

Abstract
Selective targeting of aneuploid cells is an attractive strategy for cancer treatment1. Here, we mapped the aneuploidy landscapes of ~1,000 human cancer cell lines, and analyzed genetic and chemical perturbation screens2–9 to reveal aneuploidy-associated cellular vulnerabilities. We identified and validated an increased sensitivity of aneuploid cancer cells to genetic perturbation of core components of the spindle assembly checkpoint (SAC), which ensures the proper segregation of chromosomes during mitosis10. Surprisingly, we also found aneuploid cancer cells to be less sensitive to short-term exposures to multiple SAC inhibitors. Indeed, aneuploid cancer cells became increasingly more sensitive to SAC inhibition (SACi) over time. Aneuploid cells exhibited aberrant spindle geometry and dynamics, and kept dividing in the presence of SACi, resulting in accumulating mitotic defects, and in unstable and less fit karyotypes. Therefore, although aneuploid cancer cells could overcome SACi more readily than diploid cells, their long-term proliferation was jeopardized. We identified a specific mitotic kinesin, KIF18A, whose activity was perturbed in aneuploid cancer cells. Aneuploid cancer cells were particularly vulnerable to KIF18A depletion, and KIF18A overexpression restored their response to SACi. Our study reveals a novel, therapeutically-relevant, synthetic lethal interaction between aneuploidy and the SAC.
Aneuploidy, defined as copy number changes encompassing entire chromosome-arms or whole chromosomes, is the most prevalent genetic alteration in human cancer11,12 (Supplementary Note 1). As cancer cells are almost invariably aneuploid12, whereas normal cells are (almost) always euploid13, the identification of aneuploidy-targeting drugs has been a long sought-after goal of cancer research. Whereas aneuploidy-augmented cellular vulnerabilities have been described in yeast14–17, their systematic identification remains elusive in human cancer. Large-scale studies are required to control for potentially-confounding factors, and isogenic in vitro systems are needed to validate differential dependencies and dissect them mechanistically.
Sensitivity to genetic SAC perturbation
To identify cellular vulnerabilities associated with a high degree of aneuploidy, we evaluated the aneuploidy landscapes of 997 human cancer cell lines, using published copy number profiles from the Cancer Cell Line Encyclopedia2. Each cell line was assigned an “aneuploidy score” (AS)12,18 based on the number of chromosome arms gained or lost in that cell line, relative to its basal ploidy (Fig. 1a, Extended Data Fig. 1a, and Supplementary Table 1). We then analyzed the association of aneuploidy with gene essentiality, using two distinct data sets of loss-of-function screens across 689 and 712 cell lines3,4 (Methods). Next, we performed a genome-wide comparison of the top (highly-aneuploid; median=25 chromosome-arm alterations) and bottom (near-euploid; median=3 chromosome-arm alterations) cell line quartiles, in order to identify differential vulnerabilities (Fig. 1a); specifically, we searched for genes whose depletion is more lethal in highly-aneuploid cell lines than in euploid (or near-euploid) ones.
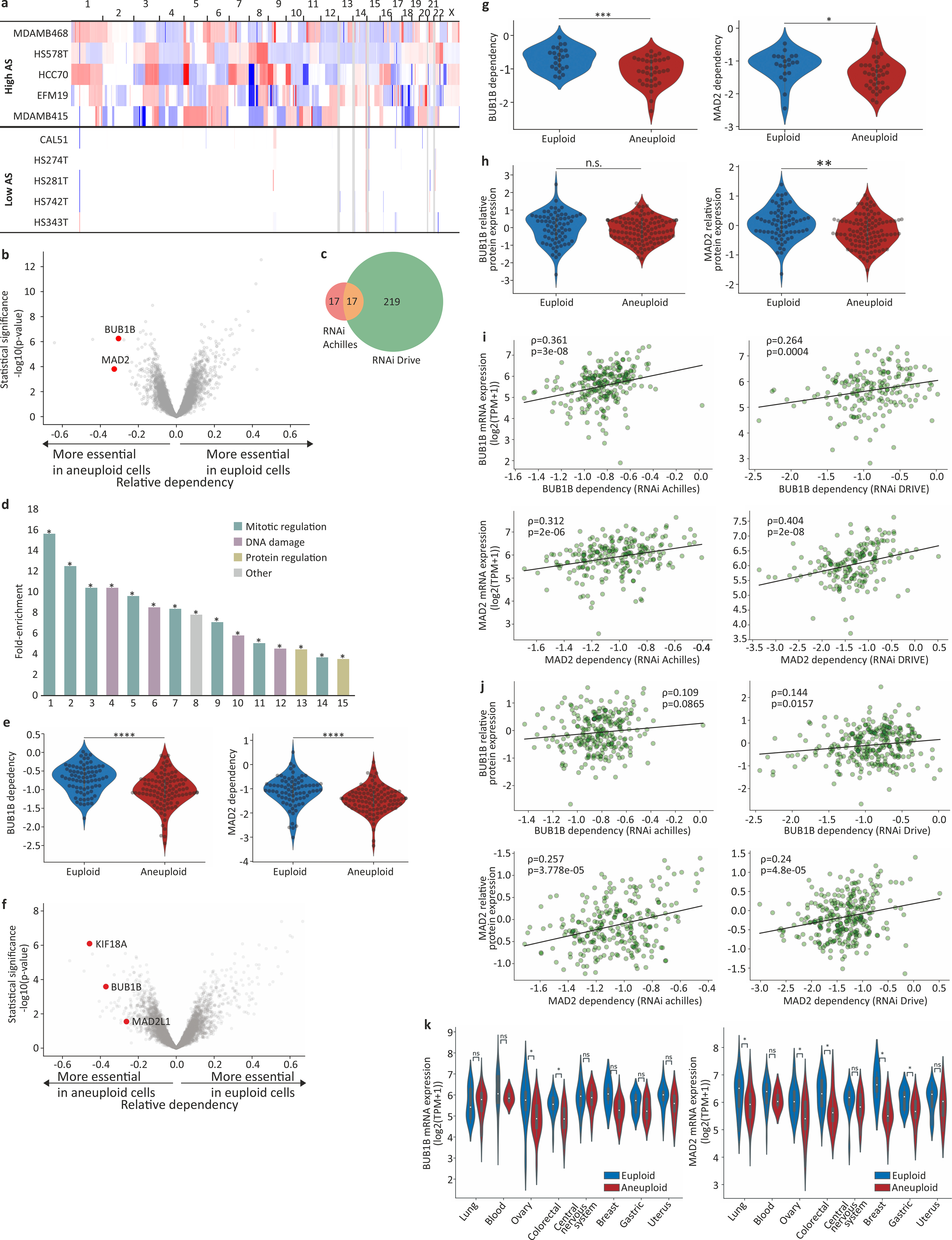
Figure 1: Differential sensitivity of aneuploid cancer cells to inhibition of the spindle assembly checkpoint.
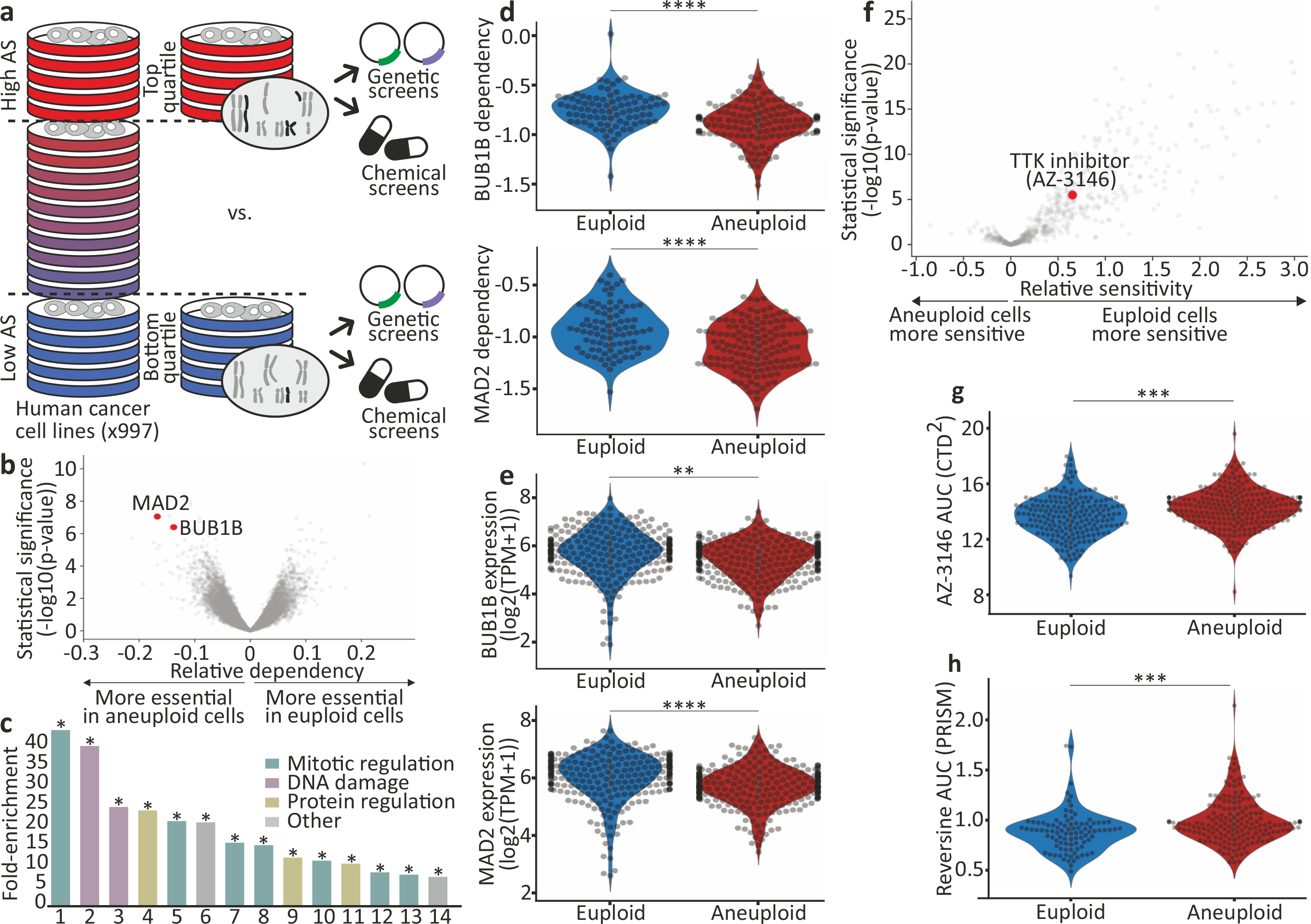
(a) Schematics of our large-scale comparison of genetic and chemical dependencies between near-euploid and highly-aneuploid cancer cell lines. Cell lines were assigned aneuploidy scores (AS), and the genetic and chemical dependency landscapes were compared between the top and bottom AS quartiles. (b) The differential genetic dependencies between the near-euploid and highly-aneuploid cancer cell lines (top vs. bottom quartiles), based on the genome-wide Achilles RNAi screen. BUB1B and MAD2 are highlighted in red. (c) The pathways enriched in the list of genes that are more essential in highly-aneuploid than in near-euploid cancer cell lines (effect size<−0.1, q<0.1) in the Achilles RNAi screen. The full list is available in Supplementary Table 3. The most enriched pathway is the SAC. *, Benjamini-corrected p<0.1; one-tailed Fisher’s exact test. (d) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (top) and MAD2 (bottom) in the Achilles RNAi screen. The more negative a value, the more essential the gene is in that cell line. ****, p=7e-07 and p=2e-07 for BUB1B and MAD2, respectively; two-tailed t-test. (e) Comparison of mRNA expression levels of BUB1B (top) and MAD2 (bottom) between near-euploid and highly-aneuploid cancer cell lines. **, p=0.001; ****, p=3e-06 for BUB1B and MAD2, respectively; two-tailed t-test. (f) The differential drug sensitivities between the near-euploid and highly-aneuploid cancer cell lines, based on the large-scale CTD2 drug screen. AZ-3146, the only SAC inhibitor in the screen, is highlighted in red. (g) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the SAC inhibitor AZ-3146 in the CTD2 drug screen. ***, p=2e-04; two-tailed t-test. (h) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the SAC inhibitor reversine, as evaluated by the PRISM assay. ***, p=3e-04; two-tailed t-test.
We identified 263 and 64 differential dependencies of highly-aneuploid cells in the RNAi-DRIVE and RNAi-Achilles datasets, respectively (Fig. 1b, Extended Data Fig. 1b,c, Supplementary Table 2, and Supplementary Note 2). The list of genes that were preferentially essential in aneuploid cancer cells was highly enriched for cell cycle-related pathways; in particular, the regulation of mitotic progression and the spindle assembly checkpoint (SAC) came up as the top preferentially-essential pathways (Fig. 1c, Extended Data Fig. 1d, and Supplementary Table 3). Two core members of the SAC (also known as the mitotic checkpoint), BUB1B (also known as BUBR1) and MAD2 (also known as MAD2L1), were at the top of the ‘hit’ list (Fig. 1b,d, Extended Data Fig. 1b,e–g, Supplementary Table 2, and Supplementary Note 3). Analysis of the Achilles-CRISPR/Cas9 data set5 confirmed that highly-aneuploid cell lines were more dependent on the SAC (p=0.003, q=0.1; for the enrichment of the GO term ‘mitotic cell cycle checkpoint’). However, the association between aneuploidy and SAC essentiality was weaker in this dataset, consistent with the inability of most mammalian cells to tolerate a complete SAC inactivation19,20. Further analysis showed that aneuploid cell lines exhibited a modest reduction in the mRNA and protein levels of both BUB1B and MAD2 (Fig. 1e and Extended Data Fig. 1h), and that lower expression levels were associated with greater sensitivity to genetic knockdown (Extended Data Fig. 1i–k, Supplementary Table 4, and Supplementary Note 4). The other pathways found to be more essential in aneuploid cells, were the proteasome and the DNA damage response (Fig. 1c and Supplementary Table 3), two cellular processes previously linked to the cellular response to aneuploidy (reviewed in 21).
We focused our downstream analyses on the SAC dependency, as it was the top differential vulnerability identified in our analysis, and also considering that: a) The SAC plays a key role in ensuring proper chromosome segregation during mitosis10; b) SAC perturbation leads to chromosomal instability, resulting in aneuploid karyotypes and frequently also in tumor formation22–27; and c) Inhibitors of the SAC regulator TTK (also known as MPS1) are currently used in clinical trials, either as single agents or in combination with chemotherapy28,29; however, no biomarkers of patients’ responses to SACi have been reported.
The degree of tumor aneuploidy is known to be associated with other genomic and cellular features, and in particular with tissue type, proliferation rate, CIN, WGD, and p53 function12,30–35. Indeed, all of these features strongly associated with the cancer cell line AS (Extended Data Fig. 2a–e). Importantly, however, the increased vulnerability of aneuploid cells to SAC perturbation remained robust when accounting for these (and additional) factors (Extended Data Fig. 3).
Sensitivity to chemical SAC perturbation
Next, we examined the association between aneuploidy and drug response, using three large-scale chemical screens6–9. Similar to the genetic analysis, we used the cell line AS for a comparison of drug sensitivity between the top and bottom aneuploidy quartiles (Fig. 1a). We found that aneuploid cell lines were more resistant to a short (3–5 days) exposure to a broad spectrum of drugs (Fig. 1f, Extended Data Fig. 4a, and Supplementary Table 5).
BUB1B and MAD2 work in concert with multiple other proteins to execute the crucial role of the SAC during mitosis36. TTK is particularly critical for SAC recruitment to unattached kinetochores and for complex formation37 (Supplementary Note 5). Aneuploid cancer cells were less sensitive than euploid cells to the three TTK inhibitors included in the analyzed chemical screens (Fig. 1f–g, Extended Data Fig. 4a,b, and Supplementary Table 5), in apparent contrast to the findings of the genetic analysis. Confirming the chemical screen results, we validated that highly-aneuploid cancer cell lines were more resistant than near-euploid cancer cell lines to the TTK inhibitor reversine38 (Extended Data Fig. 4c).
We next performed a pooled screen of barcoded cell lines, using the PRISM platform9, and examined the response to reversine in 578 adherent cancer cell lines (Supplementary Table 6). Indeed, highly-aneuploid cells were significantly more resistant than near-euploid cells to a 5-day reversine treatment (Fig. 1h, Extended Data Fig. 4d,e, and Supplementary Note 6).
SAC dependency evolves over time
Why do aneuploid cells exhibit increased sensitivity to genetic perturbation of SAC components, but reduced sensitivity to multiple TTK inhibitors? Three potential explanations may underlie this discrepancy: 1) The degree of protein inhibition and/or the target specificity may differ between genetic and pharmacological perturbations. 2) Perturbation of distinct SAC components may have differential cellular consequences. 3) The viability effect may depend on the different assay time points; drug response was evaluated following 3d-5d of SACi, whereas the response to genetic perturbations was evaluated following >14d of SACi, as these are the typical time points for chemical and genetic perturbation screens, respectively.
