


Abstract
Bottom-up proteomics analyses have been proved over the last years to be a powerful tool in the characterization of the proteome and are crucial for understanding cellular and organism behaviour. Through differential proteomic analysis researchers can shed light on groups of proteins or individual proteins that play key roles in certain, normal or pathological conditions. However, several tools for the analysis of such complex datasets are powerful, but hard-to-use with steep learning curves. In addition, some other tools are easy to use, but are weak in terms of analytical power. Previously, we have introduced ProteoSign, a powerful, yet user-friendly open-source online platform for protein differential expression/abundance analysis designed with the end-proteomics user in mind. Part of Proteosign's power stems from the utilization of the well-established Linear Models For Microarray Data (LIMMA) methodology. Here, we present a substantial upgrade of this computational resource, called ProteoSign v2, where we introduce major improvements, also based on user feedback. The new version offers more plot options, supports additional experimental designs, analyzes updated input datasets and performs a gene enrichment analysis of the differentially expressed proteins. We also introduce the deployment of the Docker technology and significantly increase the speed of a full analysis. ProteoSign v2 is available at http://bioinformatics.med.uoc.gr/ProteoSign.
Graphical Abstract
Graphical Abstract.
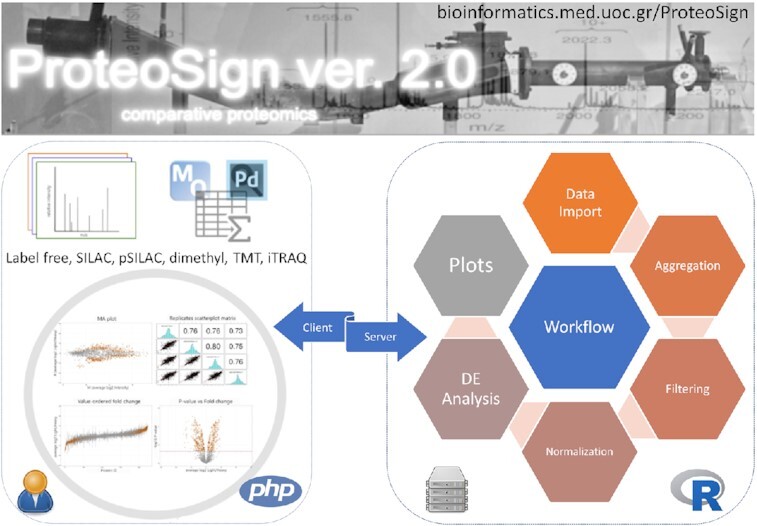
ProteoSign v2: a faster and evolved user-friendly online tool for statistical analyses of differential proteomics.
INTRODUCTION
Mass spectrometry (MS)-based quantitative proteomics is a powerful approach to study the global proteome dynamics in a cell, a tissue or an organism (1). The latest technological advances in bioanalytical chemistry, mass spectrometry and bioinformatics, allow the detection, relative quantitation and functional annotation of thousands of proteins in a single experiment in an hour using the so-called bottom-up proteomics approach (2,3). In contrast to the still developing top-down proteomics approach where intact proteins are analyzed by MS, in the widely used bottom-up approach, proteins are proteolytically digested into peptides and then separated and analysed. The peptides are analysed with MS techniques so that their accurate mass, absolute or relative abundance and amino acid sequence are determined. This information is used to deduce the abundance and primary structure of the peptides' parent proteins. A significant drawback of the bottom-up approach is its inability to unequivocally identify the different isoforms of the proteins, in contrast to the top-down proteomics approaches. However, the bottom-up approach provides better separation of peptides in both nLC and MS level resulting in a much higher coverage of the predicted proteome. Thus, the bottom-up approach is the most commonly used one in high-throughput proteomics (4). There are many available proteomic differential expression analysis tools, such as Perseus (probably the most popular one) (5), DanTE (6), Prostar (7), MsqRob (8), ProteoSign (9), MSstats (10), Rover (11), HiQuant (12), PIQMIe (13), Scaffold Q+S, ProtExA (14), StatQuant (15) etc. These tools display differences in terms of features, such as filtering, normalization, aggregation, statistical methods, types of analyses, data import/export formats, plots offered etc. They also have different user requirements (need of statistical and programming skills), experimental and software restrictions (for comparison, see (9)).
