

Abstract
Aims
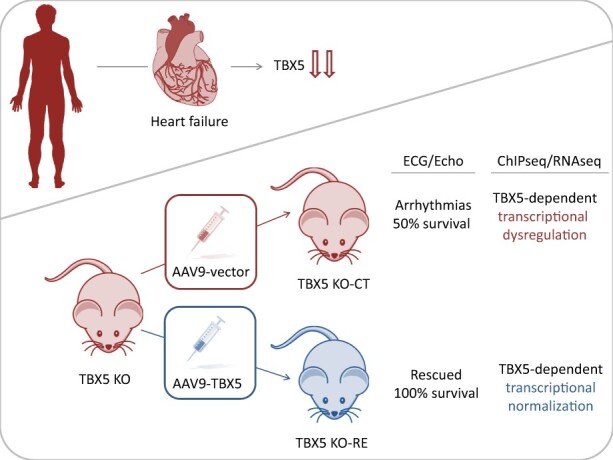
Arrhythmias and sudden cardiac death (SCD) occur commonly in patients with heart failure. We found T-box 5 (TBX5) dysregulated in ventricular myocardium from heart failure patients and thus we hypothesized that TBX5 reduction contributes to arrhythmia development in these patients. To understand the underlying mechanisms, we aimed to reveal the ventricular TBX5-dependent transcriptional network and further test the therapeutic potential of TBX5 level normalization in mice with documented arrhythmias.
Methods and results
We used a mouse model of TBX5 conditional deletion in ventricular cardiomyocytes. Ventricular (v) TBX5 loss in mice resulted in mild cardiac dysfunction and arrhythmias and was associated with a high mortality rate (60%) due to SCD. Upon angiotensin stimulation, vTbx5KO mice showed exacerbated cardiac remodelling and dysfunction suggesting a cardioprotective role of TBX5. RNA-sequencing of a ventricular-specific TBX5KO mouse and TBX5 chromatin immunoprecipitation was used to dissect TBX5 transcriptional network in cardiac ventricular tissue. Overall, we identified 47 transcripts expressed under the control of TBX5, which may have contributed to the fatal arrhythmias in vTbx5KO mice. These included transcripts encoding for proteins implicated in cardiac conduction and contraction (Gja1, Kcnj5, Kcng2, Cacna1g, Chrm2), in cytoskeleton organization (Fstl4, Pdlim4, Emilin2, Cmya5), and cardiac protection upon stress (Fhl2, Gpr22, Fgf16). Interestingly, after TBX5 loss and arrhythmia development in vTbx5KO mice, TBX5 protein-level normalization by systemic adeno-associated-virus (AAV) 9 application, re-established TBX5-dependent transcriptome. Consequently, cardiac dysfunction was ameliorated and the propensity of arrhythmia occurrence was reduced.
Conclusions
This study uncovers a novel cardioprotective role of TBX5 in the adult heart and provides preclinical evidence for the therapeutic value of TBX5 protein normalization in the control of arrhythmia.
Keywords: Sudden cardiac death, T-box 5, Arrhythmia, AAV9 in vivo re-expression, Heart failure
Graphical Abstract
1. Introduction
Cardiovascular disease (CVD) is the number one cause of death worldwide (World Health Organization factsheets 09/2016). More than 60% of all deaths due to CVDs are represented by out-of-hospital sudden cardiac death (SCD).1 Current pharmacological heart failure treatments aim at protecting the heart from neurohumoral overstimulation, which may trigger fatal arrhythmias.
T-box 5 (TBX5) is an essential transcription factor for cardiac development.2 In humans, TBX5 mutations have been shown to account for 35% of Holt–Oram Syndrome (HOS) cases.3 HOS is a rare autosomal congenital disease linked to abnormal cardiac electrophysiology and arrhythmias.4 A genome-wide association study (GWAS) revealed a strong association between alterations in the TBX5 locus and atrial fibrillation, atrioventricular block, and QRS prolongation,5 suggesting a role of TBX5 in cardiac rhythm control beyond embryonic development.
In line with the human data, heterozygous TBX5 knockout (KO) mice exhibit a similar phenotype as observed in patients with HOS, including conduction defects, heart, and limb malformations.6 Specific deletion of TBX5 in the adult ventricular conduction system (VCS) resulted in reduced Nav1.5 and CX40 expression, loss of fast conduction, arrhythmias, and SCD.7 In the adult atria, inducible TBX5 loss of function caused primary spontaneous and sustained atrial fibrillation.8 While the role of TBX5 for normal electrophysiological function in atrial tissue and the VCS is well established, it remains unclear whether TBX5 plays a critical role for cardiac homeostasis in adult ventricular cardiomyocytes (CMs).
We found that TBX5 protein abundance is significantly lower in left ventricular biopsies of patients with human ischaemic heart disease and dilated cardiomyopathies (DCMs) when compared to non-failing (NF) donor hearts. Therefore, we investigated the relevance of Tbx5 loss in adult ventricular myocardium by generating an inducible ventricular CM-specific Tbx5 KO model (vTbx5KO). Integrative analysis of RNA-sequencing and chromatin immunoprecipitation-sequencing revealed novel TBX5 transcriptional targets in the adult ventricles responsible for electrical signal propagation and cardioprotection. Finally, we tested the potential of TBX5 level normalization to rescue vTbx5KO mice from arrhythmias and SCD.
2. Methods
2.1. Animal experiments
Tbx5LDN/LDN mice9 were crossed with Myh6-MerCreMer10 mice in a C57BL/6N background. In 10–12 weeks old mice, activation of Cre-recombinase was induced by i.p. injections of tamoxifen (TX) for three subsequent days [30 mg/kg/day (Sigma Aldrich, Hamburg/Germany), dissolved in 10% Ethanol (Carl Roth, Karlsruhe/Germany), and 90% Miglyol (Caelo, Hilden/Germany)]. We named the recombined mice as vTbx5KO, Tbx5LDN/LDN mice as Flox, and Myh6-MerCreMer mice as Cre throughout the study. Irrespective of the genotype, all animals were injected with TX to control for TX -induced effects. Recombination was confirmed by reverse transcription and quantitative polymerase chain reaction (RT-(q)PCR) using a primer pair flanking exons 3 and 4 of Txb5 transcript (Supplementary material online, Table S1).
For hypertrophy induction (experiments described in Figure 2E), 2 weeks upon TX injections, osmotic minipumps (Alzet) were subcutaneously implanted in vTbx5KO or Flox mice for the delivery of angiotensin (Ang) II (Ang; 1.44 mg/kg/day for 2 weeks) as previously described.11 Saline solution was used as vehicle. Osmotic minipumps were implanted under anaesthesia consisting of Medetomidine (0.5 mg/kg body weight in 0.9% NaCl, i.p.), Midazolam (5 mg/kg body weight in 0.9% NaCl, i.p.), and Fentanyl (0.5 mg/kg body weight in 0.9% NaCl, i.p.). Anaesthesia was antagonized by Atipamezol (2.5 mg/kg body weight in 0.9% NaCl, s.c.) and Flumazenil (0.5 mg/kg body weight in 0.9% NaCl, s.c.). Anaesthesia medication was delivered by intraperitoneal injections. There is a postoperative pain therapy with Buprenorphine (0.05 mg/kg body weight, s.c.). For in vivo re-expression adeno-associated-virus (AAV) vectors were injected into the tail vein (2 × 1012 viral particles/mouse) of anaesthetized mice, 2 weeks after TX injections under 2% [vol/vol] Isoflurane inhalation.