To resolve this conundrum, we turned to isogenic models of TP53-WT, near-diploid cells and their highly-aneuploid derivatives. We induced cytokinesis failure in HCT116 and RPE1 cells, thus generating tetraploid cells that spontaneously became aneuploid39 (termed HPT, HCT116-derived Post-Tetraploid; and RPT, RPE1-derived Post-Tetraploid; Extended Data Fig. 5a–c). These otherwise-isogenic aneuploid cell lines were exposed to two TTK inhibitors, reversine and MPI-0479605. The highly-aneuploid derivatives were more resistant to both drugs in a 5-day assay (Fig. 2a and Extended Data Fig. 6a), and this could not be explained by different proliferation rates or by a general drug resistance of the aneuploid derivatives (Extended Data Fig. 6b,c). Similarly, the highly-aneuploid isogenic lines exhibited increased resistance to siRNA-mediated knockdown of BUB1B, MAD2 and TTK (Fig. 2b and Extended Data Fig. 6d). The same results were obtained with a subset of near-euploid and highly-aneuploid cancer cell lines used in the original screens (Extended Data Fig. 6e), as well as with an independent, distinct system of RPE1 cells and their aneuploid derivatives40 (Fig. 2c,d, Extended Data Fig. 5d, Extended Data Fig. 6f, and Supplementary Note 7). Therefore, aneuploid cells exhibited short-term resistance to both genetic and chemical inhibition of all three SAC components.
Figure 2: The effect of aneuploidy on cellular sensitivity to SACi in isogenic human cell lines.
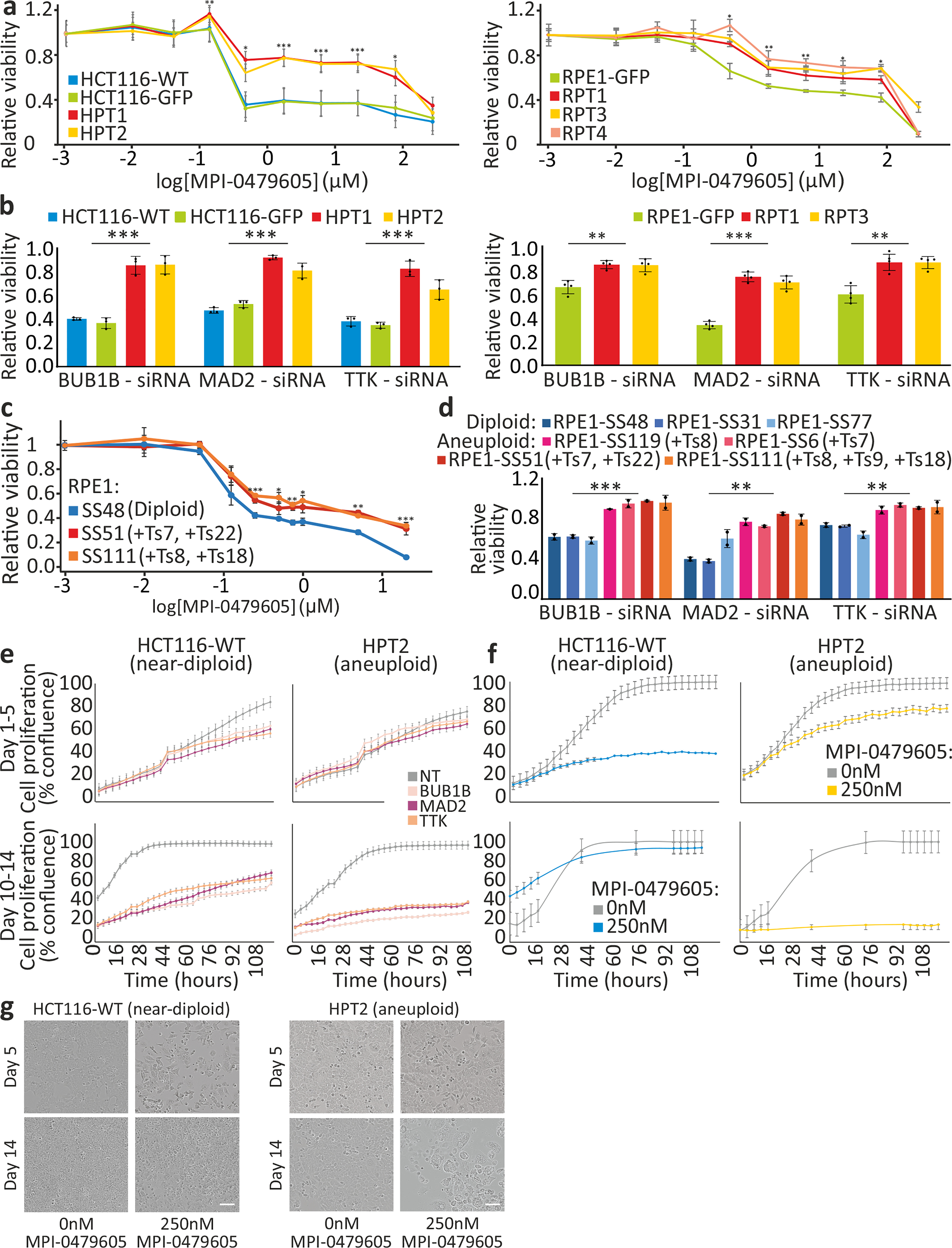
(a) Dose response curves of HCT116/HPT cells (left), or RPE1/RPT cells (right), to MPI-0479605 (120hr). EC50=0.09μM, 0.08μM, 5.02μM and 4.85μM, for HCT116-WT, HCT116-GFP, HPT1 and HPT2, respectively. EC50=0.16μM, 1.48μM, 1.52μM and 3.31μM, for RPE1-GFP, RPT1, RPT3 and RPT4, respectively. *, p<0.05; **, p<0.01; ***, p<0.001; two-tailed t-test. (b) The response of HCT116 and HPT cells (left; n=3), or RPE1 and RPT cells (right; n=4), to siRNA-mediated knockdown of BUB1B, MAD2 or TTK (72hr). Results are normalized to a non-targeting siRNA control. **, p<0.01; ***, p<0.001; two-tailed t-test. (c) Dose response curves of the near-diploid RPE1 clone SS48 and its isogenic aneuploid clones SS51 and SS111, to MPI-0479605 (120hr). EC50=0.02μM, 0.08μM and 0.04μM, for SS48, SS51 and SS111, respectively. *, p<0.05; **, p<0.01; ***, p<0.001; two-tailed t-test; n=3 for near-diploid and n=4 for aneuploid clones. (d) Response of 3 near-diploid and 4 aneuploid RPE1 clones to siRNA-mediated knockdown of BUB1B, MAD2 or TTK (72hr). Results are normalized to a non-targeting siRNA control. **, p<0.01; ***, p<0.001; two-tailed t-test. (e) Proliferation curves of HCT116 and HPT cells cultured in the presence of siRNAs against BUB1B, MAD2, TTK, or a non-targeting control siRNA. (f) Proliferation curves of HCT116 and HPT cells cultured in the presence of MPI-0479605 (250nM) or DMSO control. (g) Representative images of the cells from the experiment described in (f). Scale bar, 100μm. Calculated doubling times for (e) and (f) are presented in Extended Data Fig. 8a. In all plots, data represent the mean ± s.d. unless otherwise noted; n=3 biological replicates in all experiments unless otherwise noted.
To determine whether the differences between the genetic and chemical screens were due to the different time points of viability assessment, we followed the proliferation of HCT116 and HPT cells in response to prolonged genetic or chemical SACi. At d5, siRNA-mediated knockdown of BUB1B, MAD2 or TTK had a greater effect on the near-diploid HCT116 cells, in line with the previous viability measurements; however, by d14 of knockdown this trend reversed, and the highly-aneuploid HPT cells became more sensitive to SACi (Fig. 2e and Extended Data Fig. 7a). We observed the same reversal of relative sensitivity when we assessed long-term (d14) vs. short-term (d5) cell viability following exposure to chemical TTK inhibitors (Fig. 2f,g and Extended Data Fig. 7b,c). The same was observed with the isogenic diploid/aneuploid RPE1 clones (Extended Data Fig. 7d), and with the near-diploid and highly-aneuploid cancer cell lines (Extended Data Fig. 7e). Thus, the time point of viability assessment is critical for the results, explaining the apparent inconsistency between the genetic and chemical screens.
Cellular response to SAC inhibition
We next compared the expression changes induced by SACi in near-diploid and highly-aneuploid cells (Fig. 3a, Extended Data Fig. 8a, Supplementary Table 7, and Supplementary Note 8). The transcriptional responses to different SAC inhibitors were nearly identical within each cell line, and the two near-diploid cell lines clustered separately from the highly-aneuploid cell lines (Fig. 3b). Gene set enrichment analysis revealed that negative regulation of cell cycle and positive regulation of cell death topped the differentially-affected gene sets (Extended Data Fig. 8b,c). These findings suggest that 3d post-drug exposure, although the highly-aneuploid cells seem to be more resistant than their near-diploid counterparts, they already begin to downregulate cell cycle and upregulate cell death pathways that will ultimately lead to their elimination.
Figure 3: Transcriptional, cellular and karyotypic characterization of SACi in aneuploid cells.
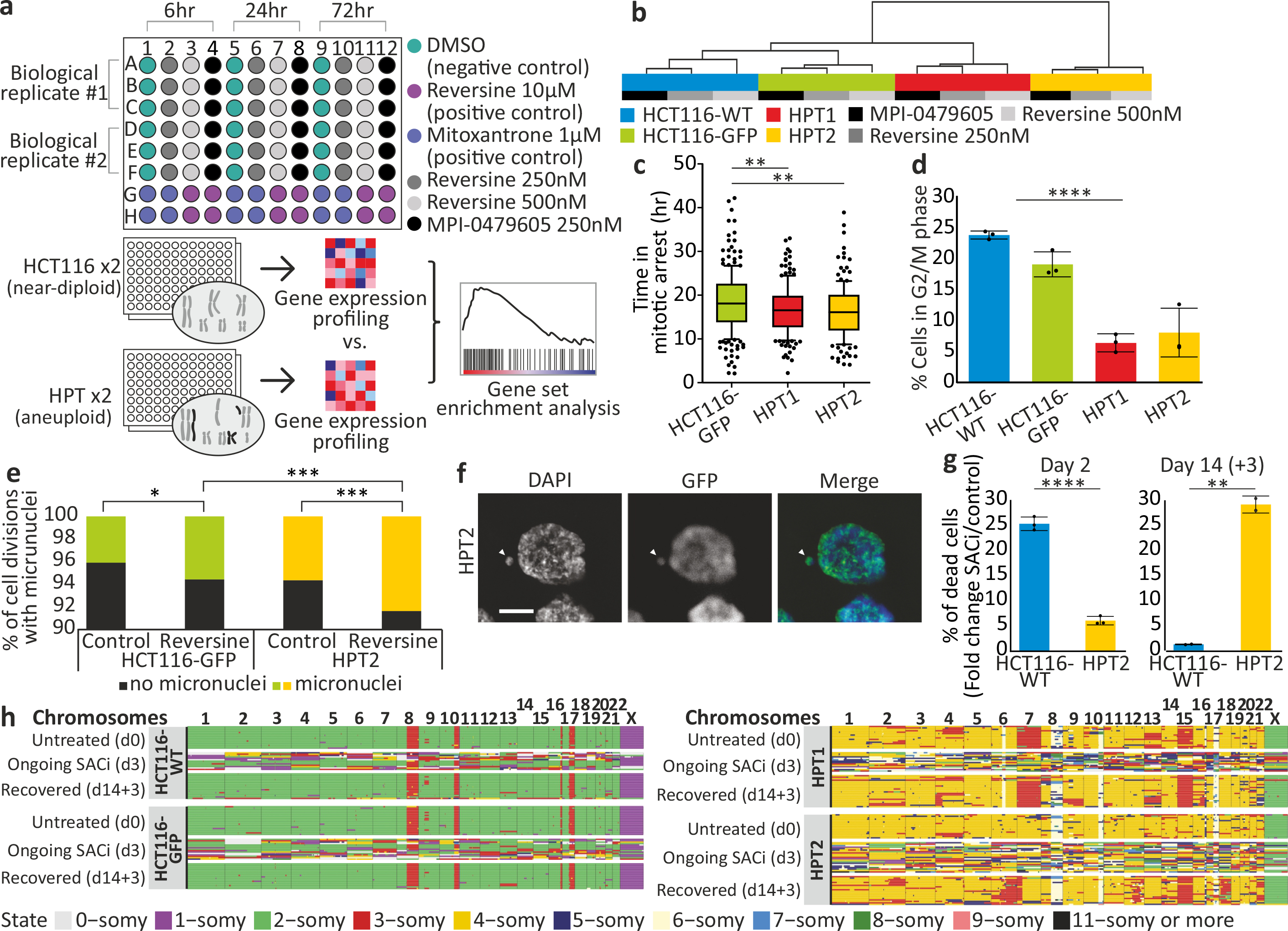
(a) Schematics of gene expression profiling. HCT116 and HPT cells were treated with two SAC inhibitors, reversine (250nM and 500nM) and MPI-0479605 (250nM), global gene expression profiles were generated at 6hr, 24hr and 72hr post-drug exposure, and gene set enrichment analysis was performed to compare the transcriptional effect of SACi. (b) Unsupervised hierarchical clustering of the 4 cell lines based on drug-induced transcriptional changes. (c) Time from mitotic arrest to division following nocodazole (200ng/mL) treatment. **, p<0.01; two-tailed t-test. Bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range; circles, individual cells. (d) Flow cytometry-based quantification of G2/M phase arrest induced by 48hr exposure to MPI-0479605 (250nM). ****, p=1e-05; two-tailed t-test. (e) The prevalence of micronuclei formation in HCT116 and HPT cells cultured under standard conditions or exposed to reversine (500nM) for 24hr. *, p=0.007; ***, p<0.001; two-tailed Fisher’s exact test. (f) Representative images of micronuclei formation in HPT2 cells exposed to reversine (500nM) for 24hr. Scale bar, 10μm. (g) Flow cytometry-based quantification of sub-G1 cell fraction induced by 48hr (left) or 14d (+3d of recovery; right) exposure to MPI-0479605 (250nM). **, p=0.002; ****, p=3e-05; two-tailed t-test. (h) Genome-wide copy number profiles of HCT116 and HPT single cells, as generated by the AneuFinder algorithm, at baseline (untreated; do), after 3d of MPI-0479605 (250nM) treatment (ongoing SACi; d3), and after recovery from 14d of treatment (recovered; d14+3). Individual cells are represented as rows, with chromosomes plotted as columns. In all plots, data represent the mean ± s.d. unless otherwise noted; n=3 biological replicates in all experiments unless otherwise noted.
Thus, we hypothesized that aneuploid cancer cells overcame SACi more readily than diploid cells, but consequently acquired severe aberrations that jeopardized their survival and proliferation. Indeed, the HPT cells overcame mitotic arrest faster after exposure to the microtubule depolymerizing drug nocodazole (Fig. 3c). When treated with a SAC inhibitor, the induction of cell cycle arrest, and the decrease in the mitotic index, were weaker in HPT cells relative to HCT116 cells (Fig. 3d and Extended Data Fig. 8d). Furthermore, SACi in HPT cells resulted in significantly more mitotic aberrations, such as multipolar cell divisions, micronuclei formation and cytokinesis failure (Fig. 3e,f and Extended Data Fig. 8e–g). Consequently, SACi initially induced more cell death in HCT116 cells than in HPT cells, but prolonged drug exposure ultimately resulted in much more cell death within the aneuploid cultures (Fig. 3g). We obtained very similar results with RPE1 cells and their highly-aneuploid RPT derivatives (Extended Data Fig. 8h–j). These results confirm that highly-aneuploid cells can overcome SACi more readily than their parental near-diploid cells, resulting in the accumulation of a variety of mitotic aberrations and eventually in their death.
Karyotype evolution after SACi
We next characterized the karyotype composition of the HCT116 and HPT cell lines before, during and after SACi. We used single-cell DNA sequencing41 to karyotype a total of 210 single cells across two near-diploid (HCT116-WT and HCT116-GFP) and two highly-aneuploid (HPT1 and HPT2) lines at three time points – prior to treatment (d0); following a short-term SACi (d3); and after recovery from a long-term exposure (d14+3).
Prior to treatment, the karyotypic heterogeneity and the degree of aneuploidy were both higher in the HPT populations, as expected (Fig. 3h and Extended Data Fig. 8k,l). Three days of SACi induced CIN in all lines, but the resultant karyotypic heterogeneity was significantly higher in HPT cells (p=2e-09; Extended Data Fig. 8k–m), in line with the increased prevalence of mitotic aberrations observed in these cells. In the near-diploid populations, the cells that survived prolonged drug treatment had the same near-diploid karyotype – and low degree of karyotypic heterogeneity – as the untreated cells (Fig. 3h and Extended Data Fig. 8k,l), suggesting that the original near-diploid karyotype was more fit than the aneuploid karyotypes induced by the drug. In contrast, in the aneuploid populations, the (fewer) surviving cells had highly-aneuploid and heterogeneous karyotypes. Several aneuploidies became more prevalent following treatment, but these events were not shared between the two HPT clones (Fig. 3h and Extended Data Fig. 8k,l). Therefore, there was no evidence for selection for one specific karyotype in the treated HPT cells.
