genetic distance (36.1), when compared to low-grade/high-grade sample comparisons (44.2) and low-grade/low-grade sample comparisons (45.2). However, these results did not reach statistical significance (p=0.21 for LG/LG vs HG/HG, p=0.26 for LG/HG vs HG/HG, two-tailed Mann-Whitney U test).
Detection of KLF4 mutations in IPMN cyst fluid
In order to determine the prevalence of KLF4 mutations in an independent cohort, we employed next generation sequencing analysis of human IPMN cyst fluid samples using the Safe Sequencing System (Safe-SeqS), a method designed to reduce sequencing errors and detect low-frequency mutations.20 Clinical and pathological features of the analyzed cyst fluid samples are presented in the online supplementary table 6. Our Safe-SeqS assay was designed to assess the previously identified hotspots in KLF4 at codons 409 and 411. The cyst fluid samples were from 63 IPMNs, including 26 low-grade and 37 high-grade IPMNs, with all diagnoses confirmed on pathological review of resected specimens. In this cohort of 63 cyst fluid samples, we identified KLF4 mutations in 19 samples (30%), including 11 samples with 1 KLF4 mutation, 7 samples with two distinct KLF4 mutations, and 1 sample with three distinct KLF4 mutations (table 1). The prevalence of multiple KLF4 mutations in our cyst fluid samples (8 of 63, 13%) is similar to that identified in whole exome sequencing (2 of 17, 12%). Hotspot mutations in KLF4 were identified in 12 of 26 (46%) low-grade IPMN cyst fluid samples, compared to 7 of 37 (19%) high-grade IPMN cyst fluid samples using Safe-SeqS (p=0.027, two-tailed Fisher’s exact test). These results confirm the high prevalence of KLF4 mutations in IPMNs as well as the enrichment of these in low-grade lesions. In addition, we identified significantly different KLF4 mutation prevalence based on histological subtype, with mutations in 10 of 22 (45%) gastric-type IPMNs but only 3 of 22 (14%) intestinal-type IPMNs (p=0.045, two-tailed Fisher’s exact test) (online supplementary table 7). The detectability of KLF4 mutations in cyst fluid samples highlights their potential utility in preoperative risk stratification of IPMNs.
Table 1:
KLF4 Mutations Identified in Cyst Fluid Samples by Safe-SeqS.
| 1st KLF4 mutation | 2nd KLF4 mutation | 3rd KLF4 mutation | p-value+ | p-value+ | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| position | MAF* | position | MAF* | position | MAF* | ||||||
| Cyst 229 | 110249341 | 0.006 | 110249348 | 0.005 | 0.46 | 0.027 | 0.19 | 0.26 | |||
| Cyst 234 | 110249348 | 0.011 | |||||||||
| Cyst 244 | 110249348 | 0.346 | |||||||||
| Cyst 318 | 110249348 | 0.027 | |||||||||
| Cyst 792 | 110249348 | 0.002 | |||||||||
| Cyst 839 | 110249348 | 0.149 | 110249341 | 0.008 | |||||||
| Cyst 840 | 110249348 | 0.014 | |||||||||
| Cyst 867 | 110249341 | 0.087 | 110249348 | 0.039 | 110249348 | 0.007 | |||||
| Cyst 903 | 110249348 | 0.013 | |||||||||
| Cyst 1307 | 110249348 | 0.213 | 110249341 | 0.079 | |||||||
| Cyst 1308 | 110249341 | 0.375 | |||||||||
| Cyst 1315 | 110249341 | 0.158 | 110249348 | 0.064 | |||||||
| Cyst 358 | 110249348 | 0.288 | 0.19 | 0.08 | |||||||
| Cyst 646 | 110249348 | 0.009 | |||||||||
| Cyst 674 | 110249348 | 0.180 | |||||||||
| Cyst 842 | 110249348 | 0.274 | 110249341 | 0.167 | |||||||
| Cyst 843 | 110249348 | 0.018 | 110249348 | 0.003 | |||||||
| Cyst 837 | 110249341 | 0.124 | |||||||||
| Cyst 1257 | 110249348 | 0.059 | 110249341 | 0.011 | |||||||
LG, low-grade, HG, high-grade;
MAF: Mutant allele frequency, reported as the mean of duplicate assays;
p-value for comparison of prevalence in LG and HG using two-tailed Fisher’s exact test
DISCUSSION
Using multi-region whole exome sequencing of IPMNs with both low-grade and high-grade components, we identified prevalent mutations in a previously unappreciated driver of pancreatic neoplasia: KLF4. Prevalent mutations at an oncogenic hotspot in KLF4 have been previously reported in meningiomas and have been implicated as a universal genetic feature of the secretory subtype of meningioma.25,26 In the pancreas, loss of heterozygosity at the KLF4 locus has been reported in PDAC, but frequent somatic mutations in this gene have not been previously reported.33 Somatic mutations at hotspot positions in KLF4 have been reported in three cases in previous whole exome sequencing studies of IPMNs, but mutations at the prevalence in our study (with somatic mutations in >50% of analyzed IPMNs) have not been previously reported.8,13 This is likely due to the enrichment of KLF4 mutations in regions of low-grade dysplasia, as previous comprehensive sequencing studies of IPMNs have focused on high-grade IPMNs or those with associated invasive carcinomas. We identified a total of 19 IPMNs previously analyzed by whole exome sequencing in the literature, the vast majority from high-grade IPMNs.8,9,11–13 KLF4 mutations were identified in three samples from two different studies, indicating a prevalence of 16% which is similar to the prevalence in high-grade IPMNs in our study. However, because KLF4 mutations occurred in one or two samples in previous studies, they could not be separated from the much larger number of passenger mutations in these studies.
