


Abstract
With the explosive growth of protein sequences, large-scale automated protein function prediction (AFP) is becoming challenging. A protein is usually associated with dozens of gene ontology (GO) terms. Therefore, AFP is regarded as a problem of large-scale multi-label classification. Under the learning to rank (LTR) framework, our previous NetGO tool integrated massive networks and multi-type information about protein sequences to achieve good performance by dealing with all possible GO terms (>44 000). In this work, we propose the updated version as NetGO 2.0, which further improves the performance of large-scale AFP. NetGO 2.0 also incorporates literature information by logistic regression and deep sequence information by recurrent neural network (RNN) into the framework. We generate datasets following the critical assessment of functional annotation (CAFA) protocol. Experiment results show that NetGO 2.0 outperformed NetGO significantly in biological process ontology (BPO) and cellular component ontology (CCO). In particular, NetGO 2.0 achieved a 12.6% improvement over NetGO in terms of area under precision-recall curve (AUPR) in BPO and around 2.6% in terms of 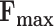 in CCO. These results demonstrate the benefits of incorporating text and deep sequence information for the functional annotation of BPO and CCO. The NetGO 2.0 web server is freely available at http://issubmission.sjtu.edu.cn/ng2/.
in CCO. These results demonstrate the benefits of incorporating text and deep sequence information for the functional annotation of BPO and CCO. The NetGO 2.0 web server is freely available at http://issubmission.sjtu.edu.cn/ng2/.
Graphical Abstract
Graphical abstract.
The workflow of NetGO 2.0 that improves large-scale protein function prediction with massive sequence, text, domain, family and network information.
INTRODUCTION
With great biomedical and pharmaceutical significance, identifying the functions of proteins can help understand life at the molecular level (1). Launched in 1998, Gene Ontology (GO) is widely used for describing the functions of genes/proteins (2). To date, GO contains 44 117 biological concepts (January 2021), covering three different domains: molecular functional ontology (MFO), biological process ontology (BPO) and cellular component ontology (CCO). Due to the development of sequencing technology, there has been an explosive growth in the number of available protein sequences. However, only a fraction of the sequences have experimentally supported functional annotations. For example, proteins with experimental GO annotations account for 0.1% or less of total sequences in UniProKB (March 2021) (3). Elucidating the functions of proteins by conducting biochemical experiments is time-consuming and expensive. Therefore, it is imperative to design high-performance AFP algorithms to fill the gap between the increasing number of protein sequences and the limited number of known functional annotations.
As part of the efforts to boost the development of effective and efficient AFP, critical assessment of functional annotation (CAFA) has been held four times to date: CAFA1 in 2010–2011, CAFA2 in 2013–2014, CAFA3 in 2016–2017 and CAFA4 in 2019–2020 (under evaluation) (4–6). Given about 100 000 protein sequences, the participants have been required to submit their predicted GO terms (relevant to target proteins) before a deadline (T0) since CAFA2. For assessing the performance of different AFP methods, CAFA uses a time-delayed evaluation process. Specifically, the organizers wait a few months (T1) to gather test proteins with newly experimental annotations as the benchmark data. There are two types of proteins in the benchmark: no-knowledge and limited-knowledge proteins. Both types of proteins receive the first experimental annotation in a target domain between T0 and T1. However, no-knowledge proteins do not have any experimental annotations before T0, while limited-knowledge proteins do before T0 in at least one of the other domains. Here we focus on no-knowledge proteins as the vast majority of proteins do not have any experimental annotations.
