


Abstract
Uncovering how transcription factors regulate their targets at DNA, RNA and protein levels over time is critical to define gene regulatory networks (GRNs) and assign mechanisms in normal and diseased states. RNA-seq is a standard method measuring gene regulation using an established set of analysis stages. However, none of the currently available pipeline methods for interpreting ordered genomic data (in time or space) use time-series models to assign cause and effect relationships within GRNs, are adaptive to diverse experimental designs, or enable user interpretation through a web-based platform. Furthermore, methods integrating ordered RNA-seq data with protein–DNA binding data to distinguish direct from indirect interactions are urgently needed. We present TIMEOR (Trajectory Inference and Mechanism Exploration with Omics data in R), the first web-based and adaptive time-series multi-omics pipeline method which infers the relationship between gene regulatory events across time. TIMEOR addresses the critical need for methods to determine causal regulatory mechanism networks by leveraging time-series RNA-seq, motif analysis, protein–DNA binding data, and protein-protein interaction networks. TIMEOR’s user-catered approach helps non-coders generate new hypotheses and validate known mechanisms. We used TIMEOR to identify a novel link between insulin stimulation and the circadian rhythm cycle. TIMEOR is available at https://github.com/ashleymaeconard/TIMEOR.git and http://timeor.brown.edu.
Graphical Abstract
Graphical Abstract.
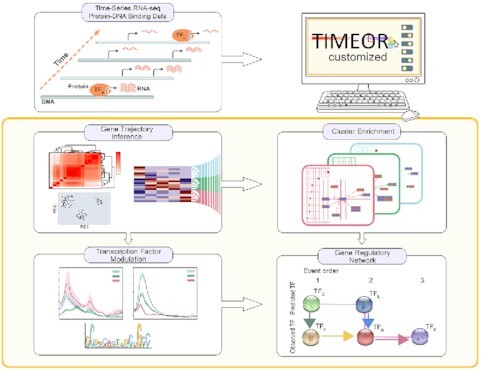
TIMEOR: novel web tool generates gene regulatory network and beyond to assign mechanism combining temporal and multi-omics data.
INTRODUCTION
Cellular responses involve a complex cascade of interacting factors which include multiple genes that influence each other at the DNA, RNA and protein levels either through direct or indirect regulation. These cascades become apparent when we alter a specific regulatory mechanism through a biological experiment allowing us to (i) construct the directed gene regulatory network (GRN), which provides a temporal model defining such cellular interactions and (ii) uncover the associated mechanism(s) (1). Most such interactions are poorly understood, with many of the genes involved remaining unidentified (2). Using multi-omics techniques, including time-series RNA-seq (3) and protein–DNA interaction data (e.g. CUT&RUN (4) and ChIP-seq (5)), biologists aim to measure changes in gene expression and transcription factor (TF) binding over time to accurately identify the regulatory mechanisms. Specifically, biologists perturb a system over time in ordered ‘case’ experiments, generally compared with the ‘control’ experiment(s) either at the first or subsequent time points. We can then determine both gene differential expression (DE), that is, the set of genes showing statistically significant quantitative changes in RNA-seq expression levels between two experiments, and evidence of direct interaction via protein–DNA binding data. The most important goal of such experiments is to reconstruct GRNs, highlighting the up and downstream candidates for future experiments or therapeutic treatment targets (6). In diseases such as cancer and diabetes, millions of dollars are spent each year to uncover better therapeutic target genes with fewer off-target effects on the patient (7). Understanding the temporal direct and indirect interactions between genes and gene products provides us the insights needed to prevent off target effects. To date, methods providing this understanding by integrating time-series and multi-omics data are urgently needed to accurately identify GRNs and assign mechanisms.
To identify key regulatory mechanisms within a biological system, raw multi-omics data must be (i) pre-processed to generate corrected data for (ii) DE (i.e. primary) analysis and finally (iii) GRNs are reconstructed (i.e. secondary) analysis. Within pre-processing, users perform quality control, alignment, read count calculation, normalization, and correction. Current pipeline methods hard-code these pre-processing steps, thus not allowing users to compare different pre-processing step methods, despite the many method options available, which can significantly impact or alter downstream results. Then, these pipeline methods summarize regulatory mechanisms in secondary analysis via (i) semi-informative gene ontology (GO) plots, showing what processes are affected and those genes involved; (ii) and/or pathways (i.e. directed gene graphs), showing directionality on a predefined network at a short time-scale with no crosstalk connections between pathways and/or (iii) networks (i.e. undirected gene graphs), thus losing directionality between genes and conflating gene and protein identity, yet gaining crosstalk between pathways and longer time-scales. However, none of these three outputs include a true GRN: a directed network highlighting how the key regulatory genes influence each other over time.
Furthermore, current pipeline methods incorporate only a subset of these stages, suffering from three additional limitations. (i) They do not accurately process and determine DE of temporal data. In fact, most pipeline methods used for time-series RNA-seq data DE analysis use categorical DE methods that do not consider time dependencies. Importantly, time-series DE methods algorithms are underutilized (8). The few DE methods that do consider temporal dynamics are not yet part of current pertinent pipeline methods which are therefore incomplete (9–14). For time-series DE methods that do exist, some can only analyze specific experimental designs or require particular pre-processed data as input (2,8,15). (ii) They do not predict the order of action of the key regulators of transcription, i.e. transcription factors (TFs) (2,14). (iii) While there are several pipelines to analyze each omic data type (RNA-seq or ChIP-seq) in isolation, joint analysis of multiple omic data types is required to uncover a GRN that models changes at different molecular levels and over time (16,17).
No end-to-end time-series and multi-omics pipeline exists that given raw omics data produces a GRN. Furthermore, a plethora of methods can be chosen within each stage, not all time-series experimental designs are the same, and downstream results heavily depend on how the RNA-seq data are processed. Most current pipeline methods do not allow users to compare the multiple possible methods, providing only one tool for each step (BioJupies (18); RSEQREP (10)) even though it is recommended to consider multiple methods for analyses such as DE (8). Furthermore, most only perform part of the analysis to infer regulatory mechanisms involved in the biological experiment (VIPER (19); BioWardrobe (20)). Also, the majority of platforms do not use multiple data-types (iDEP (21); T-REx (22)), even though we know that RNA-seq does not provide direct evidence of gene interaction. Other tools used to process such time-series and multi-omics data have to be pieced together such as Galaxy (23) and do not use a time-series DE model for time-series data such as TuxNet (13) thus disregarding time dependencies (Table 1). In Table 1, features are listed in a progressive fashion following the flow of common analysis (Pre-processing, Primary, and Secondary Analyses), and Accessibility Features, which enable users to interpret complex TF GRNs.
Table 1.