Several studies have assessed the expression levels of KLF4 in normal and neoplastic pancreas.33–35 In the normal pancreas, KLF4 was found to be localized to the nuclei of pancreatic ductal epithelial cells.33 Intriguingly, loss of KLF4 expression has been reported in a sizable proportion (>85%) of PDACs,33 whereas increased expression was observed in human and mouse acinar-to-ductal metaplasia (ADM) and pancreatic intraepithelial neoplasia (PanIN) lesions.34 We are aware of only one study to date describing KLF4 expression in IPMN.35 This study demonstrated that KLF4 expression is restricted to a small proportion of highly mucinous cells in both human and mouse IPMN specimens, potentially representing regions of low-grade dysplasia.35 Expanded analysis of KLF4 expression in larger IPMN cohorts and correlation with KLF4 mutation status are warranted to clarify the relationship between KLF4 mutation, expression, and role in tumorigenesis.
KLF4, also known as gut-enriched KLF (GKLF), is an important member of Kruppel-like transcription factor family with multiple putative functions.36 KLF4 was initially identified as a key regulator of cell fate decisions, such as cell proliferation, differentiation, and apoptosis. The identification of KLF4 as one of the four “Yamanaka factors” that can reprogram differentiated somatic cells into pluripotent stem cells (OCT3/4, SOX2, KLF4, and MYC), as well as its essential role in the maintenance of genome stability, further substantiate its role in cell fate determination.37 KLF4 has been reported to have tumor suppressive functions in several tumor types, and both experimental and clinical evidence has shown that the loss of KLF4 protein expression can cause altered cell proliferation, differentiation, and precancerous changes in adult digestive organs.36 These processes are mediated by multiple oncogenic pathways, including Wnt/β-catenin, TGF-β1, and p21WAF1/Cip1 signaling.36 Recent studies have also indicated that KLF4 is a negative regulator of epithelial-to-mesenchymal transition (EMT), revealing several critical genes as a direct transcriptional targets of KLF4, including CDH1, CDH2, VIM, CTNNB1, VEGF, and MAPK8.38
In the pancreas, the functional role of KLF4 varies at different points in tumorigenesis, with multiple studies suggesting pro-tumorigenic function in pancreatic tumor initiation and tumor-suppressive function in advanced PDAC.34 Studies of KLF4 in genetically engineered mouse models of pancreatic cancer have demonstrated overexpression of this gene in early pancreatic neoplasia, while experimental overexpression in pancreatic cancer cell lines led to cell cycle arrest and growth inhibition.35,36 These genetically engineered mouse models suggested that dysregulation of the KLF4 signaling pathway promotes PDAC progression and metastasis, but paradoxically, KLF4 ablation attenuates the formation of ADM and PanIN after pancreatic injury in the setting of mutant KRAS. Together with our data showing enrichment of KLF4 mutations in low-grade regions, this raises the intriguing hypothesis that KLF4 mutations are selected early in pancreatic tumorigenesis but are then selected against as lesions progress. Still, although the prevalence and pattern of KLF4 mutations in our study provide strong evidence that KLF4 is an oncogene in neoplastic pancreatic cysts, our sequencing data cannot provide mechanistic insights into the selective pressures in IPMNs. As such, we cannot further evaluate the hypothesis of temporal changes in selective forces with the current data. The functional impacts of hotspot KLF4 mutations in pancreatic tumorigenesis remain to be determined and represent a critical direction of future investigation, and further experimental data in model systems will be required to support or refute this hypothesis.