Figure 2.

Characterization of vTbx5KO mice cardiac function under basal and stress conditions. (A) Mating scheme for vTbx5KO mouse generation by mating Myh6-MerCreMer10 and TBX5LDN/LDN9 mice. (B) RT-(q)PCR analysis of TBX5 recombination in the ventricles and in the atria (Flox n = 5; KO = 6 biological replicates). (C) Survival curve of vTbx5KO mice shows significantly reduced lifespan as compared to control mice (Cre n = 9; Flox n = 4; KO n = 24 biological replicates). (D) vTbx5KO shows cardiac dysfunction with mildly reduced EF and SV decrease 16 weeks post-recombination (KO pre n = 19, 8w n = 18, 16w n = 12 biological replicates: the animal number decreased due to SCD). (E) Angiotensin (Ang) II-treated vTbx5KO mice show exacerbated cardiac function (EF), hypertrophic remodelling (HW/BW; heart weight/body weight, LVPWth; left ventricular posterior wall thickness) and decompensation (LVIDd; left ventricular inner diameter in diastole) as compared to Ang II-treated Flox mice (Flox vehicle n = 19; Flox Ang n = 11; KO vehicle n = 7; KO Ang n = 7 biological replicates). (F) CM Cross-sectional cell area (CSA) is increased in Ang II-treated vTbx5KO mice compared to Ang-Flox mice (Flox vehicle n = 19; Flox Ang n = 11; KO vehicle n = 12; KO Ang n = 11 biological replicates). (G) Collagen staining with picrosirius red shows that Ang II-induced fibrosis is exacerbated in vTbx5KO vs. Flox mice. Scale bar: 1 mm. Data are presented as mean ± SEM. Statistical analysis was performed by (B) one-way ANOVA with Dunnett’s multiple comparison post hoc test; (C) log-rank test (Mantel–Cox); (D–F) paired t-tests; (G) One-way ANOVA followed by Sidak’s multiple comparison post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
2.2. Echocardiography analysis
Echocardiography was performed in anaesthetized mice, under 2% [vol/vol] Isoflurane inhalation, as described previously.12 Ventricular dimensions were measured with a Visual Sonics Vevo 2100 Imaging System equipped with a 45 MHz MS-550D MicroScan transducer. The observer was unaware of genotype and treatment. All procedures were performed by the SFB 1002 service unit (S01 Disease Models) according to standard operating procedures.
2.3. Telemetric electrocardiogram analysis
Mice were implanted with telemetric electrocardiogram (ECG) transmitters ETA-F10 (Data Science International) subcutaneously as described before.13 The telemetry transmitter (model: TA-F10, Data Sciences International) was implanted under anaesthesia consisting of Medetomidine (0.5 mg/kg body weight in 0.9% NaCl, i.p.), Midazolam (5 mg/kg body weight in 0.9% NaCl, i.p.), and Fentanyl (0.5 mg/kg body weight in 0.9% NaCl, i.p.). Anaesthesia was antagonized by Atipamezol (2.5 mg/kg body weight in 0.9% NaCl, s.c.) and Flumazenil (0.5 mg/kg body weight in 0.9% NaCl, s.c.). Anaesthesia medication was delivered by intraperitoneal injections. There is a postoperative pain therapy with Buprenorphine (0.05 mg/kg body weight, s.c.). Mice were allowed to recover and stabilize for 2 weeks prior to any intervention. Twenty-four hours ECGs were recorded before KO induction with TX and 1, 2, 4, and 8 weeks after recombination. ECG recordings were analysed with Ponemah Physiology Platform 6.3 (Data Science International) using template-based analysis.
2.4. AAV vector production
AAV serotype 9 vector production and purification were done according to Jungmann et al.14 In short, the helper plasmid pDP9rs (a derivate of pDP2rs) and an AAV vector genome plasmid (either pds Tnnt2-rluc or pds- Tnnt2 -mTBX5) were co-transfected into 293 T cells resulting in AAV9-luc or AAV9-TBX5. pds- Tnnt2 -rluc contains a Renilla luciferase reporter gene and pds Tnnt2-mTBX5 contains the murine cDNA of TBX5 both under control of the −502/+42 bp human troponin-T promoter (Tnnt2).15 AAV vectors were purified using Iodixanol step gradient centrifugation and titrated as reported before.14
2.5. Electrophysiological study of isolated hearts
Hearts were excised under deep terminal anaesthesia (single intraperitoneal dose 400 mg/kg pentobarbital sodium), and the aorta was cannulated and retrogradely perfused using 37°C Krebs-Henseleit buffer (in mmol/L; NaCl 118, NaHCO3 24.88, KH2PO4 41.18, glucose 5.55, Na-pyruvate 2, MgSO4 0.83, CaCl2 1.8, KCl 4.7) equilibrated with a 95% oxygen/5% carbon dioxide gas mixture. The hearts were mounted on a vertical Langendorff apparatus (Hugo Sachs Electronic-Harvard Apparatus GmbH) and constantly perfused. An octapolar mouse electrophysiologic catheter (CIBER MOUSE; NuMED) was placed in the right atria and ventricles for atrial and ventricular pacing, respectively. Three murine monophasic action potentials were continuously and simultaneously recorded from the right ventricular free wall, septal area, and left ventricular free wall epicardium.16 Atrial S1 pacing was performed to measure activation times from right atrium to right ventricle; endocardial right ventricular pacing was performed to measure ventricular activation times and both for steady-state action potential durations. To test ventricular arrhythmia inducibility, programmed ventricular stimulation was performed using a single encroaching premature stimulus (S2). All signals were digitized and stored on digital media for offline analysis. Experiments and analyses were performed in a blinded fashion. Details of the method have been described previously.17
2.6. RNA isolation, reverse transcription, and quantitative polymerase chain reaction analysis
RNA was isolated using the NucleoSpin® RNA kit (Macherey-Nagel, Dueren/Germany) according to the manufacturer’s instructions. RT-(q)PCR were performed as described previously.18 All primer sequences used in this study are listed in Supplementary material online, Table S1.
2.7. Statistical analysis
For comparison of more than two groups, we used one-way or two-way ANOVA followed by appropriate post hoc test as indicated in the figure legends. When two groups were compared Student’s t-test was carried out. Data are presented as individual data points with mean (indicated by a horizontal line), bar graphs with standard error of the mean (SEM), or as box-and-whiskers-plots with the box extending from the 25th to 75th percentile, whiskers indicating min to max, + indicates the mean value, and the line indicates the median. Values are presented as mean ± SEM. P <0 .05 values were considered significant.
2.8. Study approval
All animal experiments were approved by the local competent authority; Lower Saxony Animal Review Board—LAVES; AZ-G15/2029) or UK national body (30/2967), respectively. All procedures conform to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
All DCM, ischaemic cardiomyopathy (ICM), and NF patient samples were collected (upon written consent) according to the Declaration of Helsinki and were approved by the local Ethics Committee at the University Medical Center Goettingen (AZ-31/9/00). NF controls derive from anonymous heart transplant donors. No additional information is available for these samples.
3. Results
3.1. Reduced ventricular TBX5 expression is associated with failing myocardium
Although the effects of TBX5 loss in congenital heart disease have been well documented in humans and rodents TBX5 regulation in heart failure was elusive.19 We examined TBX5 expression in samples obtained from the left ventricles of NF hearts and from patients with ICM and DCM; 89% of the patients presented with arrhythmias as ventricular tachycardia, atrial fibrillation, or syncope (Figure 1A). We found TBX5 protein to be of significantly lower abundance in ICM and DCM vs. NF heart muscle, suggesting a role for TBX5 beyond the context of congenital heart disease (Figure 1B).