TIMEOR provides new functionalities to uncover temporal regulatory network and associated mechanisms. Exp.: experimental, Auto.: automated, QC: quality control, co.: correlation, PCA: principal component analysis, norm.: normalization, corr.: correction, DE: differential expression, enrich.: enrichment, TF: transcription factor, GRN: gene regulatory network
| TIMEOR | BioJupies | iDEP | Biowardrobe | RSEQREP | VIPER | T-REx | TuxNet | |
|---|---|---|---|---|---|---|---|---|
| Pre-processing | ||||||||
| Adaptive defaults given exp. design | x | x | ||||||
| Auto. data process from raw .fastqs | x | x | x | x | x | x | ||
| QC, sample co., PCA, norm., corr. | x | x | x | x | x | x | x | x |
| Primary Analysis | ||||||||
| Multiple method compare options | x | x | ||||||
| Close time point DE model(s) | x | |||||||
| Unsupervised time-gene clustering | x | x | x | x | x | x | ||
| Secondary Analysis | ||||||||
| GO, network and pathway enrich. | x | x | x | x | x | x | x | |
| De novo motif analysis | x | |||||||
| Multi-omics data integration | x | x | ||||||
| TF enrichment analysis | x | x | x | |||||
| Temporal GRN construction | x | x | ||||||
| Accessibility Features | ||||||||
| Web interface | x | x | x | x | x | x | ||
| Interactive results | x | x | x | |||||
| Project management | x | x | x | x | ||||
| NUMBER FEATURES | 14 | 7 | 7 | 6 | 6 | 5 | 4 | 4 |
In summary, the field requires an end-to-end analysis pipeline method using time-series algorithms to characterize gene dynamics over time that integrates TF binding data to reconstruct GRNs as directed networks from ordered RNA-seq data. It is important that such a pipeline method enables users to compare multiple methods for RNA-seq processing steps to accommodate diverse experimental designs, while leveraging each user's expertise of the input experiment data (8). In this paper, we propose TIMEOR (Trajectory Inference and Mechanism Exploration with Omics data in R) - an adaptive, streamlined pipeline method that: (a) uses time-series models to generate informative predictions about GRNs and associated mechanisms; (b) supplies tailored methods for diverse experimental designs; (c) performs multi-omics and experimentally determined protein–protein interaction data integration and (d) provides an accessible and interactive interface to interpret temporal and multi-omics results. We present the overview of TIMEOR in Figure 1.
Figure 1.

TIMEOR enables users to interrogate and reconstruct gene regulatory networks. Blue boxes denote main user-guided stages. (A) Users specify a time-series RNA-seq data set in the Pre-process Stage in which the data are automatically retrieved, normalized, corrected, filtered and aligned to the selected organism reference genome using multiple alignment methods for comparison. Importantly, users may also input the count matrix directly, skipping to normalization and correction in step 3 of stage A. (B) The resulting count matrix is passed to Primary Analysis where multiple differential expression (DE) methods are run to produce a time-series clustermap of gene DE trajectories over time. (C) DE results are passed to Secondary Analysis for gene ontology, pathway, network, motif, protein–DNA binding data analysis, and gene regulatory network reconstruction. TF: transcription factor.
TIMEOR is the first automated interactive web (24,25) and command line time-series, multi-omics pipeline method for DE and comprehensive downstream analysis (Supplementary Figures S1 and S2). TIMEOR reconstructs interpretable and user-guided GRNs, going beyond to also assign mechanism. Specifically, TIMEOR’s RShiny web-interface leverages users’ knowledge of the biological system by providing options to users during each analysis stage (24). TIMEOR can retrieve raw sequence reads (RNA-seq .fastq files) or read count matrices, and then performs all analysis from quality control and DE to enrichment (Figure 1A, B). Next, TIMEOR integrates motif and protein–DNA binding data to define TF binding patterns and reconstructs a TF GRN (Figure 1C). The web-interface enhances reproducibility and ease for users to dedicate more time to interpreting results and planning follow-up experiments (Supplementary Figures S1 and S2). Overall, TIMEOR is an adaptive method to return GRNs from time-series RNA-seq and protein–DNA binding data which integrates motif enrichment and temporal gene interactions.
We validated TIMEOR on both simulated and real data, demonstrating that TIMEOR can predict known and novel gene relationships within TF GRNs. Using real time-series RNA-seq data collected after insulin stimulation (26), TIMEOR discovered a novel ordered cascade of TF–TF interactions, identifying a new link between insulin stimulation and a validated circadian clock GRN (27–30). TIMEOR revealed that insulin regulates several key circadian clock TFs, providing a new molecular mechanism linking a high sugar diet to sleep disruption (31–33). Overall, TIMEOR facilitates future analyses of integrated omics data to uncover novel biological and disease mechanisms from multi-omic and temporal gene expression data sets.
MATERIALS AND METHODS
The TIMEOR web-app (https://timeor.brown.edu) hosted through a partnership between Brown's Computational Biology Core (https://cbc.brown.edu) and DRSC/TRiP (https://fgr.hms.harvard.edu/tools) at Harvard Medical School Research Computing allows users to automatically and interactively analyze their time-series multi-omics data from Drosophila melanogaster, Mus musculus, and Homo sapiens (Supplementary Figures S1 and S2). The web-app and software package consist of multiple stages (Figure 1). First, the Pre-process Stage automatically chooses several methods to optimally pre-process user data and generate results after users answers six questions (Figure 1A, Supplementary Figure S1A–D). Second, in the Primary Analysis stage, DE results are compared between multiple continuous and categorical time-series methods (9,12,34). Users can also compare new results with those from a previous study to determine which DE method results to use for downstream analysis. TIMEOR then automatically clusters and plots (25) the selected gene DE trajectories over time (Figure 1B, Supplementary Figure S1E). Third, the results are sent to the Secondary Analysis stage (Figure 1C) where three categories of analysis are performed in different tabs: Enrichment, identifies the genes and gene types that are over-represented within each cluster (Supplementary Figure S2A); Factor Binding, predicts which TFs are post-transcriptionally influencing the expression of each gene cluster using motif and protein–DNA binding data (Supplementary Figure S2B, C); and Temporal Relations, identifies TFs GRN (Supplementary Figure S2D). Overall, each tab takes users through a series of exploratory results to determine the best predicted TF GRN (Supplement Method Details).
Current pipeline methods are unable to effectively uncover gene regulatory mechanisms
Compared to similar existing methods, TIMEOR fills a knowledge gap as the only pipeline method to use time-series and multi-omics data to infer GRNs and provides twice as many features to uncover GRNs and associated mechanisms from time-series and multi-omics data. These methods were chosen based on their claims to analyze time-series RNA-seq data from raw .fastq files, integrate protein–DNA binding data to elucidate direct and indirect effects, and/ or perform downstream analysis to return at least one of three aforementioned secondary analysis outputs after DE: (i) semi-informative GO plots, (ii) pathways and/ or (iii) gene networks (both directed and undirected) (10,13,18–23).
We compared these methods based on 14 features needed to infer regulatory mechanisms and GRN reconstruction from time-series and multi-omics data (Table 1, left). TIMEOR uniquely supports diverse experimental designs for time-series data, and provides multiple methods to compare for alignment, normalization, and DE, including time-series specific DE models. TIMEOR not only provides all three analysis outputs after DE as mentioned above, but also the GRN from time-series RNA-seq and protein–DNA binding data. TIMEOR ties each stage coherently within a web-interface for users to generate interpretable results, gather the project management workflow used, and use paper-ready figures (Table 1). Note that we do not consider stand-alone GRN inference methods as they do not handle pre-processing, primary analysis, and the majority of secondary analysis. They also predominantly handle single time point data, and do not fully leverage multi-omics data (Supplement Method Details).
Overall, compared to current pipeline methods, TIMEOR has five unique features: (i) adaptive default analysis methods that can be customized to each experimental design; (ii) multiple method comparisons for alignment, normalization, and DE (for distant and close time point data); and (iii) statistical, graphical and interactive results for data exploration. (iv) Within each cluster of similarly regulated genes, TIMEOR performs automated gene enrichment, pathway, network, motif and protein–DNA binding data analysis. (v) Lastly, TIMEOR merges experimentally determined gene networks (35), time-series RNA-seq and motif and protein–DNA information to reconstruct TF GRNs with directed causal interaction edges by labeling the causal interaction and regulation (activation or repression) between genes and gene products.