The distinct mutation patterns of driver genes suggest their role in specific stages of pancreatic tumorigenesis. Mutations in the hotspots of the initiating oncogenes KRAS and GNAS were most often shared among all samples from a given IPMN, suggesting that these mutations occur early in tumorigenesis. In contrast to KRAS and GNAS, TP53 mutations were not common, occurring in only 2 IPMNs and consistently limited to high-grade components. No mutations were identified in our IPMN cohort in SMAD4, despite frequent mutations in this gene in PDAC and IPMN-associated invasive carcinomas. These findings are consistent with previous studies suggesting that mutations in these genes occur very late in pancreatic tumorigenesis, and in particular that SMAD4 mutations are typically limited to invasive carcinoma.10,14,39,40
As discussed above, in contrast to these later drivers, mutations in KLF4 were uniquely enriched in regions of low-grade dysplasia, suggesting distinct selective forces on these mutations. The pattern of KLF4 mutations suggests that, due to the complexities of clonal evolution, not all driver mutations in early pancreatic tumorigenesis can be detected by studying advanced cancers, highlighting the importance of direct analysis of precursor lesions. It is also important to note that many IPMNs lacked a high-grade specific driver gene, and copy number alterations were largely shared between matched low-grade and high-grade components, raising the possibility of a non-genetic driver of progression to high-grade dysplasia in a subset of cases. Overall, our data suggest that there is not a single universal genetic pathway to high-grade dysplasia.
In total, 76 multi-region samples from 17 IPMN cases were analyzed by whole-exome sequencing in our study. In the majority of IPMNs (16/17 cases, 94%), low-grade and high-grade IPMNs shared common somatic mutations, suggesting evolution from a common ancestor. These observations raise the fundamental question of whether high-grade IPMN arises from low-grade IPMN. Because we analyzed a single resected IPMN specimen from each patient and thus observed each IPMN at a single point in time, we cannot directly observe this common ancestor of low-grade and high-grade IPMN. However, the molecular alterations predicted in the common ancestor by Treeomics are most consistent with those previously reported in low-grade IPMN.10 Thus, our data suggest that the common ancestor of the low-grade and high-grade IPMN regions we sequenced was low-grade IPMN, supporting the idea that high-grade IPMN typically arises from low-grade IPMN. Intriguingly, in addition to the shared mutations, each region we sequenced also independently accumulated a set of private mutations, even when the high-grade regions were in direct contact with low-grade regions within the same pancreatic duct (e.g., IP6, IP7). These results demonstrate independent evolution of both low-grade and high-grade regions after divergence from the common ancestor, suggesting that continued selection shapes the genetic alterations in both IPMN grades. However, the higher proportion of mutations in high-grade regions with a NCF near 1 suggests a clonal selection event that is unique to the development of high-grade dysplasia. We also identified one notable exception (IP24; 1/17 cases, 6%), in which the low-grade and high-grade components shared no somatic mutations. In this case, low-grade and high-grade regions arose as independent clones in the same duct.
Pancreatic cysts are frequently identified incidentally on abdominal imaging, creating the unique clinical problem of surveillance that balances cancer prevention with overtreatment.41 To date, several different sets of guidelines have been released for the management of pancreatic cystic neoplasms, including those that can progress to invasive carcinoma, such as IPMN and MCN.5,6,42 In these guidelines, clinical decision-making relies largely on radiographic and clinical features, augmented by biochemical and cytologic analyses of cyst content. However, currently available diagnostic tools and algorithms are still imperfect. There is a discrepancy between the pre- and post-operative diagnosis in >30% of the pancreatic lesions, and 25% of patients who undergo surgery have pancreatic cysts without malignant potential, highlighting a need for improved approaches to preoperative diagnosis.43–45 Recent studies have identified a combination of molecular and clinical features that classified cyst type with >90% sensitivity and >90% specificity.19,46 However, separating low-grade and high-grade precancerous cysts remains a challenging even with these advanced approaches. The results of our genomic analysis of IPMN progression may improve this discrimination and thus contribute to multidisciplinary assessment and management of IPMN.
More specifically, our results provide further insights into the use of molecular alterations in cyst fluid for IPMN risk stratification. SMAD4 mutations were absent in our cohort of non-invasive IPMNs, and TP53 mutations were uncommon and limited to areas of high-grade dysplasia, suggesting that alterations in these genes are specific markers for IPMNs at high risk of malignant progression or associated invasive carcinomas.19,47 In contrast, KLF4 mutations were frequently limited to regions of low-grade dysplasia in our IPMNs analyzed by multi-region sequencing of tissue samples, and mutations in this gene were significantly more prevalent in cyst fluid samples from low-grade IPMNs. These data suggest that KLF4 mutations may add to the discriminatory power of molecular cyst fluid analysis, though it is important to note that these mutations were also present in a smaller proportion of high-grade IPMNs. Thus, KLF4 mutations are not an entirely specific marker of low-grade dysplasia and will likely need to be interpreted in combination with other clinical and molecular features to accurately assess the risk of IPMN progression. Furthermore, because a single IPMN can contain both low-grade and high-grade components, the finding of a mutation suggestive of low-grade dysplasia does not rule out a higher grade component elsewhere in the IPMN. While the difference in prevalence of KLF4 mutations in low-grade and high-grade IPMNs was statistically significant, these observations suggest significant challenges to their clinical utility, and thus the clinical impact of our study should be interpreted with caution. Assessment of KLF4 mutations in larger cohorts will be required to clarify the value added by these mutations to existing risk stratification approaches. In addition, assessment of KLF4 mutation prevalence in other pancreatic precancerous lesions and cyst types will be required to interpret the implications of these mutations in biospecimens.