Figure 1.

TBX5 expression in human and mouse left ventricles. (A) Table showing the information of the individuals from whom ventricular biopsies were obtained. Patients with DCM and ICM in contrast to NF controls, 89% of them suffered from arrhythmias. (B) Representative immunoblot analysis of TBX5 in human left ventricles of DCM, ICM, and NF samples normalized to CASQ2 or GAPDH. Samples loaded on the same blot, but non-contiguously, are indicated by a black line (total sample size: NF n = 7; DCM n = 11; ICM n = 11 biological replicates). Data are presented as mean ± SEM. Statistical analysis was performed by one-way ANOVA with Dunnett’s multiple comparison post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001.
In line with previous reports showing strong expression of TBX5 in the adult murine atria and the VCS (6), we found the ventricular TBX5 transcript in the ventricles corresponded to ∼5% of the expression observed in the atria (Figure 2B, Tbx5 levels of Flox ventricles vs. Flox atria). Immunostainings demonstrated prominent TBX5 signal in atrial tissue and AV node and lower levels of TBX5 in CX43pos cells throughout the mouse ventricular myocardium (Supplementary material online, Figure S1).
Intrigued by this expression, we aimed to investigate the role of TBX5 in the adult heart and generated a conditional TX-inducible CM-specific KO mouse model (Myh6-MerCreMer/TBX5LDN/LDN) by mating two established mouse models (Figure 2A).9,10 TX-induced recombination significantly reduced TBX5 transcript in the ventricles, but not in the atria (Figure 2B), resulting in a ventricular-specific Tbx5 KO model (as vTbx5KO). Immunofluorescence analysis of the different cardiac compartments showed that after recombination TBX5 was still expressed in the VCS (AV node and Bundle of HIS) and atria (right and left) but not in the right and left ventricles (Supplementary material online, Figure S2A–D). Semi-quantification analysis further corroborated this observation and showed an insignificant reduction of TBX5 positive nuclei in vTbx5KO mice in atrial and nodal cells but a significant reduction ventricular CMs (Supplementary material online, Figure S2E).
TBX5 loss in the ventricles significantly reduced animal survival 3–4 months after recombination (Figure 2C). Sixteen weeks post-recombination, stroke volume (SV) was significantly reduced, and cardiac function as indicated by ejection fraction (EF) was deteriorated (Figure 2D) in comparison to Cre controls (Supplementary material online, Figure S3A). No fibrosis was observed in Cre control or vTbx5KO hearts as shown by picrosirius red staining (Supplementary material online, Figure S3B).
3.2. vTbx5KO leads to accelerated cardiac decompensation upon remodelling
To study the role of TBX5 upon cardiac remodelling, we induced mild hypertrophy by chronic stimulation with Ang II in vTbx5KO and TBX5LDN/LDN Cre negative (Flox) control mice. Four weeks after recombination, Flox and vTbx5KO mice did not show differences in cardiac function (EF), hypertrophic growth (HW/BW; heart weight/body weight, LVPWth; left ventricular posterior wall thickness), or fibrosis (Figure 2E–G). Ang II-treated vTbx5KO mice developed heart failure with reduced EF (Figure 2E) and an exacerbated hypertrophic and fibrotic response (Figure 2E–G). Although Ang II treatment-induced hypertrophy in both Flox and vTbx5KO mice (Figure 2E), only the latter exhibited left ventricular dilation (LVIDd; left ventricular inner diameter in diastole; Figure 2E), indicating an earlier onset of decompensation in the absence of TBX5.
3.3. TBX5 expression is essential for electrical signal propagation in the adult ventricle
To evaluate the impact of TBX5 loss on electrical signal propagation, we monitored cardiac rhythm by telemetric ECG analysis. Two weeks upon recombination, we observed significant PR and QRS prolongation (Figure3A andB) in vTbx5KO mice. Flox and Cre controls had no significant changes in ECG parameters. Eight weeks upon recombination PR and QRS interval were prolonged by 26 ± 2 and 7.5 ± 1.2 ms, respectively. In line, QT interval was significantly increased and the heart rate (HR) was decreased (Supplementary material online, Figure S3C). All vTbx5KO mice presented 2nd-degree atrioventricular blocks, in line with published data.7,8,19 We also recorded 3rd-degree AV blocks with ventricular escape rhythm, ventricular tachyarrhythmias, and occasionally asystoles (Figure 3C). The increased propensity for ventricular arrhythmia was further demonstrated by right ventricular endocardial septal S1S2 pacing in isolated hearts showing arrhythmias in 60% of the vTbx5KO vs. 23% and 38% in Flox and Cre hearts, respectively (Supplementary material online, Figure S3D).
Figure 3.

vTbx5KO mice presented with conduction defects and arrhythmia. (A) Representative ECG traces of Flox and vTbx5KO mice recorded by telemetric ECG 2 weeks upon recombination. (B) Statistical analysis of telemetric ECG measurements reveals prolonged PR and QRS intervals from 1 to 8 weeks post-rec. Line indicates Cre control mean value ± SEM 4 weeks post-rec (Cre n = 6; Flox n = 6; KO n = 7–13 biological replicates. The animal number decreased during measurements due to SCD). (C) vTbx5KO mice present with atrioventricular blocks, ventricular tachycardias, and asystoles. (D) Electrophysiological studies of isolated paced Flox, Cre, and vTbx5KO hearts show prolonged activation times from RA to RV, endocardial RV to epicardial RV, RV to septum, and RV to LV (Cre n = 10; Flox n = 8; KO n = 8 biological replicates). Data are presented as mean ± SEM. Statistical analysis was performed by (B) one-way ANOVA with Sidak’s multiple comparison post hoc test against pre-recombination data of the same group; (D) One-way ANOVA followed by Tukey’s multiple comparison post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
S1 pacing in isolated hearts with an octapolar catheter showed a prolonged electrical propagation from the RA to RV in vTbx5KO (13.5 ± 4.6 ms vs. Cre/Flox; Figure 3D), in line with the PR prolongation observed by telemetry as well as previous findings reported in a VCS-specific TBX5-KO model (6). Interestingly, we observed significant delays in electrical activation time from the RV septum to the epicardial free wall (2.5 ± 0.8 ms vs. Cre/Flox), from the RV to the ventricular septum (3.5 ± 1.4 ms vs. Cre/Flox) and from the RV endocardium to the LV of vTbx5KO hearts (4.5 ± 1.6 ms vs. Cre/Flox). These data supported the hypothesis that TBX5 is a critical and so far underappreciated control element for the electrical activation in the working myocardium.