As mentioned before, AFP can be regarded as a challenging problem of large-scale multilabel classification, where each protein is an instance with each GO term as one of its labels. The challenges are from two main aspects: the label (GO) and instance (protein). For the label side, AFP must be efficient in dealing with the scaling problem of a large number of GO terms. With >40 000 GO terms in total, each protein is usually associated with only dozens of terms. For the instance side, AFP must be effective in integrating all kinds of protein information. CAFA has demonstrated the research community’s efforts in addressing these challenges. Previously, we developed GOLabeler, the best performing method in CAFA3 for all three GO domains in terms of Fmax (7). GOLabeler addressed the above challenges by using the learning to rank (LTR) framework (8). Specifically, given a query protein, GOLabeler first ranks a small number of promising candidate GO terms that are predicted by its component methods and then returns the top-ranked GO terms as the prediction results. As a sequence-based method, GOLabeler uses information about sequence homology and protein domain/family that is derived from protein sequences by BLAST and InterProScan (9,10), respectively. By considering the importance of biological network information, NetGO (11) further integrates additional massive network information in STRING (12) under the same LTR framework of GOLabeler, resulting in the improved performance of AFP in both BPO and CCO. However, human-annotated literature information about proteins is still largely ignored in NetGO, which is actually an important resource for AFP in our previous work, DeepText2GO (13). In addition, latent sequence information extracted by deep learning based methods has demonstrated the benefits of improving performance in CCO (14), whereas NetGO relies on traditional machine learning based methods.
Under the same LTR framework of NetGO, in this work, we propose NetGO 2.0, which further incorporates both manually annotated literature information about each protein in SwissProt (3) by logistic regression and latent sequence information by recurrent neural network (RNN). Note that out of all 564 000 proteins in SwissProt, around 457 000 have manually annotated MEDLINE literature information, with only around 71 500 having experimental GO annotations. The training and test datasets were generated by following the protocol of CAFA. Our prediction results have shown that NetGO 2.0 performed significantly better than NetGO in two domains of GO: BPO and CCO, with a 12.6% improvement of AUPR in BPO, and around 2.6% of Fmax in CCO over NetGO. These results demonstrate the benefits of incorporating text and deep sequence information for the functional annotation of BPO and CCO. NetGO 2.0 has also participated in CAFA4. The preliminary results of CAFA4 reported in ISMB2020 (July 2020) show that NetGO 2.0 was ranked among the top methods.
NETGO 2.0: NEW FEATURES AND UPDATES
Overview
Figure 1 illustrates the systematic procedure of NetGO 2.0, which is similar to NetGO. As shown in the figure, the detailed processes of training and testing are described in NetGO (11). NetGO 2.0 consists of seven component methods: Naive, BLAST-KNN, LR-3mer, LR-InterPro, NetKNN, LR-Text and Seq-RNN. The first five component methods are all from NetGO, which use GO frequency, sequence homology, amino acid trigram, domain/family/motif and protein network information, respectively. Note that one component of NetGO, LR-ProFET, is absent in NetGO 2.0. It has very little effect on the overall performance. Without it, NetGO 2.0 has also improved its efficiency. Compared with NetGO, LR-Text and Seq-RNN are two newly added components of NetGO 2.0. Therefore, we describe them in the next subsection. Note that LR stands for Logistic Regression and KNN for K-nearest neighbors.
Figure 1.

The framework of NetGO 2.0 with seven steps. The top five component methods are from NetGO, while LR-Text relies on text information and Seq-RNN using RNN to extract sequence information. An offline training process consists of Steps  , while an online test process involves Steps
, while an online test process involves Steps  .
.
New components of NetGO 2.0
LR-text
Figure 2 illustrates the procedure of LR-Text, which is a component of our previous work DeepText2GO (13). For a given protein and its UniProt ID, we first obtain the corresponding text data from PubMed by issuing a query for the relevant publications manually annotated in SwissProt (3). Specifically, we use only the title and abstract of each returned article and combine all of them to form a document. For this document, we then combine its sparse TF-IDF (term frequency-inverse document frequency) representation and dense semantic representation generated by Doc2Vec (15) as the text feature of this queried protein. The reasons for this are as follows. TF-IDF preserves the word statistics while Doc2Vec captures the complex context information from the text data. As such, these two representations are complementary to each other, with focusing on different aspects of text information. For each GO term, an independent logistic regression is finally trained with the obtained text-based features of proteins in the training set for predicting the score of this GO term.
Figure 2.