Like all cancer genomics studies, our study has limitations. We analyzed a relatively small number of IPMNs. Still, this represents the largest cohort to date of IPMNs without associated cancer analyzed by whole exome sequencing, and we analyzed 76 IPMN exomes in total, a large increase in sample size from previous studies. Moreover, although we performed multi-region whole exome sequencing, we analyzed only a small proportion of the neoplastic epithelium in each case by microdissecting pathologically defined regions from 1–3 tissue blocks. The design of such multi-region sequencing studies represents a balance between the comprehensiveness of lesional sampling versus genomic analysis. In this study, we chose to comprehensively analyze the genome of the analyzed regions but not sample the whole IPMN. Of note, we employed the opposite balance in a recently published study in which we analyzed all available tissue from a cohort of IPMNs by targeted next generation sequencing of a small driver gene panel.12 The latter approach allows comprehensive assessment of genetic heterogeneity with respect to known drivers but does not allow identification of new driver genes, which is a key finding in the current study. Taken together, the two approaches provide complementary insights into pancreatic tumorigenesis via the IPMN pathway.
It is also important to note that our cohort size did not permit us to perform computational methods of driver gene assessment such as MutSigCV.48 As such, the importance of infrequently mutated genes in our study should be interpreted with caution, as the mutation prevalences have not been corrected for important confounders such as gene size, nucleotide context, and replication timing. Still, we identify KLF4 mutations at oncogenic hotspots in >50% of analyzed IPMNs, and these specific mutations are predicted to be drivers based on CHASMplus analysis. Thus, methods such as MutSigCV are not required to confirm the driver gene status of KLF4. Another important caveat in our study is that we analyzed exomes from FFPE tissue, which has been documented to contain more artifacts than sequencing data from fresh or frozen tissue.49 Although this is one possible explanation for the observation that we identified a similar number of mutations in IPMN samples to that previously identified in PDAC, an alternative explanation is also possible. Because we performed laser capture microdissection and analyzed multiple small, morphologically discrete regions of each IPMN, it is also likely that our experimental design allowed a higher sensitivity for subclonal mutations, particularly compared to bulk sequencing of paucicellular PDACs. A final caveat is that whole exome sequencing, as employed in our study, cannot identify all types of genomic alterations. Future studies using whole genome sequencing will be required to confidently place chromosomal rearrangements, chromothripsis, and whole genome doubling on the timeline of IPMN tumorigenesis.
In this study, we report comprehensive multi-region whole exome sequencing of pathologically well characterized IPMNs with both low-grade and high-grade components. This approach identified a new genetic driver of IPMN tumorigenesis and highlighted unique evolutionary processes not previously appreciated in precancerous pancreatic neoplasia. In addition, our results provide a novel biomarker that may refine risk stratification of IPMNs using cyst fluid analysis.
Supplementary Material
Supplementary Figure 1: Distribution of Somatic Mutations in LG and HG Regions in Each Analyzed IPMN. Heatmaps depicting the distribution of nonsynonymous somatic mutations among different regions in each IPMN. Black boxes indicate mutations with VAF >15%, and grey boxes indicate VAF of 5–15%. Representative images of neoplastic tissue stained by hematoxylin and eosin, with colored circles indicating the microdissected regions for sequencing analysis. Chow–Ruskey plot of shared and unique somatic mutations among regions in each IPMN.
Supplementary Figure 2: Copy Number Profile of Each Region in Analyzed IPMNs. Copy number alterations were called from whole exome sequencing using CNVkit. Blue indicates copy loss, while red denotes copy gain.
Supplementary Figure 3: Driver Mutations Ranked by Mutation Frequency. Mutated genes are ranked by the number of mutations per kb gene size. Genes with mutations in >3 IPMNs and >0.5 mutations per kb gene size are included.
