


Abstract
Increasing evidence proves the essential regulatory roles of non-coding RNAs (ncRNAs) in biological processes. However, characterizing the specific functions of ncRNAs remains a challenging task, owing to the intensive consumption of the experimental approaches. Here, we present an online platform ncFANs v2.0 that is a significantly enhanced version of our previous ncFANs to provide multiple computational methods for ncRNA functional annotation. Specifically, ncFANs v2.0 was updated to embed three functional modules, including ncFANs-NET, ncFANs-eLnc and ncFANs-CHIP. ncFANs-NET is a new module designed for data-free functional annotation based on four kinds of pre-built networks, including the co-expression network, co-methylation network, long non-coding RNA (lncRNA)-centric regulatory network and random forest-based network. ncFANs-eLnc enables the one-stop identification of enhancer-derived lncRNAs from the de novo assembled transcriptome based on the user-defined or our pre-annotated enhancers. Moreover, ncFANs-CHIP inherits the original functions for microarray data-based functional annotation and supports more chip types. We believe that our ncFANs v2.0 carries sufficient convenience and practicability for biological researchers and facilitates unraveling the regulatory mechanisms of ncRNAs. The ncFANs v2.0 server is freely available at http://bioinfo.org/ncfans or http://ncfans.gene.ac.
Graphical Abstract
Graphical abstract.
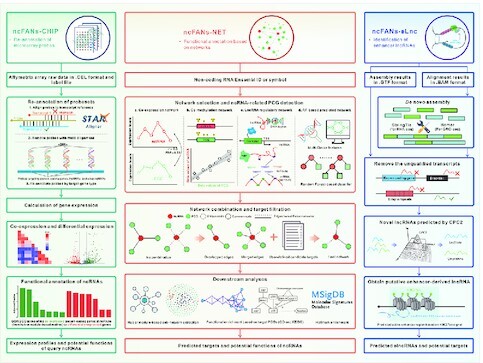
The workflow of ncFANs v2.0.
INTRODUCTION
The eukaryotic genome comprises a large portion of non-coding genes with limited protein-coding potentials. Over the past decade, non-coding RNAs (ncRNAs) such as long non-coding RNA (lncRNA), microRNA (miRNA), transfer RNA (tRNA) and small nucleolar RNA (snoRNA) have been widely verified to interact with protein-coding genes (PCGs) and participate in various biological processes through diversified molecular mechanisms (1–4). Nevertheless, compared to the relatively well-annotated functions of PCGs, much less is known about the regulatory mechanisms of ncRNA. Moreover, for the rapid growth of the newly identified ncRNAs and the disproportionate supply of experimental validation, characterizing the roles of ncRNAs remains a long way ahead and requires assistance from computational approaches.
Based on high-throughput data and some intrinsic features of ncRNAs such as structure and sequence information, some bioinformatics methods have been designed for the functional annotation of ncRNAs. For example, ncFANs (5) is the earliest online server for lncRNA functional annotation using microarray-based co-expression patterns between lncRNAs and PCGs. Besides, lncFunTK (6) integrates multi-Omics data and discovers the interactive genes of lncRNAs to evaluate the importance and predict the specific functions of lncRNAs. AnnoLnc2 (7) provides a scheme to identify new lncRNAs, annotate their functions based on lncRNA–PCG co-expression relationships and predict the subcellular localization. However, almost all tools only served lncRNAs and cannot be used for other kinds of ncRNAs. Furthermore, some of them have entered a low-support stage and even been inaccessible. Moreover, most of the currently available tools assigned the functions of the related PCGs to the query ncRNAs, but they rarely considered the condition-specific nature of the ncRNA-PCG relationships, especially the tissue and disease specificity. All these limitations urge the development of a more powerful functional annotator.
Therefore, we present ncRNA Functional ANnotation Server (ncFANs) v2.0, the updated version of our previous ncFANs (5), which supports more kinds of ncRNAs, embeds more diversified annotation methods and provides a user-friendly interface. Compared to the old version, ncFANs v2.0 comprises three functional modules (Figure 1), including the new ncFANs-NET for data-free functional annotation based on four kinds of ncRNA-PCG networks, ncFANs-eLnc for enhancer-derived lncRNA (elncRNA) identification and the enhanced ncFANs-CHIP for microarray data-based functional annotation. Our ncFANs v2.0, thus, carries sufficient practicability for biological researchers. This web server is freely available at http://bioinfo.org/ncfans or http://ncfans.gene.ac/ and opens to all users, and there is no login requirement.