The TIMEOR application: accessible and streamlined tool to infer gene regulatory networks while validating and generating testable hypotheses from temporal and multi-omics data
Pre-process stage
The first stage of the TIMEOR package is the pre-process stage where users can either load and retrieve published data sets of interest and perform preliminary analyses, or load any read count matrix of time-series data (Figure 1A). To do the former, users can find their desired time-series RNA-seq data on Gene Expression Omnibus (GEO) and upload GEO’s automatically generated SRA Run Table (36). TIMEOR will then automatically generate a corresponding metadata file for subsequent analyses. After answering six basic questions regarding the experimental design, TIMEOR establishes the tailored adaptive default methods to use for time-series RNA-seq data processing (Supplementary Figure S1A). TIMEOR then automatically: (i) retrieves and stores the raw .fastq files of interest in a unique directory created for each user; (ii) checks RNA-seq data quality; (iii) uses the adaptive defaults to compare and choose from several methods to align and calculate read counts per gene, resulting in a gene-by-replicate read count matrix (Supplementary Figure S1B). Importantly, users may also choose to directly upload their own read count matrix and associated metadata file. Then, TIMEOR provides several options to correlate (Pearson or Spearman) and perform principal component analysis (PCA) between sample replicates, both automatically output as interactive plots. From these results, users can choose from several options to normalize (upper quartile or trimmed mean of m-values) and then correct (Harman) (37) the resulting read count matrix in preparation for the next stage (Supplementary Figure S1C, D) (Supplement Method Details). Importantly, TIMEOR provides a ‘Getting Started’ first tab where input data details are explained, providing helpful tips for new users.
Primary analysis
In Primary Analysis, users leverage the adaptive default methods to perform the most appropriate gene DE analysis on their data. Alternatively, if users answered ‘yes’ to the question ‘Compare multiple methods (alignment and differential expression)’, then multiple DE methods (continuous and categorical) are automatically run, and users can visualize the output in both a table and Venn diagram illustrating overlapping differentially expressed genes at an adjusted p-value threshold input by users (9,12,34). A previous study gene list can be uploaded into TIMEOR and displayed as another category in the Venn diagram to help users determine which DE method results to use for downstream analysis. TIMEOR automatically clusters the chosen method's gene DE trajectories, while also providing suggestions for manual cluster choice (Figures 1B, S1E and S3D). These cluster results are then passed to Secondary Analysis (Supplement Method Details).
Secondary analysis
On the first tab of Secondary Analysis (Figure 1C) called Enrichment, users can toggle through each cluster to explore several types of enrichment within each cluster, including GO, pathway (38), network (35), and de novo motif discovery (39) (Supplementary Figure S2A). On the second tab, Factor Binding, TIMEOR determines observed TFs (i.e. differentially expressed genes that are TFs) and predicted TFs (i.e. TFs enriched to bind to differentially expressed genes within a cluster) (Supplementary Figure S2B) (40). TIMEOR provides four predicted TF tables: (i) Top Predicted Transcription Factors by Orthology table, (ii) Top Predicted Transcription Factors by Motif Similarity table, and their associated Top Transcription Factors Table per Method for both (iii) orthology and 4) motif similarity (Supplementary Figure S2C).
TIMEOR identifies both the observed and predicted TFs that could be temporally interacting. The observed TFs are identified leveraging AnimalTFDB (41) which lists known TFs for our organisms of interest. In order to identify predicted TFs and produce the first two tables described above, TIMEOR uses Rcistarget (40) to scan up to 21 TF prediction methods for evidence of enriched TF binding to each cluster of genes by both orthology and motif similarity. That is, each enriched motif is associated with candidate TFs based on orthologous sequences or based on similarities between annotated and unknown motifs (40,42). For both types, TIMEOR then summarizes the top four predicted TFs across all methods as follows: for each method, we first retain the ranked top four predictions. For each index 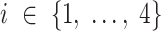 let
let 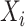 be the set of TFs that are reported in the
be the set of TFs that are reported in the 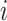 th position across the methods. For each TF in
th position across the methods. For each TF in 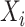 , if at least a fraction of 40% (adjustable) of methods report the same TF at position
, if at least a fraction of 40% (adjustable) of methods report the same TF at position 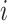 , the TF is included in the concordance set
, the TF is included in the concordance set 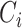 . Thus, the final output is an ordered list (
. Thus, the final output is an ordered list (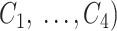 of TFs that are concordantly reported by TF prediction methods. Note that
of TFs that are concordantly reported by TF prediction methods. Note that 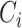 may be empty if there is no TF reported by at least 40% of methods at position
may be empty if there is no TF reported by at least 40% of methods at position 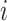 . This process is performed for each gene trajectory cluster, and for TFs predicted by orthology and motif similarity. For each of the resulting two tables (Top Predicted Transcription Factors by Orthology, and Top Predicted Transcription Factors by Motif Similarity), per cluster, TIMEOR combines the observed TFs, the top four predicted TFs, and their associated ENCODE identifiers (IDs) for all predicted TFs (43,44). All tables are available on download. Results from the Top Predicted Transcription Factors by Orthology table is displayed in TIMEOR’s web-app (Supplementary Figure S2C).
. This process is performed for each gene trajectory cluster, and for TFs predicted by orthology and motif similarity. For each of the resulting two tables (Top Predicted Transcription Factors by Orthology, and Top Predicted Transcription Factors by Motif Similarity), per cluster, TIMEOR combines the observed TFs, the top four predicted TFs, and their associated ENCODE identifiers (IDs) for all predicted TFs (43,44). All tables are available on download. Results from the Top Predicted Transcription Factors by Orthology table is displayed in TIMEOR’s web-app (Supplementary Figure S2C).
On the third tab Temporal Relations, TIMEOR uses the observed TFs per cluster and the top one predicted TF per cluster (from Top Predicted Transcription Factors by Orthology table) to determine the Temporal Relations Table between TFs, which defines a directed TF GRN (Figure 2, Supplementary Figure S2D, S2E). TIMEOR does this by harnessing the information previously gained by users traversing the Preprocessing, Primary and other Secondary Analysis stages to construct a TF GRN. This is done specifically by integrating (i) time-series RNA-seq data which TIMEOR clustered, (ii) knowledge of the observed TFs per cluster, (iii) the top one (of four) predicted TFs per cluster (listed in Top Predicted Transcription Factor by Orthology table, Supplementary Figure S2C) and (iii) STRINGdb's ‘experimentally determined’ edges (35) showing interacting TFs. TIMEOR infers interaction edges between pairs of TFs from either STRINGdb's known ‘experimentally determined’ interactions (35) or ‘predicted’: TIMEOR’s novel suggested interaction, as detailed below.
Figure 2.

Temporal Relations Table combines enables reconstruction of the transcription factor (TF) gene regulatory network. Diagram representation of algorithm separated into (i) temporal and TF information provided before Temporal Relations Table is built. (ii) TIMEOR combines five inputs, one of which is the experimentally determined interactions, to pass to TIMEOR's algorithm to (iii) reconstruct GRN (in table format). Activation is represented by +, repression is represented by –. Note we set the time frame of interaction within one time point (user question six to set adaptive default methods).
