

Abstract
Sickle cell disease (SCD) is a monogenic disorder characterized by recurrent episodes of severe bone pain, multi-organ failure, and early mortality. Although medical progress over the past several decades has improved clinical outcomes and offered cures for many affected individuals living in high-income countries, most SCD patients still experience substantial morbidity and premature death. Emerging technologies to manipulate somatic cell genomes and insights into the mechanisms of developmental globin gene regulation are generating potentially transformative approaches to cure SCD by autologous hematopoietic stem cell (HSC) transplantation. Key components of current approaches include ethical informed consent, isolation of patient HSCs, in vitro genetic modification of HSCs to correct the SCD mutation or circumvent its damaging effects, and reinfusion of the modified HSCs following myelotoxic bone marrow conditioning. Successful integration of these components into effective therapies requires interdisciplinary collaborations between laboratory researchers, clinical caregivers, and patients. Here we summarize current knowledge and research challenges for each key component, emphasizing that the best approaches have yet to be developed.
Historical perspectives
Studies of sickle cell disease (SCD), a life-threatening, multisystem genetic blood disorder that affects approximately 100,000 Americans and millions worldwide, have driven many advances in science and medicine. In 1910, Herrick described “sickle-shaped cells” in a dental student from the West Indies suffering from pain episodes and anemia (1). In 1949, Pauling and colleagues noted abnormalities in the properties of sickle hemoglobin (HbS), which were shown by Ingram in 1956 to result from altered amino acid composition, distinguishing SCD as “the first molecular disease” (2, 3). Around the same time, Neel and Beet determined SCD inheritance to be autosomal recessive (4, 5). The concept of natural selection for malaria resistance in SCD heterozygotes was proposed by Haldane in 1949 and confirmed by Allison a few years later (6, 7). It wasn’t until 1978 that Kan and Dozy reported the diagnosis of SCD by DNA analysis of fetal amniotic fluid cells, heralding a new era in genetic testing (8).
The deadly manifestations of SCD and its genetic features have been recognized for centuries in Africa, where hundreds of thousands of affected individuals are born each year. The concepts of “ọgbanje” (Ibo) and “abiku” (Yoruba), which translate to “a child destined to die and be born repeatedly to the world,” are attributed to SCD (9, 10). Medical advances have improved outcomes of affected individuals in high-income countries, but most patients continue to experience severe morbidities and premature mortality, beginning in adolescence. Now, emerging scientific discoveries are fueling innovative strategies to treat SCD via genetic manipulation of autologous hematopoietic stem cells (HSCs), promising effective cures (11, 12). However, the field has been shaken by recent reports of myeloid malignancies following lentiviral vector–mediated (LV-mediated) β-globin replacement gene therapy. Specifically, of 47 SCD patients treated with LV gene therapy in two related clinical trials (NCT02140554 and NCT04293185) over the past six years, three have been diagnosed with myelodysplastic syndrome or acute myeloid leukemia (13, 14). Here we review autologous genetic therapies for SCD, aiming to provide a balanced view of the risks and benefits of this rapidly evolving field.
Pathophysiology of SCD
SCD is caused by mutations in the HBB gene, which encodes the β-globin subunit of adult hemoglobin (HbA, α2β2) (15, 16). Most affected individuals are homozygous for a p.Glu6Val substitution resulting in the production of βS-globin, which combines with α-globin to form HbS (α2βS2). Another common form of SCD results from compound heterozygosity between HbS and HbC (p.Glu6Lys), resulting in HbSC (α2βSβC) disease. Hemoglobin S and C mutations frequently coexist with α- or β-thalassemia alleles, which can modify SCD phenotypes (17). Under hypoxic conditions, HbS or HbC form rigid polymers that cause red blood cells (RBCs) to acquire a sickle shape and initiate a complex pathophysiology that includes hemolysis, inflammation, dysregulated nitric oxide metabolism, hypercoagulation, and vasculopathy. Consequently, patients experience severe acute pain episodes, chronic pain, progressive multi-organ damage, and premature death. Common manifestations include stroke, acute lung injury (acute chest syndrome), bone avascular necrosis, and chronic heart, lung, and kidney disease. Loss of splenic function beginning in infancy predisposes patients to sepsis.
Unmet clinical needs
Medical costs for SCD in the United States exceed 1 billion dollars per year (18). Newborn screening and medical therapies have greatly improved the survival of children with SCD in high-income countries, although most patients continue to experience morbidities and die in early adulthood. In Africa, India, and the Middle East, there are millions of SCD patients who lack access to modern medical care, many of whom die before age 5. Ideally, all children with SCD would receive safe, effective curative therapy early in life to minimize organ damage. Allogeneic hematopoietic stem cell transplantation (HSCT) is the only approved cure, with an overall event-free survival of approximately 90%–95% after transplantation from HLA-matched sibling donors (19–21). Results of HSCT using alternative donor sources are promising, but many patients still experience immunological complications, such as graft rejection and graft-versus-host disease. As the risks of allogeneic HSCT remain substantial, determining which young SCD patients are most likely to benefit from this procedure is complex (22, 23). Hence, safer cures are needed. Genetic correction of autologous HSCs eliminates immune toxicities associated with allogeneic HSCT and expands patient eligibility by allowing affected individuals to be their own HSC donors. The major existing safety concern of these therapies is genotoxicities predisposing to malignant transformation; efforts are underway to understand and avoid this problem.
Autologous genetic therapies for SCD
All current protocols to treat SCD by autologous HSCT include four major steps (Figure 1): (i) the obtaining of ethical informed consent; (ii) isolation of patient CD34+ hematopoietic stem and progenitor cells (HSPCs), a complex mixture of long-term bone marrow–repopulating HSCs and committed progenitors, some of which provide early, short-term hematopoietic reconstitution after HSCT; (iii) ex vivo genetic manipulation of CD34+ cells targeting the HSC subpopulation to correct the SCD mutation or circumvent its toxicities; and (iv) myelotoxic/myeloablative conditioning to create a receptive bone marrow niche, followed by infusion of the modified HSPCs.
Figure 1. Four major steps in autologous hematopoietic stem cell (HSC) therapies to treat SCD.
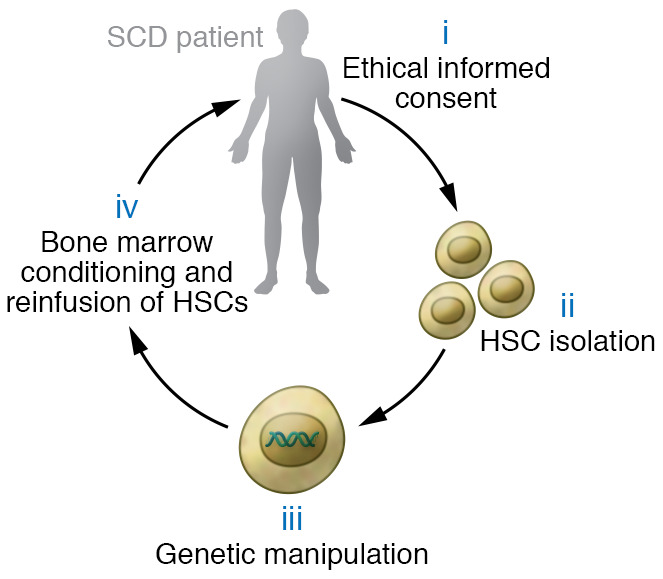
(i) Clinical researchers inform participants about the procedure, associated risks, and alternative treatments, then obtain written consent. (ii) CD34+ HSPCs are mobilized with plerixafor and isolated from blood by apheresis. (iii) HSPCs are manipulated ex vivo to correct the SCD mutation or induce HbF expression. (iv) The participant receives bone marrow conditioning with myelotoxic/myeloablative agents, followed by infusion of the modified HSPCs.
Ethical informed consent.
Translating scientific advances into SCD patient care is challenging. Participation in high-risk, potentially curative clinical trials can be influenced by patient-perceived disease burden, expected benefits, fear of toxicities, and socio-ecological factors including anticipated stress, religious beliefs, and support systems (24–26). Other barriers include lack of appropriate educational material, therapy-associated costs, and limited access to longitudinal care. A recent report by the National Academies of Science, Engineering, and Medicine (NASEM) noted that “the SCD community has developed a significant lack of trust in the health care system due to the nearly universal stigma and lack of belief in their reports of pain, a lack of trust that has been further reinforced by historical events, such as the Tuskegee experiment” (27). Many patients fear that HIV can be transmitted by lentiviral vectors (LVs), a myth that can be dispelled by better education and a trustful relationship with care providers (28, 29). Some SCD patients may overestimate the potential benefits of an experimental therapy and believe that no alternative care options exist. The participation of individuals with SCD in high-risk/high-reward clinical trials should be explored through a shared decision-making process with health care providers using culturally sensitive disease- and treatment-specific approaches tailored to the knowledge level of study participants. A multilevel decision aid for choosing disease-modifying treatments for SCD incorporates patient features (trust, SCD severity), decision characteristics (risk-benefit, urgency), physician perspectives (patient/family motivation, patient psychosocial characteristics), and environmental factors (institutional practice, insurance coverage) (30). Long-term collaborative relationships between clinical investigators, caregivers, and eligible subjects for SCD clinical trials are essential and require ongoing attention to potential communication barriers, social/cultural issues, and patient values (31). Given the uncertainties of high-risk therapeutic clinical trials, it may be valuable to conduct psychosocial assessments evaluating the emotional function, coping ability, and social support stability of potential participants to facilitate informed consent and better manage outcome expectations. Offering coping strategies and opportunities for clinical trial participants to explore potentially unexpected or less favorable outcomes enhances the shared decision-making model (26).
Isolation of HSPCs from individuals with SCD.
Most genetic therapies for SCD require collecting 4 × 106 to 15 × 106 autologous peripheral blood CD34+ HSPCs per kilogram (32, 33). Mobilization with granulocyte CSF is contraindicated because of potentially life-threatening immune cell activation (34). Until recently, bone marrow aspiration was the standard method for collecting CD34+ HSPCs from SCD patients. This strategy usually requires two to four separate harvests under general anesthesia, which can trigger serious complications (34). However, four recent studies indicate that CD34+ HSPCs can be mobilized safely in adults with SCD using plerixafor, a small molecule that inhibits interaction of the HSC chemokine receptor CXCR4 with its ligand, stromal-derived factor-1α (SDF-1α), on bone marrow niche cells (Table 1), with fewer severe adverse events in comparison with bone marrow harvesting in one study comparing both methods (35–38). Several conclusions derive from these studies. First, plerixafor mobilization of HSPCs, followed by apheresis harvesting, is generally safe and effective in SCD. The major toxicity noted was vaso-occlusive pain crisis that resolved with medical therapy. It is unclear whether this toxicity was caused by the drug, apheresis collection, or both. Second, the fraction of long-term repopulating HSCs and their suitability for genetic manipulation may be superior in plerixafor-mobilized HSPCs compared with those obtained by bone marrow aspiration (39, 40). Third, peripheral blood CD34+ cell counts peak as early as 3–6 hours after plerixafor administration, and apheresis should be initiated within this time frame. Fourth, the safest and most effective plerixafor dose appears to be 240 μg/kg, although higher doses are worth studying. Fifth, the CD34+ cell yield varies greatly between subjects. Hydroxyurea therapy was associated with reduced CD34+ cell numbers and therefore should be discontinued for at least 2–4 weeks prior to CD34+ cell mobilization and collection, and longer periods may be better. To minimize adverse events associated with plerixafor mobilization and apheresis, RBC transfusion (simple or exchange) should be initiated to maintain blood hemoglobin level of 10 g/dL with HbS fraction less than 30% after discontinuing of hydroxyurea.