Previously, we published ProteoSign, a web tool for differential and statistical analysis on quantification datasets which was published in the 2017 Nucleic Acids Research web server issue (8). It uses Linear Models for Microarray Data (LIMMA) methodology in order to statistically assess the difference in abundance of proteins between two or more proteome states. It provides descriptive statistics, plots, automated data reformatting and offers the minimum customizability in order to keep its interface simple. ProteoSign had no restriction regarding the experimental methods used as input which gives the opportunity to analyze both label-free and labeled experiments. It accepts as input proteomics quantification data produced by either MaxQuant (MQ) (16) or Proteome Discoverer PD (Thermo Scientific). The data can derive from label-free or labeled experiments, currently supporting SILAC (17), pulsed SILAC (18), iTRAQ (19), TMTs (20) and dimethyl labeling (21) whereas label-swap replication is also supported.
Here, we present a significantly updated second version of ProteoSign (v2), guided by feedback from actual users and the broader proteomics community. In this update, we have implemented new features, while still preserving the user friendliness of the tool. The new features and improvements include acceptance of updated input datasets, much faster analysis performance, support of new experimental designs, user-customised and reproduced plots, easy installation in a local server, improved documentation, deployment of the Docker technology and gene enrichment analysis of differentially expressed proteins.
NEW FEATURES AND UPDATES
General design and input data
The pipeline of ProteoSign v2 is shown in Figure 1. The frontend is written in HTML and JavaScript and consists of a welcome page, an analytical help page with examples and relevant pictures describing the whole process, and a detailed documentation. ProteoSign's backend is written in PHP and R and manages the data uploading and analysis processes, as well as the results visualization and downloading processes. It is platform independent and is fully compatible with all major browsers (Mozilla Firefox, Google Chrome, Apple Safari, Opera, etc). In its first version, ProteoSign accepted quantified differential proteomics data, produced by either Proteome Discoverer (PD) 1.3+ or MaxQuant (MQ) 1.3.0.5+. The updated version now accepts datasets also produced by Proteome Discoverer (PD) 2.4. Apart from the previous support of experimental designs, many users required support for Replication Multiplexing, i.e. experiments where biological/technical replicates or conditions are represented either as different tags or as different MS runs, a feature that was integrated in the new version of ProteoSign. Additionally, the User-Interface, help pages and documentation files have integrated user-suggested changes towards a more user-friendly environment and learning experience.
Figure 1.

ProteoSign's v2 pipeline.
Performance
There has been an extensive rewriting and optimization of the source code, in order to increase the performance (in terms of speed) of ProteoSign. Moreover, data tables (in R; package data.table) are now employed instead of dataframes. This new feature is better suited for large datasets and facilitates ProteoSign v2 to increase its running speed by 2- or 3-fold. Specifically, the features are fast aggregation of large data (e.g. 100GB in RAM), fast ordered joins, fast additions/modifications/deletions of columns by group without the use of temporary copies, column listings and faster text reading/writing. As a case study, we conducted the same analysis as in the first version of ProteoSign using the same demonstration datasets and calculated its running time. Speed improvements are presented in Table 1.
Table 1.
Running time comparison between ProteoSign and ProteoSign v2
| Data set and PRIDE ID | Data size (MB) | Conditions | Biological replicates | Technical replicates | Fractionation | Samples | Running time ProteoSign v1 (min) | Running time ProteoSign v2 (min) |
|---|---|---|---|---|---|---|---|---|
| SILAC 2-plex (MQ) PXD001909 (23) | 122 | 2 | 3 | 2 | Y | 72 | <1 | 0.35 (21 s) |
| SILAC 2-plex (MQ) large PXD000778 (24) | 787 | 2 | 4 | 6 | Y | 240 | 6 | 2 |
| SILAC 2-plex (PD) large PXD000778 (24) | 1100 | 2 | 4 | 6 | Y | 40 | 4 | <2 |
| Label-Free (MQ) large PXD004124 (25) | 1070 | 2 | 2 | 3 | Y | 108 | 7 | <4 |
| TMT (MQ) PXD002622 (26) | 62 | 2 | 5 | 0 | Y | 50 | 2 | <1 |
| TMT (PD) PXD002622 (26) | 109 | 2 | 5 | 0 | Y | 50 | 2 | <1 |
| iTRAQ (PD) PXD004869 | 684 | 4 | 2 | 0 | Y | 42 | 12 | <5 |
| pSILAC 3-plex (MQ) PXD001976 (27) | 336 | 2 | 6 | 0 | Y | 120 | 3 | <2 |
| pSILAC 3-plex (PD) PXD001976 (27) | 831 | 2 | 6 | 0 | Y | 120 | 7 | <4 |
| Dimethyl 2-plex (PD) large PXD002073 (28) | 1505 | 2 | 3 | 0 | Y | 36 | 9 | <5 |
User intervention
By adopting a visual analytics approach, in its current version, users can manually adjust several parameters and affect output results. For example, users can initially define the adjusted P-value threshold for differentially expressed proteins at any time. In addition, they have the option to disqualify proteins which were not quantified with at least a user defined number of different peptides, in at least a user defined number of biological replicates. Finally, users can load or save the parameter set of each run and quickly rerun the analysis without the need to re-enter every parameter. The latter is especially useful when planning, setting and defining an experimental design. In addition, the users can customize and reproduce any of the offered plots. The relevant R script is included and there is an extended help file describing how to run the processes in the command line.