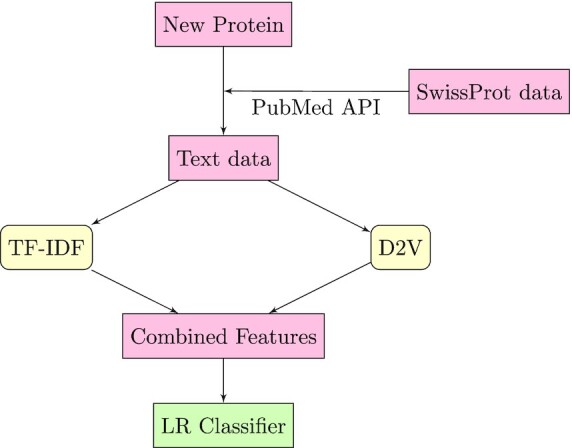
The procedure of LR-Text: the relevant publications of a given protein are first retrieved. Then, the publications are represented as TF-IDF and Doc2Vec. This new representation of the text is used for training of LR-Text.
Seq-RNN
Except for LR-Text, we also use Seq-RNN to extract the deep representation of a protein sequence. The overview of Seq-RNN is shown in Figure 3. For a given protein sequence, Seq-RNN first represents each amino acid by a semantic dense embedding. Such an embedding is then input into a BiLSTM (Bi-directional Long Short-Term Memory) (16) network to generate its hidden representations. By doing so, the representations contain context information from two directions. This is because BiLSTM captures the dependency relationships between different protein sequences. Finally, a max-pooling layer generates the overall representation of this particular protein sequence. With such a representation, Seq-RNN can predicts the probability of each GO term by using a fully connected layer. We use the binary cross-entropy as our objective function during the training process.
Figure 3.
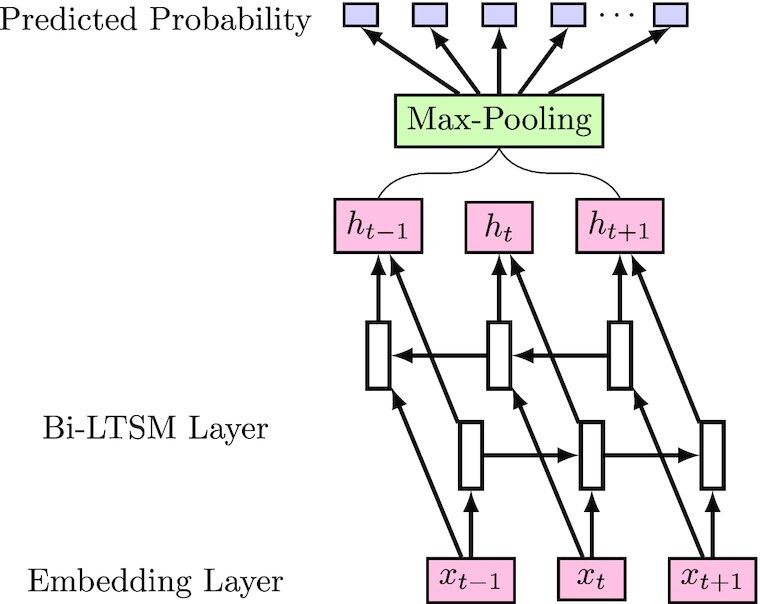
The procedure of Seq-RNN: The embedding layer first embeds an amino acid into a dense vector, BiLSTM then generates the hidden representation by capturing the context information of amino acids, and the max-pooling layer finally produces the overall representation.
New user interface
We redesigned the user interface of NetGO 2.0 to make it more user-friendly. On the ‘Server’ page, we allow users to run both NetGO and NetGO 2.0, which allows users to make comparisons. It should be noted that even if there is no text information available for a query protein, NetGO 2.0 still outperforms NetGO slightly as a result of using deep sequence representation generated by Seq-RNN. We reported the performance of NetGO 2.0 on test proteins with and without text information in the Supplementary Tables S4 and S5. For reducing the processing time, the PubMed citations annotated in SwissProt have been downloaded into our web server. Compared to NetGO, the running time of NetGO 2.0 is completely acceptable considering its performance improvement. For 1000 input proteins, the results can be returned in approximately 1.5 h by NetGO 2.0, compared with about 73 min for NetGO. Supplementary Table S6 compares the mean running times of NetGO and NetGO 2.0 for different numbers of input proteins.