Figure 2 shows a schematic of how TIMEOR integrates these to generate a TF GRN. Specifically, the observed and top one predicted TFs within the Top Predicted Transcription Factors by Orthology table (Supplementary Figure S2C) are combined with the temporal dynamics across gene trajectory clusters to form the Temporal Relations Table. This table is called ‘Temporal Relations Between Observed and Top Predicted Factors’ on TIMEOR’s website. This table defines the GRN and specifically highlights the temporal relationships between each pair of TFs. This relationship is defined as the collection of all tuples (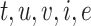 ): the time at which regulation state changes
): the time at which regulation state changes 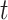 ; the source TF
; the source TF 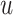 (either predicted or observed); the target TF
(either predicted or observed); the target TF 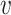 , which must be observed (as TIMEOR only reports regulation changes for those TFs we see in the data); interaction type
, which must be observed (as TIMEOR only reports regulation changes for those TFs we see in the data); interaction type 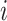 , and regulation type
, and regulation type 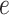 . Each interaction type
. Each interaction type 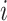 can be known, i.e. experimentally determined (reported in STRINGdb) or predicted: TIMEOR’s novel suggested interaction. The latter (predicted interaction) correspond to pairs
can be known, i.e. experimentally determined (reported in STRINGdb) or predicted: TIMEOR’s novel suggested interaction. The latter (predicted interaction) correspond to pairs 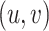 of TFs that i) change regulation state at a given time
of TFs that i) change regulation state at a given time 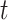 , and ii) have previously been unreported to interact. Each regulation can either be activation
, and ii) have previously been unreported to interact. Each regulation can either be activation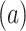 , or repression (
, or repression (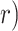 , thereby defining
, thereby defining 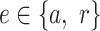 . We describe how these regulation types are assigned below using the temporal dynamics across each gene trajectory cluster. The collection of all tuples (
. We describe how these regulation types are assigned below using the temporal dynamics across each gene trajectory cluster. The collection of all tuples (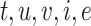 ) of all interactions comprises the Temporal Relations Table which defines the TF GRN.
) of all interactions comprises the Temporal Relations Table which defines the TF GRN.
Algorithmically, TIMEOR is first given a window of time required from users (question six, Supplementary Figure S1A). This defines the number of change-points 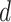 to consider for interactions between TFs. To illustrate TIMEOR’s steps let that window be 1 in this example, and follow Figure 2. Thus TIMEOR considers interactions at the transitions between timepoint
to consider for interactions between TFs. To illustrate TIMEOR’s steps let that window be 1 in this example, and follow Figure 2. Thus TIMEOR considers interactions at the transitions between timepoint 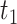 and
and 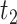 and then
and then 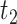 and
and 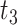 , which defines
, which defines 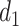 and
and 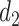 , respectively. For the first change-point
, respectively. For the first change-point 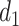 , cluster A is downregulated with observed TF
, cluster A is downregulated with observed TF 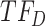 , and the top predicted TF to bind cluster A is
, and the top predicted TF to bind cluster A is 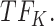 Both
Both 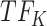 and
and 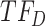 are known to interact (35). Cluster B is upregulated with observed TF
are known to interact (35). Cluster B is upregulated with observed TF 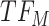 . Thus, at
. Thus, at 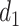 , TIMEOR infers that predicted TF
, TIMEOR infers that predicted TF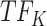 downregulates observed TF
downregulates observed TF 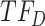 in an experimentally determined interaction, and TIMEOR predicts a novel interaction that observed TF
in an experimentally determined interaction, and TIMEOR predicts a novel interaction that observed TF 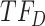 upregulates observed TF
upregulates observed TF 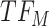 . At the next change-point
. At the next change-point 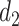 , cluster B is downregulated with observed TF
, cluster B is downregulated with observed TF 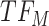 . Cluster C is activated with observed TF
. Cluster C is activated with observed TF 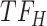 , and the top predicted TF to bind cluster C is
, and the top predicted TF to bind cluster C is 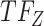 . Observed TFs
. Observed TFs 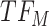 and
and 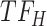 are known to interact, so TIMEOR infers that observed TF
are known to interact, so TIMEOR infers that observed TF 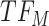 upregulates
upregulates 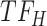 in an experimentally determined interaction. TIMEOR also infers that predicted TF
in an experimentally determined interaction. TIMEOR also infers that predicted TF 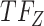 upregulates
upregulates 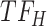 in a predicted interaction. This information is detailed in the resulting Temporal Relations Table. Examples can be seen in Figure 4C and Supplementary Figure S3E. At the bottom of this last tab TIMEOR provides the STRINGdb web-interface to facilitate users to visualize and customize the resulting temporal regulatory network. In this way, users can add and move TF nodes around, as well as add other non-TF genes to the network that were identified by TIMEOR in earlier steps (Figures 4D, 5A, Supplementary Figure S2D, S2E).
in a predicted interaction. This information is detailed in the resulting Temporal Relations Table. Examples can be seen in Figure 4C and Supplementary Figure S3E. At the bottom of this last tab TIMEOR provides the STRINGdb web-interface to facilitate users to visualize and customize the resulting temporal regulatory network. In this way, users can add and move TF nodes around, as well as add other non-TF genes to the network that were identified by TIMEOR in earlier steps (Figures 4D, 5A, Supplementary Figure S2D, S2E).
Figure 4.

TIMEOR leverages temporal and multi-omics data to identify a novel gene regulatory network. (A) TIMEOR’s clustermap (25) highlighting TIMEOR’s temporal clustering with observed TFs present in the earliest changing clusters. (B) Validation of predicted TFs using ChIP-seq data provided through TIMEOR to show binding affinity to genes across each cluster. (C) Excerpt of TIMEOR’s Temporal Relations Table showing temporal relationship between TFs. (D) Combining TIMEOR’s Temporal Relations Table, predicted TF information, and STRINGdb (35), TIMEOR outputs a gene regulatory network connecting insulin signaling and the circadian rhythm cycle.
Figure 5.

TIMEOR identifies that insulin acts as a cue for the circadian clock. (A) TIMEOR identifies a GRN comprising both observed and the top predicted TFs binding each cluster (i.e. event order). This causal interaction network (TF GRN) has a subnetwork of previously identified known circadian interactions (see top bracket) (27,30). (B) Schematic showing that TIMEOR found two new players predicted to influence circadian rhythm cycle and are induced by insulin: HNF4, which is known to regulate glucose-dependent insulin secretion, and CG32772, which could regulate the cwo gene.
In summary, TIMEOR leverages AnimalTFDB (41) to identify observed TFs within each gene trajectory cluster, and summarizes Rcistarget (40) results to identify predicted TFs (top four per gene trajectory cluster). Using just the observed TFs and top one predicted TF per gene trajectory cluster, TIMEOR then identifies which TFs could be directly interacting to form a TF GRN, via known (STRINGdb (35)) or predicted interaction edges, using the temporal dynamics between clusters within a window of time defined by users (question six Supplementary Figure S1A). This process to form a TF GRN is detailed above and in Figure 2. Thus, using time-series RNA-seq, motif and protein–DNA binding data analysis, and experimentally determined network information, TIMEOR generates a TF GRN (Figure 2, 4D, and Supplementary Figure S2D, S2E). TIMEOR outputs all results in each user's personal analysis session folder to download for future use (Supplementary Figure S2F), and provides in-depth tutorials, demonstrations, and videos (Supplementary Figure S2G).