3.4. Transcriptome and chromatin occupancy analysis reveals novel TBX5 targets in the adult ventricle
To decipher the molecular mechanisms underlying arrhythmia development and cardiac decompensation after TBX5 loss, we set to identify the ventricular TBX5 targets by integrative transcriptome and chromatin occupancy analysis. The latter showed that TBX5 binds preferably at promoter regions (<1000 bp from transcription start site, TSS), intronic regions and regions at the 5ʹ UTR of target genes (Figure 4A). TBX5 is known to bind to promoter regions as well as active enhancer regions and synergistically activate gene transcription by associating with GATA422 and NKX2.5.23 Conversely, TBX5 competes with transcriptional repressors such as TBX324 by binding at the same regions. Hence, we examined TBX5 co-occupancy with known epigenetic marks of active enhancers such as polymerase 2 (POL2) and histone H3 lysine 27 acetylation (H3K27ac)25 as well as NKX2.5, GATA4 and TBX324 using public data (Figure 4D). In total, 2046 TBX5-bound regions showed co-occupancy with H3K27ac and POL2, indicating that these are active enhancer regions.26,27 Statistical analysis showed high correlation of TBX5 occupancy with POL2, H3K27ac, NKX2.5, GATA4, and TBX3 (r = 0.78, 0.82, 0.75, 0.58, and 0.82 respectively; Figure 4E), in line with previously published data showing binding of TBX5 close to NKX2.5, GATA4, and TBX3 binding sites. To find novel transcription factors which may co-operate or compete with TBX5 for the regulation of the same genes, we performed de novo motif analysis by HOMER of TBX5-bound active enhancer regions (indicated by H3K27ac enrichment). Besides the expected NKX2.5 and GATA4 motifs, the most enriched motif was Myeloid Ecotropic Viral Integration Site 1 and 2 (Meis 1 and 2; Figure 4F). These transcription factors are important for limb formation28 as well as CM cell cycle control,29 similar to TBX5,30,31 but so far have not been linked to TBX5-dependent transcription.
Figure 4.

Chromatin Immunoprecipitation analysis of endogenous TBX5 in the adult mouse ventricle. (A) Analysis of enriched genomic locations upon TBX5-ChIP shows that TBX5 preferably binds to promoter sites, downstream of the gene body, in the 5ʹ UTR and intronic regions (CEAS package20 on Cistrome). TBX5-bound regions were annotated to genes using GREAT.21 These regions were analysed for the 10 most enriched biological process clusters (B) and the 10 most enriched human disease phenotype clusters (C) of TBX5 peaks. (D) Heatmaps showing that TBX5 bound regions are highly co-occupied by marks of active enhancers (H3K27ac, POL2),25 known cofactors (NKX2.5, GATA4) and TBX324; analysed data from previously published datasets. (E) Statistical analysis of co-occupancy showing Pearson’s correlation coefficients. The scale bar in (B) depicts normalized RPKM values and in (C) co-occupancy percentage. (F) De novo motif analysis by HOMER of total TBX5 bound regions. The scale bar in (B) depicts normalized RPKM values and in (C) co-occupancy percentage. Results are displayed in (D) as heatmaps using deeptools in Galaxy and in (E) as the statistical analysis of co-occupancy showing Pearson’s correlation coefficients.
To identify genes affected by ventricular TBX5 down-regulation, Flox controls, and vTbx5 (Supplementary material online, Figure S5A, Table S4), which may have contributed to blood clot formation observed in all investigated vTbx5KO mice (Supplementary material online, Figure S5B). KO ventricles were subjected to RNA-sequencing (n = 3/group). We identified 97 significantly down-regulated and 93 significantly up-regulated transcripts (P < 0.05 and log2FC ≥ 0.8; Figure 5A, full list in Supplementary material online, Table S2). Gene ontology (GO) analysis of the down-regulated transcripts identified an association with muscle system processes such as ‘contraction and conduction’, ‘potassium ion transmembrane transport’, and ‘muscle adaptation’ (Figure 5B, Supplementary material online, Table S3). On the other hand, GO enrichment of the up-regulated genes indicated the blood coagulation-related process ‘platelet activation’.
Figure 5.

Integrative chromatin occupancy and transcriptome analysis identified novel putative TBX5 downstream targets in the adult ventricle. (A) RNA-Sequencing results displayed in a volcano plot. Ninety-seven genes are down- and 93 are up-regulated in vTbx5KO ventricles using a cut-off of P < 0.05 and log2-fold-change of >0.8 or <−0.8. (B) Gene ontology analysis (ClueGO32,33) of biological processes of the down-regulated genes in vTbx5KO mice. (C) TBX5 co-occupancy with marks of active enhancers H3K27ac and POL2 identified 2046 putative TBX5 active enhancers. These were annotated to genes with GREAT (Gja1 and Fgf16 enhancers were not annotated automatically by GREAT, but manually identified by BED file analysis in the IGV platform34,35). Venn-diagram shows the intersection of those genes with the regulated transcripts of the RNA-Seq analysis. (D) Gene ontology analysis of biological processes for 47 ventricular TBX5 targets.
To determine the intersection of genes bound by TBX5 and transcriptionally dysregulated upon TBX5 loss, we integrated the TBX5-bound genes with regulated transcripts (RNA-Seq, log2FC > 0.8, P < 0.05) upon TBX5 loss. The analysis of 2362 TBX5 active enhancer-associated genes and 190 regulated genes resulted in intersections of 7 (out of 93) up-regulated and 47 (out of 97) down-regulated genes (Figure 5C, full list in Supplementary material online, Table S5). None of the 7 up-regulated transcripts was implicated in blood coagulation or platelet activation, indicating that genes implicated in platelet activation are not directly regulated by TBX5. In addition, we could not detect any coagulation gene cluster in the complete gene pool associated to the total 8849 TBX5 peaks. GO of the intersection of down-regulated genes bound by TBX5 included conduction and contraction (Gja1, Kcnj5, Kcng2, Cacna1g, Chrm2), cytoskeleton organization (Fstl4, Pdlim4, Emilin 2, Cmya5) as well as cardiac protection upon hypertrophic response (Fhl2, Gpr22, Fgf16; Figure 5D, Supplementary material online, Table S5) and thus rendering them putative contributors to the observed heart failure with arrhythmia phenotype in vTbx5KO mice.
3.5. Validation of ventricular TBX5 downstream targets
Transcript expression of the identified TBX5 genomic targets was significantly down-regulated upon in vTbx5KO vs. Flox and Cre controls (Figure 6A). The identified putative TBX5 enhancer regions for these genes (Figure 6B) were co-occupied by POL2, H3K27ac, TBX3, NKX2.5, and GATA4 (Supplementary material online, Figure S6–S8). Moreover, TBX5 ChIP-qPCR analysis in vTbx5KO vs. Flox ventricles for a subset of genes (Gja1, Kcnj5, Cacna1g, Chrm2, Fhl2, Fgf16, and Pdlim4) showed no enrichment in the absence of TBX5 providing proof that these newly identified genomic regions are bona fide TBX5 enhancer elements (Figure 6C).
Figure 6.

Validation of the newly identified ventricular TBX5 target genes. (A) Validation of target gene expression by RT-(q)PCR (Flox, light grey n = 13; Cre, dark grey, n = 11; vTbx5KO, red, n = 10). (B) Visualization of the corresponding TBX5 peaks related to the 47 down-regulated genes described using IGV. The TBX5-ChIP-Seq lane is displayed in red and gene features in blue. The peaks that were investigated further are indicated with green bars. Immunoblot analysis of CX43, GIRK4, and GAPDH shows a strong down-regulation of CX43 and GIRK4 in vTbx5KO mice. Representative blot of n = 6 per group. (C) ChIP-qPCR analysis of the novel TBX5 enhancer regions in vTbx5KO mice vs. Flox ventricles (n = 3 biological replicates/group). (D) Immunoblot analysis of CX43, GIRK4, and GAPDH shows a significant down-regulation of CX43 and GIRK4 in vTbx5KO mice. Representative blot of n = 6 per group. (E) CX43 expression is strongly reduced in the ventricle of vTbx5KO mice shown by immunofluorescence staining for CX43 (red) and cTNT (green). Scale bar: 50 µm. (F) Gja1 and Kcnj5 enhancer activity analysis by luciferase measurements show enhancement of luciferase expression by TBX5 co-transfection (n = 9 technical replicates/3 independent experiment). EV: expression vector pCMV2c-flag, and TBX5: pCMV2c-TBX5-flag. Data are presented as mean ± SEM. Statistical analysis was performed by (A) one-way ANOVA for each transcript followed by Tukey’s multiple comparison post hoc test; (C, F) unpaired, two-tailed Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To validate the impact of TBX5 loss on the protein expression of ventricular genes overlapping in the RNA and ChIP-Seq data, we analysed connexin 43 (CX43, encoded by Gja1) and G-Protein coupled inward rectifier potassium channel 4 (GIRK4, encoded by Kcnj5) in vTbx5KO hearts. VTbx5KO ventricles showed a sustained reduction of both proteins (Figure6D andE, Supplementary material online, Figure S9A–C).