On the ‘Result’ page, we also added the prediction scores and ranks of Seq-RNN and LR-Text. As such, users can further understand how much the new components of NetGO 2.0 help and improve the performance of AFP specifically for their queried proteins.
RESULTS
Benchmark datasets
The rules for constructing our datasets are the same as those for CAFA1 (4), CAFA2 (5) and CAFA3 (6). We collected protein sequences from UniProt (17) and obtained experimental annotations from SwissProt (3), GOA(http://http://www.ebi.ac.uk/GOA) (18) and GO(http://geneontology.org/page/download-annotations) (2). For PPI network information, we used STRING (12) version 11.0, which contains 3 123 056 667 protein interactions from 5090 organisms. Moreover, we searched for the relevant citations of a protein in MEDLINE, according to the procedure mentioned before. There are 175 610 documents in our training dataset.
Similarly, NetGO 2.0 makes predictions using three datasets.
-
Training: training for components
All data annotated in December 2018 or before.
-
LTR: training for LTR
no-knowledge and limited-knowledge proteins experimentally annotated from January 2019 to January 2020 and not before January 2019.
-
Testing: testing for competing methods
All proteins experimentally annotated between February 2020 and October 2020 and not before February 2020.
All the datasets are available at https://drive.google.com/drive/folders/1wSS-R335UcNMToMskx3dE4XcTaLvCAOc. The Supplementary Table S1 reports the numbers of proteins in the above datasets.
Performance evaluation metrics
To evaluate the prediction results, we use AUPR (area under the precision-recall curve) and Fmax as two main evaluation metrics. AUPR and Fmax are widely used for datasets with unbalanced distributions and multi-label classification. AUPR punishes false positive prediction, which is suitable for highly imbalanced data. Fmax is an official metric of CAFA. The definitions are given in the Supplementary Data.
For testing, given an input of protein sequences, all components in NetGO 2.0 compute their pair scores of proteins and candidate GO terms. With these scores, the LTR model produces an output list of GO terms in relative order. The Fmax and AUPR scores on GO labels in three domains are then generated.
Validation results
Table 1 reports the test results for NetGO 2.0 and its compared methods. The methods with achieving the best performance are highlighted in bold. The seven component methods of NetGO 2.0 are shown in the upper part of the table. It is worth to point out that the overall absolute metric scores of an AFP method may vary largely between NetGO and NetGO 2.0 for different test datasets. However, the relative performance of its different component models is stable. As an example, we can see that LR-InterPro performed best in all three branches of GO, Net-KNN was good at BPO and CCO, and BLAST-KNN was good at MFO in both NetGO and NetGO 2.0. On the other hand, a single Seq-RNN or LR-Text method did not perform well compared with Blast-KNN, LR-InterPro and Net-KNN. However, with different techniques and information sources, they become different when integrated with NetGO 2.0.
Table 1.