Table 1. Plerixafor-mediated HSC mobilization in patients with SCD.

The CD34+ cell yield after a single plerixafor dose and apheresis cycle is usually insufficient for successful autologous therapy. Several strategies to maximize the HSPC yield per collection cycle are under investigation. Inflammation may alter the properties of SCD HSPCs and impair collection and purification. Adjustment of apheresis parameters to compensate for these features can improve CD34+ cell yields, but may also interfere with subsequent purification steps by increasing RBC contamination (37, 38). New CXCR4 antagonists are under study (41–44). In non-SCD individuals, the CXCR2 agonist GROβ, given alone or with plerixafor, can efficiently mobilize a unique population of HSPCs with superior long-term repopulating capabilities (45). These new agents may improve HSPC collection from individuals with SCD.
The CD34+ population contains only a small, variable proportion of the key target cells for SCD gene therapy, which include multipotent progenitors and HSCs that provide short- and long-term hematopoietic reconstitution, respectively (46). Both the CD90+CD45RA– and CD38– subfractions of CD34+ cells are enriched for HSCs (47, 48). Enumerating these subpopulations in autologous donor CD34+ cells may estimate more accurately the number of biologically relevant target cells available for gene therapy, correlate better with engraftment levels, and provide a refined target population for genetic modification in order to simplify manufacturing and reduce associated costs.
In vitro genetic correction of HSCs.
Multiple tools and strategies exist for modifying HSCs to circumvent SCD pathologies (Figures 2 and 3): (a) gene therapy via transduction with an LV encoding an antisickling β-like globin gene driven by erythroid-specific regulatory elements; (b) creation of genetic alterations that induce RBC fetal hemoglobin (HbF) expression; or (c) direct repair of the mutant SCD codon (valine), either to normal (glutamic acid) or to a benign, nonsickling variant, such as hemoglobin G-Makassar (alanine). Tools for genome editing include zinc finger nucleases (ZFNs), transcription activator–like effector nucleases (TALENs), CRISPR/Cas9, base editors, and prime editors (49, 50). Early clinical studies to treat SCD using LVs and genome editing are under way (Table 2).
Figure 2. Tools for genetic manipulation of patient CD34+ HSPCs to treat SCD.

(A) LV gene therapy: An antisickling β-like globin gene or BCL11A shRNA flanked by erythroid regulatory elements is inserted into a replication-deficient LV that is packaged into vector particles. The LV integrates semi-randomly into the host HSPC genome and is expressed in erythroid progeny. The β-like globin forms functional hemoglobin, while the BCL11A shRNA induces γ-globin expression to raise HbF levels. (B) Genome editing: The RNA-guided Cas9 nuclease binds the DNA target site via its associated guide RNA (gRNA) and creates a precise DSB that is repaired either by NHEJ, generating insertion-deletion mutations that induce HbF; or by HDR, which utilizes a donor DNA repair template to correct the SCD codon. (C) Base editing: Catalytically impaired Cas9n fused to either a cytosine or adenosine deaminase introduces precise base pair alterations. Adenosine (A) base editors convert A:T to G:C; cytosine base editors convert C:G to T:A. Base editors are used to induce HbF or convert the SCD codon to a benign variant. (D) Prime editing: Cas9n fused to a reverse transcriptase binds the target site via base pairing with the guide portion of the associated prime editing guide RNA (pegRNA) and creates a single-stranded DNA nick. The reverse transcriptase domain uses the pegRNA template to synthesize the desired edit following the nick. Cellular DNA repair machinery removes the endogenous DNA “flap” and repairs the nick to generate a heteroduplex intermediate that is converted to the edited product by DNA repair.
Figure 3. Genetic manipulations to treat SCD.
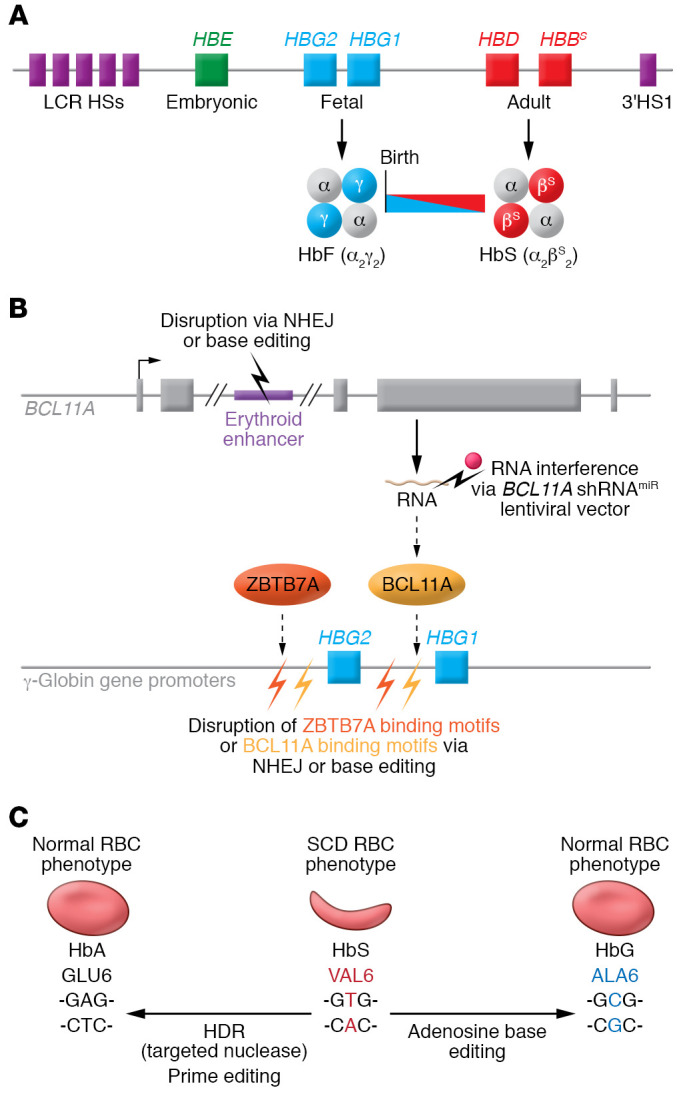
(A) The developmentally regulated β-like globin gene cluster is shown. Noncoding transcriptional regulatory regions are shown as DNase I–hypersensitive sites (HSs) at the locus control region (LCR) and 3′ to the HBB gene (3′HS1). The fetal γ-globin genes (HBG1 and HBG2) are expressed during late gestation, resulting in the production of RBC HbF. Around birth, γ-globin expression declines and is replaced by β-globin, resulting in a shift from HbF to HbA (α2β2) normally, or HbS (α2βS2) in the case of SCD. Inhibiting the γ- to β-globin switch has been a historical Holy Grail for treating SCD and β-thalassemia. (B) Induction of HbF by interfering with the expression or function of HBG1/HBG2 transcriptional repressors BCL11A or ZBTB7A. Strategies for manipulation of autologous SCD patient HSCs include disruption of an erythroid-specific BCL11A gene enhancer via genome-editing nuclease–mediated NHEJ or base editing, transduction with an LV that drives erythroid-specific expression of a BCL11A shRNA, and disruption of BCL11A or ZBTB7A binding motifs in the HBG1 and HBG2 promoters. (C) Conversion of the mutant SCD codon (valine) to normal glutamic acid can be engineered by genome-editing nuclease–mediated HDR or by prime editing. Alternatively, the SCD mutant valine codon can be converted to alanine by adenosine base editing to generate the nonsickling benign variant Hb G-Makassar.
Table 2. Autologous genetic therapies for SCD and β-thalassemia in clinical trials.

β-Globin gene addition via lentiviral vectors.
Because HSC self-renewal and differentiation require multiple rounds of cell division, therapeutic transgenes for hematopoietic disorders must be integrated into chromosomal DNA. The use of self-inactivating, HIV-based LVs for this purpose has been developed and refined extensively over more than 25 years and is now producing promising results for treating multiple blood disorders, including β-hemoglobinopathies (SCD and β-thalassemia) (11, 12, 51, 52). Importantly, LVs can accommodate the complex regulatory elements required to support high-level, erythroid-specific expression of a β-like globin transgene while maintaining the capacity to transduce HSCs efficiently (Figure 2A).
The first apparent gene therapy cure for SCD, reported in a 13-year-old boy in France in 2017, used an LV encoding the antisickling β-globin variant βA-T87Q (HGB-206, NCT02140554; ref. 53). At 15 months after therapy, the blood hemoglobin level was 11.8 g/dL with 48% HbAT87Q and 49% HbS, and the participant was symptom free. In a parallel trial conducted in the United States (HGB-206, NCT02140554), an evolution over three successive patient groups has been reported in a series of abstracts (54–58). Initially, efficacy was suboptimal owing to inefficient HSPC collection and transduction (HGB-206/NCT02140554, group A; n = 7). These problems have been gradually overcome by clinical and technical modifications in group B (n = 2), including pretherapy RBC transfusions, shifting from bone marrow–derived HSCs to plerixafor-mobilized cells, and cell manufacturing protocol refinements, including LentiBOOST (Sirion Biotech), an amphipathic membrane-modifying molecule that improves LV transduction (59–61). Seventeen individuals treated under the most recent version of the protocol (group C) with a median follow-up of 11 months became RBC transfusion free and experienced a greater than 99% reduction in severe pain events (55). The median vector copy number (VCN) was 3.8 (2.3–5.7) copies per diploid genome, resulting in a blood hemoglobin level of 11.2 (10.5–16.2) g/dL, approximately half of which was derived from the βA-T87Q transgene. Additional LV gene therapy trials for SCD have been opened recently (Table 2), and new strategies being developed to improve the efficacy of LVs are likely to improve this approach (62–65). Overall, current evidence indicates that LV gene therapy for SCD is effective, although recently raised safety concerns must be addressed (13, 14).
Genetic modifications to induce HbF expression.