To confirm regulatory activity of TBX5 on identified enhancer regions of Gja1 and Kcnj5, we performed luciferase-based activity experiments. The TBX5 enhancer region for Kcnj5 is located 1 kb downstream of the 3ʹ UTR, whereas for Gja1 is located 600 kb downstream its TSS (Figure 6B). The latter region is identified as an enhancer with a cardiac expression pattern in the mouse embryo (Enhancer Browser; https://enhancer.lbl.gov/; data set mm7436). We could not detect the TBX5-binding peak at 30 MB distance from the TSS identified previously (7) as a putative TBX5 enhancer for atrial Gja1 (Supplementary material online, Figure S9D). The newly identified enhancers were co-occupied by GATA4 and NKX2.5 (Supplementary material online, Figure S6) and contained three T-box binding elements (TBE). Co-transfection of TBX5 encoding cDNA plasmid activated the Gja1 and Kcnj5 enhancers (Figure 4F), providing proof for the direct control of Gja1 and Kcnj5 transcription by TBX5 in ventricular myocardium.
3.6. TBX5 re-expression restores related transcriptional profiles and ameliorates cardiac function and conduction in vTbx5KO mice
Our data showed that ventricular suppression of TBX5 is detrimental to the adult heart due to transcriptional dysregulation of important cardiac genes implicated in cardiac rhythm regulation. Thus, we asked whether TBX5 re-expression, after long-term reduction, could restore its transcriptional programme and rescue from arrhythmias. To address this question, we initially planned to transduce mice 2 months after recombination. However, due to the high SCD incidence in vTbx5KO under isoflurane anaesthesia (60%), we had to transduce mice 2 weeks upon recombination, when the TBX5 targets are already deregulated and arrhythmias are already present, but the mortality rate under anaesthesia is zero. AAV9 particles (2 × 1012 viral particles per mouse) containing the coding sequence of TBX5 (KO-RE, n = 13) under the Tnnt2 promoter or a control vector (KO-CT, n = 11) were used. AAV9-TBX5 re-expression was validated at the protein level by immunoblot analysis (Supplementary material online, Figure S10A). The long-term TBX5 overexpression did not alter the heart dimensions as demonstrated by CM cross-sectional area analysis, HW/BW measurements, and bright-field images of the hearts (Supplementary material online, Figure S10B–D). Six weeks upon AAV9 injections, the levels of TBX5 and its downstream targets were significantly higher in KO-RE mice as compared to the KO-CT, with most of them reaching the levels of respective Cre control (Figure 7A–C). Immunofluorescence analysis of TBX5 and CX43 in KO-CT and KO-RE (n = 3/group) showed robust re-expression of TBX5 and CX43 in the ventricles (Supplementary material online, Figure S11). Interestingly, CMs with no detectable TBX5 expression strongly expressed CX43.
Figure 7.

In vivo TBX5 re-expression rescues arrhythmic phenotype of vTbx5KO mice while restoring TBX5-mediated transcription. (A) Transcript levels of newly identified ventricular TBX5 targets were analysed by RT-(q)PCR (KO-RE, TBX5-re-expression, blue, n = 7; KO-CT, control vector, red, n = 6 biological replicates). SCN5α was used as a known TBX5 target. All transcript values are normalized to the corresponding values from Cre controls, which are depicted with a dashed line. (B) Western blot analysis of CX43 and FHL2 protein levels normalized to GAPDH (KO-RE, n = 7; KO-CT n = 6 biological replicates). (C) Representative immunofluorescence analysis of CX43 in Cre, KO-CT, and KO-RE. Note the low expression of CX43 in KO-CT ventricles and its restoration in KO-RE hearts. (D) Echocardiography analysis parameters of KO-CT (red, n = 5 biological replicates) vs. KO-RE mice (blue, n = 6 biological replicates) monitored up to 14 weeks upon AAV9 injections. (E) Heart rate variability (HRV) represented by Poincaré plots; low variability in KO-RE indicates lower incidence of arrhythmia compared to KO-CT. One thousand consecutive beats were included per mouse/plot. The Poincaré plots were statistically analysed, the standard deviation of the HRV (SD1) was clearly increased in KO-CT mice (suggestive of arrhythmias) and remained comparable to Cre control mice after AAV9- transduction (KO-RE). Dashed lines indicated Cre mean values ± SEM. Statistical analysis of SD1 from HRV analysis shows significantly lower HRV in KO-RE vs. KO-CT mice. (n = 5 biological replicates per group). (F) QRS interval in KO-RE mice is significantly lower than KO-CT at 6, 12, and 16 weeks after TBX5 AAV9 injections. (G) Survival curve of KO-RE vs. KO-CT mice (KO-CT and KO-RE n = 5 and n = 6 biological replicates, respectively). Statistical analysis was performed by (A, B, E) unpaired, two-tailed Student’s t-test; (F) two-way ANOVA followed by Sidak’s multiple comparison post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To evaluate the relevance of TBX5 normalization in cardiac function, arrhythmia, and SCD incidence upon its long-term loss, a subset of mice (n = 6 KO-CT and n = 5 KO-RE) was followed for ∼16 weeks upon AAV9 re-expression. KO-RE mice showed no signs of progressive cardiac dysfunction as observed in KO-CT in the course of 16 weeks (Figure 7D). RR tachography showed that over the following 10 weeks KO-CT severely deteriorated whereas KO-RE mice presented with a more regular RR similar to Cre controls (Figure 7E). Moreover, QRS was significantly shorter in KO-RE mice compared to KO-CT (Figure 7F) reaching the QRS levels of Cre control mice and KO-RE mice were rescued from SCD (Figure 7G).
These data provide proof of principle that normalization of TBX5 level is sufficient to recover the altered transcriptional dysregulation and protect the heart from fatal arrhythmias and pathologic remodelling by restoring TBX5-mediated transcription.
4. Discussion
TBX5 is an essential transcription factor for normal cardiac development and mutations in the TBX5 locus are linked to abnormal cardiac conduction (Holt–Oram Syndrome). The role of TBX5 in the atria and the VCS has been investigated in detail.7,8 On the contrary, due to the relatively low TBX5 levels in the ventricular myocardium, it was believed that its role in this cardiac compartment may be less important.
We have previously shown that TBX5 levels are reduced upon chronic Ang II stimulation in adult mouse hearts.11 Similarly, in a rat heart-on-a-chip model, chronic Ang II stimulation resulted in lower levels of Tbx5 and its target Scn5α37 Our current study demonstrates for the first time that ventricular TBX5 is also suppressed in human heart failure, suggesting that its loss may participate in cardiac pathologies beyond congenital heart disease.