Performance comparisons of NetGO 2.0 with its component and competing methods against testing data
| F-max | AUPR | Coverage | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MFO | BPO | CCO | MFO | BPO | CCO | MFO | BPO | CCO | |
| Naïve | 0.416 | 0.256 | 0.542 | 0.276 | 0.118 | 0.464 | 1.00 | 1.00 | 1.00 |
| Blast-KNN | 0.632 | 0.312 | 0.566 | 0.542 | 0.132 | 0.405 | 0.91 | 0.88 | 0.81 |
| LR-3mer | 0.427 | 0.258 | 0.552 | 0.317 | 0.125 | 0.478 | 1.00 | 1.00 | 1.00 |
| LR-InterPro | 0.651 | 0.325 | 0.641 | 0.623 | 0.166 | 0.587 | 1.00 | 1.00 | 1.00 |
| Net-KNN | 0.519 | 0.325 | 0.596 | 0.416 | 0.192 | 0.528 | 0.99 | 0.99 | 0.98 |
| RNN | 0.524 | 0.265 | 0.574 | 0.424 | 0.124 | 0.477 | 1.00 | 1.00 | 1.00 |
| LR-Text | 0.464 | 0.248 | 0.479 | 0.353 | 0.154 | 0.403 | 0.72 | 0.46 | 0.66 |
| DeepGOPlus | 0.620 | 0.305 | 0.620 | 0.521 | 0.115 | 0.493 | 1.00 | 1.00 | 1.00 |
| GOLabeler | 0.667 | 0.326 | 0.631 | 0.647 | 0.193 | 0.557 | 1.00 | 1.00 | 1.00 |
| NetGO | 0.674 | 0.362 | 0.646 | 0.653 | 0.239 | 0.583 | 1.00 | 1.00 | 1.00 |
| NetGO 2.0 | 0.666 | 0.366 | 0.663 | 0.655 | 0.269 | 0.593 | 1.00 | 1.00 | 1.00 |
In the lower part, NetGO 2.0 is compared with NetGO and a state-of-the-art deep learning based method, DeepGOPlus, which is a hybrid of the sequence homology based method DIAMOND and a deep learning component DeepGOCNN (14). We can see that NetGO 2.0 significantly outperformed all the compared methods in BPO and CCO. This demonstrates that BPO and CCO predictions can benefit from both deep sequence representation and text information. Note that, although NetGO shows a better performance in Fmax over MFO, NetGO 2.0 still did best in AUPR over MFO. This may be because MFO terms are related more to sequence homology and domain/family information, from which our new components benefit less. As a whole, NetGO 2.0 achieved a 12.6% improvement over NetGO in terms of AUPR in BPO and around 2.6% in terms of Fmax in CCO. Finally, the superiority of NetGO 2.0 was further validated by 100 bootstrapped datasets with a paired t-test (P-values < 0.05 for all cases except for MFO. See Supplementary Data for details). As a result, NetGO 2.0 achieved first place in the preliminary results of CAFA4 for most cases in terms of MFO, BPO and CCO. We also reported performance comparisons on testing data in terms of Smin and with the P-values separately in the Supplementary Tables S2 and S3.
THE NETGO WEB SERVER
Comparisons with recent AFP Web servers
In recent years, many research teams have launched websites that are open to the public for protein function prediction, such as COFACTOR (19), INGA 2.0 (20), DeepGOPlus (14), GOLabler (7) and NetGO (11). As shown in Table 2, we compare NetGO 2.0 with websites that have emerged in the last few years from three aspects: (i) the types of information used. All these web servers for AFP use protein sequence information, and some of them use PPI network information. Note that NetGO 2.0 is the only web server that makes use of literature information. (ii) The approach used for integration. Both COFACTOR and INGA 2.0 use a popular consensus function to integrate the prediction scores of different component methods with the assumption of their Independence. On the other hand, DeepGOPlus uses a weighted sum to combine the prediction scores of sequence homology and sequence based deep learning methods. In contrast, GOLabeler, NetGO and NetGO 2.0 rely on an advanced machine learning technique, LTR, to effectively integrate the prediction results from different components. (iii) The maximum number of sequences allowed in one job. COFACTOR accepts only one protein sequence in each job, which may be related to its high consumption of computing resources for predicting and handling protein structure information. DeepGOPlus and INGA 2.0 can accept up to 10 protein sequences in one job. In contrast, NetGO 2.0 is so powerful that it can handle up to 1000 input protein sequences.
Table 2.