Normally around birth, transcription of γ-globin switches gradually to that of β-globin, thereby shifting the production of HbF to HbA. The onset of SCD symptoms coincides with this switch, usually around 6 months of age (Figure 3A). Methods to induce HbF therapeutically for β-hemoglobinopathies have been sought for decades (66–68). This HbF therapy premise derives from a naturally occurring benign genetic condition termed hereditary persistence of fetal hemoglobin (HPFH), which results in persistently elevated HbF levels (more than 30%) in all RBCs (69, 70). Individuals who coinherit HPFH and SCD exhibit few or no SCD effects because HbF inhibits HbS polymerization (71) and γ-globin gene (HBG1 and HBG2) induction competes for the locus control region, a powerful upstream enhancer, to suppress mutant HBBS gene expression (72, 73). Two transcriptional repressors, ZBTB7A and BCL11A, participate in the developmental silencing of HBG1 and HBG2 by binding to their respective promoter cis-regulatory elements (74–76). Some HPFH variants disrupt these cis elements to inhibit repressor binding, while other variants create nearby de novo binding sites for transcriptional activators (70). Numerous approaches to induce RBC HbF therapeutically for SCD via genetic manipulation of HSCs are under investigation (Table 2 and Figure 3B).
RNA interference to silence BCL11A expression.
Transduction of normal or SCD donor CD34+ cells with a microRNA-adapted short hairpin RNA (shRNAmiR) that suppresses BCL11A expression induced high-level HbF in RBC progeny generated in vitro or in vivo after xenotransplantation into immunodeficient mice (77–79). Six subjects have been treated in a clinical trial using this approach (NCT03282656). Preliminary results with 7–29 months follow-up include RBC HbF levels of 20%–41% and hemoglobin levels of 9.3–11.4 g/dL (33). No post–gene therapy acute vaso-occlusive pain events, acute chest syndrome, or stroke were reported in any participant, although one subject continued to experience priapism for up to 8 months after gene therapy and another remains transfusion dependent for cerebral vasculopathy, albeit at a decreased transfusion frequency.
Genome editing to activate γ-globin gene expression.
Genome-editing nucleases, such as ZFNs, TALENs, and CRISPR/Cas9, introduce precisely targeted double-stranded DNA breaks (DSBs) that are subsequently resolved by endogenous repair pathways, either non-homologous end joining (NHEJ) or homology-directed repair (HDR) (Figure 2B and refs. 49, 50). The Cas9 nuclease is easiest to use because its sequence specificity is programmed by an associated guide RNA (gRNA) that binds the DNA target site via Watson-Crick base pairing, in contrast to other genome-editing nucleases that are programmed by more complicated protein engineering. In HSCs, most DSBs are resolved by NHEJ, which typically introduces base pair insertions or deletions (indels) that can disrupt DNA regulatory motifs. Using this strategy, several groups have induced RBC γ-globin transcription and HbF expression by creating indels that disable an erythroid-specific BCL11A gene enhancer (80–82). Preliminary results from a clinical study (CLIMB SCD-121, NCT03745287) are promising (Table 2 and refs. 32, 83). Two SCD participants (follow-up 3 and 12 months) maintained a hemoglobin level of around 10 g/dL with 46.8% and 42.4% HbF. Two participants (one with SCD and one with β-thalassemia) exhibited sustained high-level editing of the targeted alleles (78%–81%) in bone marrow cells at 6 and 12 months. Although longer studies in more individuals are required, these findings indicate efficient modification of long-term repopulating HSCs.
An alternative strategy to induce RBC HbF uses genome editing–mediated NHEJ to disrupt HBG1 and HBG2 promoter motifs that recruit the BCL11A or ZBTB7A repressor proteins (Figure 3B and refs. 84–86). Another clinical study (NCT04443907) is examining the safety and efficacy of Cas9-mediated disruption of a BCL11A binding site in the γ-globin gene promoters (Table 2).
It is also possible to disrupt DNA regulatory elements using base editors, engineered Cas9-directed DNA modification enzymes that introduce precise, targeted nucleotide alterations (Figure 2C and ref. 49). Base editors contain modified versions of Cas9, termed Cas9 nickase (Cas9n), fused to a nucleotide deaminase. Adenosine base editors contain Cas9n fused to a laboratory-evolved adenosine deaminase that converts targeted A:T base pairs to G:C pairs. Cytosine base editors fuse Cas9n with a cytosine deaminase to convert targeted C:G base pairs to T:A pairs. Base editors are now being used to disrupt DNA elements that silence γ-globin expression, including the BCL11A erythroid enhancer (87) and binding sites for BCL11A or ZBTB7A in the HBG1 and HBG2 genes (88, 89).
Direct repair of the mutant SCD codon.
The most desirable strategy for genetic correction of SCD is to convert the mutant codon (valine, GTG) to the normal one (glutamic acid, GAG). Codon conversion can be achieved by genome editing–mediated HDR, or prime editing (Figure 2C). Achieving high-level SCD correction by genome editing–mediated HDR is complicated by low rates of HSC correction, concomitant formation of indels that disrupt the HBB reading frame, and the requirement for an exogenous DNA repair template, which can be challenging to deliver and is potentially cytotoxic (90–94). In most preclinical studies of genome editing, human HSCs are approximated by their capacity to repopulate the bone marrow of immunodeficient mice at 16 weeks following transplantation. An early study attained 11.8% ± 3.7% allele correction by HDR and 55% ± 19% indels in bulk HbSS CD34+ HSPCs; following xenotransplantation, allele correction measured in HSCs was 2.3% ± 1.8%, with 46% ± 6% indels (95). Another early study achieved approximately 50% SCD allele correction in bulk HbSS CD34+ cells (92). Although xenotransplantation analysis of this population was not reported, the investigators achieved 3.5% HDR at the same target site in bone marrow–repopulating HSCs using a GFP-encoding DNA repair template. A more recent study reported HDR correction of 33.6% SCD alleles in bulk CD34+ cells. Four months after xenotransplantation, the correction rate declined to 23%, with approximately 30% having at least one corrected allele and 57% containing biallelic gene-disrupting indels (96). In a humanized mouse model for HbSS SCD, investigators have achieved an average of 14.8% (1%–35.4%, n = 9) allele correction in repopulating HSCs, with improvement of hemolytic anemia (97). Direct correction of the SCD codon will likely improve with protocols and technologies designed to enhance the rates of HDR in HSCs and/or select for those that are genetically corrected (98–104).
Prime editors contain Cas9n fused to an engineered reverse transcriptase that directly copies edited sequence information from a prime editing guide RNA (pegRNA) into a target DNA locus, then causes the cell to replace the original DNA sequence on both strands with the newly synthesized DNA flap (Figure 2D and refs. 49, 105). Prime editing can convert the SCD mutation to the wild-type allele at relatively high efficiencies in HEK293 cells (26%–52%; ref. 105) but requires further optimization for high-frequency targeted modification of human HSCs.
Base editors cannot create the T-to-A transversion required to revert the mutant SCD codon (valine, GTG) to wild-type (glutamic acid, GAG). However, adenine base editors can convert the mutant valine to alanine (GCG) to generate hemoglobin G-Makassar (Hb G-Makassar), a rare naturally occurring, nonsickling variant discovered in Southeast Asia (Figure 3C). Hb G-Makassar heterozygotes and one reported homozygote exhibit normal RBC indices, indicating that the variant is benign (106–109). Protein evolution strategies have developed an A base editor that can convert HbS alleles to Hb G-Makassar efficiently in HEK293 cells (110) and in mouse repopulating HSCs from subjects with SCD (111).
Genotoxicities associated with genetic modification of HSCs.
All methods to manipulate the HSC genome carry the potential for genotoxicity, the major concern being inadvertent clonal malignant transformation. Leukemia caused by γ-retroviral vector–mediated insertional activation of the LMO2 proto-oncogene was a major setback for early clinical studies of gene therapy for immunodeficiency (11, 12, 112). While the use of modified LVs markedly reduced this problem, Espinoza et al. reported dysplastic clonal hematopoiesis following LV transduction of HSPCs in a rhesus macaque model (113). This study identified two factors that increased the risk for malignancy: a high VCN (nine) and a potent murine stem cell virus (MSCV) constitutively active promoter-enhancer in the LV long terminal repeat. The LV used in the study was a hybrid vector that likely affected the insertional profile, contributing to the malignancy. None of the LVs used in clinical trials for β-hemoglobinopathies use the MSCV element (11). In the HGB-207 (NCT02906202) clinical trial for β-thalassemia, one participant treated with LV gene therapy developed a dominant HSC clone with insertional activation of HMGA2, a DNA-binding protein associated with benign and malignant tumor formation (114). Hematopoiesis remained stable over 15 months, and there have been no subsequent reports on this patient. It remains to be determined whether the myeloid malignancies recently reported in two individuals with SCD gene therapy are related to LV insertion (13, 14).
To our knowledge, no clinically relevant adverse event attributable to LV integration has been reported in any clinical trial for any indication (12). Nonetheless, preclinical safety studies of all LVs must determine integration sites after transduction in vitro, and assess for clonal dominance after transplantation of transduced cells in animal models (112). In human LV gene therapy trials, long-term (15 years) clinical evaluations and longitudinal determinations of vector integration sites in purified hematopoietic lineages must be included to assess clonal dominance as part of safety monitoring. These studies may also offer insights into the biology of hematopoietic differentiation and clonal succession (115, 116). It should be possible to enhance such studies by analyzing single cells, which is now being done to map clonal trajectories in malignant hematopoiesis (117).
Genome-editing nucleases may induce several genotoxicities, although it is too early to know the clinical impact (118–120). First, genome-editing proteins can create unintended off-target DSBs followed by formation of small indels, usually in DNA regions with homology to the targeted site. Second, on- and off-target DSBs can create kilobase-scale local DNA rearrangements or deletions, including loss of the entire chromosomal arm telomeric to the DSB. Moreover, DSBs can activate the TP53 tumor suppressor protein, leading to cell cycle delay, apoptosis, and selective pressure for TP53 gene loss, which can promote malignant transformation (121–125). Methods to investigate potential genotoxicities include computational algorithms based on homology to the on-target nucleotide sequence, whole genome sequencing, and molecular cloning approaches that enrich for genomic segments with genome editing–induced DSBs, followed by next-generation sequencing (118–120). Standard karyotyping approaches and/or more sensitive high-throughput sequencing–based methods, such as Uni-Directional Targeted Sequencing (UDiTaS), may detect translocations and chromosomal rearrangements caused by genome editing (126). Current methods can detect genome-editing off-target mutations at sensitivities from 0.01% to 1%. Theoretically, oncogenic mutations could occur at lower rates. Moreover, the current regulatory standard for tumorigenicity is to evaluate a patient-sized dose of genome-edited HSPCs in immunodeficient mice. As the natural life span of these mice is less than 1 year, this assay may fail to detect oncogenic mutations that require longer time frames for clonal expansion. Hence, a major challenge for therapeutic genome editing is to develop more sensitive detection methods for genotoxicities, including cell-based approaches to assess oncogenic risks associated with specific gene targeting protocols.