Supplementary Figure 4: IPMN Phylogenies Constructed Using Treeomics. Treeomics generated phylogenetic trees from all nonsynonymous mutations identified in each IPMN region. Potential driver gene mutations (including those identified in previous pancreatic cancer genomics studies, as well as those mutated in >3 IPMNs and >0.5 mutations per kb gene size in the current study) are indicated by their gene name on the lineage in which they occur. Numbers indicate the number of nonsynonymous somatic mutations occurring in each trunk or branch.
Supplementary Table 1: Clinical Characteristics of IPMN Patient Cohort
Supplementary Table 2: Summary of Sample Characteristics for Multi-region Whole Exome Sequencing
Supplementary Table 3: Summary of Whole Exome Sequencing Data
Supplementary Table 4: Somatic Mutations Identified in Whole Exome Sequencing
Supplementary Table 5: Focal Copy Number Alterations Identified in Whole Exome Sequencing
Supplementary Table 6: Summary of Patient and Sample Characteristics for Cyst Fluid Analysis
Supplementary Table 7: KLF4 Mutations Identified in Cyst Fluid Samples According to IPMN Subtype
Summary Box.
1. What is already known about this subject?
Intraductal papillary mucinous neoplasms (IPMNs) are the most common neoplastic cysts in the pancreas and can progress to invasive pancreatic adenocarcinoma.
Comprehensive sequencing of small IPMN cohorts has identified driver genes in advanced lesions, and targeted multi-region sequencing has demonstrated genetic heterogeneity in IPMNs. However, comprehensive multi-region genomic analysis of IPMNs is required to elucidate the evolutionary features of neoplastic progression.
2. What are the new findings?
Multi-region whole exome sequencing revealed that KLF4 hotspot mutations (K409 and S411) were identified in >50% of the analyzed IPMNs, and these mutations were more frequently detected in regions with low-grade dysplasia than with high-grade dysplasia.
KLF4 mutations can be identified in IPMN cyst fluid samples using the Safe Sequencing System (Safe-SeqS), and these mutations are significantly more prevalent in cyst fluid from IPMNs with low-grade dysplasia.
Phylogenetic analyses demonstrated diverse patterns of tumor initiation and progression, suggesting that there is not a single universal genetic pathway into high-grade dysplasia in IPMNs.
3. How might it impact on clinical practice in the foreseeable future?
These results underscore the potential of KLF4 hotspot mutations as a novel biomarker for pancreatic cyst risk stratification, as KLF4 mutations are predominantly detected in low-grade IPMNs.
Both inter-patient and inter-region genetic heterogeneity in IPMNs should be considered during the development of molecular approaches for pancreatic cyst assessment.
ACKNOWLEDGEMENTS
The authors thank Dr. Ralph Hruban for helpful discussions and pathological expertise in the histological subtyping of IPMNs for this study. The authors thank Dr. Bert Vogelstein, Janine Ptak, Natalie Silliman, Joy Schaeffer, Lisa Dobbyn, and Maria Popoli for expert technical assistance.
The authors acknowledge the following sources of funding: NIH/NCI P50 CA62924; NIH/NIDDK K08 DK107781; Sol Goldman Pancreatic Cancer Research Center; Buffone Family Gastrointestinal Cancer Research Fund; Carol S. and Robert M. Long Pancreatic Cancer Research Fund; Kaya Tuncer Career Development Award in Gastrointestinal Cancer Prevention; AGA-Bernard Lee Schwartz Foundation Research Scholar Award in Pancreatic Cancer; Sidney Kimmel Foundation for Cancer Research Kimmel Scholar Award; AACR-Incyte Corporation Career Development Award for Pancreatic Cancer Research; American Cancer Society Research Scholar Grant RSG-18-143-01-CSM; Emerson Collective Cancer Research Fund; Rolfe Pancreatic Cancer Foundation; Joseph C Monastra Foundation; The Gerald O Mann Charitable Foundation (Harriet and Allan Wulfstat, Trustees); Susan Wojcicki and Denis Troper; Lustgarten Foundation for Pancreatic Cancer Research; CAMS Innovation Fund for Medical Sciences 2016-I2M-1-001 and 2019-I2M-1-001; Virginia and D.K. Ludwig Fund for Cancer Research; Sol Goldman Sequencing Facility at Johns Hopkins; Howard Hughes Medical Institute; Associazione Italiana Ricerca Cancro (grant number: 12182)
Abbreviations:
- ADM
acinar-to-ductal metaplasia
- CHASMplus
Cancer-specific High-throughput Annotation of Somatic Mutations plus
- FFPE
formalin-fixed paraffin-embedded
- gnomAD
Genome Aggregation Database
- HG
high-grade
- IPMN
Intraductal papillary mucinous neoplasm
- LG
low-grade
- NCF
neoplastic cell fraction
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- Safe-SeqS
Safe Sequencing System
- TCGA
The Cancer Genome Atlas
Footnotes
ETHICS APPROVAL
This study was approved by Institutional Review Board of The Johns Hopkins Hospital. Patients or the public were not involved in the design, conduct, reporting, or dissemination plans of our research.