We conclude that SACi induces lower levels of CIN in surviving near-diploid cells, enabling the selection of cells that preserve the original near-diploid karyotype; in contrast, highly-aneuploid cells experience a higher degree of CIN, and cannot readily select for a fit karyotype that would enable their long-term survival.
Spindle alterations in aneuploid cells
In order to study the molecular underpinning of the differential response to SACi, we analyzed the changes in spindle proteins in the HPT and RPT cells compared to their parental cell lines. The mRNA and protein expression levels of one specific mitotic kinesin, KIF18A, were reduced in the HPT cells (Fig. 4a and Extended Data Fig. 9a,b). Interestingly, depletion of KIF18A was shown to alter the spindle geometry, making the spindle longer and wider42,43, and KIF18A knockdown was reported to significantly decrease the kinetochore-microtubule stability in HCT116 cells44. The HPT cells exhibited similarly altered spindle geometry: spindle length, width and angle all significantly increased in the HPT cells compared to their near-diploid parental cells (Fig. 4b,c and Extended Data Fig. 9c). These structural changes were associated with alterations in spindle activity: microtubule polymerization rate, EB1α-tubulin co-localization and microtubule-kinetochore attachments were significantly reduced in the HPT cells (Fig. 4d and Extended Data Fig. 9d,e). Thus, highly-aneuploid cells exhibited altered spindle geometry and dynamics.
Figure 4: Altered spindle geometry and dynamics, and increased dependency on the mitotic kinesin KIF18A, in aneuploid cancer cells.
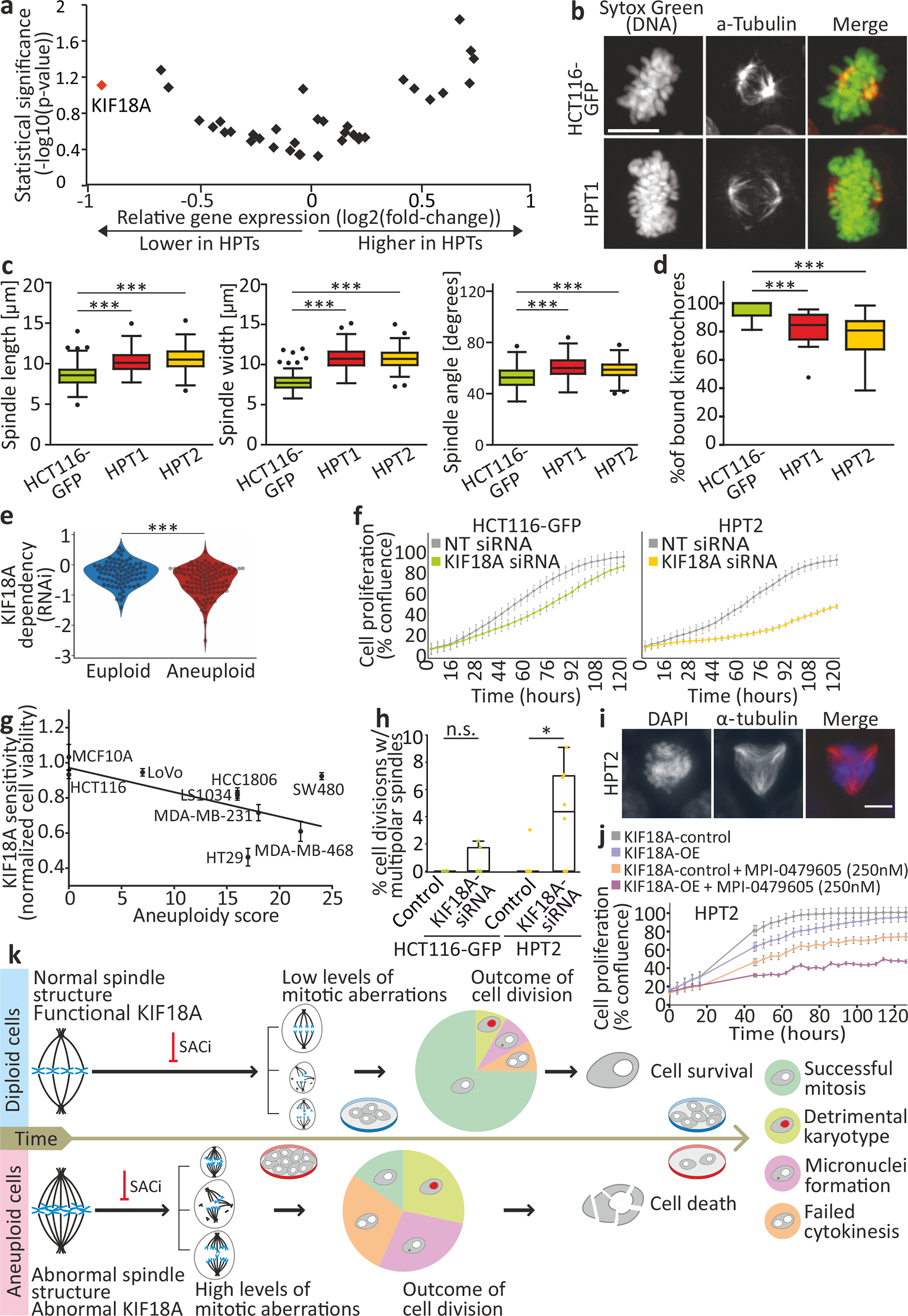
(a) Differential mRNA expression levels of mitotic kinesins between HCT116 and HPT cell lines. KIF18A is highlighted in red. (b) Imaging of metaphase spindle in HCT116-GFP and HPT1 cells, Scale bar, 10μm. (c) Spindle length (left), width (middle), and angle (right) in HCT116 and HPT cells. The definitions of length, width and angle are shown in Extended Data Fig. 9c. ***, p<0.001; two-tailed t-test. (d) The percentage of spindle microtubule-bound kinetochores in HCT116 and HPT cells. ***, p<0.001; two-tailed t-test. (e) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the RNAi-DRIVE dataset. The more negative a value, the more essential the gene is. ***, p=3e-04; two-tailed t-test. (f) Proliferation curves of HCT116-GFP and HPT2 cells cultured in the presence of a KIF18A-targeting siRNA, or a non-targeting control siRNA. (g) The sensitivity of cancer cell lines to the knockdown of KIF18A as a function of their AS. Spearman’s ρ = −0.66 (p=0.026; one-tailed test). (h) The prevalence of cell divisions with multipolar spindles in HCT116 and HPT2 cells treated with KIF18A-targeting siRNAs or a non-targeting control siRNA. n.s., p>0.05; *, p=0.03; two-tailed t-test. (i) Representative images of multipolar spindles in HPT2 cells following siRNA-mediated KIF18A knockdown. Scale bar, 10μm. (j) Proliferation curves of HPT2 cells before and after over-expression of KIF18A (KIF18A-OE), in the absence or presence of MPI-0479605 (250nM). KIF18A-OE increases the inhibitory effect of SACi. (k) A model of the evolving response of aneuploid cancer cells to SACi. For more details, see Supplementary Note 12. In all plots, data represent the mean ± s.d. unless otherwise noted; n=3 biological replicates in all experiments. In all box plots: bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range; circles, individual data points.
Therefore, we hypothesized that aneuploid cells might also be more dependent on KIF18A function. To test this hypothesis, we turned back to our large-scale genomic analysis of cancer cell lines (Supplementary Table 2). Indeed, highly-aneuploid cancer cells were significantly more dependent on KIF18A knockdown and knockout compared to near-euploid cancer cells (Fig. 4e, Extended Data Fig. 9f,g, Extended Data Fig. 10a–i, and Supplementary Note 9), although there was no significant difference in KIF18A mRNA and protein expression levels between the groups (Extended Data Fig. 9h–k, Supplementary Note 10, and Supplementary Table 8). KIF18A was the only differentially essential kinesin in our analysis (out of 42 kinesins tested), and ranked #11 overall on the list of genes most preferentially essential in aneuploid cancer cells in the RNAi-DRIVE data set (Supplementary Table 2).
We confirmed that the aneuploid cells were more sensitive to KIF18A depletion using siRNA-mediated knockdown in HCT116 and HPT cells (Fig. 4f and Extended Data Fig. 9l–n). Moreover, the sensitivity to KIF18A depletion45 strongly correlated with AS across a panel of nine cancer cell lines (Spearman’s ρ=−0.66, p=0.026; Fig. 4g). Live-cell imaging identified a modest mitotic delay in HPT cells following siRNA-mediated KIF18A KD (Extended Data Fig. 9o), followed by a significant increase in multipolar cell divisions (Fig. 4h,i) and micronuclei formation (Extended Data Fig. 9p); in contrast, KIF18A depletion in the near-diploid HCT116 cells did not lead to similarly severe aberrations (Fig. 4h and Extended Data Fig. 9o,p). We obtained very similar results with the RPE1 cells and their highly-aneuploid RPT derivatives (Extended Data Fig. 10j–n). In line with these results, two recent manuscripts recently reported an increased sensitivity to KIF18A perturbation in chromosomally-unstable aneuploid cell lines45, as well as in aneuploid WGD+ cell lines46.
Lastly, we examined whether the observed differential sensitivities of aneuploid cells to SACi and to KIF18A depletion were functionally related. We overexpressed KIF18A in HPT cells (Extended Data Fig. 9q) and examined their sensitivity to SACi. Whereas KIF18A overexpression alone had minimal effect on cell viability and proliferation, it sensitized the aneuploid cells to short-term SACi (Fig. 4j and Extended Data Fig. 9r). This ‘phenotypic rescue’ experiment demonstrates a causal link between KIF18A and the cellular sensitivity to SACi. Further study is required to elucidate the nature of this interaction at the molecular level.
Discussion
The potential of targeting aneuploid cells to selectively kill cancer cells remains unfulfilled. Here, we assigned AS to ~1,000 cancer cell lines, performed a comprehensive analysis of large-scale genetic and chemical perturbation screens, and identified increased dependency of aneuploid cancer cells on the SAC core members, BUB1B and MAD2 (Supplementary Note 11). Using a subset of ten cancer cell lines, as well as three model systems of isogenic near-diploid and highly-aneuploid cell lines, we confirmed the increased vulnerability of aneuploid cells to SACi. Transcriptional profiling, flow cytometry, scDNAseq and imaging-based analyses of mitosis revealed an altered response of aneuploid cells to SACi. Finally, we found the mitotic kinesin gene KIF18A to be preferentially essential in aneuploid cells, and functionally related to their increased dependency on the SAC activity.
Our findings reveal that aneuploid cells can initially overcome SACi more readily than diploid cells; however, the resultant aberrant cells exhibit severe viability and proliferation defects (Fig. 4k and Supplementary Note 12). These findings may have several important implications for the clinical use of TTK inhibitors, as they suggest that aneuploidy may serve as a biomarker for predicting drug response to this class of drugs, highlight the value of testing such drugs over a longer time course, and identify a potential need to develop selective inhibitors of BUB1B and MAD2 (Supplementary Note 13). In addition, the increased sensitivity of aneuploid cancer cells to KIF18A inhibition is interesting per se, given the attempts to develop highly-selective and bioactive KIF18A inhibitors47 (Supplementary Notes 13–15).
Lastly, our large-scale analyses revealed additional candidate vulnerabilities that deserve experimental validation (e.g., increased sensitivity to proteasome inhibition; Supplementary Table 3). Furthermore, our characterization of aneuploidy profiles and scores across the CCLE lines (Supplementary Table 1) will be useful for the identification of additional genomic features and cellular vulnerabilities associated with high degree of aneuploidy or with specific recurrent aneuploidies. To facilitate further interrogation of this resource, we have integrated the cell line aneuploidy profiles and scores into the DepMap portal (www.depmap.org/portal/). We hope that this study will thus pave the way for the routine integration of aneuploidy status in the genomic analysis of cancer dependencies.
Methods
Aneuploidy score assignment
Aneuploidy was quantified by estimating the total number of arm-level gains and losses for each cell line, based on the published ABSOLUTE copy number data of the CCLE dataset2. The median total modal copy number (sum of allelic copy numbers) across segments was estimated for each chromosome arm (weighted for segment length), and compared to the cell line’s background ploidy in order to call the chromosome-arm copy number status (gain/loss/neutral). AS was defined as the total number of chromosome arms that were gained/lost. The cell lines with bottom-quartile AS (corresponding to cell lines with a median of 3 chromosome-arm copy number changes; min = 0, max = 7) were defined as the “near-euploid” group, and the cell lines with the top-quartile AS (corresponding to cell lines with a median of 25 chromosome-arm copy number changes; min = 22, max =36) were defined as the “highly-aneuploid” group.
Association of aneuploidy with genomic and phenotypic features
Cell line doubling time measurements were obtained from Tsherniak et al3. The mutation calls and mRNA expression levels were obtained from the CCLE mutation and gene expression data sets (19q4 DepMap release; CCLE_mutations.csv and CCLE_expression_full.csv, respectively)2. The genetic perturbation datasets used were the gene_effect files from RNAi Achilles53, RNAi DRIVE53, and CRISPR Achilles (19q4 DepMap release). RNAi data is available at https://doi.org/10.6084/m9.figshare.6025238.v4 and CRISPR, mutation, and expression data is available at https://doi.org/10.6084/m9.figshare.11384241.v2. The chemical perturbation datasets used were the PRISM Repurposing Secondary Screen9, CTD2 54,55, and GDSC56. Normalized protein abundance measurements across cell lines were obtained from Nusinow et al.57 Cell line microsatellite instability was determined using next-generation sequencing and PCR-based phenotyping, obtained from Chan et al.58 Cell lines were split into two groups: the upper and lower quartiles of AS. Genes that were preferentially dependent in highly-aneuploid compared to near-euploid cell lines were identified using linear modelling performed in parallel across genes using the R package Limma59. The difference in mean dependency between the groups was evaluated for each gene, and associated P-values were derived from empirical-Bayes-moderated t-statistics. Q values were computed using the Benjamini–Hochberg method60. This process was repeated with various features of the cell lines (cell lineage, HET70 score50 or doubling time) included as a covariate. To remove the effects of confounding variables (cell lineage, HET70 or doubling time), we fit linear regression models (Scikit-learn)61 and computed the residuals, maintaining the across-cell line average dependency scores fixed. To test mRNA expression as a predictor of genetic perturbation of BUB1B and MAD2, linear regression models were fit using the lm function from R Stats Package62, including lineage annotations as co-variates.
Association between common essential genes and drug response
Chemical perturbation log-fold change data from the PRISM Repurposing Primary Screen9 were correlated with the respective annotated drug targets in the genetic perturbation data. Log-fold change data were calculated relative to DMSO, and ComBat was used to correct for experimental confounders, as described in Corsello et al.9 Common essential genes were defined as on DepMap (www.depmap.org/portal/).
Functional enrichment analysis
The list of differentially-essential genes between the near-euploid and highly-aneuploid groups (effect size<−0.1, q<0.1) was subjected to a DAVID functional annotation enrichment analysis49, focusing on the GO Biological Process gene sets. The full list of genes included in each screen was used as background.
Reversine biomarker analysis
The scikit-learn’s RandomForestRegressor61 was used to predict Reversine AUC values for 502 cell lines. The input features were (19Q4 release): RNA-Seq expression data for both protein-coding and non-coding regions (CCLE_expression_full.csv); mutation statuses, broken into three binary matrices: damaging, hotspot and other (CCLE_mutations.csv); and gene level copy number (CCLE_gene_cn.csv). Data are available at https://doi.org/10.6084/m9.figshare.11384241.v2. Like in Dempster et al., we used tenfold cross-validation, filtered features to the 1,000 having the highest Pearson correlation with the Reversine AUC values in the training set, and reported accuracy via Pearson correlation between the measured AUC values and the complete set of out-of-sample predictions63. To estimate feature importance values we retrained the model on all the samples and used RandomForestRegressor’s feature_importances_attribute. This attribute is a measure of the average contribution of a feature to decreasing the variance when splitting values at nodes.
Generation of isogenic near-diploid and highly-aneuploid cell lines
The HPT and RPT aneuploid derivatives were generated from the near-diploid human colorectal cancer cell line HCT116, and from the human immortalized retinal pigment epithelium cell line RPE1, respectively. The cells were treated with dihydrocytochalasin D for 18hr, washed in DMEM and cloned by limiting dilution to obtain single cell clones within 30 days. Individual clones were then screened by flow cytometry for DNA content and near-tetraploid cell lines were validated via metaphase spreads. Selected clones were further characterized by multicolor FISH karyotyping and by SNP array profiling, which showed that the cell lines were not stable as tetraploid, but were chromosomally unstable, and quickly became highly-aneuploid, mostly through chromosome loss. The characterized cell lines were expanded and stored in liquid nitrogen. Cells were propagated for a maximum of 5 additional passages prior to carrying out the experiments. Further description is available in the original paper reporting their derivation39. Chromosome count, based on a standard karyotypic analysis of the HCT116 and HPT cell lines, is shown in Extended Data Fig. 5b,c.
To generate cell lines harboring stable aneuploid karyotypes (and euploid controls), RPE1-hTERT cells were transiently treated with the TTK inhibitor reversine (500nM, 24hr) to induce random chromosome gains and losses (aneuploid population), or with a vehicle control (for the euploid population). After drug wash-out, euploid and aneuploid populations were single-cell sorted in 384 well plates (FACSAria, BD Biosciences), expanded, and their karyotypes assessed by bulk DNA sequencing. The karyotypic analysis of the RPE1-SS clones is shown in Extended Data Fig. 5d.