Approaches to reduce off-target DSBs by enhancing the specificity of genome-editing nucleases have focused mainly on CRISPR/Cas9 systems, which are more versatile and adaptable than ZFNs and TALENS (118, 119). The specificity of Cas9-mediated DSBs may be increased via at least five methods: using dimeric versions that require two distinct adjacent gRNA-programmed binding domains to install a DSB; developing Cas9 variants with reduced catalytic activity at regions of DNA with partial homology to the on-target site; modifying the structure or length of the programming gRNA; employing short gRNAs as decoys against potential off-target loci; and using phage-derived proteins that antagonize CRISPR/Cas9 nuclease activity (118, 119, 127–129).
In contrast to standard genome-editing nucleases and LVs, base editors act through mechanisms that are independent of DSBs, facilitating more precise DNA modifications and reducing some genotoxicities (49, 124, 130). However, base editors also carry unique potential to create undesired modifications. At target sites, base editors can produce “bystander” edits of nearby A or C nucleotides or induce low-frequency DSBs with resultant indels. Base editors can also induce low-level off-target deamination of A or C nucleotides in DNA or RNA through Cas-dependent and -independent mechanisms (131, 132). Several laboratories have created variants of adenine and cytosine base editors by altering their Cas9 and/or deaminase domains (131–143).
Similar to base editors, prime editing does not act through the creation of DSBs. Early studies indicate that prime editing is less prone to off-target modifications than is conventional Cas9 nuclease with the same gRNAs (105, 144–146).
Bone marrow conditioning and infusion regimens
Myelotoxic chemotherapy and/or irradiation before infusion of genetically modified autologous HSCs eliminates resident HSPCs that compete for the hematopoietic niche, facilitating engraftment. Most autologous HSCT protocols use high-dose myeloablative conditioning to promote full replacement of bone marrow with corrected HSCs (32, 33, 53, 147, 148). Major toxicities include multi-organ damage, infertility, and myeloid neoplasms (149). One individual who received LV gene therapy with high-dose busulfan for SCD developed myelodysplastic syndrome after approximately 3 years (13). As the malignant clone did not harbor LV DNA, transformation was attributed to busulfan conditioning. The myelodysplastic syndrome transformed to acute myeloid leukemia, and the patient subsequently underwent induction chemotherapy followed by haploidentical transplantation. Unfortunately, relapsed disease ultimately led to his death. Genotoxic agents like busulfan create mutations that can synergize with preexisting germline or somatic cancer susceptibility mutations, and/or an abnormal bone marrow microenvironment, to induce malignant evolution (150).
Because normal or genetically corrected RBCs survive longer in the circulation than SCD RBCs, therapeutic effects may be gained by establishing partial bone marrow chimerism with normal or HbAS HSCs (151–153). Sub-ablative conditioning has been incorporated into one LV gene therapy clinical trial for SCD (154) and may reduce HSCT toxicities for all SCD patients. However, a recent report raises caution (155). Three of 76 adult SCD patients who received allogeneic HSCT with reduced-intensity conditioning developed graft rejection followed by myeloid leukemia. Two individuals studied further were found to harbor somatic TP53 mutations in bone marrow HSPCs prior to HSCT. Most likely, outgrowth of these preleukemic clones was favored by low-intensity conditioning, similar to what has been observed with HSCT for myelodysplastic syndrome or acute myeloid leukemia (156, 157). Thus, it may be prudent to screen adults with SCD for somatic mutations associated with clonal hematopoiesis before autologous HSCT.
Reduced-toxicity HSCT conditioning regimens using antibodies against HSPC cell surface receptors are under study for treating refractory leukemia (158) and may also be effective for autologous HSCT. Unconjugated anti-CD117 (c-KIT) antibody is being tested as a non-genotoxic conditioning agent for allogeneic HSCT in infants with severe combined immunodeficiency, with promising early data (159, 160). Killing of anti-CD117–bound HSPCs is inhibited by their expression of CD47 and its interaction with signal regulatory protein-α (SIRPα) on the surface of immune effector cells (161). Thus, coadministration of anti-CD47 may enhance the efficacy of anti-CD117 for pre-HSCT conditioning (162). Studies in mice and nonhuman primates indicate that bone marrow conditioning with toxin-linked antibodies against CD45 or CD117 can facilitate donor HSC engraftment with minimal toxicity (163–167). Reduced-toxicity bone marrow conditioning with radioisotope-linked antibodies has been examined in allogeneic HSCT for hematological malignancies (168–172). Similar approaches using α-emitters with high linear energy transfer and short path lengths (40–90 μm) (173, 174), such as actinium-225 and astatine-211, may provide reduced-toxicity conditioning in autologous HSCT for nonmalignant blood disorders, including SCD.
Preclinical endpoints that predict therapeutic responses
Ideally, genetic correction of autologous SCD patient HSCs will generate 100% nonsickling RBCs with normal circulatory life span, eliminate disease symptoms, and arrest end-organ damage. Modification of all HSCs is not required to achieve this goal because corrected RBCs have a survival advantage in the circulation. The clinical benefits of any autologous HSCT for SCD depend on the fraction of modified HSCs in the bone marrow and the relative survival advantage conferred to RBC progeny, which can be modeled mathematically (151, 175). In general agreement with these models, allogeneic HSCT studies with normal or SCD-heterozygous donors indicate that chimerism as low as 20% can result in 100% circulating donor RBCs (151–153, 176). By this analogy, heterozygous or homozygous correction of the mutant SCD codon via HDR must occur in at least 20% of repopulating HSCs. The same may hold true for base editor conversion of HbS to Hb G-Makassar, assuming that the HbGS heterotetramer is nonsickling under normal physiological conditions.
Lentiviral vector gene therapy.
Variables that predict successful LV gene therapy for SCD include the fraction of modified human HSCs, expression levels of the β-like globin transgene, its antisickling properties, and its ability to outcompete endogenously expressed βS-globin for binding to α-globin during Hb assembly. Clinical trial data using the BB305 βA-T87Q-globin LV (HGB-206, NCT02140554) demonstrate a relatively high VCN in the preinfusion CD34+ cell product (mean 3.8 copies per diploid genome), which may be required for full therapeutic efficacy (177). This VCN generates approximately 16–20 pg βA-T87Q per cell in an immortalized erythroid line harboring an engineered βS mutation (178). Interestingly, this study showed that expression of the βA-T87Q transgene caused VCN-dependent reductions in endogenous βS-globin mRNA and protein.
Genetic induction of HbF.
Predicting the therapeutic requirements for γ-globin gene induction is complex because the protective effects of HbF on SCD can be partial and the levels required to inhibit SCD pathologies are likely organ specific. For example, high HbF is associated with longer life span, reduced pain episodes, and fewer leg ulcers (179, 180). A protective role for HbF against cerebrovascular disease is less clear. Several studies show that silent cerebral infarcts or ischemic strokes associate with low HbF levels (181–183). However, these associations were not replicated in two large, multicenter studies (184, 185).
Genetic modifications that induce HbF pancellularly are likely to attenuate SCD-related morbidities more effectively than those that induce HbF heterocellularly (179, 180). Many patients with SCD in India and parts of the Middle East carry the Arab-Indian haplotype at the β-like globin locus, which is associated with approximately 20% HbF expressed heterocellularly. These individuals experience SCD-related morbidities, although later in life compared with patients in the United States and other regions where the Arab-Indian haplotype is less common (186, 187). In contrast, individuals with SCD and HPFH possessing more than 30% HbF distributed pancellularly appear to be symptom free (179, 188, 189). Thus, while the protective thresholds for HbF induction may vary across different organ systems, genetic manipulations that induce more than 30% pancellularly should produce substantial clinical benefits and may induce cures. For comparison, six individuals who received autologous HSCs that were transduced with LV encoding erythroid-expressed BCL11A shRNA exhibited a median of 30.5% (range, 20.4%–41.3%) HbF in RBC lysates, with 70.8% (range, 58.9%–93.6%) of individual RBCs expressing HbF, as detected by immunoflow cytometry (F cells) (33). One individual who received autologous HSCs harboring Cas9-disrupted BCL11A erythroid enhancer has maintained 42%–49% HbF expressed pancellularly in RBCs at 15 months after therapy (32). All of these patients have exhibited markedly reduced pain episodes and improved laboratory parameters of hemolysis. It will be important to correlate laboratory studies with detailed multiorgan assessments at 5 and 10 years on these and future gene therapy research participants.
Induction of HbF inhibits RBC sickling by reducing the effective concentration of HbS, which is the primary determinant of its polymerization at low oxygen tension (71). It is estimated that 9–12 pg HbF per RBC can block HbS polymerization in venous capillary beds (179). The fraction of RBCs that achieve this HbF threshold is poorly predicted by standard clinical tests such as percentage HbF in RBC lysates and detection of F cells with anti-HbF antibodies. Moreover, these tests imprecisely predict SCD severity. Thus, improved analytical methods to quantify HbF and HbS concentrations in individual RBCs and to measure their propensity for sickling under physiological oxygen concentrations should enhance preclinical testing of genetic strategies to treat SCD (190–195).
Perspectives and future
While studies of SCD over the past 50 years have benefited medical science, it may be argued that affected patients have fared less well. The 2020 NASEM report notes that “the health care needs of individuals living with SCD have been neglected by the U.S. and global health care systems, causing them and their families to suffer” (27). Now we are poised to develop new potentially curative therapies based on autologous HSCT. Enabling technologies are advancing rapidly and the future is promising. However, the field is in its infancy, the best strategies remain unknown, and each step (Figure 1) requires further optimization. Moreover, advances are also occurring in alternative donor allogeneic HSCT, including the use of haploidentical donors with non-myeloablative bone marrow conditioning (196–198). Ensuring that patients and families are fully informed about the potential risks and benefits of different curative approaches represents a major ethical challenge (19, 199).
Recent reports of SCD patients developing myeloid neoplasms after undergoing LV gene therapy remind us that experimental treatments come with unknown risks. Of 47 patients treated in the largest LV gene therapy trial to date, three have been diagnosed with myeloid malignancies. One of these has been attributed to busulfan conditioning, and studies are under way to investigate the etiologies of the two cases reported more recently (13, 14). Leukemia has not been reported in several hundred individuals who have been treated with LV gene therapy for indications other than SCD, including 63 β-thalassemia patients who received the same vector used for SCD in separate clinical trials. Thus, SCD itself may predispose to gene therapy–related myeloid malignancies, possibly by enhancing the rate of preleukemic somatic mutations acquired before treatment (155, 200) (discussed in “Bone marrow conditioning and infusion regimens” above) and/or by creating an abnormal bone marrow microenvironment (150, 201). Similar risks could also apply to newer LV gene therapy or genome editing protocols. A promising trial of LV-mediated posttranscriptional silencing of BCL11A was also paused out of an abundance of caution, though no such events have been described in the nine individuals treated to date (Table 2 and ref. 202). These recent observations highlight the need for close long-term clinical and molecular monitoring, beginning before the initiation of gene therapy.