Figure 1.

The workflows of three functional modules of ncFANs v2.0. ncFANs v2.0 is composed of ncFANs-CHIP for microarray data-based functional annotation, ncFANs-NET for data-free functional annotation based on pre-built networks and ncFANs-eLnc for identification of enhancer-derived lncRNAs.
RESULTS
ncFANs-NET for pre-built network-based functional annotation
ncFANs-NET is a new module designed for the data-free functional annotation of human ncRNAs based on four kinds of pre-built ncRNA-PCG networks (Figure 2), including the normal tissue- and cancer-specific co-expression network, co-methylation network, general lncRNA-centric regulatory network, and random forest-based lncRNA-PCG interactive network. Users only need to input the query ncRNAs and set the parameters of each chosen network. Notably, the co-expression and co-methylation networks support five kinds of ncRNAs (see the table at http://ncfans.gene.ac/statistics/), while the other two networks are dedicated to lncRNAs. As our previous studies have proposed these networks (5,8–12), the construction workflows will be briefly described as below:
Figure 2.

Data architecture of ncFANs v2.0, including the pre-built networks of ncFANs-NET, the pre-annotated enhancers of ncFANs-eLnc and the supported array type of ncFANs-CHIP. The full lists of these data can be fetched at http://ncfans.gene.ac/statistics/.
Co-expression network. The strategy adopted to construct the co-expression network is similar to that we previously proposed (9). First, RNA-seq data of 54 normal tissues and 33 cancer types were downloaded from GTEx (13) and the TCGA portal (https://www.cancer.gov/tcga). The sample sizes and gene numbers are recorded at http://ncfans.gene.ac/statistics/. Next, the R package WGCNA (14) was used to calculate the Spearman correlation coefficients (Rho), Fisher’s asymptotic P-value, and topological overlap measure (TOM) (15). A pair of genes with adjusted P-value (FDR correction) <0.05, Rho and TOM greater than user-defined cutoffs will be co-expressed. Apart from the condition-specific networks, users can also choose the overall normal or cancer network, which is defined as the pairs of genes co-expressed in a certain proportion of tissues or cancer types.
Co-methylation network. Recent studies proposed that gene pair’s co-methylation patterns always indicated their similar molecular functions and thus can be used for ncRNA functional annotation (8,16–18). Our strategy for co-methylation network construction is shown in the following. For normal tissues, a total of 40 human methylation profiles based on Bisulfite-seq were collected from the MethBank database (19) and classified by methylation types and genomic regions. For each gene, the methylation level in the specific condition of methylation type and the genomic region was calculated. For cancers, Illumina 450K array data were downloaded from the TCGA database (https://www.cancer.gov/tcga) of which the probes targeting the promoter regions (±2 kb around transcription start site) were reserved for further analyses. Next, the transcripts with at least three consistent probes in the promoter regions (the average Spearman correlation coefficients among the consistent probes should be >0.2) were considered to be valid and reliable records. Moreover, the average beta values of the probes targeting the same promoter were assigned as the gene methylation levels. If a gene has multiple detectable isoforms and alternative promoters, only the maximum methylation level was assigned to the gene. Finally, the correlations between the gene methylation levels were calculated using the R package WGCNA (14) as described in the above section.
LncRNA-centric regulatory network (LncRnet). LncRnet is composed of the lncRNA-PCG relationships detected by multi-Omics data and software, which is also proposed by our team (10,11,20). To construct the lncRNA-centric regulatory network, we investigated the lncRNA-PCG relationships in aspects of transcription factor (TF) binding activities, competing endogenous RNA (ceRNA) mechanism, binding activities of RNA-binding proteins (RBPs) and lncRNA–DNA triplexes. The specific workflows of obtaining these relationships are similar to that used in the published study (10). Briefly, for lncRNA–TF interaction, a total of 11 356 ChIP-seq datasets involved in 1354 TFs were obtained from the Cistrome database (21). The ncRNAs with TF peaks located in the promoter regions were considered to be regulated. For the ceRNA mechanism, 2656 mature human miRNAs sequences were downloaded from the miRBase database (22). Next, the lncRNA–miRNA and mRNA–miRNA interactions predicted by miRanda (23) with default parameters were reserved to investigate the competing possibility and strength between lncRNAs and mRNAs, which were assessed by the hypergeometric test and MuTaME scores as previously described (10). For RBP-binding activities, both experimental and computational methods are supported. In experimental method, the lncRNA–RBP interactions identified by CLIP-seq data were collected from POSTAR2 database (24), in which 216 RBPs and 12 963 lncRNAs are involved. In computational method, the sequences of 1803 RBPs annotated in GO:0003723 were obtained from the Uniprot database (25), of which the binding possibility to lncRNAs was predicted by lncPro (26) with default parameter. For lncRNA–DNA triplex, we provided the information predicted by Triplexator (27), which merely serves as a reference for users and is not incorporated into the network construction due to the limited reliability. Finally, all obtained lncRNA–PCG relationships were integrated with the protein–protein interactions (PPIs) from the STRING database (28) to construct the overall interaction network.