Comparisons of NetGO 2.0 with recent web servers in AFP
| Web Server | Feature/Component | Consensus approach | Maximum number of sequences in one job |
|---|---|---|---|
| COFACTOR (19) | Protein structure; Sequence; Protein–protein interaction (PPI) networks; | Consensus function | 1 |
| INGA 2.0 (20) | Homology; Domain architectures; PPI networks; | Consensus function | 10 |
| DeepGOPlus (14) | Sequence and motif-based function information; | Weighted sum | 10 |
| GOLabeler (7) | GO term frequency; Sequence-based information; | Learning to rank (LTR) | 1000 |
| NetGO (11) | GO term frequency; Sequence-based information; PPI networks | LTR | 1000 |
| NetGO 2.0 | GO term frequency; Sequence-based information; PPI networks; Deep pattern in sequence; Related literature; | LTR | 1000 |
Case study
The CAAT box DNA-binding protein NFYB-1 (Uniprot ID: O17286) is a nuclear transcription factor subunit in Caenorhabditis elegans. Given its source information, the previous version of NetGO predicts that GO term anatomical structure development (GO:0048856) ranks 23rd in the BPO ordering list (http://issubmission.sjtu.edu.cn/ng2/result/1615431733), while in the NetGO 2.0 results, the GO term ranks 19th (http://issubmission.sjtu.edu.cn/ng2/result/1615403345). In more detail, the candidate GO term ranks 64th, 55th, 7th and 6th in the results of BLAST-KNN, LR-InterPro, NetKNN and LR-Text, respectively. From this, we can see that NetGO 2.0, incorporating literature information, performs better for the target protein. Specifically, this GO term describes the development of the anatomical structure. The literature (PMID:23933492) associated with protein NFYB-1 shows T-box gene expression plays an essential role in the development of pharyngeal precursors and body wall muscles (21). Therefore, our NetGO 2.0 utilizes this literature information to build a stronger correlation between protein NFYB-1 and the GO term as shown in Figure 4. Supplementary Table S7 shows the ground truth GO terms in BPO of target protein O17286, as well as top 20 predictions of NetGO and NetGO 2.0.
Figure 4.

A comparison of prediction results for the CAAT box DNA-binding protein NFYB-1 (Uniprot:O17286) over BPO between NetGO and NetGO 2.0. We only show the GO terms in top three levels of BPO, which are associated with top 20 GO terms by NetGO and NetGO 2.0, respectively. The GO terms with prediction scores higher than 0.6 are shown with colors ([1.0, 0.9), [0.9, 0.8), [0.8, 0.7), [0.7, 0.6), [0.6, 0.0]).
CONCLUSION
In this paper, we have presented NetGO 2.0, a web server for large scale protein function prediction by using massive sequence, text, domain/family and network information. By using additional protein text annotation and deep sequence representations in its new component methods, NetGO 2.0 outperformed its predecessor, NetGO, especially in BPO and CCO. The superior performance of NetGO 2.0 shows that (i) the use of additional information is helpful for AFP; (ii) neural networks can further extract high order information hidden in the sequence and (iii) the LTR framework can integrate new information and methods well.
The upgraded NetGO 2.0 web server still maintains its high performance and stability. It will provide more benefits to biomedical practitioners.
Supplementary Material
Contributor Information
Shuwei Yao, School of Computer Science, Fudan University, Shanghai 200433, China; Institute of Science and Technology for Brain-Inspired Intelligence, Fudan University, Shanghai 200433, China.
Ronghui You, School of Computer Science, Fudan University, Shanghai 200433, China; Institute of Science and Technology for Brain-Inspired Intelligence, Fudan University, Shanghai 200433, China.
Shaojun Wang, Institute of Science and Technology for Brain-Inspired Intelligence, Fudan University, Shanghai 200433, China.
Yi Xiong, Department of Bioinformatics and Biostatistics, Shanghai Jiao Tong University, Shanghai 200240, China.
Xiaodi Huang, School of Computing and Mathematics, Charles Sturt University, Albury, NSW 2640, Australia.