RESULTS
Evaluation of TIMEOR on simulated data
To determine TIMEOR’s robustness to recover TF GRNs, we simulated temporal expression patterns in Homo sapiens for genes known to interact with each other. Specifically, we used Polyester (45) to simulate four RNA-seq expression cascading activation patterns over two biological replicates of six time points for 63 genes at ∼20× sequencing coverage (Supplementary Figure S3E). We used the first time point as the control for the subsequent five time points. To simulate adequate background gene expression, we sampled and analyzed two biological replicates from a real time-series RNA-seq experiment of Fusobacterium nucleatum-stimulated human gingival fibroblasts control samples (GEO: GSE118691) taken at 2, 6, 12, 24, and 48 h (46,47). We simulated a constant RNA-seq expression of one-fold across all six time points for all those genes that were active in at least one time point. To assess TIMEOR’s performance in inferring the ground truth GRN, we varied the percent concordance of the top TF identified by each method. For a concordance value of 35% and above, TIMEOR recovered simulated TF GRNs with perfect precision (Figure 3A). As expected, at low percent concordance between TF prediction methods TIMEOR predicts other TF interactions which were not simulated. Furthermore, TIMEOR achieved perfect recall for all percent concordance thresholds except at high percent concordance (65% and above) which led to one ground truth TF to drop out. This simulation highlights TIMEOR’s ability to infer accurate temporal relations between TFs by integrating time-series RNA-seq, protein–DNA binding, motif data, and experimentally determined gene interaction information (Supplement Result Details).
Figure 3.

TIMEOR accurately recovers known and novel mechanisms and genomic relationships. (A) Precision by percent concordance curve of TIMEOR’s ability to recover a simulated Homo sapiens TF GRN where the percent concordance between TF prediction methods ranges from 2 to 85%. (B) TIMEOR’s clustermap (25) of significant genes’ trajectories using z-score to denote change in downregulation (blue) and upregulation (red). (C) Without clustering, we recapitulate previous findings (26) and illustrate through GO analysis. Note TIMEOR uses Benjamini–Hochberg adjusted p-value correction. (D) TIMEOR’s pathway analysis for largest cluster b, which identifies the same ribosome biogenesis pathway as Zirin et al. (26). (E) TIMEOR highlights cluster e snoRNA pseudogenes and 28SrRNA pseudogenes which are enriched (hypergeometric test). (F) TIMEOR identified Lobe, which interacts directly with MYC (47). This interaction was not identified by prior analysis even though MYC was the focus of the previous study (26).
Evaluation of TIMEOR on real data
We next ran TIMEOR on an RNA-seq time-series experiment where Drosophila SR2+ cells were incubated with insulin. RNA-seq was performed on 10 consecutive time points every 20 minutes with three biological replicates (26). Previously, Zirin et al. found that the MYC TF regulates tRNA synthetases and ribosome biogenesis genes which enhance growth of cells in which MYC is overexpressed. In what follows, we describe how TIMEOR recapitulated these previous findings and generated novel insights by analyzing each gene trajectory cluster separately (Figure 3), and then jointly, thereby uncovering temporal dynamics among observed and predicted TFs by merging gene trajectory cluster information (Figure 4). TIMEOR thus enabled us to formulate a new biological hypothesis regarding insulin stimulating the circadian rhythm cycle (Figure 5).
Using time-series data from Drosophila cells before and after insulin stimulation, TIMEOR recapitulates previous findings (26) and discovers novel insights into insulin signaling. In primary analysis, TIMEOR compared results from three different DE methods: DESeq2 (34), Next maSigPro (12), and ImpulseDE2 (9). ImpulseDE2 showed the most significant overlap with the list of 1211 differentially expressed genes from Zirin et al. (p-value = 5.33e–127 using the hypergeometric test) and the highest overlap with other methods (Supplementary Figure S3C). Zirin et al. filtered and followed up on 33 of these 1211 genes to create a highly specific set. When TIMEOR overlapped these three methods with those 33 genes, ImpulseDE2 again showed the highest overlap with the previous study (p-value = 7.9e–137 using the hypergeometric test) and other methods. GO analysis within TIMEOR was consistent with the findings from Zirin et al. (26) which showed enrichment for genes regulating ribosome biogenesis (Figure 3C).
Next, TIMEOR clustered (25) the DE data from ImpulseDE2 into six clusters using Euclidean distance between gene trajectories and Ward's method to relate clusters of genes (Figure 3B). TIMEOR then generated the pathway, network, and GO analysis for each cluster. We found that cluster b, the largest cluster, showed enrichment in the ribosome biogenesis (Figure 3D) pathway which was previously identified (Figure 3C). Moreover, TIMEOR’s clustering analysis (Figure 3B) showed that there were five additional clusters with different temporal patterns that were not identified by Zirin et al.
Importantly, TIMEOR determined that new information may be gained by considering the dynamics of gene expression patterns when analyzing time-series experiments. TIMEOR found that the other gene trajectories represented by clusters a, c, d, e and f each reveal new insights about insulin signaling (Figure 3B). Clusters a and e contained novel genes identified by TIMEOR that did not overlap with the previous study (26): snoRNA and 28SrRNA pseudogenes. For these sets of non-coding genes, most pathway and GO analysis do not work, but TIMEOR is able to highlight these genes, which are enriched through a hypergeometric significance test (Figure 3E). These 28SrRNA-Psi may encode ribosomal RNA fragments that have been mislabeled as pseudogenes, which is consistent with the previous identification of ribosome biogenesis genes (48–50). Cluster a only contained two genes, one snoRNA and one 28SrRNA pseudogene and interestingly these genes followed a similar trajectory to cluster e genes that differed only late in the time-series. Cluster f contains the group of genes that are repressed earliest in the regulatory cascade, including Lobe, which was not identified as differentially expressed in the previous study even though it interacts directly with MYC, which was studied as a key regulator (Figure 3F) (51).
Our main task is to uncover how each gene cluster influences the expression of the other gene clusters through TFs by generating a TF GRN. To this end, TIMEOR integrates temporal dynamics between gene trajectory clusters with predicted TFs and STRINGdb's experimentally determined network to temporally relate both observed (Figure 4A arrows) and predicted TFs (Figures 4B and S2C) within each cluster in a Temporal Relations Table (Figure 4C, Supplementary Figure S2D, S3E). TIMEOR identified the circadian rhythm TFs pdp1 and cwo as observed TFs which change in gene expression in response to insulin early in the regulatory cascade (Figure 4A). In the earliest regulated cluster f (repressed over time), TIMEOR identified the observed TF gene cg32772, followed in temporal order by cwo in cluster d and pdp1 in cluster c.
Next, TIMEOR leveraged motif analysis and ChIP-seq data to uncover and validate predicted TF regulators for each cluster (Figure 4B). TIMEOR identified the available ENCODE data for all predicted TFs, as ChIP-seq data were not available after insulin stimulation. Using TIMEOR’s list of predicted TF ENCODE IDs, we chose the most comparable ChIP-seq data to input into TIMEOR’s predicted TF average binding profiles (Figure 4B). As predicted by the Top Predicted Transcription Factors Table by Orthology, HNF4 showed the strongest binding affinity to the early repressed cluster f genes (gene body) and to the promoters of the next activated cluster (cluster d genes).
Then, we examined the two most popular TF prediction methods JASPAR (52) and HOMER (53) and found that CYC ranks as the second most likely TF to bind to genes in cluster d (Supplementary Figure S3F). As predicted, when examining TF binding, CYC bound most strongly to the promotor of cluster d genes. CYC also showed strong binding within the gene body of genes in cluster f, which is the cluster repressed just before cluster d is activated. Moreover, CYC is known to bind to the genes encoding or physically associated with most of the observed TFs and most of the other top predicted TFs (27,30,35). Importantly, CYC bound to the promotor of the observed TF gene cwo in cluster d. All of this suggests that CYC is the most enriched to bind cluster d (27). CWO was an observed TF in cluster d and bound strongly to the transcription start sites (TSS) of genes in that cluster (Figure 4B), as well as cluster c where there is a known interaction with pdp1 (27,30). In the next activated cluster, MYC showed strong binding to the later expressed genes in cluster b as predicted in the Top Predicted Transcription Factors by Orthology table. Therefore, average binding profiles in combination with TF prediction results (Supplementary Figure S3F) provide support that the predicted TFs identified by TIMEOR are likely to be involved in regulating insulin signaling.