Random forest-based network (RF-based network). In this part, the lncRNA–PCG relationships were obtained based on our recently published method (12). According to the user-defined cutoff, ncFANs-NET will extract the qualified lncRNA–PCG interactions and corresponding PPIs from the STRING database (28) to construct the overall RF-based network.
After obtaining the relationships between ncRNAs and PCGs, ncFANs-NET will annotate the functions of ncRNAs using hub- and module-based methods described by Liao et al. (9). In the hub-based methods, ncFANs-NET selected the directly connected PCGs of the hub ncRNAs in the network and assigned the enriched functions of these PCGs to ncRNAs. The functional enrichment analyses, including gene ontology (GO), KEGG pathway and MSigDB hallmark enrichment (29) were performed using our self-made scripts (see https://github.com/zhangyw0713/FunctionEnrichment). In the module-based methods, SPICi (30) in the unweighted mode and with default parameter replaced the previous Markov cluster algorithm (MCL) to extract the close-connected modules from the overall network, owing to the overwhelming performance and speed. According to the assumption that the genes with similar functions tend to be concentrically distributed, the functions of ncRNAs can be represented by the enriched functions of PCGs in the same module.
To explicitly demonstrate the usage of ncFANs-NET, we performed a case study based on three differentially expressed ncRNAs (lncRNA PVT1, lncRNA LINC00265 and snoRNA AL356356.1) in human colorectal cancer (CRC). First, 3 query ncRNAs and 4234 differentially expressed PCGs in CRC which were identified by our previous study (11) and served as candidate targets were input (Figure 3A). Next, four kinds of networks were chosen (Figure 3A) of which the co-expression and co-methylation networks can be cancer-specific and thus set as TCGA-COAD. The other parameters are shown in Figure 3B. After receiving the launch instruction from the ‘Run’ button, ncFANs-NET started the network-based functional annotation and returned three parts of results. In the first part (Figure 4A), ncFANs-NET provided the basic information of the query ncRNAs especially the expression levels indicating their biological importance in the tissues or cancers. Next, the functional annotation results based on the selected networks were displayed in the second part (Figure 4B). For example, in the result based on the co-expression network, query ncRNAs-related network was extracted and visualized in a force-directed layout as shown in Figure 4B. Moreover, 2 modules and 3 ncRNA hubs were respectively identified. As we can see, lncRNA LINC00265 and snoRNA AL356356.1 were simultaneously clustered in module 1, indicating their similar regulatory functions in molecular biosynthesis in CRC. Notably, the only significantly enriched hallmark ‘mitotic splendid’ implied their potential role in mitosis and cell division.
Figure 3.

Input interface of ncFANs-NET. Users need to (A) input the query ncRNAs and optional target PCGs set and choose the background networks. The parameters of the networks are shown in (B).
Figure 4.

Output interface of ncFANs-NET. (A) Users are first provided with the basic information of the query ncRNAs. (B andC) And they can switch to view the annotated functions of the ncRNAs based on the selected networks. (D) Finally, the ncRNA-PCG relationships obtained from different networks can be intersected or merged to re-annotate the functions.