Shanfeng Zhu, Institute of Science and Technology for Brain-Inspired Intelligence, Fudan University, Shanghai 200433, China; Key Laboratory of Computational Neuroscience and Brain-Inspired Intelligence (Fudan University), Ministry of Education, Shanghai 200433, China; MOE Frontiers Center for Brain Science, Fudan University, Shanghai 200433, China; Zhangjiang Fudan International Innovation Center, Shanghai 200433, China; Shanghai Key Lab of Intelligent Information Processing, Fudan University, Shanghai 200433, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [61872094 to S.Z., 61832019 to Y.X.]; Shanghai Municipal Science and Technology Commission [2018SHZDZX01, 2017SHZDZX01]; ZJ Lab; Shanghai Center for Brain Science and Brain-Inspired Technology; 111 Project [B18015 to S.Y., R.Y., S.W.]; Information Technology Facility, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences.
Conflict of interest statement. None declared.
REFERENCES
- 1. Weaver R.F. Molecular Biology (WCB Cell & Molecular Biology). 2011; McGraw-Hill Education. [Google Scholar]
- 2. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.et al.. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boutet E., Lieberherr D., Tognolli M., Schneider M., Bansal P., Bridge A.J., Poux S, Bougueleret L., Xenarios I.. Edwards D. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. Plant Bioinformatics: Methods and Protocols. 2016; NY: Springer; 23–54. [DOI] [PubMed] [Google Scholar]
- 4. Radivojac P., Clark1 W.T., Oron T.R., Schnoes A.M., Wittkop T., Sokolov A., Graim K., Funk C., Verspoor K., Ben-Hur A.et al.. A large-scale evaluation of computational protein function prediction. Nat. Methods. 2013; 10:221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang Y., Oron T.R., Clark W.T., Bankapur A.R., D’Andrea D., Lepore R., Funk C.S., Kahanda I., Verspoor K.M., Ben-Hur A.et al.. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016; 17:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou N., Jiang Y., Bergquist T.R., Lee A.J., Kacsoh B.Z., Crocker A.W., Lewis K.A., Georghiou G., Nguyen H.N., Hamid M.N.et al.. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 2019; 20:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. You R., Zhang Z., Xiong Y., Sun F., Mamitsuka H., Zhu S.. GOLabeler: improving sequence-based large-scale protein function prediction by learning to rank. Bioinformatics. 2018; 34:2465–2473. [DOI] [PubMed] [Google Scholar]
- 8. Li H. A short introduction to learning to rank. IEICE Trans. 2011; 94-D:1854–1862. [Google Scholar]
- 9. Altschul S., Madden T., Schäffer A., Zhang J., Zhang Z., Miller W., Lipman D.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G.et al.. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2015; 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. You R., Yao S., Xiong Y., Huang X., Sun F., Mamitsuka H., Zhu S.. NetGO: improving large-scale protein function prediction with massive network information. Nucleic Acids Res. 2019; 47:W379–W387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P.et al.. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017; 45:D326–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. You R., Huang X., Zhu S.. DeepText2GO: improving large-scale protein function prediction with deep semantic text representation. Methods. 2018; 145:82–90. [DOI] [PubMed] [Google Scholar]
- 14. Kulmanov M., Hoehndorf R.. DeepGOPlus: improved protein function prediction from sequence. Bioinformatics. 2020; 36:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Le Q., Mikolov T.. Distributed representations of sentences and documents. Proceedings of the 31st International Conference on Machine Learning. 2014; Bejing: 1188–1196. [Google Scholar]
- 16. Graves A., Schmidhuber J.. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Networks. 2005; 18:602–610. [DOI] [PubMed] [Google Scholar]
- 17. U. Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huntley R., Sawford T., Mutowo-Meullenet P., Shypitsyna A., Bonilla C., Martin M., O’Donovan C.. The GOA database: gene ontology annotation updates for 2015. Nucleic Acids Res. 2015; 43:1057–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang C., Freddolino P.L., Zhang Y.. COFACTOR: improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 2017; 45:W291–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piovesan D., Tosatto S.C.. INGA 2.0: improving protein function prediction for the dark proteome. Nucleic Acids Res. 2019; 47:W373–W378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Milton A.C., Packard A.V., Clary L., Okkema P.G.. The NF-Y complex negatively regulates Caenorhabditis elegans tbx-2 expression. Dev. Biol. 2013; 382:38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.