Cluster d nicely demonstrates how users can leverage TIMEOR’s four predicted TF tables to identify temporal relationships between TFs (Supplementary Figure S2C, S3F). Each TF prediction method is run individually on each cluster of genes, hence not integrating information across clusters. Thus, TIMEOR enables users to view results across all clusters and generates new testable hypotheses through its four predicted TF tables and GRN. The Top Predicted Transcription Factors by Motif Similarity table reported CG9272 as the most likely TF to bind cluster d and ChIP-seq results showed strong binding to the gene body of genes in cluster d (Supplementary Figure S3G). In the Top Predicted Transcription Factors by Orthology table, TIMEOR reported TBP as the top predicted TF to bind (Supplementary Figure S3F). While there are known (i.e. experimentally determined) interactions between TBP and the observed TF CG32772, and between TBP and the predicted TF HNF4 in cluster f (35) (Figure 4D), TBP had an indistinguishable average binding profile across most clusters likely because it is a basal TF (Supplementary Figure S3H). Therefore, TIMEOR generated a testable hypothesis that TBP functions with CG32772 and HNF4 to modulate gene regulation, while also providing strong evidence leveraging the four predicted TF tables and protein–DNA data, to identify CYC as the most likely predicted TF to bind cluster d (Figure 4D, Supplementary Figure S3F).
Integrating the observed and predicted TFs within TIMEOR’s Temporal Relations Table, TIMEOR augmented the experimentally determined STRINGdb network to predict a new GRN stimulated by insulin which has the following steps (Figures 4D and 5): step 1: The top predicted TF HNF4 which binds to cluster f represses the observed TF encoding gene cg32772. Step 2: The CYC TF and repression of the CG32772 TF activate the cwo gene in the next activated cluster d. Step 3: The CWO TF activates to the pdp1 gene in the next activated cluster c. Two of these predicted interactions have been experimentally validated in other contexts related to the circadian rhythm cycle: (i) the CYC TF is known to activate the cwo gene (28,30); (ii) the CWO TF is known to bind to the pdp1 gene (27,30). In this way, TIMEOR predicted a new GRN stimulated by insulin.
CYC, CWO and PDP1 are essential TFs which regulate the circadian rhythm cycle and function together in the temporal order identified by TIMEOR (27,28,30). TIMEOR infers a model in which the TF CYC binds to and activates the cwo gene producing the CWO TF which influences the expression of cluster c (Figure 5). Furthermore, the CWO TF binds to the gene encoding the observed TF pdp1 in cluster c, and this information is thus highlighted by directed arrows in the GRN (Figure 5A). Consistent with our predicted GRN, Zhou et al. (30) and Fathallah-Shaykh et al. (27) experimentally determine that CYC functions earlier than CWO, which functions earlier than PDP1 during the regulation of circadian rhythms (Figure 5B).
In contrast to the circadian rhythm genes, little is known about observed TF CG32772 other than that it contains a zinc finger domain (54). Given the temporal dynamics showing that CG32772 expression was repressed prior to the activation of the cwo gene, TIMEOR predicted that the loss of CG32772 activates the expression of the observed cwo gene. Therefore, CG32772 is predicted to act as a repressor of the cwo gene, which is a novel and testable hypothesis. For example, the CG32772 gene could be depleted to define its role in insulin signaling. Overall TIMEOR linked insulin signaling to a known circadian GRN and identified a new predicted TF that can be investigated in the future (Figure 5).
DISCUSSION
Novel, accessible and reproducible pipeline method integrating time-series RNA-seq and multi-omics data
Here, we presented Trajectory Inference and Mechanism Exploration with Omics data in R (TIMEOR). This pipeline method provides several advances including being the first accessible, adaptive and streamlined pipeline method to infer temporal dynamics between genes by integrating multi-omics data, including time-series RNA-seq and protein–DNA binding data (ChIP-seq or CUT&RUN) from multiple experimental designs. TIMEOR can compare multiple alignment, normalization and DE methods automatically and provide options for users to choose, which results to use downstream. Our method provides multiple close and distant time point DE methods and performs automatic gene trajectory clustering while also enabling users to manually cluster their data. TIMEOR performs gene trajectory cluster enrichment by providing GO, network, pathway and motif analysis. Then, TIMEOR integrates protein–DNA binding data to infer which predicted TFs (reported by TIMEOR) could be influencing the expression of each cluster. TIMEOR even provides ENCODE IDs for all of the potential protein–DNA interaction datasets it identifies. The web-interface for TIMEOR provides users with the ability to interact, explore, and test variants of multiple types of analysis without coding skills. Importantly, TIMEOR enables users to infer temporal relations between TFs and their gene targets through integration of time-series and multi-omics data with experimentally determined gene interaction networks.
TIMEOR enables researchers to run new analyses while also being able to reproduce results from past studies in a simple to use interface. Future work could include providing an RNA-seq read trimming method, read mapping methods, and DE methods for users (55). Further, many comparative studies suggest taking a union of the DE results between methods for a more robust set of genes in downstream analysis, thus we would like to provide this functionality to users (8). We plan to provide more interactive features such as integrating visNetwork (56) to visualize networks, and integrate methods to examine temporal differential splicing and isoform expression. Furthermore, we would also like to add a set of methods specifically targeted for single-cell RNA-seq analysis. Lastly, there are several stand-alone GRN reconstruction methods, which we would like to integrate into TIMEOR (Supplement Method Details). Our modular framework enables addition of novel and existing methods from pre-processing to GRN reconstruction in future versions.
Insulin acts as a cue for the circadian clock
We used TIMEOR to identify TF GRNs that are activated by insulin stimulation with two goals: (i) to uncover the dynamic changes in molecular function after insulin stimulation; (ii) to identify the order of action of TFs in this process by integrating temporal RNA-seq data, motif analysis, ChIP-seq data, and experimentally determined gene interaction networks. Overall, TIMEOR recapitulated the findings from a previous study (26) while also determining that insulin signaling activates the transcription of TFs that regulate the circadian rhythm cycle (Figure 5A). More research is needed to understand this relationship, although several studies suggest that the circadian clock coordinates the daily rhythm in human glucose metabolism (33,57).
Insulin resistance is a major contributor to the development of Type 2 Diabetes, caused by disrupting insulin, which alters metabolism (Figure 5B). The circadian rhythm cycle regulates several daily processes including metabolizing glucose to be used as energy and regulating insulin sensitivity (31,33). Disrupting the circadian rhythm cycle (by staying up late or shift work) dysregulates metabolism, which can cause weight gain and Type 2 diabetes (58). Therefore, it is known that disrupting the circadian clock can dysregulate insulin signaling. However, TIMEOR found a novel link, which shows that insulin directly regulates TFs that are known to alter the circadian clock such as cyc, cwo and pdp1. TIMEOR identified two new TFs that are predicted to influence the circadian rhythm cycle and are induced by insulin: (i) HNF4 is known to regulate glucose-dependent insulin secretion (59) and (ii) CG32772 could be regulating the cwo gene. Further validation experiments will be required to test these new promising hypotheses.