Cell culture
HCT116, RPE1 cells, their aneuploid derivatives HPT and RPT, MDAMB468, A101D, EN, VMCUB1, CAL51 and SW48 were cultured in DMEM (Life Technologies) with 10% fetal bovine serum (Sigma-aldrich) and 1% penicillin-streptomycin-glutamine (Life Technologies). SH10TC, NCIH1693, MHHNB11 and PANC0813 were cultured in RPMI-1640 (Life Technologies) with 10% fetal bovine serum (Sigma-aldrich) and 1% penicillin-streptomycin-glutamine (Life Technologies). PANC0813 medium was supplemented with 10units/mL human recombinant insulin (Sigma-Aldrich), and MHHNB11 medium was supplemented with MEM Non-Essential Amino Acids (Sigma-Aldrich). Cells were incubated at 37°C, 5% CO2 and passaged twice a week using Trypsin-EDTA (0.25%) (Life Technologies). Cells were tested for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza), according to the manufacturer’s instructions.
PRISM screening
The PRISM screen was performed as described in Corsello et al.9. Briefly, barcoded cell lines were pooled (25 cell lines per pool) based on doubling time and frozen into assay-ready vials. Vials were thawed and one pool was immediately plated in 384-well plate at 1,250 cells per well in triplicate. 24hr later, cells were plated onto assay ready plates containing 8 different concentrations of reversine (3 fold-dilutions ranging from 0.9nM to 20μM) or control DMSO. 5d later, cells were lysed, and lysate plates were then pooled for amplification and barcode measurement. Viability values were calculated by taking the median fluorescence intensity of beads corresponding to each cell line barcode, and normalizing them by the median of DMSO control treatments. High-quality viability measurements could be generated for 530 cell lines. Dose response curves were calculated by fitting four-parameter curves to viability data for each compound and cell line using the R package “drc” and fixing the upper asymptote of the logistic curves to 1; the area under the dose response curve (AUC) values were calculated using a normalized integral, as discussed in Corsello et al.9 (Supplementary Table 6).
Cell growth rate analysis
Kinetic cell proliferation assays were monitored using the IncuCyte S3 Live Cell Analysis System (Essen Bioscience). 96-well plates were incubated at 37°C, 5% CO2. Four, non-overlapping planes of view phase contrast images were captured using a 10x objective, with data collected every 4hr for the duration of each experiment. IncuCyte Base Software was used to calculate average confluence. Population doublings were calculated using the formula Tdoubling = (log2(ΔT))/(log(c2) − log(c1)), where c1 and c2 are the minimum and maximum percentage confluence during the linear growth phase, respectively, and ΔT was the time elapsed between c1 and c2. Cell masking in representative images was done for visualization purposes, using ilastik image analysis software51.
Drug response assays
For drug experiments, cells were plated at 1 × 104 cells per well and treated with compounds 24hr later. MPI-0479605 was purchased from MedChem Express (Princeton, NJ, USA), reversine and mitoxantrone were purchased from Sigma-Aldrich (Saint-Louis, MO, USA, and). For prolonged drug exposure (14d or longer), cells were split as necessary, so that all control cells were split at the same time, and all treated cells were split at the same time. Cells were allowed to recover before drugs were replenished, and day count was based on the total number of days in the presence of the drug (e.g., d14 corresponds to 14 days of drug exposure). Following incubation with the drug, viability was assessed either by live-cell imaging using the IncuCyte S3 Live Cell Analysis System (Essen Bioscience) or using CellTiter-Glo (Promega) or Crystal Violet staining (Sigma). Luminescence and absorbance were quantified using an Envision Plate Reader (PerkinElmer). Experiments were performed in triplicates that were averaged and normalized to negative (DMSO-matched) control. EC50 values were calculated in GraphPad Prism using an asymmetric (five parameters) nonlinear regression model.
Cell transfections
Cells were seeded in 100μL of medium in black, clear bottom 96-well plates (Corning 3904) excluding edge wells at 5 × 103 cells per well a day prior to transfections. For siRNA experiments, cells were transfected with 25nM siRNA against BUB1B, MAD2L1, TTK, KIF18A, or a non-targeting control (Dharmacon ON-TARGETplus SMARTpool) in triplicate using DharmaFECT 1 Transfection Reagent (Dharmacon) as per the manufacturer’s protocol. For prolonged siRNA exposure (14d or longer), cells were split as necessary, so that all control cells were split at the same time, and all treated cells were split at the same time. Cells were allowed to recover before siRNAs were replenished, and day count was based on the total number of days in the presence of the siRNA (e.g., d14 corresponds to 14 days of siRNA exposure). For KIF18A overexpression experiments, cells were transfected with 100 ng pMX229, a gift from Linda Wordeman (Addgene plasmid #23002), using TransIT®-LT1 Transfection Reagent (Mirus). For combination experiments with SAC inhibitors, cells were transfected and treated with drugs simultaneously. Following incubation with the siRNAs, the overexpressing vector and/or the drugs, viability was assessed either by live-cell imaging using the IncuCyte S3 Live Cell Analysis System (Essen Bioscience) or using CellTiter-Glo (Promega). Luminescence was measured using an Envision Plate Reader (PerkinElmer). Experiments were performed in triplicates that were averaged and normalized to negative (DMSO-matched) control.
RNA Extraction and Real-Time quantitative PCR analysis.
RNA was extracted from cells using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s protocol. For gene expression analysis, cDNA was generated from 1μg of RNA with the iScript cDNA synthesis kit (Bio-Rad) as per the manufacturer’s protocol. Using the QuantiTect SYBR Green PCR kit, 100ng of cDNA was amplified according to the manufacturer’s instructions with primers targeting BUB1B (catalog no. QT00008701), MAD2 (catalog no. QT00094955), TTK (catalog no. QT00035168), KIF18A (catalog no. QT00042455), or GAPDH (catalog no. QT00273322) as an endogenous control (Quantitect Primer Assay, Qiagen). Data analysis was performed with the QuantStudio 6 and 7 Flex Real-Time PCR System Software v1.0 (Applied Biosystems, Life Technologies) using the ΔΔCt method.
Western blotting
Processed total cell lysates were separated by SDS-PAGE. Protein size was estimated using ‘PrecisionPlus All Blue’ or ‘PrecisionPlus Kaleidoscope’ protein markers (BioRad). Separated proteins were then transferred to a methanol-activated polyvinylidene difluoride membrane (PVDF, Roche) using wet transfer Mini-PROTEAN II electrophoresis system (BioRad), or to nitrocellulose membrane (BioRad) using Trans-Blot Turbo electrophoresis system (BioRad). Membranes were blocked in 5% skim milk (Fluka) in Tris-buffered saline with 0.05% Tween20 (TBST), decorated with respective primary antibodies diluted in blocking solution overnight at 4°C with gentle agitation. Further, the membranes were rinsed for 30 min with TBST with a triple buffer exchange, incubated with HRP-conjugated secondary antibodies (R&D Systems), followed by triple TBST wash, chemiluminescence using ECLplus kit and detection either on ECL hyperfilm (GE Healthcare), on X-ray hyperfilm processor MI-5 (Medical Index) or using Fujifilm Luminescent Image Analyzer (LAS-3000 Lite) system (Fujifilm). Protein band quantification was carried out using ImageJ (National Institutes of Health, http://rsb.info.nih.gov/ij/). The following primary antibodies were used: anti-Kif18A rabbit64 (1:500), affinity-purified polyclonal antibody raised against an N-terminal GST-tagged fragment (Kif18AbN), a gift from Dr. Thomas Mayer, University of Konstanz, Germany; anti-Kif18A rabbit (1:5,000), Bethyl Laboratories (catalog no. A301–080A); anti-GAPDH goat (1:1,000), Abcam (catalog no. ab9483); anti-α-Tubulin mouse (1:2,000), Sigma (catalog no. T6199). Uncropped scans of all gels are shown in Supplementary Fig. 4.
Transcriptional profiling
Cells were exposed to reversine (at 250nM or 500nM) or to MPI-0479605 (250nM) and transcriptional profiling was performed at 6hr, 24hr and 72hr post drug exposure. DMSO was used as a negative control, and 1μM mitoxantrone and 10μM reversine were used as positive cytotoxic controls. The L1000 expression-profiling assay was performed as previously described52,65. First, mRNA was captured from cell lysate using an oligo dT-coated 384-well Magnefy microspheres. The lysate was then removed, and a reverse-transcription mix containing Superscript IV reverse transcriptase was added. The plate was washed and a mixture containing both upstream and downstream probes for each gene was added. Each probe contained a gene-specific sequence, along with a universal primer site. The upstream probe also contained a microbead-specific barcode sequence. The probes were annealed to the cDNA over a 6-h period, and then ligated together to form a PCR template. After ligation, Platinum Taq and universal primers were added to the plate. The upstream primer was biotinylated to allow later staining with streptavidin–phycoerythrin. The PCR amplicon was then hybridized to Luminex microbeads via the complimentary, probe-specific barcode on each bead. After overnight hybridization, the beads were washed and stained with streptavidin–phycoerythrin to prepare them for detection in Luminex FlexMap 3D scanners. The scanners measured each bead independently and reported the bead color and identity and the fluorescence intensity of the stain. A deconvolution algorithm converted these raw fluorescence intensity measurements into median fluorescence intensities for each of the 978 measured genes, producing the GEX level data. These GEX data were then normalized based on an invariant gene set, and then quantile-normalized to produce QNORM level data. An inference model was applied to the QNORM data to infer gene expression levels for a total of 10,174 features (Supplementary Table 7). Per-strain gene expression signatures were calculated using a weighted average of the replicates, for which the weights are proportional to the Spearman correlation between the replicates. These signatures were then queried against the reference dataset Touchstone (GEO accession # GSE92742)52 to assess similarity. The top 100 up- and downregulated genes in each signature were compared to the reference data, yielding a rank-ordered list of most similar reference signatures.
For downstream analyses (unsupervised clustering and GSEA), differential gene expression profiles were computed for the L1000 profiles. In order to maximize the expression signal, differential expression was computed jointly using profiles measured at 24hr and 72hr for each cell line and drug treatment. Specifically, log-fold-change was estimated between drug-treated profiles at 24 and 72 hours and DMSO-treated profiles at 24 and 72 hours for each experimental condition. This estimation was carried out using the ‘limma-trend’ pipeline59, in which p-values were estimated based on empirical-Bayes moderated t-statistics. Unsupervised hierarchical clustering was performed on these differential expression profiles using complete-linkage clustering, as implemented in the R function ‘hclust’. Pearson correlation was used as a similarity measure between the expression profiles. For analysis of gene set enrichment of transcriptional response signatures, enrichment was measured using the original GSEA method66 (based on the estimated log-fold-change), which estimates the concentration of each gene set in the list of up- and downregulated genes. We used the GSEA implementation in the R package ‘fgsea’67. The collection of gene sets used was ‘Biological Processes’ gene set collection from MSigDB v6.268.
The analysis of the mRNA expression levels of mitotic kinesins was based on microarray-based transcriptional profiling of HCT116 and HPT cells (GEO accession # GSE47830)69.
Microscopy
Cells were grown on plain glass or FBN-coated or gelatin-coated coverslips. For analysis, cells were either fixed in cold methanol followed by 4% paraformaldehyde, blocked with 10% FBS in PBS-T, or cold methanol containing 1% paraformaldehyde, blocked with 20% goat serum in antibody diluting buffer (Abdil; TBS, pH 7.4, 1% BSA, 0.1% Triton X-100, and 0.1% sodium azide) before incubating with the specified primary antibodies. Coverslips were mounted onto slides using Prolong Gold anti-fade mounting medium with DAPI (Molecular Probes). Images were acquired with a microscope (Axio Imager Z1; Carl Zeiss) equipped with CSU22 unit (Yokogawa Corporation of America) and CoolSnap HQ2 camera (Photometrics) controlled by SlideBook software or a Ti-E inverted microscope (Nikon Instruments) with a Clara cooled charge-coupled device (CCD) camera, Spectra-X light engine (Lumencore) (Andor) controlled by NIS Elements software (Nikon Instruments). Imaging of z stacks with 0.3 – 0.7 μm steps covering the entire volume of the mitotic apparatus were collected with a Plan-Apochromatic 1.40 NA 60× or 100x immersion oil objective lens. Live-cell imaging of cells in CO2-independent media (Gibco) utilized Nikon Plan Apo 20X or 40X DIC N2 0.75 NA objectives and an environmental chamber at 37°C.
Mitotic arrest assay
Cells were seeded in black 96-well plates two days prior to imaging and treated with Nocodazole at a concentration of 200 ng/ml. Imaging was performed with a 6-min time-lapse for 50h with GFP (1000 ms exposure) and DIC (200 ms exposure) using 20x air objective. Image analysis was performed using Slidebook 6 software (Intelligent Imaging Innovations).
Microtubule regrowth assay
Microtubule regrowth assay was performed as previously described70. The cells were incubated with 1μg/ml nocodazole for 3h and placed in ice for 1hr to depolymerize microtubules. Microtubule regrowth was analyzed after transfer to drug-free medium at 37°C. Cells were washed in PHEM buffer and depolymerized tubulin was removed with 0.2% Triton in PHEM buffer for 1 min. The cells were then washed in 1×PBS and fixed in 3.7% formaldehyde for 15 min. An immunofluorescence assay for β-tubulin and pericentrin was performed after permeabilization in 0.5% Triton and blocking in PBTA. Quantification of mean β-tubulin fluorescence intensity in the region of the centrosome was measured by ImageJ in a circle of constant diameter across all samples around the centrosome. At least 40 cells were analyzed in each sample of three independent biological experiments.
Microtubule dynamics by EB3 tracking
The cells transfected with EB3-EGFP were seeded in 96-well glass bottom plate. 24hr later, the VS83 was added for 18hr. Spinning disk confocal microscope with an incubator box was used for the microscopy. Live cell 60 s movies were taken using a spinning disk confocal microscope with a 100x objective, z-stacks 400 nm, time resolution 400 ms. The mean velocity was calculated as the instantaneous velocity between at least three consecutive time as v = mean distance (micrometers)/time (seconds).
Quantitative analysis of spindle angle and length
Images were collected by taking z stacks with a step size of 0.3 μm covering the entire volume of the mitotic spindle. Fluorescence signal quantification in the spindle was performed using the SlideBook software. Distances were measured after defining the position of the two poles and correcting for projection errors.
Quantification of multipolar spindles, micronuclei and unsuccessful cytokinesis
Multipolar spindles and micronuclei were counted in cells labeled with antibodies against α-tubulin and γ-tubulin, as well as DAPI. The percentage of mitotic cells with spindles containing more than two poles and the percentage of interphase cells with micronuclei are reported. The percentage of cells that exited mitosis as a single cell was determined from live imaging of cells using DIC and reported as those that fail cytokinesis.
Karyotyping
Low-pass whole-genome sequencing (LP-WGS)
Genomic DNA was extracted using Qiagen extraction kit (Qiagen), amplified and barcoded using Nextera reagents (Illumina). Whole-genome amplified DNA samples were purified with 1.5X SPRI beads in an automated setup. Post purification, Illumina libraries were made using the Illumina Nextera XT kit. Samples were pooled, quantified by qPCR, and sequenced on HiSeq2000 on Single End flowcell lanes. Sequence reads were trimmed to 40 nucleotides and aligned to the mouse (mm9) or human (hg19) reference genomes using the BWA (0.7.12) backtrack algorithm. HMMcopy (0.1.1)71 was used to detect copy number alterations by estimating DNA copy number in 500-kb bins controlling for mappability and GC content (calculated by HMMcopy gcCounter). CNV analyses were performed using the method described by Knouse et al.72, running HMMcopy with evalue=0.995, and a dnacopy (1.50.1) run with alpha=0.0001.
G-banding
The cells were treated with 50 ng/ml microtubule-depolymerizing drug colchicine (Serva) for 4.5hr, then centrifuged with a table-top centrifuge, swollen in 75 mM KCl in a 37°C water bath for 15 minutes, fixed with Carnoy solution (75% methanol and 25% acetic acid) and spread on a wet glass slide with a glass Pasteur pipette. The slides were dried at 42°C and stained with Giemsa dye (Fluka). The slides were imaged with an inverted microscope with 100x objective; 30 – 50 metaphases were scored for each cell line.