To investigate the impact of ventricular TBX5 loss in the adult hearts, we generated a TBX5 KO mouse model using the Myh6-MerCreMer mouse line (vTbx5KO), which resulted in significant loss of TBX5 in the working myocardium but not in atria or VCS. Although the underlying mechanism remains elusive, genetically modified mice mated with Myh6-MerCreMer have been reported to present inefficient recombination in the atria.38–40 At baseline, vTbx5KO mice presented with progressive dysfunction similar to what is observed in TBX5 haploinsufficient mice.7,41 Ang II-treated vTbx5KO showed an exacerbated function and hypertrophy as compared to Ang II Flox controls, which is in line with the observation that HOS patients with different mutations of TBX5 leading to haploinsufficiency suffer from cardiac hypertrophy.42 Moreover, vTbx5KO presented with AV blocks and tachycardias as it was reported for HOS patients with TBX5 loss of function.42
Since vTbx5KO mice still showed prominent TBX5 expression in the conduction system, we investigated whether the impact of TBX5 loss in the working myocardium would lead to phenotype of similar severity as the one observed in VCS-specific TBX5 deletion.7 Interestingly, the delay in electrical signal propagation in the ventricles (QRS) was 70% more pronounced as compared to the VCS TBX5KO model.7 To validate that the exacerbated delay in ventricular cardiac conduction in the vTbx5KO vs. the published conduction-specific model is due to the loss of TBX5 in the ventricular myocardium, we performed S1 pacing in isolated Flox, Cre, and vTbx5KO hearts. We observed significantly longer activation times in the ventricular myocardium of vTbx5KO mice validating that loss of TBX5 in the myocardium contributes to an exacerbated electrical signal propagation delay. In line with these data, we observed earlier onset and higher propensity of SCD in vTbx5KO mice compared to the previous described VCS TBX5KO model,7 strongly supporting the importance of TBX5 in the whole ventricle. Moreover, the global and homogeneous suppression of TBX5 and its targets in the working myocardium suggest that it is rather unlikely that the observed phenotype is the result of heterogenous TBX5 expression in the ventricle.
To investigate the effect of ventricular TBX5 loss on target expression, we performed integrative genome-wide chromatin occupancy in wild-type mice and comparative transcriptome analysis of vTbx5KO vs. Flox mice. VCS-specific TBX5 deletion has been shown to result in Nav1.5 (aka Scn5α) and CX40 down-regulation.7 Our transcriptomic analysis revealed regulation of known TBX5 targets such as Scn5α but not at the same level as novel ventricular targets including Gja1, Kcnj5, and Chrm2 in the vTbx5KO ventricles, which were not affected in the conduction-specific TBX5 KO model, strongly supporting a compartment-specific action of TBX5 in the adult myocardium.
Kcnj5 is translated into a G-protein activated inward rectifier potassium channel subunit 4 (GIRK4) of the cardiac muscarinic channel (IKAch). In the atria and AV node, acetylcholine released from the parasympathetic nerves activates CHRM2, which in turn activates IKAch, allowing cell hyperpolarization and thus heart rate deceleration.43 The role of both GIRK4 and CHRM2 in the ventricles is not well studied because of the dogma supporting the absence of parasympathetic innervation in the ventricular myocardium (for review see Ref.44). However, several studies recently demonstrated the opposite45,46 and further showed that parasympathetic denervation results in ventricular arrhythmias which can be rescued by addition of acetylcholine. Moreover, there is a number of in vivo studies in which vagal nerve stimulation reduced arrhythmias post-MI and even normalized CX43 expression (for review see Ref.44). Importantly, Kcnj5 mutations in humans result in decreased expression of GIRK4 in the ventricle, delay of ventricular conduction, and repolarization leading to long QT syndrome 13 (LQTS13).47Chrm2 mutations lead to familial DCM with high incidence of SCD, ventricular arrhythmias, and AV blocks.48 In line with these observations, vTbx5KO mice show reduced Kcnj5 and Chrm2 expression, a delay in ventricular conduction, spontaneous VTs and SCD. Hence, we show evidence supporting the contribution of parasympathetic nerves in ventricular arrhythmia development and indicate a role for TBX5-mediated regulation of CHRM2 and GIRK4 in the ventricles.
Moreover, the observed electrical signal propagation delay in vTbx5KO myocardium may be partially attributed to the massive ventricular CX43 loss. CX43 is the predominant connexin that is responsible for electrical coupling of the working myocardium.49 CX43 KO in adult mouse hearts leads to QRS prolongation and SCD.50 Although CX40 has been identified as the molecular target of TBX5 in the VCS, it has been recently shown that TBX5 KO in the atria leads to CX43 repression at the transcription level.8 We now identify the TBX5 enhancer region of Gja1 and show that ventricular TBX5 KO results in significant repression of CX43 protein levels in the adult working myocardium. Similar to our KO model, DCM and ICM hearts presented with arrhythmias express significantly lower CX43 levels,51 strongly suggesting a role for TBX5 in this pathophysiological mechanism.
Since we found that ventricular TBX5 levels are particularly low in heart failure patients, we tested the potential of TBX5 re-expression to protect the heart from arrhythmia and remodelling after long-term TBX5 loss. The most important question was, if the re-expression of a single transcription factor would be able to reduce the already developed arrhythmias by re-establishing the transcriptional balance in vTbx5KO mice. Therefore, we initially administered TBX5 packaged in AAV9 2 months after TX injections. However, we did not anticipate that 60% of the vTbx5KO mice would die under isoflurane anaesthesia. This observation raised the point of anaesthesia-induced SCD in vTbx5KO mice and led us to inject the mice 2 weeks after TX injections. At this timepoint, CX43 and other conduction-related proteins were already significantly reduced, and the mice already developed AV blocks and QRS prolongation. Although the mice already developed a phenotype, they were not susceptible to anaesthesia-induced SCD.
In contrast to a previous report in which CM-specific overexpression of an isoform of TBX5 leads to cardiac hypertrophy,52 in our model, TBX5 normalization did not alter the cardiac dimensions. TBX5 re-expression restored TBX5-dependent transcription related to electrical signal propagation and cardioprotection. TBX5 targets identified in vTbx5KO mice as Gja1, Kcnj5, and Chrm2 as well as commonly identified targets Scn5α were partially normalized (as compared to the respective Cre control). Interestingly, co-immunostaining of TBX5 and its target CX43 showed that not all CX43 positive CMs expressed TBX5 suggesting a cell-non-autonomous regulation of CX43. However, we cannot rule out that low, undetectable by immunofluorescence levels of TBX5 may still be present and drive transcription. Importantly, the transcriptional restoration by TBX5 normalization significantly reduced heart rate variability and QRS intervals to levels similar to Cre control mice and rescued the mice from SCD.
To the best of our knowledge, the current study provides the first proof-of-concept for the therapeutic potential of TBX5-related transcription normalization as a mean to reduce arrhythmia and SCD propensity. Finally, it suggests that re-expression of key transcription factors which are dysregulated in heart failure can alleviate transcriptional imbalance and reverse already developed pathological phenotypes.