The notion that individuals with SCD may be uniquely susceptible to gene therapy–related myeloid malignancies raises concerns about hydroxyurea as a potential contributor. Hydroxyurea is antimetabolite that has been used to treat SCD since the 1980s. Some studies indicate that hydroxyurea can be mutagenic in vitro and enhance the rate of myelodysplastic syndrome/acute myeloid leukemia transformation in patients with myeloproliferative disorders (203). However, these studies have remained inconclusive, and a World Health Organization expert panel concluded that hydroxyurea is not classifiable as to its carcinogenicity to humans (203). Moreover, long-term studies have shown that hydroxyurea therapy reduces morbidity and mortality in SCD without increasing the rates of accumulated mutations or cancer (204). It has recently been reported that individuals with SCD are at increased risk for myeloid malignancies, yet no further increase was noted in the data after hydroxyurea’s FDA approval (205). We conclude that while the potential for chronic hydroxyurea administration to influence long-term outcomes of gene therapy should be investigated, current evidence indicates that the benefits of hydroxyurea therapy outweigh the risks for most patients.
Current autologous HSCT therapies for SCD are technologically complex, expensive, and high-risk. We believe that the development of reduced-toxicity bone marrow conditioning regimens to be used in conjunction with autologous HSCT represents a high-priority challenge. As safe and effective approaches become established, it will be important to streamline manufacturing and reduce costs in order to disseminate cures more broadly. Initially, such therapies will occur in high-income countries where advanced tertiary care centers provide cell manufacturing and medical support. Delivery of autologous HSCT therapies to the vast majority of individuals with SCD, who reside mainly in low- and middle-income countries, will be facilitated by reduced-toxicity in vivo approaches that selectively modify HSCs via intravenous or bone marrow injection of targeted delivery vehicles such as engineered nanoparticles or nonintegrating viral vectors (206–208). The NIH and the Gates Foundation are collaborating to develop gene-based cures for SCD on a global scale (209). We are confident that this endeavor will succeed, although it will require time and advancing technologies. In the meantime, autologous HSCT for SCD must be considered as an essential component of a larger, multifaceted effort that also includes widespread newborn screening, institution of preventative therapies such as immunization, prophylactic penicillin and hydroxyurea, better drugs, allogeneic HSCT, and optimized infrastructures for delivering both basic and advanced medical care.
Author contributions
PAD, AS, JSP, YZ, JFT, and MJW performed background research, wrote sections of the manuscript and edited the document. MJW organized the manuscript and coordinated the writing. PAD and AS contributed equally to the authorship of this work; their order was assigned alphabetically.
Acknowledgments
We apologize to all the colleagues whose contributions we could not properly acknowledge in this Review because of length restrictions. We thank SCD patients for participating in the research described here. The authors’ research on genetic therapies for SCD is supported by NIH grants F32 DK118822 (to PAD), K01 HL125495 (to JSP), and P01 HL053749 (to MJW); the American Society of Hematology Scholar Award (to AS); Doris Duke Sickle Cell Disease/Advancing Cures grant 2017093 (to MJW and AS); the Assisi Foundation of Memphis (to MJW); and St. Jude/American Lebanese Syrian Associated Charities (ALSAC). We thank Michael Debaun, Greg Newby, and David Liu for helpful comments on the manuscript.
Version 1. 04/15/2021
Electronic publication
Footnotes
Conflict of interest: MJW is a former advisor for and first-tier equity holder in Beam Therapeutics; a second-tier advisory board member for Forma Therapeutics, Cellarity Inc., and Novartis; and a second-tier consultant for Graphite Bio. AS is the St. Jude Children’s Research Hospital site principal investigator of clinical trials for genome editing of sickle cell disease sponsored by Vertex Pharmaceuticals Incorporated/CRISPR Therapeutics (NCT03745287, titled A Safety and Efficacy Study Evaluating CTX001 in Subjects With Severe Sickle Cell Disease) and by Novartis (NCT04443907, titled Study of Safety and Efficacy of Genome-Edited Hematopoietic Stem and Progenitor Cells in Sickle Cell Disease). The industry sponsors provide funding for the clinical trial, which includes salary support. AS also is a second-tier consultant for Spotlight Therapeutics. JFT is an investigator on the bluebird bio Inc. clinical trial HGB-206 (NCT02140554, titled A Study Evaluating the Safety and Efficacy of bb1111 in Severe Sickle Cell Disease).
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(8):e146394.https://doi.org/10.1172/JCI146394.
Contributor Information
Phillip A. Doerfler, Email: Phillip.Doerfler@stjude.org.
Akshay Sharma, Email: akshay.sharma@stjude.org.
Jerlym S. Porter, Email: jerlym.porter@stjude.org.
Yan Zheng, Email: yan.zheng@stjude.org.
References
- 1.Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. JAMA. 2014;312(10):1063. doi: 10.1001/jama.2014.11011. [DOI] [PubMed] [Google Scholar]
- 2.Pauling L, Itano HA, et al. Sickle cell anemia a molecular disease. Science. 1949;110(2865):543–548. doi: 10.1126/science.110.2865.543. [DOI] [PubMed] [Google Scholar]
- 3.Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956;178(4537):792–794. doi: 10.1038/178792a0. [DOI] [PubMed] [Google Scholar]
- 4.Beet EA. The genetics of the sickle-cell trait in a Bantu tribe. Ann Eugen. 1949;14(4):279–284. doi: 10.1111/j.1469-1809.1947.tb02402.x. [DOI] [PubMed] [Google Scholar]
- 5.Neel JV. The inheritance of sickle cell anemia. Science. 1949;110(2846):64–66. doi: 10.1126/science.110.2846.64. [DOI] [PubMed] [Google Scholar]
- 6.Allison AC. Protection afforded by sickle-cell trait against subtertian malareal infection. Br Med J. 1954;1(4857):290–294. doi: 10.1136/bmj.1.4857.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haldane JBS. The rate of mutation of human genes. Hereditas. 1949;35(S1):267–273. [Google Scholar]
- 8.Kan YW, Dozy AM. Antenatal diagnosis of sickle-cell anaemia by D.N.A. analysis of amniotic-fluid cells. Lancet. 1978;2(8096):910–912. doi: 10.1016/s0140-6736(78)91629-x. [DOI] [PubMed] [Google Scholar]
- 9.Onwubalili JK. Sickle-cell anaemia: an explanation for the ancient myth of reincarnation in Nigeria. Lancet. 1983;2(8348):503–505. doi: 10.1016/s0140-6736(83)90524-x. [DOI] [PubMed] [Google Scholar]
- 10.Nzewi E. Malevolent ogbanje: recurrent reincarnation or sickle cell disease? Soc Sci Med. 2001;52(9):1403–1416. doi: 10.1016/S0277-9536(00)00245-8. [DOI] [PubMed] [Google Scholar]
- 11.Magrin E, et al. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood. 2019;134(15):1203–1213. doi: 10.1182/blood.2019000949. [DOI] [PubMed] [Google Scholar]
- 12.Naldini L. Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol Med. 2019;11(3):e9958. doi: 10.15252/emmm.201809958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsieh MM, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4(9):2058–2063. doi: 10.1182/bloodadvances.2019001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. bluebird bio Inc. bluebird bio announces temporary suspension on phase 1/2 and phase 3 studies of lentiglobin gene therapy for sickle cell disease (bb1111). News release. February 16, 2021. https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-announces-temporary-suspension-phase-12-and-phase-3.
- 15.Kato GJ, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi: 10.1038/nrdp.2018.10. [DOI] [PubMed] [Google Scholar]
- 16.Piel FB, et al. Sickle cell disease. N Engl J Med. 2017;376(16):1561–1573. doi: 10.1056/NEJMra1510865. [DOI] [PubMed] [Google Scholar]
- 17.Serjeant GR, Vichinsky E. Variability of homozygous sickle cell disease: the role of alpha and beta globin chain variation and other factors. Blood Cells Mol Dis. 2018;70:66–77. doi: 10.1016/j.bcmd.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 18. Steiner CA, Miller JL. Sickle cell disease patients in U.S. hospitals, 2004: Statistical Brief #21. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality; 2006. [PubMed] [Google Scholar]
- 19.de la Fuente J, et al. The role of haematopoietic stem cell transplantation for sickle cell disease in the era of targeted disease-modifying therapies and gene editing. Lancet Haematol. 2020;7(12):e902–e911. doi: 10.1016/S2352-3026(20)30283-0. [DOI] [PubMed] [Google Scholar]
- 20.Eapen M, et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol. 2019;6(11):e585–e596. doi: 10.1016/S2352-3026(19)30154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gluckman E, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017;129(11):1548–1556. doi: 10.1182/blood-2016-10-745711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeBaun MR, Clayton EW. Primum non nocere: the case against transplant for children with sickle cell anemia without progressive end-organ disease. Blood Adv. 2017;1(26):2568–2571. doi: 10.1182/bloodadvances.2017007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fitzhugh CD, Walters MC. The case for HLA-identical sibling hematopoietic stem cell transplantation in children with symptomatic sickle cell anemia. Blood Adv. 2017;1(26):2563–2567. doi: 10.1182/bloodadvances.2017007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khemani K, et al. Experiences and decision making in hematopoietic stem cell transplant in sickle cell disease: patients’ and caregivers’ perspectives. Biol Blood Marrow Transplant. 2018;24(5):1041–1048. doi: 10.1016/j.bbmt.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 25.Stevens EM, et al. Mistrust of pediatric sickle cell disease clinical trials research. Am J Prev Med. 2016;51(1 suppl 1):S78–S86. doi: 10.1016/j.amepre.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho HL, et al. Motivations and decision-making of adult sickle cell patients in high-risk clinical research. Biol Blood Marrow Transplant. 2020;26(6):1225–1232. doi: 10.1016/j.bbmt.2020.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. National Academies of Sciences, Engineering, Medicine. McCormick M, et al., eds. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. National Academies Press; 2020. [PubMed] [Google Scholar]