Single cell DNA sequencing (scDNAseq)
For single nuclei isolation, cell pellets were re-suspended in lysis buffer (1M tris-HCl pH7.4, 5M NaCl, 1M CaCl2, 1M MgCl2, 7.5% BSA, 10% NP-40, ultra-pure water, 10 mg/ml Hoechst 33358, 2mg/ml propidium iodide) and kept on ice in the dark for 15 min to facilitate lysis. G1 single nuclei, as assessed by PI and Hoechst staining, were sorted into 96-well plates on a BD FacsJAZZ cell sorter (BD Biosciences), and stored in −80°C until further analysis. For single-cell library preparation, single nuclei were lysed and DNA was barcoded, followed by automated library preparation (Bravo Automated Liquid Handling Platform, Agilent Technologies), as previously described73. Pooled single-cell libraries were sequenced using a NextSeq 500 machine (Illumina; up to 77 cycles; single-end). The generated data were subsequently demultiplexed using sample-specific barcodes and changed into fastq files using bcl2fastq (Illumina; version 1.8.4). Reads were aligned to the human reference genome (GRCh38/hg38) using Bowtie2 (version 2.2.4)74. Duplicate reads were marked with BamUtil (version 1.0.3)75. The aligned read data (bam files) were analyzed with AneuFinder (Version 1.14.0)41. Following GC correction and blacklisting of artefact-prone regions (extreme low or high coverage in control samples), libraries were analyzed using the dnacopy and edivisive copy number calling algorithms with variable width bins (binsize: 1 Mb; stepsize: 500 kb) and breakpoint refinement (R= 20, confint = 0.95; other settings as default). A minimum concordance of 95% between the results of the two algorithms was required. Libraries with less than five reads per bin per chromosome copy (~30,000 reads for a diploid genome) were discarded. Samples with a near-tetraploid DNA content were analyzed with the developer version of AneuFinder (Version 1.7.4; from GitHub): the min.ground.ploidy parameter was set to either 3 or 3.5 and the max.ground.ploidy parameter to 4.5, 5.0 or 5.5. The minimum and maximum ground ploidy values were determined with the results that were previously obtained with the standard (Bioconductor) version of AneuFinder. Results were subsequently curated as described above, except using a minimum concordance of 90%. Aneuploidy and heterogeneity scores were calculated as previously described41. Overall, high-quality karyotypes were generated for 210 single cells.
Flow cytometry
For cell cycle and cell death analyses, cells were trypsinized and incubated in cold PBS supplemented with 5% fetal calf serum (Sigma-Aldrich; PBS-FACS). DNA was stained either by propidium iodide (PI) or by Hoechst. For PI staining, cells were fixed in cold 70% ethanol, added dropwise while vortexing, and incubated on ice for 30 minutes. Cells were centrifuged and pellets were washed twice with PBS-FACS. 50 μl RNase A solution (100 μg/ml in PBS) was added to the pellet, followed by staining with 400 μl PI solution (50 μg/ml in PBS) per million cells. Cells were incubated for 10’ at 25°C. For Hoechst staining, pellets were incubated in the dark with 10 mg/ml Hoechst 33358 for 15’ at 4°C. Data acquisition was performed using the CytoFLEX flow cytometer (Beckman Coulter) or the BD FacsJAZZ cell sorter (BD Biosciences). Data analysis was performed using the Kaluza Analysis software 2.1 (Beckman Coulter). Gating strategy: An SSC-A/FSC-A gate was set in order to exclude cell debris, and an FSC-A/FSC-H gate was then set in order to exclude doublets. Cell cycle phases were determined manually using linear gating based on the 2N and 4N peaks of the histogram. Cell death was assessed by quantifying the fraction of cells in the subG1 population, and mitotic arrest was assessed by quantifying the fraction of cells in the G2/M population. Gating strategy is shown in Supplementary Fig. 3.
Statistical analyses
The two-sided t-test was used to compare single gene dependency and expression between the near-euploid and highly-aneuploid cancer cell lines. The two-sided Fisher’s Exact test was used for calculating the significance of the overlap of hits for the genetic perturbation datasets. The statistical analyses of all microscopy experiments were performed in GraphPad Prism. t-test was used to determine the significance of differences between the means of two groups. Fisher’s Exact test was used to determine the significance of differences in the prevalence of categorical events between groups.
Extended Data
Extended Data Figure 1: Increased sensitivity of aneuploid cancer cells to genetic inhibition of the spindle assembly checkpoint.
(a) Copy number profiles of 5 representative breast cancer cell lines from the highly-aneuploid cell line group (top quartile of AS) and 5 representative breast cancer cell lines from the near-euploid cell line group (bottom quartile of AS). (b) A volcano plot showing the differential genetic dependencies between the near-euploid and highly-aneuploid cancer cell lines (top vs. bottom quartiles), based on the genome-wide DRIVE RNAi screen4. BUB1B and MAD2, core members of the SAC, are highlighted in red. (c) A Venn diagram showing the overlap of the differentially-dependent genes (q<0.25) between the Achilles and DRIVE RNAi screens. ****, p=1e-16, two-tailed Fisher’s exact test. (d) The pathways enriched in the list of genes that are more essential in near-euploid than in highly-aneuploid cancer cell lines (effect size<−0.1, q<0.1) in the DRIVE RNAi screen, based on DAVID functional annotation enrichment analysis49. The full list is available in Supplementary Table 3. *, Benjamini-corrected p-value<0.1; one-tailed Fisher’s Exact Test. (e) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (left) and MAD2 (right) in the DRIVE RNAi screen. The more negative a value, the more essential the gene is in that cell line. ****, p=2e-06 and p=1e-04 for BUB1B and MAD2, respectively; two-tailed t-test. (f) A volcano plot showing the differential genetic dependencies between the near-euploid and highly-aneuploid cancer cell lines (top vs. bottom 10% of cell lines), based on the genome-wide DRIVE RNAi screen4. BUB1B, MAD2 and KIF18A are highlighted in red. (g) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (left) and MAD2 (right) in the DRIVE RNAi screen (top vs. bottom 10% of cell lines). The more negative a value, the more essential the gene is in that cell line. *, p=0.037; ***, p=5e-04; two-tailed t-test. (h) Comparison of protein expression levels of BUB1B (left) and MAD2 (right) between near-euploid and highly-aneuploid cancer cell lines. n.s., p>0.05; **, p=0.001; for BUB1B and MAD2, respectively; two-tailed t-test. (i) The correlations between the mRNA expression levels of BUB1B (top) and MAD2 (bottom) and the genetic dependency on these genes in the Achilles (left) and DRIVE (right) RNAi screens. Spearman’s ρ=0.36 (p=3e-08), 0.31 (p=2e-06), 0.26 (p=4e-04) and 0.40 (p=2e-08), respectively. (j) The correlations between the protein expression levels of BUB1B (top) and MAD2 (bottom) and the genetic dependency on these genes in the Achilles (left) and DRIVE (right) RNAi screens. Spearman’s ρ=0.11 (p=0.09), 0.26 (p=4e-05), 0.14 (p=0.016) and 0.24 (p=5e-05), respectively. (k) The mRNA expression levels of BUB1B (left) and MAD2 (right) in near-euploid and highly-aneuploid cancer cell lines across multiple cell lineages. *, p<0.05; two-tailed t-test.
Extended Data Figure 2: Genomic and phenotypic features associated with the degree of aneuploidy in human cancer cell lines.
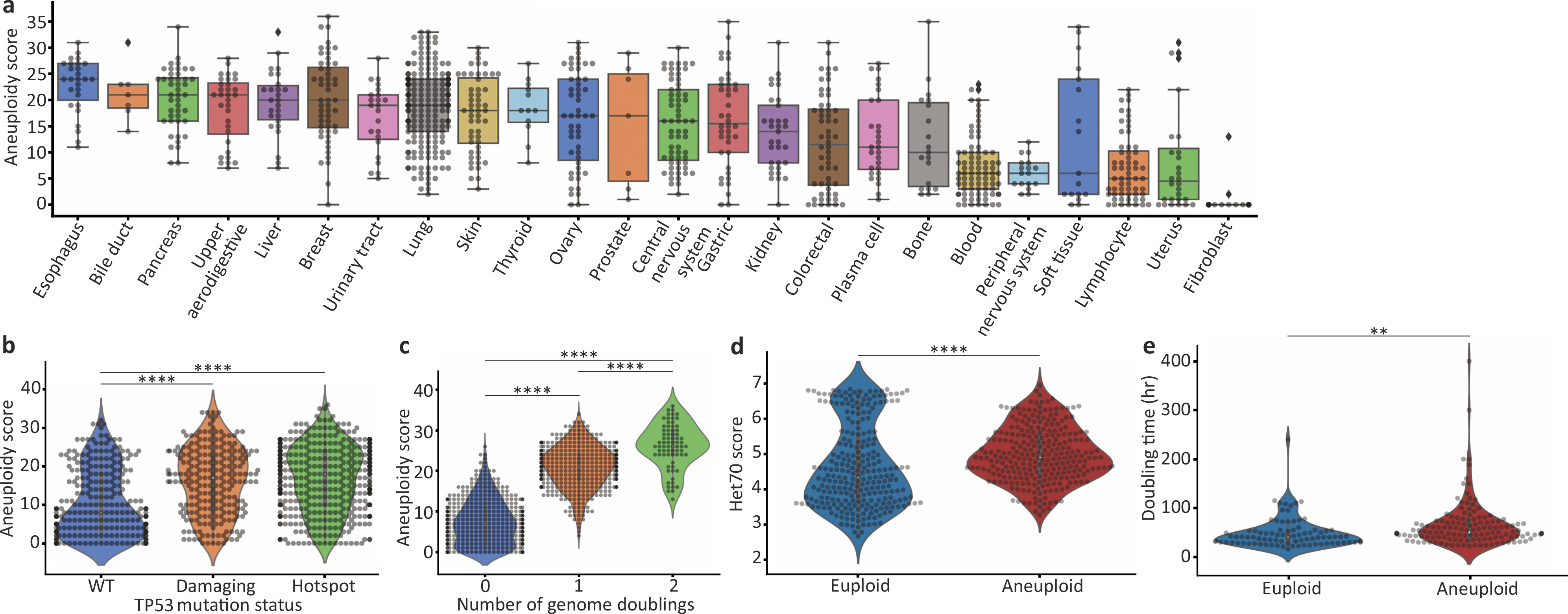
(a) The AS distribution across 23 cancer types. Bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range, individual cell lines. (b) Comparison of AS between cancer cell lines with distinct TP53 mutation status (based on CCLE annotations)2. ****, p=6e-15 and p=1e-22 for the comparisons between TP53-WT and ‘damaging’ and TP53-WT and ‘hotspot’ mutations, respectively; two-tailed t-test. (c) Comparison of AS between cancer cell with distinct genome doubling (WGD) status. ****, p=1e-192, p=2e-96 and p=6e-13 for the comparisons between WGD=0 and WGD=1, WGD=0 and WGD=2, and WGD=1 and WGD=2, respectively; two-tailed t-test. (d) Comparison of the HET70 score, a measure of karyotypic instability50, between the near-diploid and highly-aneuploid cell line groups. ****, p=2e-08; two-tailed t-test. (e) Comparison of doubling time between the near-diploid and highly-aneuploid cell line groups. **, p=0.005; two-tailed t-test.
Extended Data Figure 3: Increased sensitivity of aneuploid cancer cells to SACi remains significant when associated genomic and phenotypic features are controlled for.
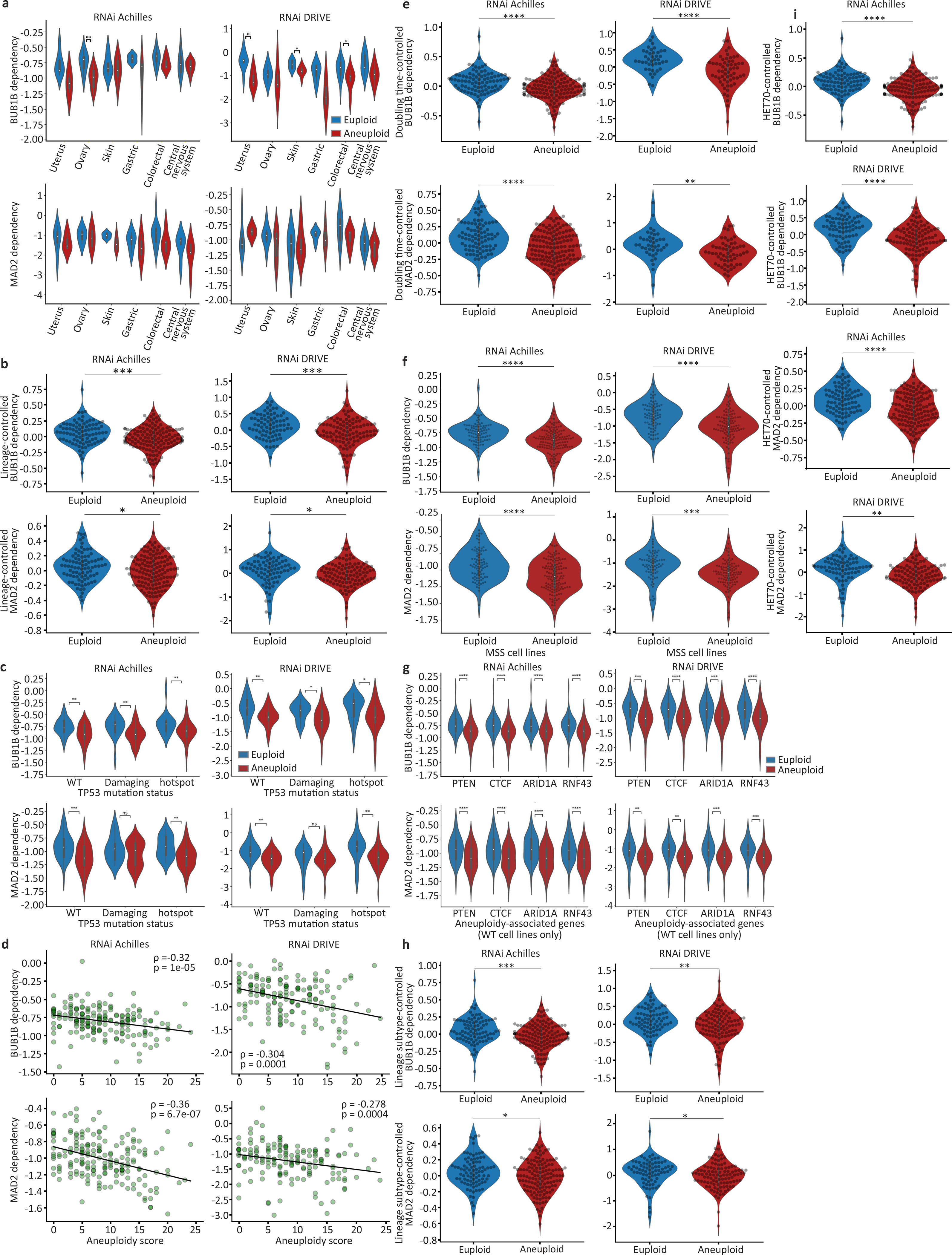
(a) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (top) and MAD2 (bottom) in the Achilles (left) and DRIVE (right) RNAi screens across multiple cell lineages. *, p<0.05; **, p<0.01; two-tailed t-test. (b) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (top) and MAD2 (bottom) in the Achilles (left) and DRIVE (right) RNAi screens, after accounting for lineage-specific differences in gene dependency scores using linear regression. ***, p=2e-04; * p=0.013; for RNAi-Achilles BUB1B and MAD2 dependencies, respectively; ***, p=5e-04; *, p=0.044; RNAi-DRIVE BUB1B and MAD2 dependencies, respectively; one-tailed t-test. (c) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (top) and MAD2 (bottom) in the Achilles (left) and DRIVE (right) RNAi screens, across TP53 mutation classes. *, p<0.05; **, p<0.01, ***, p<0.001; two-tailed t-test. (d) The correlations between AS and the dependency on BUB1B (top) and MAD2 (bottom) in the Achilles (left) and DRIVE (right) RNAi screens, for cell lines that have not undergone whole-genome duplication (i.e., cell lines with basal ploidy of n=2). Spearman’s ρ = −0.32 (p=1e-05), −0.36 (p=7e-07), −0.30 (p=1e-04) and −0.28 (p=4e-04), respectively. (e) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (top) and MAD2 (bottom) in the Achilles (left) and DRIVE (right) RNAi screens, after removing the effect of doubling time on gene dependency scores using linear regression. ****, p=1e-05 and p=9e-07, for RNAi-Achilles BUB1B and MAD2 dependencies, respectively; ****, p=1e-07; **, p=0.002; for RNAi-Achilles BUB1B and MAD2 dependencies, respectively; two-tailed t-test. (f) The sensitivity of near-euploid and highly-aneuploid cancer cell lines without microsatellite instability (MSS lines only) to the knockdown of BUB1B (top) and MAD2 (bottom) in the Achilles (left) and DRIVE (right) RNAi screens. ****, p=7e-07 and p=2e-07, for RNAi-Achilles BUB1B and MAD2 dependencies, respectively; ****, p=6e-07, for RNAi-DRIVE BUB1B dependency; ***, p=1e-04; two-tailed t-test. (g) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (top) and MAD2 (bottom) in the Achilles (left) and DRIVE (right) RNAi screens, in cell lines that are WT for the 4 genes most selectively mutated in aneuploid human tumors (after TP53)12. **, p<0.01, ***, p<0.001; ****, p<1e-04; two-tailed t-test. (h) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (top) and MAD2 (bottom) in the Achilles (left) and DRIVE (right) RNAi screens, after removing the effect of lineage subtype on gene dependency scores using linear regression. ***, p=4e-04; * p=0.015, for RNAi-Achilles BUB1B and MAD2 dependencies, respectively; **, p=0.002; * p=0.045, for RNAi-DRIVE BUB1B and MAD2 dependencies, respectively; one-tailed t-test. (i) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of BUB1B (top two plots) and MAD2 (bottom two plots) in the Achilles (top) and DRIVE (bottom) RNAi screens, after removing the effect of HET70 scores on gene dependency scores using linear regression. ****, p=9e-07, p=8e-06 and p=5e-07 for RNAi-Achilles BUB1B, RNAi-Achilles MAD2 and RNAi-DRIVE BUB1B dependencies, respectively; **, p=0.001; two-tailed t-test.