Moreover, the results based on other networks can be viewed via the left navigation bar (Figure 4C), which provides more clues for the complex functions of ncRNAs. Also, the ncRNA–PCG relationships predicted by different kinds of networks can be optionally merged or intersected. The PCG–PCG relationships can also be re-defined to obtain a more comprehensive or reliable result (Figure 4D).
ncFANs-eLnc for the identification of elncRNAs
ncFANs-eLnc provides identification of elncRNAs, which possibly exert enhancer-like function. In this module, users need to upload either the de novo assembled transcriptome in GTF format (highly recommended) or the alignment results of RNA-seq or GRO-seq data in BAM format. For alignment results, owing to storage and network issues, we set a threshold on the BAM file size (<200 mb), which should be suitable for the elncRNA identification on a certain chromosome (e.g. chr1) or some segments of interests. If users want to analyze the whole transcriptome data, they are encouraged to perform de novo assembly locally by using our strategy (see the commands in FAQ page at http://ncfans.gene.ac/help/) and then upload the GTF file instead. Next, users either provide the enhancer regions or select the pre-annotated cell- and tissue-specific enhancers (Figure 2). Our strategy adopted to annotate the enhancers is modified from that previously proposed (31). First, H3K27ac modification ChIP-seq data were collected from the Cistrome database (21). Peaks entirely located within blacklisted regions or promoters which were defined as the ±2 kb regions around transcription start sites were filtered out. Considering that the ChIP-seq peaks could be narrower than the elncRNA transcription region, we selected the ±3 kb regions around the center of these tentative enhancers as potential elncRNA-transcribing enhancers. After receiving the defined transcriptome and enhancers, ncFANs-eLnc first removes the transcripts overlapped with the known coding genes, blacklisted regions and simple repeats and then employ CPC2 (32) with default parameters to identify the novel ncRNAs. Novel lncRNAs with transcription start site located in enhancers are defined as elncRNAs.
In the output interface of ncFANs-eLnc, the details of the results, including the chromatin coordinates of the elncRNAs, the enhancer locations and the putative targets, are provided (Figure 5). The genes adjacent to the elncRNAs (±1 Mb) are defined as the putative targets and the known lncRNAs overlapped with the novel elncRNAs are also extracted, which can be viewed and downloaded by clicking on the blue buttons in the penultimate and last columns. Moreover, if the enhancer regions uploaded by users are overlapped with the promoters of the annotated lncRNAs, ncFANs-eLnc will also return the information of these lncRNAs which can be accessed by clicking on the button shown in Figure 5. Besides, the genome browser based on JBrowse (33) is provided for users to directly view the location of elncRNAs and enhancers.
Figure 5.

Output interface of ncFANs-eLnc. A table showing the information of the identified elncRNAs and enhancers were provided.
ncFANs-CHIP for microarray data-based functional annotation
ncFANs-CHIP retains the original function of our previous ncFANs (5), which predicts the functions of ncRNAs based on the re-annotated microarray data. The workflow is shown in Figure 1. Specifically, we first collected the description files in CDF format of 19 human and 16 mouse Affymetrix microarrays (Figure 2, see the full list at http://ncfans.gene.ac/statistics/). The probes of these arrays were re-annotated to PCGs and 6 kinds of ncRNAs (see the table at http://ncfans.gene.ac/statistics/) using the modified pipeline (5,9). The modification is that we used the new transcriptome reference sequences from GENCODE mouse vm24 and human v33 (34), and the mapper STAR (35) to replace the previous BLASTn (36). Of note, the tRNA reference sequences were collected from GtRNAdb 2.0 (37). Next, when users upload their raw microarray data in CEL format and define the array type, the gene expression values will be calculated based on the corresponding re-annotated probes. The differential expression analysis and the local coding-noncoding co-expression network construction with multiple flexible parameters such as fold change, P-value, correlation coefficient and TOM are optional for users (see the input interface at http://ncfans.gene.ac/ncFansChip/). If the co-expression network construction is chosen, the functional annotation will automatically launch and the functions of ncRNAs will be predicted using the hub- and module-based methods as described in ncFANs-NET.