- 28.Strong H, et al. Patient perspectives on gene transfer therapy for sickle cell disease. Adv Ther. 2017;34(8):2007–2021. doi: 10.1007/s12325-017-0587-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Persaud A, Bonham VL. The role of the health care provider in building trust between patients and precision medicine research programs. Am J Bioeth. 2018;18(4):26–28. doi: 10.1080/15265161.2018.1431327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bakshi N, et al. Proponent or collaborative: physician perspectives and approaches to disease modifying therapies in sickle cell disease. PLoS One. 2017;12(7):e0178413. doi: 10.1371/journal.pone.0178413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraft SA, et al. Beyond consent: building trusting relationships with diverse populations in precision medicine research. Am J Bioeth. 2018;18(4):3–20. doi: 10.1080/15265161.2018.1431322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frangoul H, et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N Engl J Med. 2021;384(3):252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 33.Esrick EB, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med. 2021;384(3):205–215. doi: 10.1056/NEJMoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fitzhugh CD, et al. Granulocyte colony-stimulating factor (G-CSF) administration in individuals with sickle cell disease: time for a moratorium? Cytotherapy. 2009;11(4):464–471. doi: 10.1080/14653240902849788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boulad F, et al. Safety and efficacy of plerixafor dose escalation for the mobilization of CD34(+) hematopoietic progenitor cells in patients with sickle cell disease: interim results. Haematologica. 2018;103(5):770–777. doi: 10.3324/haematol.2017.187047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esrick EB, et al. Successful hematopoietic stem cell mobilization and apheresis collection using plerixafor alone in sickle cell patients. Blood Adv. 2018;2(19):2505–2512. doi: 10.1182/bloodadvances.2018016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lagresle-Peyrou C, et al. Plerixafor enables safe, rapid, efficient mobilization of hematopoietic stem cells in sickle cell disease patients after exchange transfusion. Haematologica. 2018;103(5):778–786. doi: 10.3324/haematol.2017.184788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchida N, et al. Safe and efficient peripheral blood stem cell collection in patients with sickle cell disease using plerixafor. Haematologica. 2020;105(10):e497. doi: 10.3324/haematol.2019.236182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uchida N, et al. Bone marrow as a hematopoietic stem cell source for gene therapy in sickle cell disease: evidence from rhesus and SCD patients. Hum Gene Ther Clin Dev. 2017;28(3):136–144. doi: 10.1089/humc.2017.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leonard A, et al. Bone marrow characterization in sickle cell disease: inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br J Haematol. 2019;186(2):286–299. doi: 10.1111/bjh.15902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karpova D, et al. Mobilization of hematopoietic stem cells with the novel CXCR4 antagonist POL6326 (balixafortide) in healthy volunteers-results of a dose escalation trial. J Transl Med. 2017;15(1):2. doi: 10.1186/s12967-016-1107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abraham M, et al. Single dose of the CXCR4 antagonist BL-8040 induces rapid mobilization for the collection of human CD34+ cells in healthy volunteers. Clin Cancer Res. 2017;23(22):6790–6801. doi: 10.1158/1078-0432.CCR-16-2919. [DOI] [PubMed] [Google Scholar]
- 43.Peled A, et al. The high-affinity CXCR4 antagonist BKT140 is safe and induces a robust mobilization of human CD34+ cells in patients with multiple myeloma. Clin Cancer Res. 2014;20(2):469–479. doi: 10.1158/1078-0432.CCR-13-1302. [DOI] [PubMed] [Google Scholar]
- 44.Domingues MJ, et al. New agents in HSC mobilization. Int J Hematol. 2017;105(2):141–152. doi: 10.1007/s12185-016-2156-2. [DOI] [PubMed] [Google Scholar]
- 45.Hoggatt J, et al. Rapid mobilization reveals a highly engraftable hematopoietic stem cell. Cell. 2018;172(1–2):191–204. doi: 10.1016/j.cell.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pang WW, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. 2011;108(50):20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Humbert O, et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci Transl Med. 2019;11(503):eaaw3768. doi: 10.1126/scitranslmed.aaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masiuk KE, et al. Improving gene therapy efficiency through the enrichment of human hematopoietic stem cells. Mol Ther. 2017;25(9):2163–2175. doi: 10.1016/j.ymthe.2017.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anzalone AV, et al. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 50.Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578(7794):229–236. doi: 10.1038/s41586-020-1978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bank A, et al. Gene transfer. A potential approach to gene therapy for sickle cell disease. Ann N Y Acad Sci. 1989;565:37–43. doi: 10.1111/j.1749-6632.1989.tb24147.x. [DOI] [PubMed] [Google Scholar]
- 52.Kohn DB. Gene therapy for blood diseases. Curr Opin Biotechnol. 2019;60:39–45. doi: 10.1016/j.copbio.2018.11.016. [DOI] [PubMed] [Google Scholar]
- 53.Ribeil JA, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017;376(9):848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 54.Kanter J, et al. Outcomes for initial patient cohorts with up to 33 months of follow-up in the Hgb-206 phase 1 trial. Blood. 2018;132(suppl 1):1080. doi: 10.1182/blood-2018-99-113477. [DOI] [Google Scholar]
- 55.Kanter J, et al. Outcomes in patients treated with lentiglobin for sickle cell disease (SCD) gene therapy: updated results from the phase 1/2 HGB-206 group C study. EHA Library. 2020;295102:S282 [Google Scholar]
- 56.Kanter J, et al. Resolution of sickle cell disease manifestations in patients treated with lentiglobin gene therapy: updated results from the phase 1/2 Hgb-206 group C study. Blood. 2019;134(suppl 1):990. doi: 10.1182/blood-2019-128894. [DOI] [Google Scholar]
- 57.Kanter J, et al. Interim results from a phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease. Blood. 2017;130(suppl 1):527. doi: 10.1182/blood.V130.Suppl_1.527.527. [DOI] [Google Scholar]
- 58.Tisdale JF, et al. Current results of lentiglobin gene therapy in patients with severe sickle cell disease treated under a refined protocol in the phase 1 Hgb-206 study. Blood. 2018;132(suppl 1):1026. doi: 10.1182/blood-2018-99-113480. [DOI] [Google Scholar]
- 59.Jang Y, et al. Optimizing lentiviral vector transduction of hematopoietic stem cells for gene therapy. Gene Ther. 2020;27(12):545–556. doi: 10.1038/s41434-020-0150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Masiuk KE, et al. PGE2 and poloxamer synperonic F108 enhance transduction of human HSPCs with a β-globin lentiviral vector. Mol Ther Methods Clin Dev. 2019;13:390–398. doi: 10.1016/j.omtm.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schott JW, et al. Enhancing lentiviral and alpharetroviral transduction of human hematopoietic stem cells for clinical application. Mol Ther Methods Clin Dev. 2019;14:134–147. doi: 10.1016/j.omtm.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morgan RA, et al. Creating new β-globin-expressing lentiviral vectors by high-resolution mapping of locus control region enhancer sequences. Mol Ther Methods Clin Dev. 2020;17:999–1013. doi: 10.1016/j.omtm.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morgan RA, et al. Improved titer and gene transfer by lentiviral vectors using novel, small β-globin locus control region elements. Mol Ther. 2020;28(1):328–340. doi: 10.1016/j.ymthe.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poletti V, et al. Pre-clinical development of a lentiviral vector expressing the anti-sickling βAS3 globin for gene therapy for sickle cell disease. Mol Ther Methods Clin Dev. 2018;11:167–179. doi: 10.1016/j.omtm.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ozog S, et al. Resveratrol trimer enhances gene delivery to hematopoietic stem cells by reducing antiviral restriction at endosomes. Blood. 2019;134(16):1298–1311. doi: 10.1182/blood.2019000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paikari A, Sheehan VA. Fetal haemoglobin induction in sickle cell disease. Br J Haematol. 2018;180(2):189–200. doi: 10.1111/bjh.15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Orkin SH, Bauer DE. Emerging genetic therapy for sickle cell disease. Annu Rev Med. 2019;70:257–271. doi: 10.1146/annurev-med-041817-125507. [DOI] [PubMed] [Google Scholar]
- 68.Lettre G, Bauer DE. Fetal haemoglobin in sickle-cell disease: from genetic epidemiology to new therapeutic strategies. Lancet. 2016;387(10037):2554–2564. doi: 10.1016/S0140-6736(15)01341-0. [DOI] [PubMed] [Google Scholar]
- 69.Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci. 1998;850:38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x. [DOI] [PubMed] [Google Scholar]
- 70.Wienert B, et al. Wake-up sleepy gene: reactivating fetal globin for β-hemoglobinopathies. Trends Genet. 2018;34(12):927–940. doi: 10.1016/j.tig.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 71.Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017;129(20):2719–2726. doi: 10.1182/blood-2017-02-765891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bartman CR, et al. Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol Cell. 2016;62(2):237–247. doi: 10.1016/j.molcel.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Palstra RJ, et al. The beta-globin nuclear compartment in development and erythroid differentiation. Nat Genet. 2003;35(2):190–194. doi: 10.1038/ng1244. [DOI] [PubMed] [Google Scholar]
- 74.Martyn GE, et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet. 2018;50(4):498–503. doi: 10.1038/s41588-018-0085-0. [DOI] [PubMed] [Google Scholar]
- 75.Masuda T, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351(6270):285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu N, et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018;173(2):430–442. doi: 10.1016/j.cell.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guda S, et al. miRNA-embedded shRNAs for lineage-specific BCL11A knockdown and hemoglobin F induction. Mol Ther. 2015;23(9):1465–1474. doi: 10.1038/mt.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brendel C, et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Invest. 2016;126(10):3868–3878. doi: 10.1172/JCI87885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brendel C, et al. Preclinical evaluation of a novel lentiviral vector driving lineage-specific BCL11A knockdown for sickle cell gene therapy. Mol Ther Methods Clin Dev. 2020;17:589–600. doi: 10.1016/j.omtm.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu Y, et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med. 2019;25(5):776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chang KH, et al. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol Ther Methods Clin Dev. 2017;4:137–148. doi: 10.1016/j.omtm.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Canver MC, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527(7577):192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frangoul H, et al. Safety and efficacy of CTX001 in patients with transfusion-dependent β-thalassemia and sickle cell disease: early results from the climb THAL-111 and climb SCD-121 studies of autologous CRISPR-CAS9-modified CD34+ hematopoietic stem and progenitor cells. Blood. 2020;136(suppl 1):3–4. [Google Scholar]
- 84.Traxler EA, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. 2016;22(9):987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Metais JY, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3(21):3379–3392. doi: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weber L, et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. 2020;6(7):eaay9392. doi: 10.1126/sciadv.aay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zeng J, et al. Therapeutic base editing of human hematopoietic stem cells. Nat Med. 2020;26(4):535–541. doi: 10.1038/s41591-020-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chu SH, et al. Adenine base editing of the sickle allele in CD34+ hematopoietic stem and progenitor cells eliminates hemoglobin S. Blood. 2020;136(suppl 1):47. doi: 10.1182/blood-2020-141805. [DOI] [Google Scholar]
- 89.Mayuranathan T, et al. Adenosine base editing of γ-globin promoters induces fetal hemoglobin and inhibit erythroid sickling. Blood. 2020;136(suppl 1):21–22. doi: 10.1182/blood-2020-141498. [DOI] [Google Scholar]
- 90.Hoban MD, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125(17):2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Park SH, et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47(15):7955–7972. doi: 10.1093/nar/gkz475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dever DP, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539(7629):384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pickar-Oliver A, Gersbach CA. The next generation of CRISPR-Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20(8):490–507. doi: 10.1038/s41580-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang H, et al. Methods favoring homology-directed repair choice in response to CRISPR/Cas9 induced-double strand breaks. Int J Mol Sci. 2020;21(18):6461. doi: 10.3390/ijms21186461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.DeWitt MA, et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med. 2016;8(360):360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.DeWitt MA, et al. Gene correction using CRISPR/Cas9: IND-enabling studies to support a clinical trial of a CRISPR/homology-directed repair treatment for sickle cell disease. Mol Ther. 2020;28(4 suppl 1):1–592. [Google Scholar]