SUMMARY
Overall, TIMEOR is a new adaptive method capitalizing on cause-and-effect modeling to analyze time-series RNA-seq and protein–DNA binding data. TIMEOR’s automated pipeline structure facilitates in depth analysis of input data while supporting reproducibility and providing the best tools for a given experimental design. Importantly, TIMEOR integrates time-series and multi-omics data to model temporal dynamics between TFs in an accessible and flexible framework for user exploration and to enable targeted follow-up biological experiments.
DATA AVAILABILITY
TIMEOR is an open source package available on Github (https://github.com/ashleymaeconard/TIMEOR) through Docker, and on the web at https://timeor.brown.edu. Extensive documentation and support can be found at TIMEOR’s Github page, including video instruction and demonstration tutorials. Scripts for simulations available on Github (https://github.com/ashleymaeconard/TIMEOR_sims). Supplementary Data are available at NAR online.
In this study, we reanalyzed data generated and available on GEO with accession numbers GSE118691, and SRP190499. We also reanalyzed ChIP-seq data with GEO accession numbers ENCFF491LHJ, ENCFF829HXS, ENCFF082KKV, ENCFF680FFM, ENCFF553PBY and ENCFF145ATU.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Brown's Computational Biology Core and Harvard's DRSC/TRiP Functional Genomics Resources for hosting TIMEOR. We thank the Larschan, Lawrence, and Perrimon lab members for their advice. We thank the Adelman-Bender-Cole-Kuroda-Larschan joint lab meeting group for providing insights during presentations about this work. We thank Megan Gura and Dr. Anastasia Murthy for contributed ideas and manuscript comments.
Author contributions: A.M.C. led the project, created back and front-end methods, created simulation, gathered data, led the data analysis, generated all tables, figures, created collaborations and wrote manuscript. N.G. assisted in initially building the outline of two tabs of the front-end. Y.H. provided informatics support and ideas. N.P. provided informatics support, the main data used in manuscript, and ideas. R.S. reviewed A.M.C.’s simulation and methods development. C.L. advised A.M.C. on modeling the data and methods development. E.L. advised A.M.C. on modeling the data, methods development, interpreting the biological mechanisms, and manuscript writing.
Notes
Present address: Ashley Mae Conard, Computer Science, Brown University, Providence, RI 02912, USA.
Present address: Nathaniel Goodman, Computer Science, Brown University, Providence, RI 02912, USA.
Present address: Yanhui Hu, Genetics, Harvard Medical School, Boston, MA 02115, USA.
Present address: Norbert Perrimon, Genetics, Harvard Medical School, Boston, MA 02115, USA.
Present address: Ritambhara Singh, Computer Science, Brown University, Providence, RI 02912, USA.
Present address: Charles Lawrence, Applied Math, Brown University, Providence, RI 02912, USA.
Present address: Erica Larschan, Molecular Biology, Brown University, Providence, RI 02912, USA.
Contributor Information
Ashley Mae Conard, Computer Science Department, Brown University, Providence, RI 02912, USA; Center for Computational and Molecular Biology, Brown University, Providence, RI 02912, USA.
Nathaniel Goodman, Computer Science Department, Brown University, Providence, RI 02912, USA.
Yanhui Hu, Department of Genetics, Harvard Medical School, Boston, MA 02115, USA; Director of Bioinformatics DRSC/TRiP Functional Genomics Resources, Harvard Medical School, Boston, MA 02115, USA.
Norbert Perrimon, Department of Genetics, Harvard Medical School, Boston, MA 02115, USA; Howard Hughes Medical Institute, Harvard Medical School, Boston, MA 02115, USA.
Ritambhara Singh, Computer Science Department, Brown University, Providence, RI 02912, USA; Center for Computational and Molecular Biology, Brown University, Providence, RI 02912, USA.
Charles Lawrence, Center for Computational and Molecular Biology, Brown University, Providence, RI 02912, USA; Applied Math Department, Brown University, Providence, RI 02912, USA.
Erica Larschan, Center for Computational and Molecular Biology, Brown University, Providence, RI 02912, USA; Department of Molecular Biology, Cell Biology and Biochemistry, Brown University, Providence, RI 02912, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [5R35GM126994 to E.L.]; National Science Foundation Graduate Research Fellowship (to A.M.C.); Center for Computational and Molecular Biology (to A.M.C.); Institutional Development Award [P20GM109035] from the National Institute of General Medical Sciences of the National Institute of Health for Computational Biology Core support. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chai L.E., Loh S.K., Low S.T., Mohamad M.S., Deris S., Zakaria Z.. A review on the computational approaches for gene regulatory network construction. Comput. Biol. Med. 2014; 48:55–65. [DOI] [PubMed] [Google Scholar]
- 2. Sahraeian S.M.E., Mohiyuddin M., Sebra R., Tilgner H., Afshar P.T., Au K.F., Bani Asadi N., Gerstein M.B., Wong W.H., Snyder M.P.et al.. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis. Nat. Commun. 2017; 8:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Z., Gerstein M., Snyder M.. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009; 10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skene P.J., Henikoff S.. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 2017; 6:e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson D.S., Mortazavi A., Myers R.M., Wold B.. Genome-wide mapping of in vivo protein–DNA interactions. Science. 2007; 316:1497–1502. [DOI] [PubMed] [Google Scholar]
- 6. Brent M.R. Past roadblocks and new opportunities in transcription factor network mapping. Trends Genet. 2016; 32:736–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Florez J.C. Mining the genome for therapeutic targets. Diabetes. 2017; 66:1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spies D., Renz P.F., Beyer T.A., Ciaudo C.. Comparative analysis of differential gene expression tools for RNA sequencing time course data. Brief. Bioinformatics. 2019; 20:288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fischer D.S., Theis F.J., Yosef N.. Impulse model-based differential expression analysis of time course sequencing data. Nucleic Acids Res. 2018; 46:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jensen T.L., Frasketi M., Conway K., Villarroel L., Hill H., Krampis K., Goll J.B.. RSEQREP: RNA-Seq reports, an open-source cloud-enabled framework for reproducible RNA-Seq data processing, analysis, and result reporting. [version 2; peer review: 2 approved]. F1000Res. 2017; 6:2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michna A., Braselmann H., Selmansberger M., Dietz A., Hess J., Gomolka M., Hornhardt S., Blüthgen N., Zitzelsberger H., Unger K.. Natural cubic spline regression modeling followed by dynamic network reconstruction for the identification of radiation-sensitivity gene association networks from time-course transcriptome data. PLoS One. 2016; 11:e0160791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nueda M.J., Tarazona S., Conesa A.. Next maSigPro: updating maSigPro bioconductor package for RNA-seq time-series. Bioinformatics. 2014; 30:2598–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spurney R.J., Van den Broeck L., Clark N.M., Fisher A.P., de Luis Balaguer M.A., Sozzani R.. tuxnet: a simple interface to process RNA sequencing data and infer gene regulatory networks. Plant J. 2020; 101:716–730. [DOI] [PubMed] [Google Scholar]
- 14. Wani N., Raza K.. Integrative approaches to reconstruct regulatory networks from multi-omics data: a review of state-of-the-art methods. Comput. Biol. Chem. 2019; 83:107120. [DOI] [PubMed] [Google Scholar]
- 15. Spies D., Ciaudo C.. Dynamics in transcriptomics: advancements in RNA-seq time course and downstream analysis. Comput. Struct. Biotechnol. J. 2015; 13:469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbosa S., Niebel B., Wolf S., Mauch K., Takors R.. A guide to gene regulatory network inference for obtaining predictive solutions: underlying assumptions and fundamental biological and data constraints. Biosystems. 2018; 174:37–48. [DOI] [PubMed] [Google Scholar]
- 17. Marbach D., Costello J.C., Küffner R., Vega N.M., Prill R.J., Camacho D.M., Allison K.R.DREAM5 Consortium DREAM5 Consortium Kellis M., Collins J.J.et al.. Wisdom of crowds for robust gene network inference. Nat. Methods. 2012; 9:796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Torre D., Lachmann A., Ma’ayan A.. BioJupies: automated generation of interactive notebooks for RNA-Seq data analysis in the cloud. Cell Syst. 2018; 7:556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cornwell M., Vangala M., Taing L., Herbert Z., Köster J., Li B., Sun H., Li T., Zhang J., Qiu X.et al.. VIPER: visualization pipeline for RNA-seq, a snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinformatics. 2018; 19:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kartashov A.V., Barski A.. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 2015; 16:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ge S.X., Son E.W., Yao R.. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics. 2018; 19:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Jong A., van der Meulen S., Kuipers O.P., Kok J.. T-REx: transcriptome analysis webserver for RNA-seq Expression data. BMC Genomics. 2015; 16:663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A.et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018; 46:W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang W., Cheng J., Allaire J.J., Xie Y., McPherson J.. 2020; shiny: Web Application Framework for R. R package version 1.4.0.2https://CRAN.R-project.org/package=shiny.