Docker
All of ProteoSign v2 components along with the necessary dependencies are packed in a docker image. The docker image allows running ProteoSign v2 without installing any mandatory external libraries. This way, one can run ProteoSign v2 locally on any operating system and access it via her/his local web browser, overcoming network bandwidth issues.
Gene enrichment analysis
A new feature of ProteoSign v2 is its ability to perform gene enrichment analysis to functionally annotate significantly enriched groups consisting of differentially expressed proteins. Methods applied to the analysis originate from g:Profiler2 (22), an R package providing an R interface to g:Profiler, a web server for functional interpretation of gene lists. The query set of proteins consists of Uniprot Accession Numbers used to characterize each one of the proteins found to be expressed differentially. Incorporating an external server such as g:Profiler, analysis can be extended to a plethora of different organisms and enriched terms from several databases. The significance threshold for the detection of enriched terms and the correction method for multiple testing, are set by default values to 0.05 and Set Counts and Sizes (gSCS) respectively. Results can be viewed in an interactive table and are exported in a csv format. Detailed instructions are provided in the help pages.
Local server installation
The source code of ProteoSign v2 has been rewritten so that in its new form, it can be installed in a local server following a few simple steps (see new documentation page). Regardless of the server's system, ProteoSign can now be downloaded, installed and configured easily even from non-experts. A local server installation will provide much higher data transmission speeds, laboratory specific configuration and also the ability to incorporate ProteoSign as part of a custom pipeline.
DISCUSSION
Proteosign v2 is an updated user-friendly web server application for automated differential and statistical analysis on high-throughput proteomic quantification datasets. Novel features enable users to submit additional experimental designs (replication multiplexing), analyze various input datasets and perform gene enrichment analysis to extract differentially expressed proteins. Finally, the deployment of Docker technology along with a much faster performance (2–3×) due to significant code improvements are of great importance. Main differences between ProteoSign v1 and v2 are summarized in Table 2.
Table 2.
ProteoSign version 1 versus version 2
| Feature | ProteoSign v1 | ProteoSign v2 |
|---|---|---|
| Aggregation | X | X |
| Filtering | X | X |
| Normalization | X | X |
| Differential analysis (based on LIMMA) | X | X |
| Generation of various data plots | X | X (plus Venn diagrams) |
| Enrichment analysis | X | |
| Docker image | X | |
| Support of Proteome Discoverer (PD) 2.4 | X | |
| Ability to install to Local Server | X | |
| Support for Replication Multiplexing | X | |
| User defined parameters | X | |
| Higher speed performance | X |
SERVER INFORMATION
ProteoSign v2 is a web application that runs on Apache 2 web server hosted on a Dell PowerEdge R720xd server machine. The server runs Ubuntu (kernel 3.2) and has 128 GB RAM and comes with two Intel Xeon E5–2650 processors clocked at 2GHz.
DATA AVAILABILITY
ProteoSign v2 is freely available at http://bioinformatics.med.uoc.gr/ProteoSign and the source at https://github.com/ananas14/ProteoSign. The docker image can be pulled for docker github https://hub.docker.com/r/mpaltsai/proteosign (command: docker pull mpaltsai/proteosign).
ACKNOWLEDGEMENTS
We would like to thank ProteoSign's users for their valuable comments and interaction with the development team.