Conflict of Interest
LDW receives research support from Applied Materials. The other authors declare no conflict of interest.
REFERENCES
- 1.Basturk O, Hong SM, Wood LD, et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. The American journal of surgical pathology 2015;39(12):1730–41. doi: 10.1097/pas.0000000000000533 [published Online First: 2015/11/13] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moris M, Bridges MD, Pooley RA, et al. Association Between Advances in High-Resolution Cross-Section Imaging Technologies and Increase in Prevalence of Pancreatic Cysts From 2005 to 2014. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association 2016;14(4):585–93.e3. doi: 10.1016/j.cgh.2015.08.038 [published Online First: 2015/09/16] [DOI] [PubMed] [Google Scholar]
- 3.Laffan TA, Horton KM, Klein AP, et al. Prevalence of unsuspected pancreatic cysts on MDCT. AJR American journal of roentgenology 2008;191(3):802–7. doi: 10.2214/ajr.07.3340 [published Online First: 2008/08/22] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lermite E, Sommacale D, Piardi T, et al. Complications after pancreatic resection: diagnosis, prevention and management. Clinics and research in hepatology and gastroenterology 2013;37(3):230–9. doi: 10.1016/j.clinre.2013.01.003 [published Online First: 2013/02/19] [DOI] [PubMed] [Google Scholar]
- 5.Tanaka M, Fernandez-Del Castillo C, Kamisawa T, et al. Revisions of international consensus Fukuoka guidelines for the management of IPMN of the pancreas. Pancreatology : official journal of the International Association of Pancreatology (IAP) [et al] 2017;17(5):738–53. doi: 10.1016/j.pan.2017.07.007 [published Online First: 2017/07/25] [DOI] [PubMed] [Google Scholar]
- 6.Vege SS, Ziring B, Jain R, et al. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148(4):819–22; quize12–3. doi: 10.1053/j.gastro.2015.01.015 [published Online First: 2015/03/26] [DOI] [PubMed] [Google Scholar]
- 7.Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Science translational medicine 2011;3(92):92ra66. doi: 10.1126/scitranslmed.3002543 [published Online First: 2011/07/22] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proceedings of the National Academy of Sciences of the United States of America 2011;108(52):21188–93. doi: 10.1073/pnas.1118046108 [published Online First: 2011/12/14] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furukawa T, Kuboki Y, Tanji E, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Scientific reports 2011;1:161. doi: 10.1038/srep00161 [published Online First: 2012/02/23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amato E, Molin MD, Mafficini A, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. The Journal of pathology 2014;233(3):217–27. doi: 10.1002/path.4344 [published Online First: 2014/03/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felsenstein M, Noe M, Masica DL, et al. IPMNs with co-occurring invasive cancers: neighbours but not always relatives. Gut 2018;67(9):1652–62. doi: 10.1136/gutjnl-2017-315062 [published Online First: 2018/03/04] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer CG, Beleva Guthrie V, Braxton AM, et al. Intraductal Papillary Mucinous Neoplasms Arise From Multiple Independent Clones, Each With Distinct Mutations. Gastroenterology 2019;157(4):1123–37.e22. doi: 10.1053/j.gastro.2019.06.001 [published Online First: 2019/06/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Omori Y, Ono Y, Tanino M, et al. Pathways of Progression From Intraductal Papillary Mucinous Neoplasm to Pancreatic Ductal Adenocarcinoma Based on Molecular Features. Gastroenterology 2019;156(3):647–61.e2. doi: 10.1053/j.gastro.2018.10.029 [published Online First: 2018/10/21] [DOI] [PubMed] [Google Scholar]
- 14.Hosoda W, Chianchiano P, Griffin JF, et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. The Journal of pathology 2017;242(1):16–23. doi: 10.1002/path.4884 [published Online First: 2017/02/12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 2013;31(3):213–9. doi: 10.1038/nbt.2514 [published Online First: 2013/02/12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reiter JG, Makohon-Moore AP, Gerold JM, et al. Reconstructing metastatic seeding patterns of human cancers. Nature communications 2017;8:14114. doi: 10.1038/ncomms14114 [published Online First: 2017/02/01] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer cell 2017;32(2):185–203.e13. doi: 10.1016/j.ccell.2017.07.007 [published Online First: 2017/08/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47–52. doi: 10.1038/nature16965 [published Online First: 2016/02/26] [DOI] [PubMed] [Google Scholar]