After a task is submitted, the results will be returned in several minutes, or users can retrieve the results on the homepage using the allocated task ID. Here, we provided a case study based on the Affymetrix HG-U133A_2 array data GSE16919 in human embryonic stem cells (hESC) to exemplify the output. The parameters are default. As shown in Figure 6A, ncFANs-CHIP returned the gene composition of the expression profile calculated from the re-annotated probes. Besides, the results of the differential expression analysis and co-expression network results were also returned as we chose the options. For the differential expression analysis (Figure 6B), the detailed information of 275 up-regulated and 196 down-regulated genes in the nicotinamide-treated hESCs, such as fold change and adjusted P-values, was displayed along with the buttons for downloading the full lists. Furthermore, the bar plots showing the enriched functions of the up- or down-regulated genes in the nicotinamide-induced hESC differentiation alteration can be viewed and saved via clicking on the yellow buttons. For the co-expression network construction in hESCs (Figure 6C), ncFANs-CHIP provided the basic information of the network, such as the included ncRNA and PCG numbers, the hubs and the modules with at least one ncRNA, and the enriched functions of the PCGs. In this study, as the network was too large to view (nodes >1000 or edges >10000), users were encouraged to download the network in Cytoscape (38) file format to visualize it locally. Moreover, the information of the modules and hubs extracted from the overall co-expression network and used for ncRNA functional annotation follow (Figure 6C). In these two parts, the ncRNAs, the directly connected PCGs in the hub/module, and their potential functions are listed. For example, in this study, we detected the lncRNA DLEU2L, MIR22HG, AL450384.2 were clustered together in module 1, and the connected PCGs were enriched in the regulation of histone methylation, which indicated the possibly similar functions of these three lncRNAs in hESC differentiation through influencing histone methylation.
Figure 6.

Output interface of ncFANs-CHIP. ncFANs-CHIP returns (A) the number of ncRNAs and PCGs of re-annotated microarray data, (B) the information of the differentially expressed genes and (C) the results of functional annotation based on the co-expression network.
DISCUSSION
As the successor of ncFANs, ncFANs v2.0 inherits the original features of microarray-based functional annotation and is invested with prominent enhancement in the diversity of annotation methods and applicability for more kinds of ncRNAs, along with a user-friendly and interactive interface. The main improvements include but are not limited to the below items:
ncFANs v2.0 embeds a new module, ncFANs-NET, to provide data-free functional annotation, in which only ncRNAs and the background networks should be defined.
ncFANs-NET provides multiple kinds of annotation methods based on the pre-built networks, including the condition-specific co-expression and co-methylation networks and the versatile LncRnet and RF-based networks. Significantly, the condition-specific networks enable more accurate identification of the connected PCGs and functional annotation.
ncFANs-eLnc is the first one-stop platform for elncRNA identification.
Compared to the previous version, ncFANs-CHIP supports more types of microarray data and more kinds of ncRNAs including snoRNAs, rRNAs, miRNAs and so on (see the table at http://ncfans.gene.ac/statistics/).
Despite of the enhanced functions of our new ncFANs v2.0, there are still some shortcomings. In the future, we will put continuous effort into improving it in the following aspects. First, more species will be supported in all three modules to broaden the application fields. Moreover, the module for local co-expression network construction and functional annotation based on user-defined sequencing data will be developed. Next, in ncFANs-NET, we will keep updating the disease-specific networks, which should not be confined to cancer but include more diseases with sufficient data, such as Alzheimer’s disease and HIV infection. In ncFANs-eLnc, the sets of the pre-annotated enhancer regions will be expanded to support more kinds of tissues and cell lines. It is expected that our ncFANs will benefit from our efforts and users’ feedback, thus becoming increasingly powerful and popular.
DATA AVAILABILITY
The ncFANs v2.0 server is publicly available at http://bioinfo.org/ncfans or http://ncfans.gene.ac.
Contributor Information
Yuwei Zhang, The Affiliated Hospital of Medical School of Ningbo University, Ningbo, Zhejiang, 315000, China; Department of Preventative Medicine, Zhejiang Provincial Key Laboratory of Pathological and Physiological Technology, School of Medicine, Ningbo University, Ningbo, Zhejiang, 315000, China.
Dechao Bu, Pervasive Computing Research Center, Institute of Computing Technology, Chinese Academy of Sciences, Beijing, 100190, China.
Peipei Huo, Luoyang Branch of Institute of Computing Technology, Chinese Academy of Sciences, Henan, 471000, China.
Zhihao Wang, Luoyang Branch of Institute of Computing Technology, Chinese Academy of Sciences, Henan, 471000, China.
Hao Rong, The Affiliated Hospital of Medical School of Ningbo University, Ningbo, Zhejiang, 315000, China; Department of Preventative Medicine, Zhejiang Provincial Key Laboratory of Pathological and Physiological Technology, School of Medicine, Ningbo University, Ningbo, Zhejiang, 315000, China.