- 97.Wilkinson AC, et al. Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat Commun. 2021;12(1):686. doi: 10.1038/s41467-021-20909-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kalkan BM, et al. Development of gene editing strategies for human β-globin (HBB) gene mutations. Gene. 2020;734:144398. doi: 10.1016/j.gene.2020.144398. [DOI] [PubMed] [Google Scholar]
- 99.Pattabhi S, et al. In vivo outcome of homology-directed repair at the HBB gene in HSC using alternative donor template delivery methods. Mol Ther Nucleic Acids. 2019;17:277–288. doi: 10.1016/j.omtn.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Romero Z, et al. Editing the sickle cell disease mutation in human hematopoietic stem cells: comparison of endonucleases and homologous donor templates. Mol Ther. 2019;27(8):1389–1406. doi: 10.1016/j.ymthe.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shin JJ, et al. Controlled cycling and quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Rep. 2020;32(9):108093. doi: 10.1016/j.celrep.2020.108093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wienert B, et al. Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Nat Commun. 2020;11(1):2109. doi: 10.1038/s41467-020-15845-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lattanzi A, et al. Optimization of CRISPR/Cas9 delivery to human hematopoietic stem and progenitor cells for therapeutic genomic rearrangements. Mol Ther. 2019;27(1):137–150. doi: 10.1016/j.ymthe.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Charlesworth CT, et al. Priming human repopulating hematopoietic stem and progenitor cells for Cas9/sgRNA gene targeting. Mol Ther Nucleic Acids. 2018;12:89–104. doi: 10.1016/j.omtn.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anzalone AV, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blackwell RQ, et al. Hemoglobin G Makassar: beta-6 Glu leads to Ala. Biochim Biophys Acta. 1970;214(3):396–401. doi: 10.1016/0005-2795(70)90297-7. [DOI] [PubMed] [Google Scholar]
- 107.Mohamad AS, et al. Human hemoglobin G-Makassar variant masquerading as sickle cell anemia. Hematol Rep. 2018;10(3):7210. doi: 10.4081/hr.2018.7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sangkitporn S, et al. Hb G Makassar (beta 6:Glu-Ala) in a Thai family. J Med Assoc Thai. 2002;85(5):577–582. [PubMed] [Google Scholar]
- 109.Viprakasit V, et al. Hb G-Makassar [β6(A3)Glu→Ala; codon 6 (G A G→G C G)]: molecular characterization, clinical, and hematological effects. Hemoglobin. 2002;26(3):245–253. doi: 10.1081/HEM-120015028. [DOI] [PubMed] [Google Scholar]
- 110.Miller SM, et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol. 2020;38(4):471–481. doi: 10.1038/s41587-020-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yen JS, et al. Base editing eliminates the sickle cell mutation and pathology in hematopoietic stem cells derived erythroid cells. Blood. 2020;136(suppl 1):13–14. doi: 10.1182/blood-2020-139016. [DOI] [Google Scholar]
- 112.Fischer A, Hacein-Bey-Abina S. Gene therapy for severe combined immunodeficiencies and beyond. J Exp Med. 2020;217(2):e20190607. doi: 10.1084/jem.20190607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Espinoza DA, et al. Aberrant clonal hematopoiesis following lentiviral vector transduction of HSPCs in a Rhesus Macaque. Mol Ther. 2019;27(6):1074–1086. doi: 10.1016/j.ymthe.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cavazzana-Calvo M, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467(7313):318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scala S, et al. Dynamics of genetically engineered hematopoietic stem and progenitor cells after autologous transplantation in humans. Nat Med. 2018;24(11):1683–1690. doi: 10.1038/s41591-018-0195-3. [DOI] [PubMed] [Google Scholar]
- 116.Six E, et al. Clonal tracking in gene therapy patients reveals a diversity of human hematopoietic differentiation programs. Blood. 2020;135(15):1219–1231. doi: 10.1182/blood.2019002350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miles LA, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587(7834):477–482. doi: 10.1038/s41586-020-2864-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim D, et al. Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu Rev Biochem. 2019;88:191–220. doi: 10.1146/annurev-biochem-013118-111730. [DOI] [PubMed] [Google Scholar]
- 119.Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet. 2016;17(5):300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cheng Y, Tsai SQ. Illuminating the genome-wide activity of genome editors for safe and effective therapeutics. Genome Biol. 2018;19(1):226. doi: 10.1186/s13059-018-1610-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Haapaniemi E, et al. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24(7):927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 122.van den Berg J, et al. A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res. 2018;46(19):10132–10144. doi: 10.1093/nar/gky786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Enache OM, et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat Genet. 2020;52(7):662–668. doi: 10.1038/s41588-020-0623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ihry RJ, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24(7):939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 125.Zuccaro MV, et al. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell. 2020;183(6):1650–1664. doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
- 126.Giannoukos G, et al. UDiTaS, a genome editing detection method for indels and genome rearrangements. BMC Genomics. 2018;19(1):212. doi: 10.1186/s12864-018-4561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liang M, et al. AcrIIA5 suppresses base editors and reduces their off-target effects. Cells. 2020;9(8):E1786. doi: 10.3390/cells9081786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Coelho MA, et al. CRISPR GUARD protects off-target sites from Cas9 nuclease activity using short guide RNAs. Nat Commun. 2020;11(1):4132. doi: 10.1038/s41467-020-17952-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Naeem M, et al. Latest developed strategies to minimize the off-target effects in CRISPR-Cas-mediated genome editing. Cells. 2020;9(7):1608. doi: 10.3390/cells9071608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kosicki M, et al. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36(8):765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhou C, et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571(7764):275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 132.Grunewald J, et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569(7756):433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jin S, et al. Rationally designed APOBEC3B cytosine base editors with improved specificity. Mol Cell. 2020;79(5):728–740. doi: 10.1016/j.molcel.2020.07.005. [DOI] [PubMed] [Google Scholar]
- 134.Jeong YK, et al. Current status and challenges of DNA base editing tools. Mol Ther. 2020;28(9):1938–1952. doi: 10.1016/j.ymthe.2020.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee S, et al. Single C-to-T substitution using engineered APOBEC3G-nCas9 base editors with minimum genome- and transcriptome-wide off-target effects. Sci Adv. 2020;6(29):eaba1773. doi: 10.1126/sciadv.aba1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zuo E, et al. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat Methods. 2020;17(6):600–604. doi: 10.1038/s41592-020-0832-x. [DOI] [PubMed] [Google Scholar]
- 137.Tan J, et al. Expanding the genome-targeting scope and the site selectivity of high-precision base editors. Nat Commun. 2020;11(1):629. doi: 10.1038/s41467-020-14465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liang P, et al. Off-target effects of cytidine base editor and adenine base editor: what can we do? J Genet Genomics. 2019;46(11):509–512. doi: 10.1016/j.jgg.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 139.Doman JL, et al. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat Biotechnol. 2020;38(5):620–628. doi: 10.1038/s41587-020-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu Y, et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat Commun. 2020;11(1):2052. doi: 10.1038/s41467-020-15887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kim D, et al. Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat Biotechnol. 2019;37(4):430–435. doi: 10.1038/s41587-019-0050-1. [DOI] [PubMed] [Google Scholar]
- 142.Liang P, et al. Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat Commun. 2019;10(1):67. doi: 10.1038/s41467-018-07988-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim D, et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol. 2017;35(5):475–480. doi: 10.1038/nbt.3852. [DOI] [PubMed] [Google Scholar]
- 144.Kim DY, et al. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res. 2020;48(18):10576–10589. doi: 10.1093/nar/gkaa764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. doi: 10.1101/2020.11.07.372748. Gao P, et al. Prime editing in mice reveals the essentiality of a single base in driving tissue-specific gene expression [preprint]. Posted on bioRxiv November 8, 2020. [DOI] [PMC free article] [PubMed]
- 146.Schene IF, et al. Prime editing for functional repair in patient-derived disease models. Nat Commun. 2020;11(1):5352. doi: 10.1038/s41467-020-19136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Marktel S, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent β-thalassemia. Nat Med. 2019;25(2):234–241. doi: 10.1038/s41591-018-0301-6. [DOI] [PubMed] [Google Scholar]
- 148.Thompson AA, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 149.Ciurea SO, Andersson BS. Busulfan in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2009;15(5):523–536. doi: 10.1016/j.bbmt.2008.12.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.McNerney ME, et al. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17(9):513–527. doi: 10.1038/nrc.2017.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fitzhugh CD, et al. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood. 2017;130(17):1946–1948. doi: 10.1182/blood-2017-03-772392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Walters MC, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001;7(12):665–673. doi: 10.1053/bbmt.2001.v7.pm11787529. [DOI] [PubMed] [Google Scholar]
- 153.Abraham A, et al. Unrelated umbilical cord blood transplantation for sickle cell disease following reduced-intensity conditioning: results of a phase I trial. Biol Blood Marrow Transplant. 2017;23(9):1587–1592. doi: 10.1016/j.bbmt.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 154.Malik P, et al. Gene therapy for sickle cell anemia using a modified gamma globin lentivirus vector and reduced intensity conditioning transplant shows promising correction of the disease phenotype. Blood. 2018;132(suppl 1):1021. doi: 10.1182/blood-2018-99-119591. [DOI] [Google Scholar]
- 155.Ghannam JY, et al. Baseline TP53 mutations in adults with SCD developing myeloid malignancy following hematopoietic cell transplantation. Blood. 2020;135(14):1185–1188. doi: 10.1182/blood.2019004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wong TN, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hourigan CS, et al. Impact of conditioning intensity of allogeneic transplantation for acute myeloid leukemia with genomic evidence of residual disease. J Clin Oncol. 2020;38(12):1273–1283. doi: 10.1200/JCO.19.03011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Williams BA, et al. Antibody therapies for acute myeloid leukemia: unconjugated, toxin-conjugated, radio-conjugated and multivalent formats. J Clin Med. 2019;8(8):1261. doi: 10.3390/jcm8081261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Agarwal R, et al. Non-genotoxic anti-CD117 antibody conditioning results in successful hematopoietic stem cell engraftment in patients with severe combined immunodeficiency. Blood. 2019;134(suppl 1):800. doi: 10.1182/blood-2019-126239. [DOI] [Google Scholar]