Contributor Information
Evangelos Theodorakis, Division of Basic Sciences, University of Crete Medical School, Heraklion 71110, Greece; Department of Informatics, Technical University of Munich, Boltzmannstr. 3, 85748 Garching, Germany.
Andreas N Antonakis, Division of Basic Sciences, University of Crete Medical School, Heraklion 71110, Greece.
Ismini Baltsavia, Division of Basic Sciences, University of Crete Medical School, Heraklion 71110, Greece.
Georgios A Pavlopoulos, Institute for Fundamental Biomedical Research, BSRC “Alexander Fleming”, 34 Fleming Street, 16672 Vari, Greece.
Martina Samiotaki, Institute for Bioinnovation, BSRC “Alexander Fleming”, 34 Fleming Street, 16672 Vari, Greece.
Grigoris D Amoutzias, Bioinformatics Laboratory, Department of Biochemistry and Biotechnology, University of Thessaly, Larisa 41500, Greece.
Theodosios Theodosiou, Division of Basic Sciences, University of Crete Medical School, Heraklion 71110, Greece.
Oreste Acuto, Sir William Dunn School of Pathology, University of Oxford, South Parks Road, Oxford OX13RE, UK.
Georgios Efstathiou, Division of Basic Sciences, University of Crete Medical School, Heraklion 71110, Greece; Sir William Dunn School of Pathology, University of Oxford, South Parks Road, Oxford OX13RE, UK.
Ioannis Iliopoulos, Division of Basic Sciences, University of Crete Medical School, Heraklion 71110, Greece.
FUNDING
This work has been supported by the project “ELIXIR-GR: Managing and Analysing Life Sciences Data" (MIS: 5002780), co-financed by Greece and the European Union - European Regional Development Fund. Funding for open access charge: “ELIXIR-GR: Managing and Analysing Life Sciences Data" (MIS: 5002780), co-financed by Greece and the European Union - European Regional Development Fund.
Conflict of interest statement. None declared.
REFERENCES
- 1. Anderson N.L., Anderson N.G.. Proteome and proteomics: new technologies, new concepts, and new words. Electrophoresis. 1998; 19:1853–1861. [DOI] [PubMed] [Google Scholar]
- 2. Meier F., Geyer P.E., Winter Virreira, Cox S., Mann M.. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods. 2018; 15:440–448. [DOI] [PubMed] [Google Scholar]
- 3. Bian Y., Zheng R., Bayer F.P., Wong C., Chang Y.-C., Meng C., Zolg D.P., Reinecke M., Zecha J., Wiechmann S.et al.. Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC-MS/MS. Nat. Commun. 2020; 11:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coorssen J.R., Yergey A.L.. Proteomics is analytical chemistry: fitness-for-purpose in the application of top-down and bottom-up analyses. Proteomes. 2015; 3:440–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J.. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016; 13:731–740. [DOI] [PubMed] [Google Scholar]
- 6. Polpitiya A.D., Qian W.-J., Jaitly N., Petyuk V.A., Adkins J.N., Camp D.G., Anderson G.A., Smith R.D.. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinforma. Oxf. Engl. 2008; 24:1556–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wieczorek S., Combes F., Lazar C., Giai Gianetto Q., Gatto L., Dorffer A., Hesse A.-M., Couté Y., Ferro M., Bruley C.et al.. DAPAR & ProStaR: software to perform statistical analyses in quantitative discovery proteomics. Bioinforma. Oxf. Engl. 2017; 33:135–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goeminne L.J.E., Gevaert K., Clement L.. Peptide-level robust ridge regression improves estimation, sensitivity, and specificity in data-dependent quantitative label-free shotgun proteomics. Mol. Cell. Proteomics MCP. 2016; 15:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Efstathiou G., Antonakis A.N., Pavlopoulos G.A., Theodosiou T., Divanach P., Trudgian D.C., Thomas B., Papanikolaou N., Aivaliotis M., Acuto O.et al.. ProteoSign: an end-user online differential proteomics statistical analysis platform. Nucleic Acids Res. 2017; 45:W300–W306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi M., Chang C.-Y., Clough T., Broudy D., Killeen T., MacLean B., Vitek O.. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinforma. Oxf. Engl. 2014; 30:2524–2526. [DOI] [PubMed] [Google Scholar]
- 11. Colaert N., Helsens K., Impens F., Vandekerckhove J., Gevaert K.. Rover: a tool to visualize and validate quantitative proteomics data from different sources. Proteomics. 2010; 10:1226–1229. [DOI] [PubMed] [Google Scholar]