Extended Data Figure 4: Reduced sensitivity of aneuploid cancer cells to chemical inhibition of the spindle assembly checkpoint.
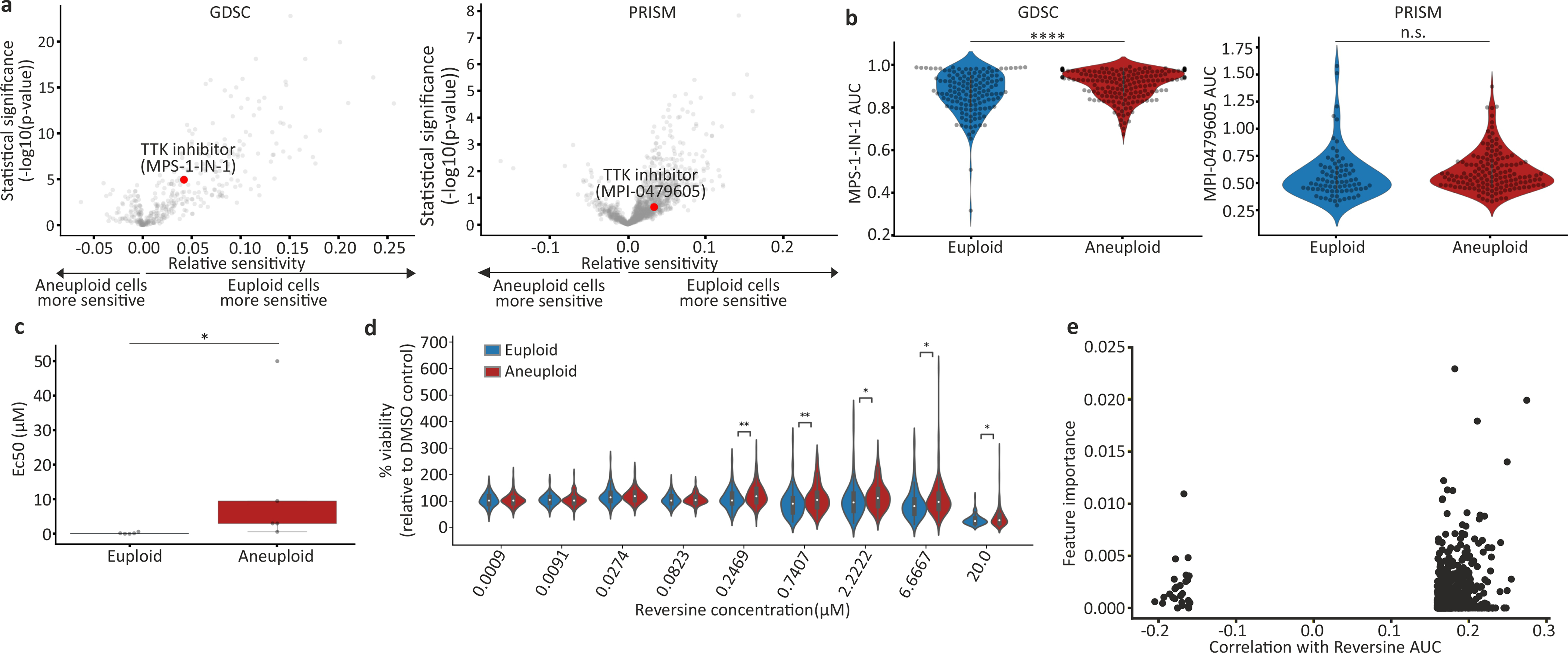
(a) Volcano plots showing the differential drug sensitivities between the near-euploid and highly-aneuploid cancer cell lines, based on the large-scale GDSC6 and PRISM screens9. MPS1-IN-1 and MPI-0479605, the only SAC inhibitors included in each screen, respectively, are highlighted in red. (b) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the SAC inhibitors MPS1-IN-1 and MPI-0479605 in the GDSC (left) and PRISM (right) screens. ****, p=1e-0.5; n.s., p=0.23; two-tailed t-test. (c) Experimental validation of the response of 5 near-euploid (CAL51, EN, MHHNB11, SW48 and VMCUB1) and 5 highly-aneuploid (MDAMB468, NCIH1693, PANC0813, SH10TC and A101D) cell lines to 72hr exposure to the SAC inhibitor reversine. *, p=0.016, two-tailed Wilcoxon rank-sum test; n=5 cell lines in each group. Bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range. (d) Comparison of the sensitivity to reversine between near-euploid and highly-aneuploid cancer cell lines subjected to the PRISM cell viability assay, confirming the reduced sensitivity of highly-aneuploid cells to a 120hr exposure to SAC inhibitors. n.s., p>0.05; *, p<0.05; **, p<0.01; two-tailed t-test. (e) An association analysis failed to identify a genomic biomarker of reversine sensitivity. Shown are the top 1000 genomic features identified by our model (see Methods). No feature stands out in terms of importance and/or correlation, and the overall predictive value is poor.
Extended Data Figure 5: Isogenic model systems of near-diploid and aneuploid cell lines.
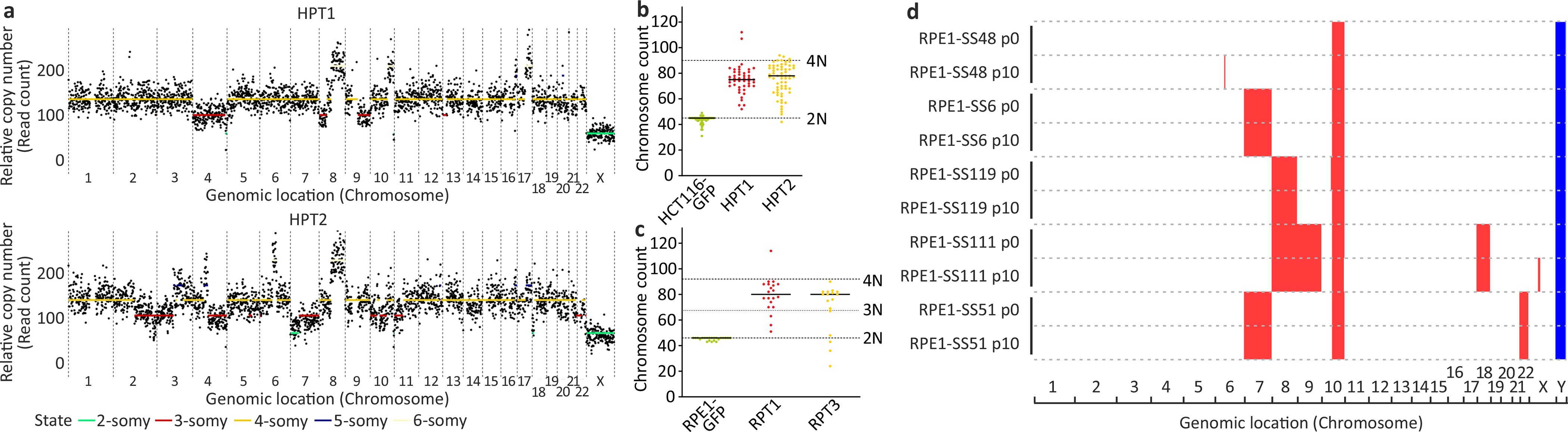
(a) scDNAseq-based copy number profiling of the HPT1 and HPT2 aneuploid cell lines. (b) Karyotyping-based chromosome count of the near-diploid HCT116 cells and their highly-aneuploid HPT derivatives. Each dot represents a metaphase spread. Average chromosome number: n=45, n=75 and n=78, for HCT116-GFP, HPT1 and HPT2, respectively. (c) Karyotyping-based chromosome count of the near-diploid RPE1 cells and their highly-aneuploid RPT derivatives. Each dot represents a metaphase spread. Average chromosome number: n=46, n=80 and n=76.5, for RPE1-GFP, RPT1 and RPT2, respectively. (d) Low-pass whole-genome sequencing-based karyotyping of near-diploid and aneuploid RPE1 clones. No karyotypic changes have been observed between passage 0 (p0) and passage 10 (p10) of each clone. Red, large (>5Mb) gains (log2CN>0.3); blue, large (>5Mb) losses (log2CN<−0.3).
Extended Data Figure 6: The effect of aneuploidy on cellular sensitivity to SACi in isogenic human cell lines.
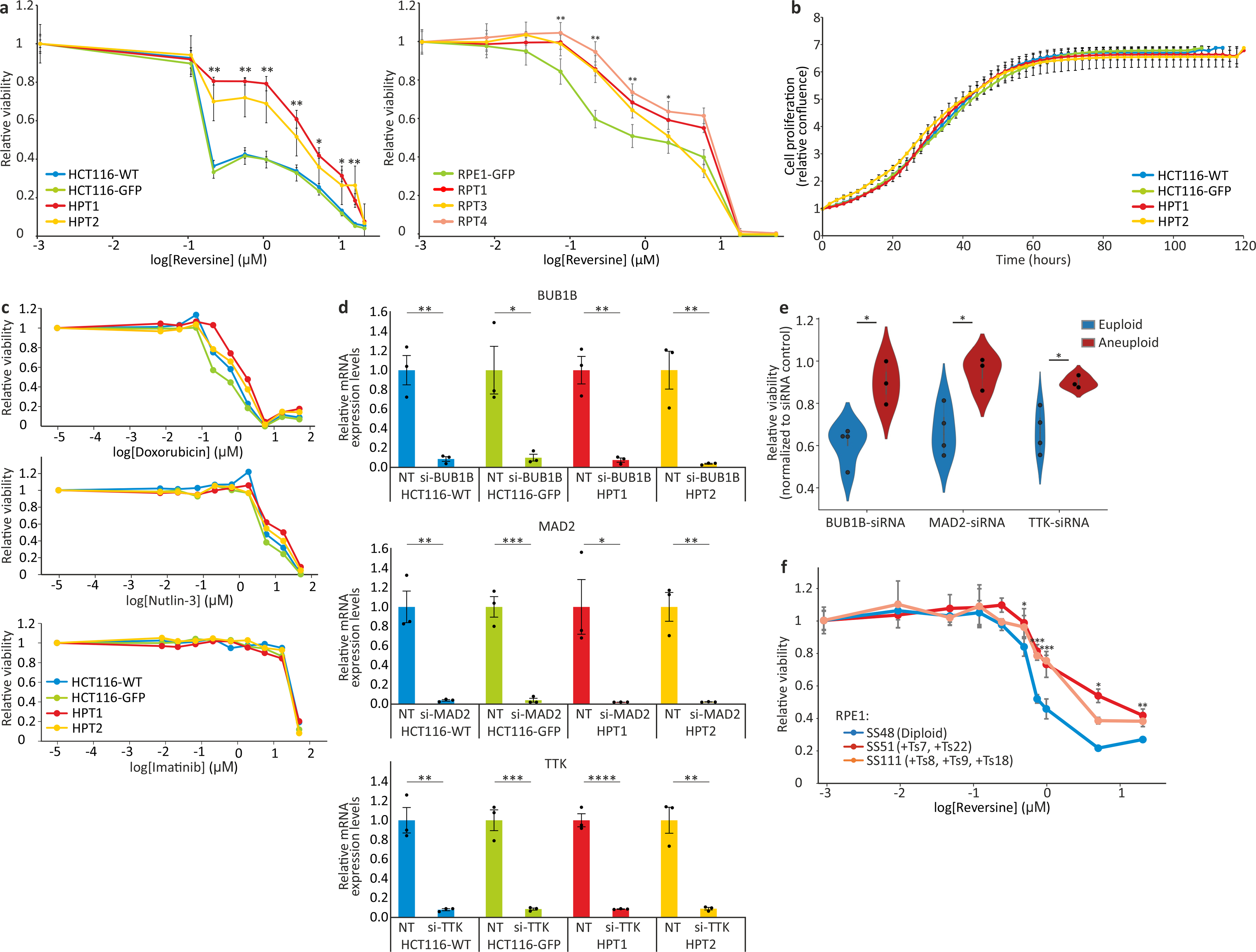
(a) Left: dose response curves of the response of near-diploid HCT116 cells and their highly-aneuploid derivatives HPT cells, to the SAC inhibitor reversine following 120hr of drug exposure. EC50 = 0.11μM, 0.11μM, 2.32μM and 1.06μM, for HCT116-WT, HCT116-GFP, HPT1 and HPT2, respectively. Right: dose response curves of the response of near-diploid RPE1 cells and their highly-aneuploid derivatives RPT cells, to the SAC inhibitor reversine following 120hr of drug exposure. EC50 = 0.13μM, 1.82μM, 0.57μM and 2.07μM, for RPE1-GFP, RPT1, RPT3 and RPT4, respectively. *, p<0.05; **, p<0.01; ***, p<0.001; two-tailed t-test. Data represent the mean ± s.d.; n=3 biological replicates. (b) Time-lapse imaging-based proliferation curves of HCT116 and HPT cells under standard culture conditions. Data represent the mean ± s.d.; n=3 biological replicates. (c) Dose response curves of the response of HCT116 and HPT cells to three drugs with unrelated mechanisms of action. Doxorubicin EC50 = 0.61μM, 0.32μM, 1.2μM and 0.89μM; Nutlin-3 EC50 = 11.88μM, 19.28μM, 15.26μM and 65.11μM; Imatinib EC50 = 17.94μM, 19.08μM, 18.77μM and 23.31μM; for HCT116-WT, HCT116-GFP, HPT1 and HPT2, respectively. (d) Relative mRNA expression levels of BUB1B, MAD2 and TTK, confirming successful siRNA-mediated knockdown of each gene in all cell lines. *, p=0.011, p=0.012 for HCT116-GFP and HPT1, respectively; **, p=0.0019, p=0.0015, p=0.0039 for BUB1B in HCT116-WT, HPT1 and HPT2, respectively; p=0.0021, p=0.0013 for MAD2 in HCT116-WT and HPT2, respectively; p=0.0011, p=0.0012 for TTK in HCT116-WT and HPT2, respectively; ***, p=0004, p=0.0005 for MAD2 and TTK in HCT116-GFP, respectively; ****, p=9e-05; one-tailed t-test; n=3 biological replicates. Data represent the mean ± s.e.m. (e) The relative viability of 4 near-diploid (CAL51, EN, MHHNB11, VMCUB1) and 3 highly-aneuploid (MDAMB468, PANC0813, SH0TA) cancer cell lines following 72hr of siRNA-mediated knockdown of 3 SAC components: BUB1B, MAD2 and TTK. Results are normalized to a non-targeting siRNA control. *, p=0.010, p=0.016, and p=0.015, for BUB1B, MAD2 and TTK, respectively; two-tailed t-test. Error bars, s.d. (f) Dose response curves of the response of the near-diploid RPE1 clone SS48 and its isogenic aneuploid clones SS51 (+Ts7, +Ts22) and SS111 (+Ts8, +Ts9, +Ts18), to the SAC inhibitor reversine following 120hr of drug exposure. EC50 = 0.66μM, 1.03μM and 1.03μM, for SS48, SS51 and SS111, respectively *, p<0.05; **, p<0.01; ***, p<0.001; two-tailed t-test. Data represent the mean ± s.d.; n=3 biological replicates.
Extended Data Figure 7: Time-dependent increased sensitivity of aneuploid cancer cells to genetic and chemical SACi.