Yanguo Li, Institute of Drug Discovery Technology, Ningbo University, Ningbo, Zhejiang, 315000, China.
Jingjia Liu, Ningbo Institute of Life and Health Industry, University of Chinese Academy of Sciences, Ningbo, Zhejiang, 315000, China.
Meng Ye, The Affiliated Hospital of Medical School of Ningbo University, Ningbo, Zhejiang, 315000, China.
Yang Wu, Pervasive Computing Research Center, Institute of Computing Technology, Chinese Academy of Sciences, Beijing, 100190, China.
Zheng Jiang, Department of Colorectal Surgery, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China.
Qi Liao, The Affiliated Hospital of Medical School of Ningbo University, Ningbo, Zhejiang, 315000, China; Department of Preventative Medicine, Zhejiang Provincial Key Laboratory of Pathological and Physiological Technology, School of Medicine, Ningbo University, Ningbo, Zhejiang, 315000, China.
Yi Zhao, Pervasive Computing Research Center, Institute of Computing Technology, Chinese Academy of Sciences, Beijing, 100190, China.
FUNDING
CAMS Innovation Fund for Medical Sciences (CIFMS) [2019-I2M-2-002, 2017-I2M-1-006]; Natural Science Foundation of Zhejiang Province [LY21C060002]; Fundamental Research Funds for the Provincial Universities of Zhejiang [SJLZ2021001]; National Natural Science Foundation of China [31970630]; Zhejiang Key Laboratory of Pathophysiology [201812]; Zhejiang Provincial Research Center for Cancer Intelligent Diagnosis and Molecular Technology [JBZX-202003]; K. C. Wong Magna Fund in Ningbo University. Funding for open access charge: National Natural Science Foundation of China [31970630].
Conflict of interest statement. None declared.
REFERENCES
- 1. Fabbri M., Girnita L., Varani G., Calin G.A.. Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 2019; 29:1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singh I., Contreras A., Cordero J., Rubio K., Dobersch S., Gunther S., Jeratsch S., Mehta A., Kruger M., Graumann J.et al.. MiCEE is a ncRNA-protein complex that mediates epigenetic silencing and nucleolar organization. Nat. Genet. 2018; 50:990–1001. [DOI] [PubMed] [Google Scholar]
- 3. Slack F.J., Chinnaiyan A.M.. The role of non-coding RNAs in oncology. Cell. 2019; 179:1033–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teng X., Chen X., Xue H., Tang Y., Zhang P., Kang Q., Hao Y., Chen R., Zhao Y., He S.. NPInter v4.0: an integrated database of ncRNA interactions. Nucleic Acids Res. 2020; 48:D160–D165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liao Q., Xiao H., Bu D., Xie C., Miao R., Luo H., Zhao G., Yu K., Zhao H., Skogerbo G.et al.. ncFANs: a web server for functional annotation of long non-coding RNAs. Nucleic Acids Res. 2011; 39:W118–W124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou J., Huang Y., Ding Y., Yuan J., Wang H., Sun H.. lncFunTK: a toolkit for functional annotation of long noncoding RNAs. Bioinformatics. 2018; 34:3415–3416. [DOI] [PubMed] [Google Scholar]
- 7. Ke L., Yang D.C., Wang Y., Ding Y., Gao G.. AnnoLnc2: the one-stop portal to systematically annotate novel lncRNAs for human and mouse. Nucleic Acids Res. 2020; 48:W230–W238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liao Q., He W., Liu J., Cen Y., Luo L., Yu C., Li Y., Chen S., Duan S.. Identification and functional annotation of lncRNA genes with hypermethylation in colorectal cancer. Gene. 2015; 572:259–265. [DOI] [PubMed] [Google Scholar]
- 9. Liao Q., Liu C., Yuan X., Kang S., Miao R., Xiao H., Zhao G., Luo H., Bu D., Zhao H.et al.. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011; 39:3864–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y., Tao Y., Ji H., Li W., Guo X., Ng D.M., Haleem M., Xi Y., Dong C., Zhao J.et al.. Genome-wide identification of the essential protein-coding genes and long non-coding RNAs for human pan-cancer. Bioinformatics. 2019; 35:4344–4349. [DOI] [PubMed] [Google Scholar]
- 11. Zhang Y., Tao Y., Li Y., Zhao J., Zhang L., Zhang X., Dong C., Xie Y., Dai X., Zhang X.et al.. The regulatory network analysis of long noncoding RNAs in human colorectal cancer. Funct. Integr. Genomics. 2018; 18:261–275. [DOI] [PubMed] [Google Scholar]