- 19.Springer S, Masica DL, Dal Molin M, et al. A multimodality test to guide the management of patients with a pancreatic cyst. Science translational medicine 2019;11(501) doi: 10.1126/scitranslmed.aav4772 [published Online First: 2019/07/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinde I, Wu J, Papadopoulos N, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proceedings of the National Academy of Sciences of the United States of America 2011;108(23):9530–5. doi: 10.1073/pnas.1105422108 [published Online First: 2011/05/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tie J, Cohen JD, Wang Y, et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut 2019;68(4):663–71. doi: 10.1136/gutjnl-2017-315852 [published Online First: 2018/02/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science (New York, NY) 2018;359(6378):926–30. doi: 10.1126/science.aar3247 [published Online First: 2018/01/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature communications 2015;6:6744. doi: 10.1038/ncomms7744 [published Online First: 2015/04/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tokheim C, Karchin R. CHASMplus Reveals the Scope of Somatic Missense Mutations Driving Human Cancers. Cell systems 2019;9(1):9–23.e8. doi: 10.1016/j.cels.2019.05.005 [published Online First: 2019/06/17] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark VE, Erson-Omay EZ, Serin A, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science (New York, NY) 2013;339(6123):1077–80. doi: 10.1126/science.1233009 [published Online First: 2013/01/26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reuss DE, Piro RM, Jones DT, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta neuropathologica 2013;125(3):351–8. doi: 10.1007/s00401-013-1093-x [published Online First: 2013/02/14] [DOI] [PubMed] [Google Scholar]
- 27.Sancho R, Jandke A, Davis H, et al. F-box and WD repeat domain-containing 7 regulates intestinal cell lineage commitment and is a haploinsufficient tumor suppressor. Gastroenterology 2010;139(3):929–41. doi: 10.1053/j.gastro.2010.05.078 [published Online First: 2010/07/20] [DOI] [PubMed] [Google Scholar]
- 28.Myant KB, Cammareri P, Hodder MC, et al. HUWE1 is a critical colonic tumour suppressor gene that prevents MYC signalling, DNA damage accumulation and tumour initiation. EMBO molecular medicine 2017;9(2):181–97. doi: 10.15252/emmm.201606684 [published Online First: 2016/12/23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piazza R, Valletta S, Winkelmann N, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nature genetics 2013;45(1):18–24. doi: 10.1038/ng.2495 [published Online First: 2012/12/12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nature genetics 2013;45(8):942–6. doi: 10.1038/ng.2696 [published Online First: 2013/07/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuboki Y, Fischer CG, Beleva Guthrie V, et al. Single-cell sequencing defines genetic heterogeneity in pancreatic cancer precursor lesions. The Journal of pathology 2019;247(3):347–56. doi: 10.1002/path.5194 [published Online First: 2018/11/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makohon-Moore AP, Zhang M, Reiter JG, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nature genetics 2017;49(3):358–66. doi: 10.1038/ng.3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zammarchi F, Morelli M, Menicagli M, et al. KLF4 is a novel candidate tumor suppressor gene in pancreatic ductal carcinoma. The American journal of pathology 2011;178(1):361–72. doi: 10.1016/j.ajpath.2010.11.021 [published Online First: 2011/01/13] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei D, Wang L, Yan Y, et al. KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer cell 2016;29(3):324–38. doi: 10.1016/j.ccell.2016.02.005 [published Online First: 2016/03/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collet L, Ghurburrun E, Meyers N, et al. Kras and Lkb1 mutations synergistically induce intraductal papillary mucinous neoplasm derived from pancreatic duct cells. Gut 2020;69(4):704–14. doi: 10.1136/gutjnl-2018-318059 [published Online First: 2019/06/04] [DOI] [PubMed] [Google Scholar]
- 36.Ghaleb AM, Yang VW. Krüppel-like factor 4 (KLF4): What we currently know. Gene 2017;611:27–37. doi: 10.1016/j.gene.2017.02.025 [published Online First: 2017/02/27] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131(5):861–72. doi: 10.1016/j.cell.2007.11.019 [published Online First: 2007/11/24] [DOI] [PubMed] [Google Scholar]
- 38.Tiwari N, Meyer-Schaller N, Arnold P, et al. Klf4 is a transcriptional regulator of genes critical for EMT, including Jnk1 (Mapk8). PloS one 2013;8(2):e57329. doi: 10.1371/journal.pone.0057329 [published Online First: 2013/03/02] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy SJ, Hart SN, Lima JF, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 2013;145(5):1098–109.e1. doi: 10.1053/j.gastro.2013.07.049 [published Online First: 2013/08/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iacobuzio-Donahue CA, Klimstra DS, Adsay NV, et al. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: comparison with conventional ductal adenocarcinomas. The American journal of pathology 2000;157(3):755–61. doi: 10.1016/s0002-9440(10)64589-0 [published Online First: 2000/09/12] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Huijgevoort NCM, Del Chiaro M, Wolfgang CL, et al. Diagnosis and management of pancreatic cystic neoplasms: current evidence and guidelines. Nature reviews Gastroenterology & hepatology 2019;16(11):676–89. doi: 10.1038/s41575-019-0195-x [published Online First: 2019/09/19] [DOI] [PubMed] [Google Scholar]