- 12. Bryan K., Jarboui M.-A., Raso C., Bernal-Llinares M., McCann B., Rauch J., Boldt K., Lynn D.J.. HiQuant: rapid postquantification analysis of large-scale MS-generated proteomics data. J. Proteome Res. 2016; 15:2072–2079. [DOI] [PubMed] [Google Scholar]
- 13. Kuzniar A., Kanaar R.. PIQMIe: a web server for semi-quantitative proteomics data management and analysis. Nucleic Acids Res. 2014; 42:W100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minadakis G., Sokratous K., Spyrou G.M.. ProtExA: a tool for post-processing proteomics data providing differential expression metrics, co-expression networks and functional analytics. Comput. Struct. Biotechnol. J. 2020; 18:1695–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Breukelen B., van den Toorn H.W.P., Drugan M.M., Heck A.J.R.. StatQuant: a post-quantification analysis toolbox for improving quantitative mass spectrometry. Bioinforma. Oxf. Engl. 2009; 25:1472–1473. [DOI] [PubMed] [Google Scholar]
- 16. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 17. Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., Mann M.. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics MCP. 2002; 1:376–386. [DOI] [PubMed] [Google Scholar]
- 18. Schwanhäusser B., Gossen M., Dittmar G., Selbach M.. Global analysis of cellular protein translation by pulsed SILAC. Proteomics. 2009; 9:205–209. [DOI] [PubMed] [Google Scholar]
- 19. Ross P.L., Huang Y.N., Marchese J.N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S.et al.. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics MCP. 2004; 3:1154–1169. [DOI] [PubMed] [Google Scholar]
- 20. Thompson A., Schäfer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A.K.A., Hamon C.. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003; 75:1895–1904. [DOI] [PubMed] [Google Scholar]
- 21. Hsu J.-L., Chen S.-H.. Stable isotope dimethyl labelling for quantitative proteomics and beyond. Philos. Transact. A Math. Phys. Eng. Sci. 2016; 374:20150364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J.. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019; 47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carter D.M., Westdorp K., Noon K.R., Terhune S.S.. Proteomic identification of nuclear processes manipulated by cytomegalovirus early during infection. Proteomics. 2015; 15:1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian R., Alvarez-Saavedra M., Cheng H.-Y.M.. Uncovering the proteome response of the master circadian clock to light using an AutoProteome system. Mol. Cell. Proteomics. 2011; 10:M110.007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suárez-Cortés P., Sharma V., Bertuccini L., Costa G., Bannerman N.-L., Rosa Sannella A., Williamson K., Klemba M., Levashina E.A., Lasonder E.et al.. Comparative proteomics and functional analysis reveal a role of Plasmodium falciparum osmiophilic bodies in malaria parasite transmission. Mol. Cell. Proteomics. 2016; 15:3243–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stewart P.A., Parapatics K., Welsh E.A., Müller A.C., Cao H., Fang B., Koomen J.M., Eschrich S.A., Bennett K.L., Haura E.B.. A pilot proteogenomic study with data integration identifies MCT1 and GLUT1 as prognostic markers in lung adenocarcinoma. PloS One. 2015; 10:e0142162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hünten S., Kaller M., Drepper F., Oeljeklaus S., Bonfert T., Erhard F., Dueck A., Eichner N., Friedel C.C., Meister G.et al.. p53-regulated networks of protein, mRNA, miRNA, and lncRNA expression revealed by integrated pulsed stable isotope labeling with amino acids in cell culture (pSILAC) and next generation sequencing (NGS) analyses. Mol. Cell. Proteomics MCP. 2015; 14:2609–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elkon R., Loayza-Puch F., Korkmaz G., Lopes R., van Breugel P.C., Bleijerveld O.B., Altelaar A.F.M., Wolf E., Lorenzin F., Eilers M.et al.. Myc coordinates transcription and translation to enhance transformation and suppress invasiveness. EMBO Rep. 2015; 16:1723–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
ProteoSign v2 is freely available at http://bioinformatics.med.uoc.gr/ProteoSign and the source at https://github.com/ananas14/ProteoSign. The docker image can be pulled for docker github https://hub.docker.com/r/mpaltsai/proteosign (command: docker pull mpaltsai/proteosign).