- 12. Zhang Y., Yi T., Ji H., Zhao G., Xi Y., Dong C., Zhang L., Zhang X., Zhao J., Liao Q.. Designing a general method for predicting the regulatory relationships between long noncoding RNAs and protein-coding genes based on multi-omics characteristics. Bioinformatics. 2020; 36:2025–2032. [DOI] [PubMed] [Google Scholar]
- 13. Consortium G.T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020; 369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langfelder P., Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ravasz E., Somera A.L., Mongru D.A., Oltvai Z.N., Barabasi A.L.. Hierarchical organization of modularity in metabolic networks. Science. 2002; 297:1551–1555. [DOI] [PubMed] [Google Scholar]
- 16. Akulenko R., Helms V.. DNA co-methylation analysis suggests novel functional associations between gene pairs in breast cancer samples. Hum. Mol. Genet. 2013; 22:3016–3022. [DOI] [PubMed] [Google Scholar]
- 17. Ma X., Yu L., Wang P., Yang X.. Discovering DNA methylation patterns for long non-coding RNAs associated with cancer subtypes. Comput. Biol. Chem. 2017; 69:164–170. [DOI] [PubMed] [Google Scholar]
- 18. Wei Y., Dong S., Zhu Y., Zhao Y., Wu C., Zhu Y., Li K., Xu Y.. DNA co-methylation analysis of lincRNAs across nine cancer types reveals novel potential epigenetic biomarkers in cancer. Epigenomics. 2019; 11:1177–1190. [DOI] [PubMed] [Google Scholar]
- 19. Li R., Liang F., Li M., Zou D., Sun S., Zhao Y., Zhao W., Bao Y., Xiao J., Zhang Z.. MethBank 3.0: a database of DNA methylomes across a variety of species. Nucleic Acids Res. 2018; 46:D288–D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y., Tao Y., Liao Q.. Long noncoding RNA: a crosslink in biological regulatory network. Brief. Bioinform. 2018; 19:930–945. [DOI] [PubMed] [Google Scholar]
- 21. Zheng R., Wan C., Mei S., Qin Q., Wu Q., Sun H., Chen C.H., Brown M., Zhang X., Meyer C.A.et al.. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019; 47:D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kozomara A., Birgaoanu M., Griffiths-Jones S.. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019; 47:D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Betel D., Koppal A., Agius P., Sander C., Leslie C.. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010; 11:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu Y., Xu G., Yang Y.T., Xu Z., Chen X., Shi B., Xie D., Lu Z.J., Wang P.. POSTAR2: deciphering the post-transcriptional regulatory logics. Nucleic Acids Res. 2019; 47:D203–D211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mann C.M., Muppirala U.K., Dobbs D. Computational prediction of RNA-protein interactions. Methods Mol. Biol. 2017; 1543:169–185. [DOI] [PubMed] [Google Scholar]
- 27. Buske F.A., Bauer D.C., Mattick J.S., Bailey T.L.. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012; 22:1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P.et al.. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019; 47:D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P.. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang P., Singh M.. SPICi: a fast clustering algorithm for large biological networks. Bioinformatics. 2010; 26:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Z., Lee J.H., Ruan H., Ye Y., Krakowiak J., Hu Q., Xiang Y., Gong J., Zhou B., Wang L.et al.. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. Nat. Commun. 2019; 10:4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang Y.J., Yang D.C., Kong L., Hou M., Meng Y.Q., Wei L., Gao G.. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017; 45:W12–W16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buels R., Yao E., Diesh C.M., Hayes R.D., Munoz-Torres M., Helt G., Goodstein D.M., Elsik C.G., Lewis S.E., Stein L.et al.. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 2016; 17:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boratyn G.M., Thierry-Mieg J., Thierry-Mieg D., Busby B., Madden T.L.. Magic-BLAST, an accurate RNA-seq aligner for long and short reads. BMC Bioinform. 2019; 20:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chan P.P., Lowe T.M.. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016; 44:D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T.. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The ncFANs v2.0 server is publicly available at http://bioinfo.org/ncfans or http://ncfans.gene.ac.