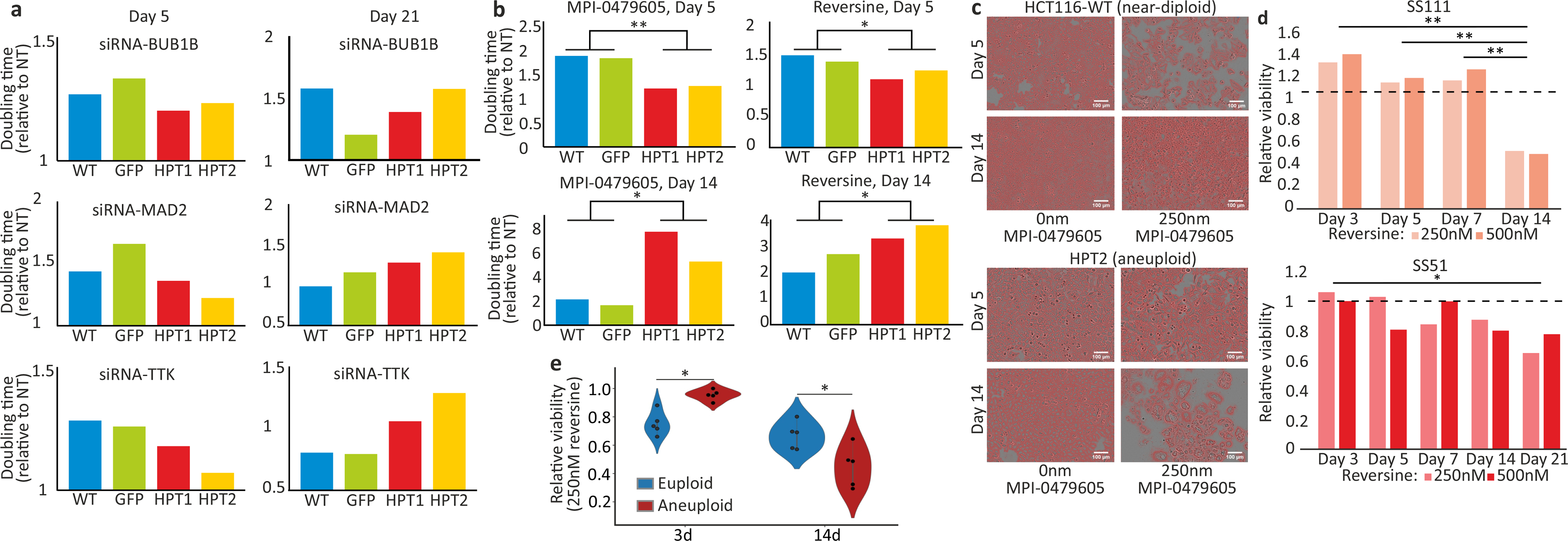
(a) Comparison of the doubling times of HCT116 and HPT cells exposed to siRNAs against BUB1B, MAD2 or TTK. The drug effect of SACi is stronger in the near-diploid HCT116 cells at d5, but is stronger in the highly-aneuploid HPT cells at d21. (b) Comparison of the doubling times of HCT116 and HPT cells exposed to the SAC inhibitors MPI-0479605 or reversine. The drug effect of SACi is stronger in the near-diploid HCT116 cells at d5, but at d21 it becomes stronger in the highly-aneuploid HPT cells. *, p=0.034, p=0.046 and p=0.049 for MPI-0479605 and reversine at d5 and d14, respectively; **, p=0.0015; one-tailed t-test; n=2 independent cell lines. (c) Representative images of cells from the drug experiment (same images as in Fig. 2g), with cell masking performed using the image analysis software ilastik51. Scale bar, 100μm. (d) The relative viability of the aneuploid RPE1 clones, SS111 and SS51, following reversine exposure. The viability effect was normalized to the effect of the drug in the near-diploid RPE1 clone, SS48. The drug effect of SACi is comparable during the first week of drug exposure, but the highly-aneuploid cells become significantly more sensitive with time. *, p=0.045, **, p=0.002, p=0.001 and p=0.005 for the comparisons between d3 and d14, d5 and d14 and d7 and d14, respectively; two-tailed t-test. (e) The relative viability of 5 near-euploid (CAL51, EN, MHHNB11, SW48 and VMCUB1) and 5 highly-aneuploid (MDAMB468, NCIH1693, PANC0813, SH10TC and A101D) cell lines to 72hr and 14d exposure to the SAC inhibitor reversine. *, p=0.012 and p=0.037, for 3d and 14d time points, respectively; two-tailed Wilcoxon rank-sum test.
Extended Data Figure 8: Transcriptional and cellular characterization of SACi in aneuploid cells.
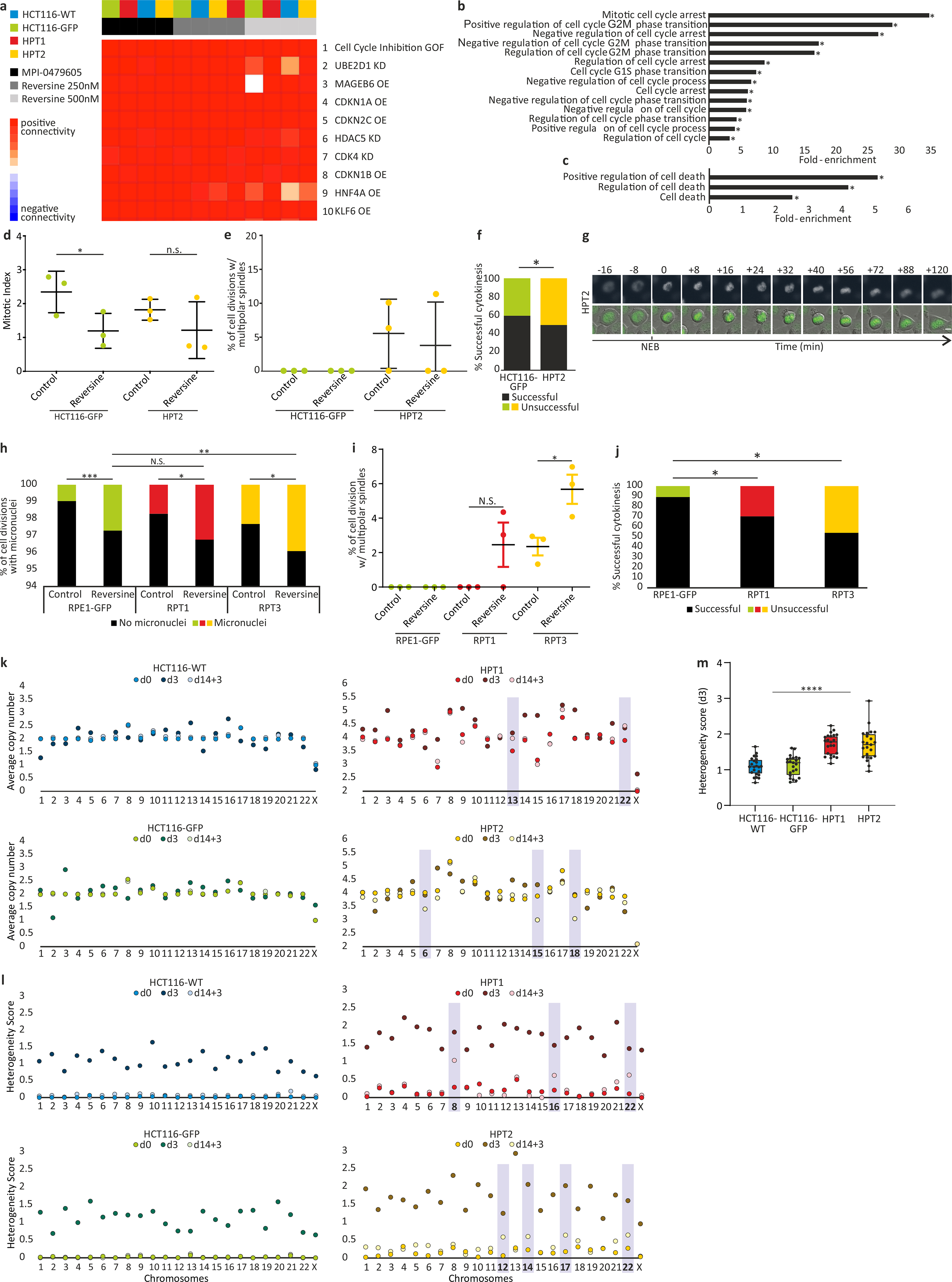
(a) The top 10 results of a Connectivity Map (CMap) query52 of the transcriptional response of HCT116 and HPT cells to the SAC inhibitors, reversine (250nM and 500nM) and MPI-0479605 (250nM). The top connection is “Cell cycle inhibition”, correctly identifying the expected mechanism of action of these compounds. GOF, gain of function; OE, over-expression; KD, knockdown. (b) Functional enrichment of gene sets related to cell cycle regulation. Shown are the gene sets that were significantly more affected by SACi in the highly-aneuploid HPT1 and HPT2 cells than in the nearly-diploid HCT116-WT and HCT116-GFP cells. *, p<0.05, one-tailed Fisher’s exact test. (c) Functional enrichment of gene sets related to cell death. Shown are the gene sets that were significantly more affected by SACi in the highly-aneuploid HPT1 and HPT2 cells than in the nearly-diploid HCT116-WT and HCT116-GFP cells. *, p<0.05, one-tailed Fisher’s exact test. (d) The mitotic index of HCT116 and HPT cells cultured under standard conditions or exposed to the SAC inhibitor reversine (500nM) for 24hr. *, p=0.035; n.s., p=0.17; two-tailed t-test; Error bars, s.d.; n=3 biological replicates. (e) Imaging-based quantification of the prevalence of cell divisions with multipolar spindles in HCT116 and HPT cell lines cultured under standard conditions or treated with reversine (500nM) for 24hr; n=3 biological replicates. Error bars, s.d. (f) The prevalence of premature mitotic exit (cytokinesis failure) in HCT116 and HPT cells exposed to the SAC inhibitor reversine (500nM) for 24hr. *, p=0.047; two-tailed Fisher’s exact test. (g) Representative images of premature mitotic exit in HPT2 cells exposed to reversine (500nM). T=0 defines nuclear envelope breakdown (NEB). Scale bar, 10μm. (h) The prevalence of micronuclei formation in RPE1 and RPT cells cultured under standard conditions or exposed to the SAC inhibitor reversine (500nM) for 24hr. n.s., p>0.05; *, p=0.013 and p=0.015 for the differences between the treated and untreated RPT1 and RPT3 cells, respectively; **, p=0.004; ***, p<0.0002; two-tailed t-test. (i) The prevalence of cell divisions with multipolar spindles in RPE1 and RPT cells cultured under standard conditions or exposed to the SAC inhibitor reversine (500nM) for 24hr. n.s., p>0.05; *, p=0.028; two-tailed t-test. Error bars, s.d. (j) The prevalence of premature mitotic exit (cytokinesis failure) in RPE1 and RPT cells exposed to the SAC inhibitor reversine (500nM) for 24hr. *, p=0.044 and p=0.019 for the comparisons between RPE1 and RPT1 or RPT3, respectively; two-tailed t-test. (k) Chromosomal copy number states of HCT116 and HPT cells at each of the 3 time points that were sequenced by scDNAseq. Differences between the pre-treated (d3) and post-treated (d14+3) populations are highlighted. (l) Chromosomal heterogeneity scores of the HCT116 and HPT cells at each of the 3 time points. Highly-heterogeneous chromosomes in the post-treated populations (d14+3) are highlighted; n=23 chromosomes. (m) Comparison of the chromosomal heterogeneity scores between the near-diploid HCT116 cells and the highly-aneuploid HPT cells. Bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range; circles, individual chromosomes. ****, p=2e-09; two-tailed t-test.
Extended Data Figure 9: Increased sensitivity of aneuploid cancer cells to perturbation of the mitotic kinesin KIF18A.
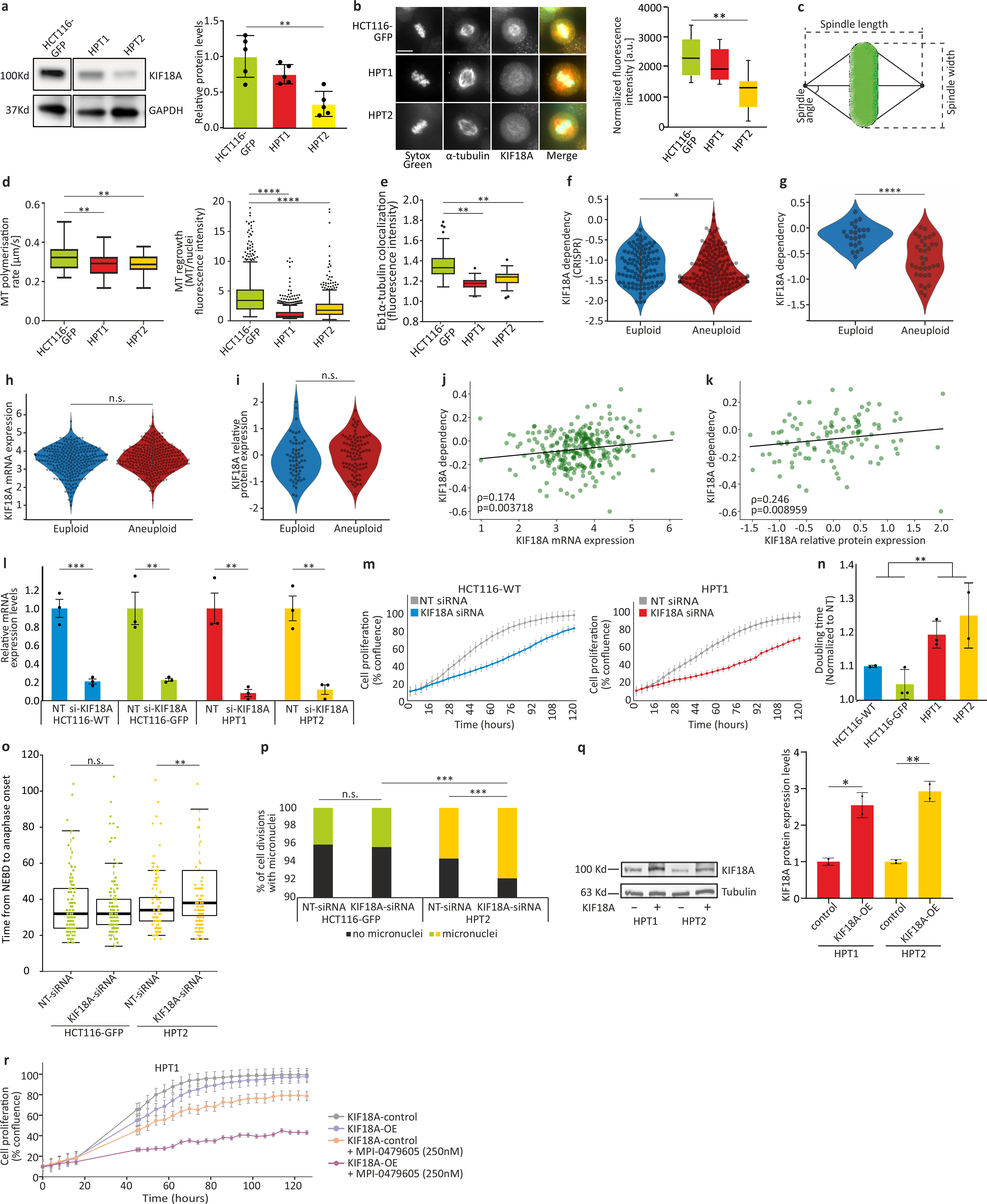
(a) Left: Western blot of KIF18A protein expression levels in HCT116 and HPT cell lines. Right: Quantification of KIF18A expression levels (normalized to GAPDH). **, p=0.002; two-tailed t-test; n=5 biological replicates. (b) Left: Imaging kinetochore-bound KIF18A protein levels in HCT116-GFP, HPT1 and HPT2 cells, Scale bars, 10μm. Right: Immunofluorescence-based quantification of KIF18A protein levels. **, p<0.01, two-tailed t-test. Bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range. (c) Schematics of the definitions of spindle length, width and angle. (d) Left: Imaging-based quantification of microtubule polymerization rate in HCT116 and HPT cells cultured under standard conditions. Right: Imaging-based quantification of microtubule regrowth following complete depolymerization in HCT116 and HPT cells. Bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range; circles, individual cell lines. *, p<0.05; **, p<0.01; ****, p<1e-4; two-tailed t-test. (e) Imaging-based quantification of EB1α-tubulin co-localization in HCT116 and HPT cells cultured under standard conditions. **, p<0.01. Bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range. (f) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockout of KIF18A in the CRISPR-Achilles dataset. The more negative a value, the more essential the gene is in that cell line. *, p=0.034; two-tailed t-test. (g) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the DRIVE RNAi screen (top vs. bottom 10% of cell lines). The more negative a value, the more essential the gene is in that cell line. ****, p=3e-06; two-tailed t-test. (h) Comparison of the mRNA expression levels of KIF18A between near-euploid and highly-aneuploid cancer cell lines. n.s., p>0.05; two-tailed t-test. (i) Comparison of the protein expression levels of KIF18A between near-euploid and highly-aneuploid cancer cell lines. n.s., p>0.05; two-tailed t-test. (j) The correlation between KIF18A mRNA expression and the genetic dependency on this gene in the Achilles-RNAi screen. Spearman’s ρ = 0.17 (p=0.004) (k) The correlation between KIF18A protein expression and the genetic dependency on this gene in the Achilles-RNAi screen. Spearman’s ρ=0.25 (p=0.009). (l) Relative mRNA expression levels of KIF18A, confirming successful siRNA-mediated KD in all cell lines 72hr post-transfection. **, p=0.006, p=0.003 and p=0.002 for HCT116-GFP, HPT1 and HPT2, respectively; ***, p=0.0007; one-tailed t-test. (m) Proliferation curves of HCT116 and HPT1 cells cultured in the presence of a KIF18A-targeting siRNA, or a non-targeting control siRNA. (n) Comparison of the doubling times of HCT116 and HPT cells following siRNA-mediated KIF18A knockdown. **, p=0.001; two-tailed t-test. (o) Time-lapse imaging-based quantification of the time from nuclear envelope breakdown (NEBD) to anaphase onset in HCT116 and HPT cell lines exposed to non-targeting or KIF18A-targeting siRNAs for 72hr. n.s, p>0.05; **, p=0.003; two-tailed t-test. Bar, median; box, 25th and 75th percentile; whiskers, 1.5 X interquartile range; circles, individual cell lines. (p) The prevalence of micronuclei formation in HCT116 and HPT cells exposed to non-targeting or KIF18A-targeting siRNAs for 72hr. n.s., p>0.05; ***, p<0.001; two-tailed Fisher’s exact test. (q) Relative protein expression levels of KIF18A, confirming successful KIF18 overexpression in the highly-aneuploid HPT1 and HPT2 cell lines 48hr post-transfection. Left: Western blot of KIF18A protein expression levels in HPT1 and HPT2 before and after KIF18A overexpression. Right: Quantification of KIF18A expression levels (normalized to α-Tubulin). *, p=0.013, **, p=0.005; one-tailed t-test; n=2 biological replicates. In all bar plots and line plots, data represent the mean ± s.d. unless otherwise noted; n=3 biological replicates unless otherwise noted. (r) Proliferation curves of HPT1 cells before and after over-expression of KIF18A (KIF18A-OE), in the absence or presence of MPI-0479605 (250nM).