- 25. Sievert C Interactive Web-Based Data Visualization with R, plotly, and shiny. 2020; Chapman and Hall/CRC; https://plotly-r.com. [Google Scholar]
- 26. Zirin J., Ni X., Sack L.M., Yang-Zhou D., Hu Y., Brathwaite R., Bulyk M.L., Elledge S.J., Perrimon N.. Interspecies analysis of MYC targets identifies tRNA synthetases as mediators of growth and survival in MYC-overexpressing cells. Proc. Natl Acad. Sci. U.S.A. 2019; 116:14614–14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fathallah-Shaykh H.M. Dynamics of the Drosophila circadian clock: theoretical anti-jitter network and controlled chaos. PLoS One. 2010; 5:e11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kadener S., Stoleru D., McDonald M., Nawathean P., Rosbash M.. Clockwork Orange is a transcriptional repressor and a new Drosophila circadian pacemaker component. Genes Dev. 2007; 21:1675–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maury E. Off the clock: from circadian disruption to metabolic disease. Int. J. Mol. Sci. 2019; 20:1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou J., Yu W., Hardin P.E.. CLOCKWORK ORANGE enhances PERIOD mediated rhythms in transcriptional repression by antagonizing E-box binding by CLOCK-CYCLE. PLoS Genet. 2016; 12:e1006430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qian J., Scheer F.A.J.L.. Circadian system and glucose metabolism: implications for physiology and disease. Trends Endocrinol. Metab. 2016; 27:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sharp B., Paquet E., Naef F., Bafna A., Wijnen H.. A new promoter element associated with daily time keeping in Drosophila. Nucleic Acids Res. 2017; 45:6459–6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stenvers D.J., Scheer F.A.J.L., Schrauwen P., la Fleur S.E., Kalsbeek A.. Circadian clocks and insulin resistance. Nat. Rev. Endocrinol. 2019; 15:75–89. [DOI] [PubMed] [Google Scholar]
- 34. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C.et al.. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013; 41:D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Edgar R., Domrachev M., Lash A.E.. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oytam Y., Sobhanmanesh F., Duesing K., Bowden J.C., Osmond-McLeod M., Ross J.. Risk-conscious correction of batch effects: maximising information extraction from high-throughput genomic datasets. BMC Bioinformatics. 2016; 17:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luo W., Brouwer C.. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013; 29:1830–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bailey T.L., Elkan C.. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:28–36. [PubMed] [Google Scholar]
- 40. Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J.et al.. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017; 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu H., Miao Y.R., Jia L.H., Yu Q.Y., Zhang Q., Guo A.Y.. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019; 47:D33–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Verfaillie A., Imrichová H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Atak Z.K.. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014; 10:e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K.et al.. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018; 46:D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Joly Beauparlant C., Lemacon A., Fournier E., Droit A. 2020; ENCODExplorer: A compilation of ENCODE metadata. R package version 2.14.0.
- 45. Frazee A.C., Jaffe A.E., Langmead B., Leek J.T.. Polyester: simulating RNA-seq datasets with differential transcript expression. Bioinformatics. 2015; 31:2778–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kang W., Jia Z., Tang D., Zhao X., Shi J., Jia Q., He K., Feng Q.. Time-course transcriptome analysis for drug repositioning in Fusobacterium nucleatum-infected human gingival fibroblasts. Front. Cell Dev. Biol. 2019a; 7:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang W., Jia Z., Tang D., Zhang Z., Gao H., He K., Feng Q.. Fusobacterium nucleatum facilitates apoptosis, ROS generation, and inflammatory cytokine production by activating AKT/MAPK and NF-κB signaling pathways in human gingival fibroblasts. Oxid. Med. Cell. Longev. 2019b; 2019:1681972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Berk A.J. Discovery of RNA splicing and genes in pieces. Proc. Natl Acad. Sci. U.S.A. 2016; 113:801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sharp P.A. Five easy pieces. Science. 1991; 254:663. [DOI] [PubMed] [Google Scholar]
- 50. Sharp P.A. The centrality of RNA. Cell. 2009; 136:577–580. [DOI] [PubMed] [Google Scholar]
- 51. Wang Y.-H., Huang M.-L.. Reduction of Lobe leads to TORC1 hypoactivation that induces ectopic Jak/STAT signaling to impair Drosophila eye development. Mech. Dev. 2009; 126:781–790. [DOI] [PubMed] [Google Scholar]
- 52. Sandelin A., Alkema W., Engström P., Wasserman W.W., Lenhard B.. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004; 32:D91–D94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tweedie S., Ashburner M., Falls K., Leyland P., McQuilton P., Marygold S.FlyBase Consortium . FlyBase: enhancing Drosophila gene ontology annotations. Nucleic Acids Res. 2009; 37:D555–D559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Almende B.V., Thieurmel B., Robert T.. 2019; visNetwork: Network Visualization using ‘vis. js’ Library. R package version 2.0. 8.
- 57. Boden G., Ruiz J., Urbain J.L., Chen X.. Evidence for a circadian rhythm of insulin secretion. Am. J. Physiol. 1996; 271:E246–E252. [DOI] [PubMed] [Google Scholar]
- 58. James S.M., Honn K.A., Gaddameedhi S., Van Dongen H.P.A.. Shift work: disrupted circadian rhythms and sleep-implications for health and well-being. Curr. Sleep Med. Rep. 2017; 3:104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Barry W.E., Thummel C.S.. The Drosophila HNF4 nuclear receptor promotes glucose-stimulated insulin secretion and mitochondrial function in adults. Elife. 2016; 5:e11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TIMEOR is an open source package available on Github (https://github.com/ashleymaeconard/TIMEOR) through Docker, and on the web at https://timeor.brown.edu. Extensive documentation and support can be found at TIMEOR’s Github page, including video instruction and demonstration tutorials. Scripts for simulations available on Github (https://github.com/ashleymaeconard/TIMEOR_sims). Supplementary Data are available at NAR online.
In this study, we reanalyzed data generated and available on GEO with accession numbers GSE118691, and SRP190499. We also reanalyzed ChIP-seq data with GEO accession numbers ENCFF491LHJ, ENCFF829HXS, ENCFF082KKV, ENCFF680FFM, ENCFF553PBY and ENCFF145ATU.