- 160.Agarwal R, et al. Toxicity-free hematopoietic stem cell engraftment achieved with anti-CD117 monoclonal antibody conditioning. Biol Blood Marrow Transplant. 2019;25(3):S92. doi: 10.1016/j.bbmt.2018.12.172. [DOI] [Google Scholar]
- 161.Chhabra A, et al. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci Transl Med. 2016;8(351):351ra105. doi: 10.1126/scitranslmed.aae0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Marjon KD, et al. An all antibody approach for conditioning bone marrow for hematopoietic stem cell transplantation with anti-cKIT and anti-CD47 in non-human primates. Blood. 2019;134(suppl 1):4428. doi: 10.1182/blood-2019-131490. [DOI] [Google Scholar]
- 163.Czechowicz A, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun. 2019;10(1):617. doi: 10.1038/s41467-018-08201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gao C, et al. Nongenotoxic antibody-drug conjugate conditioning enables safe and effective platelet gene therapy of hemophilia A mice. Blood Adv. 2019;3(18):2700–2711. doi: 10.1182/bloodadvances.2019000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li Z, et al. Hematopoietic chimerism and donor-specific skin allograft tolerance after non-genotoxic CD117 antibody-drug-conjugate conditioning in MHC-mismatched allotransplantation. Nat Commun. 2019;10(1):616. doi: 10.1038/s41467-018-08202-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Palchaudhuri R, et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat Biotechnol. 2016;34(7):738–745. doi: 10.1038/nbt.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tisdale JF, et al. A single dose of CD117 antibody drug conjugate enables autologous gene-modified hematopoietic stem cell transplant (gene therapy) in nonhuman primates. Blood. 2019;134(suppl 1):610. doi: 10.1182/blood-2019-125968. [DOI] [Google Scholar]
- 168.Scheinberg DA, et al. A phase I trial of monoclonal antibody M195 in acute myelogenous leukemia: specific bone marrow targeting and internalization of radionuclide. J Clin Oncol. 1991;9(3):478–490. doi: 10.1200/JCO.1991.9.3.478. [DOI] [PubMed] [Google Scholar]
- 169.Burke JM, et al. Cytoreduction with iodine-131-anti-CD33 antibodies before bone marrow transplantation for advanced myeloid leukemias. Bone Marrow Transplant. 2003;32(6):549–556. doi: 10.1038/sj.bmt.1704201. [DOI] [PubMed] [Google Scholar]
- 170.Matthews DC, et al. Phase I study of (131)I-anti-CD45 antibody plus cyclophosphamide and total body irradiation for advanced acute leukemia and myelodysplastic syndrome. Blood. 1999;94(4):1237–1247. [PubMed] [Google Scholar]
- 171.Mawad R, et al. Radiolabeled anti-CD45 antibody with reduced-intensity conditioning and allogeneic transplantation for younger patients with advanced acute myeloid leukemia or myelodysplastic syndrome. Biol Blood Marrow Transplant. 2014;20(9):1363–1368. doi: 10.1016/j.bbmt.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kornblit B, et al. Conditioning with α-emitter based radioimmunotherapy in canine allogeneic hematopoietic cell transplantation. Chimerism. 2012;3(2):40–42. doi: 10.4161/chim.20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Burtner CR, et al. 211Astatine-conjugated monoclonal CD45 antibody-based nonmyeloablative conditioning for stem cell gene therapy. Hum Gene Ther. 2015;26(6):399–406. doi: 10.1089/hum.2015.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jurcic JG. Radioimmunotherapy for hematopoietic cell transplantation. Immunotherapy. 2013;5(4):383–394. doi: 10.2217/imt.13.11. [DOI] [PubMed] [Google Scholar]
- 175.Altrock PM, et al. Mathematical modeling of erythrocyte chimerism informs genetic intervention strategies for sickle cell disease. Am J Hematol. 2016;91(9):931–937. doi: 10.1002/ajh.24449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Andreani M, et al. Quantitatively different red cell/nucleated cell chimerism in patients with long-term, persistent hematopoietic mixed chimerism after bone marrow transplantation for thalassemia major or sickle cell disease. Haematologica. 2011;96(1):128–133. doi: 10.3324/haematol.2010.031013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kanter J, et al. Outcomes in patients treated with lentiglobin for sickle cell disease (SCD) gene therapy: updated results from the phase 1/2 HGB-206 group c study. EHA Library. 2020;295102:S282 [Google Scholar]
- 178.Demirci S, et al. βT87Q-globin gene therapy reduces sickle hemoglobin production, allowing for ex vivo anti-sickling activity in human erythroid cells. Mol Ther Methods Clin Dev. 2020;17:912–921. doi: 10.1016/j.omtm.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Steinberg MH. Fetal hemoglobin in sickle cell anemia. Blood. 2020;136(21):2392–2400. doi: 10.1182/blood.2020007645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87(8):795–803. doi: 10.1002/ajh.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Calvet D, et al. Low fetal hemoglobin percentage is associated with silent brain lesions in adults with homozygous sickle cell disease. Blood Adv. 2017;1(26):2503–2509. doi: 10.1182/bloodadvances.2017005504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sommet J, et al. Clinical and haematological risk factors for cerebral macrovasculopathy in a sickle cell disease newborn cohort: a prospective study. Br J Haematol. 2016;172(6):966–977. doi: 10.1111/bjh.13916. [DOI] [PubMed] [Google Scholar]
- 183.van der Land V, et al. Risk factor analysis of cerebral white matter hyperintensities in children with sickle cell disease. Br J Haematol. 2016;172(2):274–284. doi: 10.1111/bjh.13819. [DOI] [PubMed] [Google Scholar]
- 184.DeBaun MR, et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood. 2012;119(16):3684–3690. doi: 10.1182/blood-2011-05-349621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ohene-Frempong K, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294. [PubMed] [Google Scholar]
- 186.Habara AH, et al. Fetal hemoglobin in sickle cell anemia: the Arab-Indian haplotype and new therapeutic agents. Am J Hematol. 2017;92(11):1233–1242. doi: 10.1002/ajh.24872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Adekile A. The genetic and clinical significance of fetal hemoglobin expression in sickle cell disease. Med Princ Pract. doi: 10.1159/000511342. [published online Semptember 4, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ngo DA, et al. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br J Haematol. 2012;156(2):259–264. doi: 10.1111/j.1365-2141.2011.08916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123(4):481–485. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 190.Man Y, et al. Standardized microfluidic assessment of red blood cell mediated microcapillary occlusion: association with clinical phenotype and hydroxyurea responsiveness in sickle cell disease. Microcirculation. doi: 10.1111/micc.12662. [published online October 6, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Xu T, et al. Characterization of red blood cell microcirculatory parameters using a bioimpedance microfluidic device. Sci Rep. 2020;10(1):9869. doi: 10.1038/s41598-020-66693-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Valdez JM, et al. A microfluidic platform for simultaneous quantification of oxygen-dependent viscosity and shear thinning in sickle cell blood. APL Bioeng. 2019;3(4):046102. doi: 10.1063/1.5118212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Francis AT, et al. Direct quantification of single red blood cell hemoglobin concentration with multiphoton microscopy. Anal Chem. 2020;92(18):12235–12241. doi: 10.1021/acs.analchem.0c01609. [DOI] [PubMed] [Google Scholar]
- 194.Chaudhury A, et al. Single-cell modeling of routine clinical blood tests reveals transient dynamics of human response to blood loss. Elife. 2019;8:e48590. doi: 10.7554/eLife.48590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Di Caprio G, et al. High-throughput assessment of hemoglobin polymer in single red blood cells from sickle cell patients under controlled oxygen tension. Proc Natl Acad Sci U S A. 2019;116(50):25236–25242. doi: 10.1073/pnas.1914056116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bolanos-Meade J, et al. Effect of increased dose of total body irradiation on graft failure associated with HLA-haploidentical transplantation in patients with severe haemoglobinopathies: a prospective clinical trial. Lancet Haematol. 2019;6(4):e183–e193. doi: 10.1016/S2352-3026(19)30031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.de la Fuente J, et al. Haploidentical bone marrow transplantation with post-transplantation cyclophosphamide plus thiotepa improves donor engraftment in patients with sickle cell anemia: results of an international learning collaborative. Biol Blood Marrow Transplant. 2019;25(6):1197–1209. doi: 10.1016/j.bbmt.2018.11.027. [DOI] [PubMed] [Google Scholar]
- 198.Fitzhugh CD, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv. 2017;1(11):652–661. doi: 10.1182/bloodadvances.2016002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Brodsky RA, DeBaun MR. Are genetic approaches still needed to cure sickle cell disease? J Clin Invest. 2020;130(1):7–9. doi: 10.1172/JCI133856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Leonard A, Tisdale JF. A pause in gene therapy: reflecting on the unique challenges of sickle cell disease. Mol Ther. doi: 10.1016/j.ymthe.2021.03.010. [published online March 19, 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Park SY, et al. Pathologic angiogenesis in the bone marrow of humanized sickle cell mice is reversed by blood transfusion. Blood. 2020;135(23):2071–2084. doi: 10.1182/blood.2019002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. National Heart, Lung, and Blood Institute. Statement on NHLBI decision to pause the Pilot and Feasibility Study of Hematopoietic Stem Cell Gene Transfer for Sickle Cell Disease. https://www.nhlbi.nih.gov/education-and-awareness/sickle-cell/statement-study-gene-transfer-scd Updated February 22, 2021. Accessed March 8, 2021.
- 203.[No authors listed] Other pharmaceutical agents. IARC Monogr Eval Carcinog Risks Hum. 2000;76:345–486. [PMC free article] [PubMed] [Google Scholar]
- 204.Yawn BP, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–1048. doi: 10.1001/jama.2014.10517. [DOI] [PubMed] [Google Scholar]
- 205.Brunson A, et al. Increased risk of leukemia among sickle cell disease patients in California. Blood. 2017;130(13):1597–1599. doi: 10.1182/blood-2017-05-783233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li C, et al. Targeted integration and high-level transgene expression in AAVS1 transgenic mice after in vivo HSC transduction with HDAd5/35++ vectors. Mol Ther. 2019;27(12):2195–2212. doi: 10.1016/j.ymthe.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Mangeot PE, et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat Commun. 2019;10(1):45. doi: 10.1038/s41467-018-07845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wang H, et al. In vivo hematopoietic stem cell gene therapy ameliorates murine thalassemia intermedia. J Clin Invest. 2019;129(2):598–615. doi: 10.1172/JCI122836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cohen J, Kaiser J. Gates and NIH join forces on HIV and sickle cell diseases. Science. 2019;366(6465):558–559. doi: 10.1126/science.366.6465.558-b. [DOI] [PubMed] [Google Scholar]
- 210. Kwiatkowski JL, et al. Long-term efficacy and safety of betibeglogene autotemcel gene therapy for the treatment of transfusion-dependent β-thalassemia: results in patients with up to 6 years of follow-up. Paper presented at: 62nd ASH Annual Meeting and Exposition; December 5–8, 2020; USA. https://ash.confex.com/ash/2020/webprogram/Paper135850.html Accessed March 29, 2021.
- 211.Smith AR, et al. Preliminary results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent beta thalassemia. Blood. 2019;134(suppl 1):3544. doi: 10.1182/blood-2019-125743. [DOI] [Google Scholar]