Data availability
Raw and processed RNA- and ChIP-Seq results have been submitted to GEO under the accession number GSE101615.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
F.S.R., A.B., L.M.I., A.R., F.S., C.N, A.J., L.C.Z., and M.P.Z. performed experiments and analysed data. F.S.R with the help of A.R. and M.P.Z. carried out most of the experiments and analysed data. F.S.R. established the endogenous TBX5-ChIP in adult heart tissue and with L.M.I. analysed the ChIP-Seq data. F.S. under the supervision of L.F. performed and analysed electrophysiological measurements in the isolated heart. O.J.M. together with L.C.Z. and M.P.Z. designed the AAV part of the study. A.J. under the supervision of O.J.M. designed and provided AAV9-TBX5 and control viral particles. M.D. under the supervision of A.E.A. established the telemetry operations and ECG measurements. K.T. collected and provided human material. F.S.R., L.M.I., L.C.Z., and M.P.Z. designed research and wrote the article. W.H.Z. reviewed the data and provided intellectual input. All authors were involved in revising the article.
Supplementary Material
Acknowledgements
The authors would like to especially thank Prof. Benoit Bruneau (University of California, San Francisco) for kindly providing us with the Tbx5LDN/LDN mice. Moreover, we thank Petra Tucholla, Silvia Bierkamp, Ines Müller, and Nashitha Kabir for the excellent technical assistance, SFB 1002 service unit S01 Disease Models for echocardiography measurements and analysis as well as NGS-Integrative Genomics (NIG) Facility, Institute Human Genetics, University Medical Center Goettingen. Finally, we would like to thank Dr Tim Meyer for his input in the analysis of heart rate variability.
Conflict of interest: The University Medical Center Goettingen has filed a patent covering the potential of TBX5 re-expression to rescue from arrhythmias. M.P.Z., L.C.Z., and W.H.Z. are listed as inventors.
Funding
This work was supported by the Startup grant of University Medical Center Goettingen; the German Foundation of Heart Research to M.P.Z.; the German Center for Cardiovascular Disease (DZHK) to M.P.Z. and O.J.M.; the Deutsche Forschungsgemeinschaft (DFG) grant ZE900-3 and SFB 1002 TP C07 to L.C.Z.; the British Heart Foundation (FS/13/43/30324) and the Foundation Leducq to L.F. M.P.Z. and W.H.Z. are supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2067/1- 390729940. W.H.Z. is supported by DZHK; DFG ZI 708/10-1, SFB 1002 TP C04/S1, SFB 937 A18; IRTG 1816 RP12; and the Foundation Leducq.
Time for primary review: 32 days
Translational perspective
Cardiovascular disease (CVD) is the number one cause of death worldwide (WHO factsheets 09/2016). Although more than 60% of CVD-related deaths are due to out-of-hospital sudden cardiac death (SCD), we have little insight in the mechanisms underlying SCD pathophysiology. Our data show a link between TBX5 dysregulation and arrhythmia occurrence in patients. To test the therapeutic potential of TBX5, we normalized TBX5 levels in a mouse model with TBX5 dysregulation, which developed arrhythmias and SCD. TBX5 normalization re-established TBX5 target gene expression and more importantly, rescued the arrhythmia phenotype. Altogether, we provide proof-of-concept for the therapeutic potential of TBX5 expression restoration against arrhythmia and SCD.
References
- 1. Adabag AS, Luepker RV, Roger VL, Gersh BJ.. Sudden cardiac death: epidemiology and risk factors. Nat Rev Cardiol 2010;7:216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Horb ME, Thomsen GH.. Tbx5 is essential for heart development. Development 1999;126:1739–1751. [DOI] [PubMed] [Google Scholar]
- 3. Cross SJ, Ching YH, Li QY, Armstrong-Buisseret L, Spranger S, Lyonnet S, Bonnet D, Penttinen M, Jonveaux P, Leheup B, Mortier G, Van Ravenswaaij C, Gardiner CA.. The mutation spectrum in Holt-Oram syndrome. J Med Genet 2000;37:785–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, Renault B, Kucherlapati R, Seidman JG, Seidman CE.. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet 1997;15:30–35. [DOI] [PubMed] [Google Scholar]
- 5. Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, Gudjonsson SA, Jonasdottir A, Mathiesen EB, Njolstad I, Nyrnes A, Wilsgaard T, Hald EM, Hveem K, Stoltenberg C, Lochen ML, Kong A, Thorsteinsdottir U, Stefansson K.. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet 2010;42:117–122. [DOI] [PubMed] [Google Scholar]
- 6. Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG.. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001;106:709–721. [DOI] [PubMed] [Google Scholar]
- 7. Arnolds DE, Liu F, Fahrenbach JP, Kim GH, Schillinger KJ, Smemo S, McNally EM, Nobrega MA, Patel VV, Moskowitz IP.. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J Clin Invest 2012;122:2509–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nadadur RD, Broman MT, Boukens B, Mazurek SR, Yang X, van den Boogaard M, Bekeny J, Gadek M, Ward T, Zhang M, Qiao Y, Martin JF, Seidman CE, Seidman J, Christoffels V, Efimov IR, McNally EM, Weber CR, Moskowitz IP.. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci Transl Med 2016;8:354ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mori AD, Zhu Y, Vahora I, Nieman B, Koshiba-Takeuchi K, Davidson L, Pizard A, Seidman JG, Seidman CE, Chen XJ, Henkelman RM, Bruneau BG.. Tbx5-dependent rheostatic control of cardiac gene expression and morphogenesis. Dev Biol 2006;297:566–586. [DOI] [PubMed] [Google Scholar]
- 10. Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD.. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 2001;89:20–25. [DOI] [PubMed] [Google Scholar]
- 11. Baurand A, Zelarayan L, Betney R, Gehrke C, Dunger S, Noack C, Busjahn A, Huelsken J, Taketo MM, Birchmeier W, Dietz R, Bergmann MW.. Beta-catenin downregulation is required for adaptive cardiac remodeling. Circ Res 2007;100:1353–1362. [DOI] [PubMed] [Google Scholar]
- 12. Zafiriou MP, Noack C, Unsold B, Didie M, Pavlova E, Fischer HJ, Reichardt HM, Bergmann MW, El-Armouche A, Zimmermann WH, Zelarayan LC.. Erythropoietin responsive cardiomyogenic cells contribute to heart repair post myocardial infarction. Stem Cells 2014;32:2480–2491. [DOI] [PubMed] [Google Scholar]
- 13. Vettel C, Lindner M, Dewenter M, Lorenz K, Schanbacher C, Riedel M, Lammle S, Meinecke S, Mason FE, Sossalla S, Geerts A, Hoffmann M, Wunder F, Brunner FJ, Wieland T, Mehel H, Karam S, Lechene P, Leroy J, Vandecasteele G, Wagner M, Fischmeister R, El-Armouche A.. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ Res 2017;120:120–132. [DOI] [PubMed] [Google Scholar]
- 14. Jungmann A, Leuchs B, Rommelaere J, Katus HA, Muller OJ.. Protocol for efficient generation and characterization of adeno-associated viral vectors. Hum Gene Ther Methods 2017;28:235–246. [DOI] [PubMed] [Google Scholar]
- 15. Werfel S, Jungmann A, Lehmann L, Ksienzyk J, Bekeredjian R, Kaya Z, Leuchs B, Nordheim A, Backs J, Engelhardt S, Katus HA, Muller OJ.. Rapid and highly efficient inducible cardiac gene knockout in adult mice using AAV-mediated expression of Cre recombinase. Cardiovasc Res 2014;104:15–23. [DOI] [PubMed] [Google Scholar]
- 16. Fabritz L, Kirchhof P, Franz MR, Eckardt L, Monnig G, Milberg P, Breithardt G, Haverkamp W.. Prolonged action potential durations, increased dispersion of repolarization, and polymorphic ventricular tachycardia in a mouse model of proarrhythmia. Basic Res Cardiol 2003;98:25–32. [DOI] [PubMed] [Google Scholar]