- 42.Jacobson BC, Baron TH, Adler DG, et al. ASGE guideline: The role of endoscopy in the diagnosis and the management of cystic lesions and inflammatory fluid collections of the pancreas. Gastrointestinal endoscopy 2005;61(3):363–70. doi: 10.1016/s0016-5107(04)02779-8 [published Online First: 2005/03/11] [DOI] [PubMed] [Google Scholar]
- 43.Del Chiaro M, Segersvärd R, Pozzi Mucelli R, et al. Comparison of preoperative conference-based diagnosis with histology of cystic tumors of the pancreas. Annals of surgical oncology 2014;21(5):1539–44. doi: 10.1245/s10434-013-3465-9 [published Online First: 2014/01/05] [DOI] [PubMed] [Google Scholar]
- 44.de Pretis N, Mukewar S, Aryal-Khanal A, et al. Pancreatic cysts: Diagnostic accuracy and risk of inappropriate resections. Pancreatology : official journal of the International Association of Pancreatology (IAP) [et al] 2017;17(2):267–72. doi: 10.1016/j.pan.2017.01.002 [published Online First: 2017/01/25] [DOI] [PubMed] [Google Scholar]
- 45.Valsangkar NP, Morales-Oyarvide V, Thayer SP, et al. 851 resected cystic tumors of the pancreas: a 33-year experience at the Massachusetts General Hospital. Surgery 2012;152(3 Suppl 1):S4–12. doi: 10.1016/j.surg.2012.05.033 [published Online First: 2012/07/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Springer S, Wang Y, Dal Molin M, et al. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology 2015;149(6):1501–10. doi: 10.1053/j.gastro.2015.07.041 [published Online First: 2015/08/09] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu J, Sadakari Y, Shindo K, et al. Digital next-generation sequencing identifies low-abundance mutations in pancreatic juice samples collected from the duodenum of patients with pancreatic cancer and intraductal papillary mucinous neoplasms. Gut 2017;66(9):1677–87. doi: 10.1136/gutjnl-2015-311166 [published Online First: 2016/07/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013;499(7457):214–18. doi: 10.1038/nature12213 [published Online First: 2013/06/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clinical chemistry 2015;61(1):64–71. doi: 10.1373/clinchem.2014.223040 [published Online First: 2014/11/26] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Distribution of Somatic Mutations in LG and HG Regions in Each Analyzed IPMN. Heatmaps depicting the distribution of nonsynonymous somatic mutations among different regions in each IPMN. Black boxes indicate mutations with VAF >15%, and grey boxes indicate VAF of 5–15%. Representative images of neoplastic tissue stained by hematoxylin and eosin, with colored circles indicating the microdissected regions for sequencing analysis. Chow–Ruskey plot of shared and unique somatic mutations among regions in each IPMN.
Supplementary Figure 2: Copy Number Profile of Each Region in Analyzed IPMNs. Copy number alterations were called from whole exome sequencing using CNVkit. Blue indicates copy loss, while red denotes copy gain.
Supplementary Figure 3: Driver Mutations Ranked by Mutation Frequency. Mutated genes are ranked by the number of mutations per kb gene size. Genes with mutations in >3 IPMNs and >0.5 mutations per kb gene size are included.
Supplementary Figure 4: IPMN Phylogenies Constructed Using Treeomics. Treeomics generated phylogenetic trees from all nonsynonymous mutations identified in each IPMN region. Potential driver gene mutations (including those identified in previous pancreatic cancer genomics studies, as well as those mutated in >3 IPMNs and >0.5 mutations per kb gene size in the current study) are indicated by their gene name on the lineage in which they occur. Numbers indicate the number of nonsynonymous somatic mutations occurring in each trunk or branch.
Supplementary Table 1: Clinical Characteristics of IPMN Patient Cohort
Supplementary Table 2: Summary of Sample Characteristics for Multi-region Whole Exome Sequencing
Supplementary Table 3: Summary of Whole Exome Sequencing Data
Supplementary Table 4: Somatic Mutations Identified in Whole Exome Sequencing
Supplementary Table 5: Focal Copy Number Alterations Identified in Whole Exome Sequencing
Supplementary Table 6: Summary of Patient and Sample Characteristics for Cyst Fluid Analysis
Supplementary Table 7: KLF4 Mutations Identified in Cyst Fluid Samples According to IPMN Subtype