Extended Data Figure 10: Increased sensitivity of aneuploid cells to KIF18A inhibition.
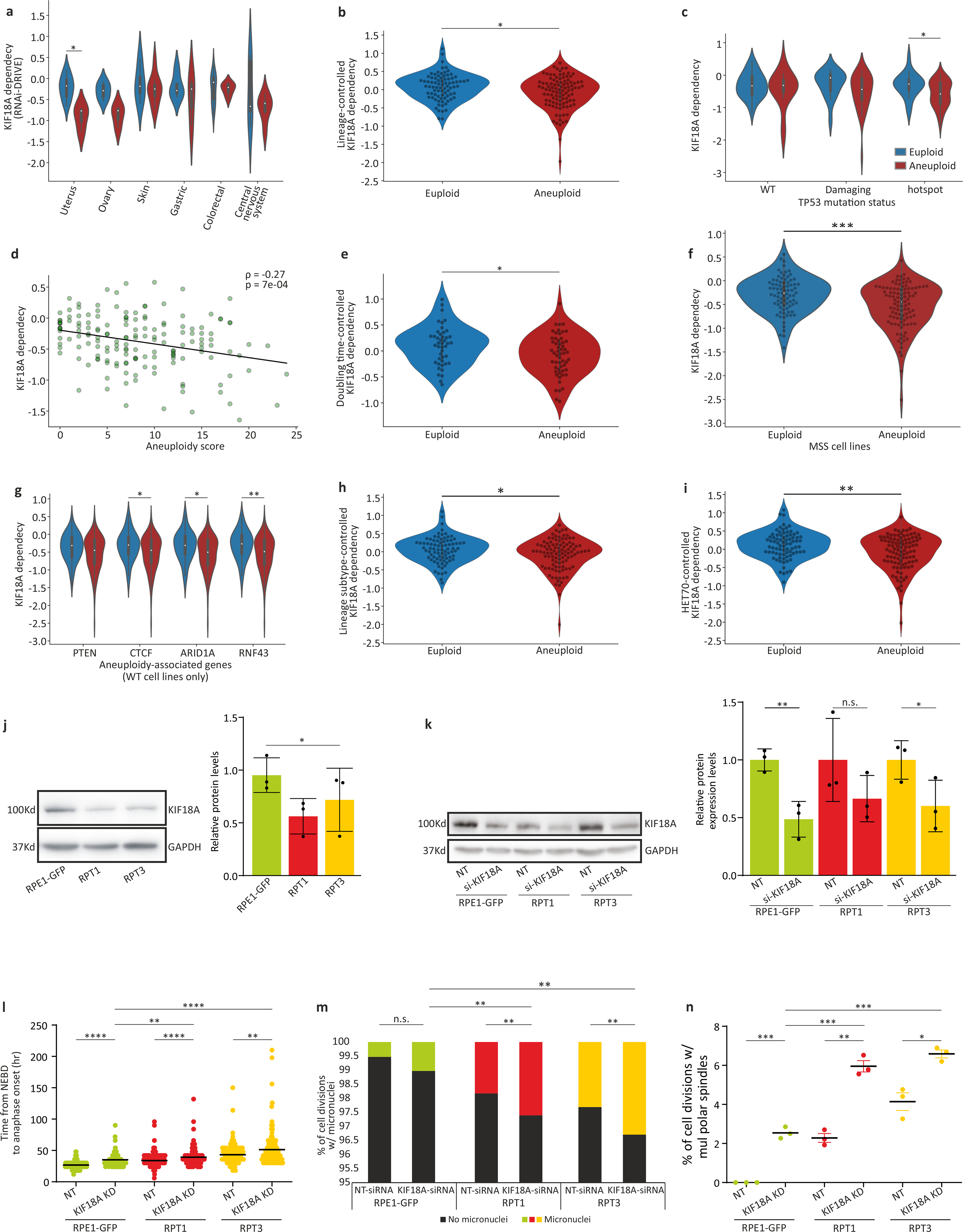
(a) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the DRIVE RNAi screen across multiple cell lineages. *, p=0.022; two-tailed t-test. (b) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the DRIVE RNAi screen, after accounting for lineage-specific differences in gene dependency scores using linear regression. *, p=0.012; two-tailed t-test. (c) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the DRIVE RNAi screen, across TP53 mutation classes. *, p=0.026; two-tailed t-test. (d) The correlations between AS and the dependency on KIF18A in the DRIVE RNAi screen, for cell lines that have not undergone whole-genome duplication (i.e., cell lines with basal ploidy of n=2). Spearman’s ρ = −0.27 (p=7e-04). (e) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the DRIVE RNAi screen, after removing the effect of doubling time on gene dependency scores using linear regression. *, p=0.022; two-tailed t-test. (f) The sensitivity of near-euploid and highly-aneuploid cancer cell lines without microsatellite instability (MSS lines only) to the knockdown of KIF18A in the DRIVE RNAi screen. ***, p=3e-04; two-tailed t-test. (g) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the DRIVE RNAi screen, in cell lines that are WT for the 4 genes most selectively mutated in aneuploid human tumors (after TP53)12. *, p=0.021 and p=0.02, for CTCF and ARID1A, respectively; **, p=0.004; two-tailed t-test. (h) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the DRIVE RNAi screen, after removing the effect of lineage subtype on gene dependency scores using linear regression. *, p=0.024; two-tailed t-test. (i) The sensitivity of near-euploid and highly-aneuploid cancer cell lines to the knockdown of KIF18A in the DRIVE RNAi screen, after removing the effect of HET70 scores on gene dependency scores using linear regression. **, p=0.003; two-tailed t-test. (j) Left: Western blot of KIF18A protein expression levels in RPE1 and RPT cell lines. Right: Quantification of KIF18A expression levels (normalized to GAPDH). *, p-0.023; one-tailed t-test. Data represent the mean ± s.d.; n=3 biological replicates. (k) Relative protein expression levels of KIF18A, confirming successful KIF18 knockdown in the RPE1 and RPT cell lines 72hr post-transfection. Left: Western blot of KIF18A protein expression levels in RPE1, RPT1 and RPT3 before and after siRNA-mediated KIF18A knockdown. Right: Quantification of KIF18A expression levels (normalized to α-Tubulin). *, p=0.034, **,p=0.004; one-tailed t-test. Data represent the mean ± s.d.; n=3 biological replicates. (l) Time-lapse imaging-based quantification of the time from nuclear envelope breakdown (NEBD) to anaphase onset in RPE1 and RPT cell lines exposed to non-targeting or KIF18A-targeting siRNAs for 72hr. **, p<0.01; ****, p<1e-04; two-tailed t-test. (m) The prevalence of micronuclei formation in HCT116 and HPT cells exposed to non-targeting or KIF18A-targeting siRNAs for 72hr. n.s., p>0.05; **, p<0.01; ***, p<0.001; two-tailed Fisher’s exact test. (n) Imaging-based quantification of the prevalence of cell divisions with multipolar spindles in HCT116 and HPT cell lines treated with non-targeting control or KIF18A-targeting siRNAs for 72hr. *, p<0.05; **, p<0.01; ***, p<0.001; two-tailed t-test; Error bars, s.d.; n=3 biological replicates.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Jordan Bryan, Jennifer Roth and Sam Bender for assistance with PRISM; Aviad Cherniack and Mustafa Kocak for helpful discussions; Dave Lam and Oana Enache for assistance with L1000 assay; Anastasya Y. Kuznetsova for the initial characterization of the HPT and RPT cell lines; Hilda van den Bos for assistance with cell sorting; and Sharon Tsach for assistance with figure preparation. This work was supported by the Azrieli Foundation (U.B.-D.), the Richard Eimert Research Fund on Solid Tumors (U.B.-D.), the Tel-Aviv University Cancer Biology Research Center (U.B.-D.), the Israel Cancer Association (U.B.-D.), and the DoD CDMRP career development award (CA191148 to U.B.-D.). Work in the Santaguida lab is supported by grants from the Italian Association for Cancer Research (MFAG 2018 - ID. 21665), Ricerca Finalizzata (GR-2018–12367077), Fondazione Cariplo, the Rita-Levi Montalcini program from MIUR, and the Italian Ministry of Health. Work in the Stumpff lab is supported by a Susan G Komen grant (CCR16377648 to J.S.), an NIH grant (GM121491 to J.S.), and a DoD PRCRP Horizon Award (W81XWH-17–1-0371 to H.L.H.M.). We dedicate this work to the memory of Angelika Amon, a giant of science, who pioneered aneuploidy research and was an inspiration to us all.
Footnotes
Competing Interests
T.R.G. is a consultant to GlaxoSmithKline and is a founder of Sherlock Biosciences. R.B. own shares in Ampressa and receives grant funding from Novartis. A.J.B. receives funding from Merck, Bayer and Novartis, and is an advisor to Earli and Helix Nano and a co-founder of Signet Therapeutics. The other authors declare no competing interests.
Additional Information
Supplementary Information is available for this paper. Correspondence and requests for materials should be addressed to ubendavid@tauex.tau.ac.il.
Code availability
The code used to generate and/or analyze the data are publicly available, or available upon request.
Data availability
All datasets are available within the article, its Supplementary Information, or from the corresponding authors upon request. Cell line aneuploidy profiles and scores are available at the DepMap portal (www.depmap.org/portal/). The analyzed CCLE genomic data is available in https://doi.org/10.6084/m9.figshare.11384241.v2. LP-WGS data have been deposited to SRA with BioProject accession number PRJNA672256.
References
- 1.Ben-David U & Amon A Context is everything: aneuploidy in cancer. Nat Rev Genet (2019) doi: 10.1038/s41576-019-0171-x. [DOI] [PubMed] [Google Scholar]
- 2.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature (2019) doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsherniak A et al. Defining a Cancer Dependency Map. Cell 170, 564–576 e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McDonald ER et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell (2017) doi: 10.1016/j.cell.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Aguirre AJ et al. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. (2016) doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang W et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. (2013) doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basu A et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell (2013) doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garnett MJ et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature (2012) doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corsello SM et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer (2020) doi: 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musacchio A & Salmon ED The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8, 379–393 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Zack TI et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45, 1134–1140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor AM et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 33, 676–689 e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knouse KA, Wu J, Whittaker CA & Amon A Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. U. S. A. (2014) doi: 10.1073/pnas.1415287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oromendia AB, Dodgson SE & Amon A Aneuploidy causes proteotoxic stress in yeast. Genes Dev. (2012) doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torres EM et al. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science (80-. ). 317, 916–924 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Hwang S et al. Serine-Dependent Sphingolipid Synthesis Is a Metabolic Liability of Aneuploid Cells. Cell Rep 21, 3807–3818 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storchová Z et al. Genome-wide genetic analysis of polyploidy in yeast. Nature (2006) doi: 10.1038/nature05178. [DOI] [PubMed] [Google Scholar]
- 18.Ben-David U et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. (2017) doi: 10.1038/ng.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kops GJPL, Foltz DR & Cleveland DW Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. U. S. A. (2004) doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wild T et al. The Spindle Assembly Checkpoint Is Not Essential for Viability of Human Cells with Genetically Lowered APC/C Activity. Cell Rep. (2016) doi: 10.1016/j.celrep.2016.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chunduri NK & Storchova Z The diverse consequences of aneuploidy. Nat Cell Biol 21, 54–62 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Baker DJ, Jin F, Jeganathan KB & van Deursen JM Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 16, 475–486 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricke RM, Jeganathan KB & van Deursen JM Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol 193, 1049–1064 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foijer F et al. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc Natl Acad Sci U S A 111, 13427–13432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foijer F et al. Deletion of the MAD2L1 spindle assembly checkpoint gene is tolerated in mouse models of acute T-cell lymphoma and hepatocellular carcinoma. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sotillo R et al. Mad2 Overexpression Promotes Aneuploidy and Tumorigenesis in Mice. Cancer Cell (2007) doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sotillo R, Schvartzman JM, Socci ND & Benezra R Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464, 436–440 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mason JM et al. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc Natl Acad Sci U S A 114, 3127–3132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pauer LR et al. Phase I study of oral CI-994 in combination with carboplatin and paclitaxel in the treatment of patients with advanced solid tumors. Cancer Invest. (2004) doi: 10.1081/CNV-200039852. [DOI] [PubMed] [Google Scholar]
- 30.Bielski CM et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet 50, 1189–1195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carter SL et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30, 413–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hieronymus H et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith JC & Sheltzer JM Systematic identification of mutations and copy number alterations associated with cancer patient prognosis. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soto M, Garcia-Santisteban I, Krenning L, Medema RH & Raaijmakers JA Chromosomes trapped in micronuclei are liable to segregation errors. J Cell Sci 131, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang CZ et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burgess A, Rasouli M & Rogers S Stressing mitosis to death. Frontiers in Oncology (2014) doi: 10.3389/fonc.2014.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dominguez-Brauer C et al. Targeting Mitosis in Cancer: Emerging Strategies. Molecular Cell (2015) doi: 10.1016/j.molcel.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 38.Santaguida S, Tighe A, D’Alise AM, Taylor SS & Musacchio A Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol (2010) doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuznetsova AY et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 14, 2810–2820 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santaguida S et al. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev Cell 41, 638–651 e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bakker B et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. (2016) doi: 10.1186/s13059-016-0971-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weaver LN et al. Kif18A uses a microtubule binding site in the tail for plus-end localization and spindle length regulation. Curr. Biol. (2011) doi: 10.1016/j.cub.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Czechanski A et al. Kif18a is specifically required for mitotic progression during germ line development. Dev. Biol. (2015) doi: 10.1016/j.ydbio.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wordeman L, Decarreau J, Vicente JJ & Wagenbach M Divergent microtubule assembly rates after short-versus long-term loss of end-modulating kinesins. Mol. Biol. Cell (2016) doi: 10.1091/mbc.E15-11-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marquis C et al. Chromosomally unstable tumor cells specifically require KIF18A for proliferation. bioRxiv 2020.06.18.159327 (2020) doi: 10.1101/2020.06.18.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quinton RJ et al. Whole genome doubling confers unique genetic vulnerabilities on tumor cells. bioRxiv 2020.06.18.159095 (2020) doi: 10.1101/2020.06.18.159095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braun J et al. Synthesis and biological evaluation of optimized inhibitors of the mitotic kinesin Kif18A. ACS Chem. Biol (2015) doi: 10.1021/cb500789h. [DOI] [PubMed] [Google Scholar]
- 48.van den Bos H et al. Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. (2016) doi: 10.1186/s13059-016-0976-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang DW et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. (2007) doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheltzer JM A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally unstable cancer cells and informs clinical prognosis. Cancer Res 73, 6401–6412 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berg S et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods (2019) doi: 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- 52.Subramanian A et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell (2017) doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McFarland JM et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat Commun 9, 4610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seashore-Ludlow B et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. (2015) doi: 10.1158/2159-8290.CD-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rees MG et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol (2016) doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iorio F et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell (2016) doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nusinow DP et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell (2020) doi: 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan EM et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature (2019) doi: 10.1038/s41586-019-1102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritchie ME et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. (2015) doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (1995) doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 61.Pedregosa F et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res (2011). [Google Scholar]
- 62.R. R core team (2018). R A Lang. Environ. Stat. Comput. R Found. Stat. Comput Vienna, Austria. URL http//www.R-project.org (2018). [Google Scholar]
- 63.Dempster JM et al. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 720243 (2019) doi: 10.1101/720243 %J bioRxiv. [DOI] [Google Scholar]
- 64.Mayr MI et al. The Human Kinesin Kif18A Is a Motile Microtubule Depolymerase Essential for Chromosome Congression. Curr. Biol (2007) doi: 10.1016/j.cub.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 65.Ben-David U et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature (2018) doi: 10.1038/s41586-018-0409-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Subramanian A et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A (2005) doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sergushichev AA An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv (2016) doi: 10.1101/060012. [DOI] [Google Scholar]
- 68.Liberzon A et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics (2011) doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Durrbaum M et al. Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genomics 15, 139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee K & Rhee K PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. J. Cell Biol (2011) doi: 10.1083/jcb.201106093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lai D & Shah S HMMcopy: copy number prediction with correction for GC and mappability bias for HTS data. R Packag. version (2012). [Google Scholar]
- 72.Knouse KA, Wu J & Amon A Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. (2016) doi: 10.1101/gr.198937.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van den Bos H et al. Quantification of aneuploidy in mammalian systems. in Methods in Molecular Biology (2019). doi: 10.1007/978-1-4939-8931-7_15. [DOI] [PubMed] [Google Scholar]
- 74.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods (2012) doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jun G, Wing MK, Abecasis GR & Kang HM An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Res. (2015) doi: 10.1101/gr.176552.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.