- 17. Wittkopper K, Fabritz L, Neef S, Ort KR, Grefe C, Unsold B, Kirchhof P, Maier LS, Hasenfuss G, Dobrev D, Eschenhagen T, El-Armouche A.. Constitutively active phosphatase inhibitor-1 improves cardiac contractility in young mice but is deleterious after catecholaminergic stress and with aging. J Clin Invest 2010;120:617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zafiriou MP, Zelarayan LC, Noack C, Renger A, Nigam S, Siafaka-Kapadai A.. Hepoxilin A(3) protects beta-cells from apoptosis in contrast to its precursor, 12-hydroperoxyeicosatetraenoic acid. Biochim Biophys Acta 2011;1811:361–369. [DOI] [PubMed] [Google Scholar]
- 19. Mori AD, Bruneau BG.. TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr Opin Cardiol 2004;19:211–215. [DOI] [PubMed] [Google Scholar]
- 20. Shin H, Liu T, Manrai AK, Liu XS.. CEAS: cis-regulatory element annotation system. Bioinformatics 2009;25:2605–2606. [DOI] [PubMed] [Google Scholar]
- 21. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010;28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D.. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003;424:443–447. [DOI] [PubMed] [Google Scholar]
- 23. Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I.. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet 2001;28:276–280. [DOI] [PubMed] [Google Scholar]
- 24. van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, T Hoen PA, Bakkers J, Barnett P, Christoffels VM.. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J Clin Invest 2012;122:2519–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He A, Gu F, Hu Y, Ma Q, Ye LY, Akiyama JA, Visel A, Pennacchio LA, Pu WT.. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat Commun 2014;5:4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spicuglia S, Vanhille L.. Chromatin signatures of active enhancers. Nucleus 2012;3:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010;107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Capdevila J, Tsukui T, Esteban CR, Zappavigna V, Belmonte JCI.. Control of vertebrate limb outgrowth by the proximal factor Meis2 and distal antagonism of BMPs by Gremlin. Mol Cell 1999;4:839–849. [DOI] [PubMed] [Google Scholar]
- 29. Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA.. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013;497:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goetz SC, Brown DD, Conlon FL.. TBX5 is required for embryonic cardiac cell cycle progression. Development 2006;133:2575–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agarwal P, Wylie JN, Galceran J, Arkhitko O, Li C, Deng C, Grosschedl R, Bruneau BG.. Tbx5 is essential for forelimb bud initiation following patterning of the limb field in the mouse embryo. Development 2003;130:623–633. [DOI] [PubMed] [Google Scholar]
- 32. Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J.. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009;25:1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bindea G, Galon J, Mlecnik B.. CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics 2013;29:661–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP.. Integrative genomics viewer. Nat Biotechnol 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thorvaldsdottir H, Robinson JT, Mesirov JP.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013;14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Visel A, Minovitsky S, Dubchak I, Pennacchio LA.. VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res 2007;35:D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horton RE, Yadid M, McCain ML, Sheehy SP, Pasqualini FS, Park SJ, Cho A, Campbell P, Parker KK.. Angiotensin II induced cardiac dysfunction on a chip. PLoS One 2016;11:e0146415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen T, Aneas I, Sakabe N, Dirschinger RJ, Wang G, Smemo S, Westlund JM, Cheng H, Dalton N, Gu Y, Boogerd CJ, Cai CL, Peterson K, Chen J, Nobrega MA, Evans SM.. Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. J Clin Invest 2011;121:4640–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT.. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med 2007;13:970–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schoger E, Carroll KJ, Iyer LM, McAnally JR, Tan W, Liu N, Noack C, Shomroni O, Salinas G, Groß J, Herzog N, Doroudgar S, Bassel-Duby R, Zimmermann W-H, Zelarayán LC.. CRISPR-mediated activation of endogenous gene expression in the postnatal heart. Circ Res 2020;126:6–24. [DOI] [PubMed] [Google Scholar]
- 41. Zhu Y, Gramolini AO, Walsh MA, Zhou YQ, Slorach C, Friedberg MK, Takeuchi JK, Sun H, Henkelman RM, Backx PH, Redington AN, Maclennan DH, Bruneau BG.. Tbx5-dependent pathway regulating diastolic function in congenital heart disease. Proc Natl Acad Sci U S A 2008;105:5519–5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boogerd CJ, Dooijes D, Ilgun A, Mathijssen IB, Hordijk R, van de Laar IM, Rump P, Veenstra-Knol HE, Moorman AF, Barnett P, Postma AV.. Functional analysis of novel TBX5 T-box mutations associated with Holt-Oram syndrome. Cardiovasc Res 2010;88:130–139. [DOI] [PubMed] [Google Scholar]
- 43. Bettahi I, Marker CL, Roman MI, Wickman K.. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem 2002;277:48282–48288. [DOI] [PubMed] [Google Scholar]
- 44. Brack KE, Winter J, Ng GA.. Mechanisms underlying the autonomic modulation of ventricular fibrillation initiation–tentative prophylactic properties of vagus nerve stimulation on malignant arrhythmias in heart failure. Heart Fail Rev 2013;18:389–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ulphani JS, Cain JH, Inderyas F, Gordon D, Gikas PV, Shade G, Mayor D, Arora R, Kadish AH, Goldberger JJ.. Quantitative analysis of parasympathetic innervation of the porcine heart. Heart Rhythm 2010;7:1113–1119. [DOI] [PubMed] [Google Scholar]
- 46. Jungen C, Scherschel K, Eickholt C, Kuklik P, Klatt N, Bork N, Salzbrunn T, Alken F, Angendohr S, Klene C, Mester J, Klocker N, Veldkamp MW, Schumacher U, Willems S, Nikolaev VO, Meyer C.. Disruption of cardiac cholinergic neurons enhances susceptibility to ventricular arrhythmias. Nat Commun 2017;8:14155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, Olesen SP, Rasmussen HB, Ellinor PT, Gao L, Lin X, Li L, Wang L, Xiao J, Liu Y, Liu Y, Zhang S, Liang D, Peng L, Jespersen T, Chen YH.. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet 2010;86:872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang L, Hu A, Yuan H, Cui L, Miao G, Yang X, Wang L, Liu J, Liu X, Wang S, Zhang Z, Liu L, Zhao R, Shen Y.. A missense mutation in the CHRM2 gene is associated with familial dilated cardiomyopathy. Circ Res 2008;102:1426–1432. [DOI] [PubMed] [Google Scholar]
- 49. van Veen TA, van Rijen HV, van Kempen MJ, Miquerol L, Opthof T, Gros D, Vos MA, Jongsma HJ, de Bakker JM.. Discontinuous conduction in mouse bundle branches is caused by bundle-branch architecture. Circulation 2005;112:2235–2244. [DOI] [PubMed] [Google Scholar]
- 50. Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI.. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res 2001;88:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kostin S, Rieger M, Dammer S, Hein S, Richter M, Klovekorn WP, Bauer EP, Schaper J.. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol Cell Biochem 2003;242:135–144. [PubMed] [Google Scholar]
- 52. Georges R, Nemer G, Morin M, Lefebvre C, Nemer M.. Distinct expression and function of alternatively spliced Tbx5 isoforms in cell growth and differentiation. Mol Cell Biol 2008;28:4052–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed RNA- and ChIP-Seq results have been submitted to GEO under the accession number GSE101615.