


Abstract
Prediction of protein–RNA interactions is important to understand post-transcriptional events taking place in the cell. Here we introduce catRAPID omics v2.0, an update of our web server dedicated to the computation of protein–RNA interaction propensities at the transcriptome- and RNA-binding proteome-level in 8 model organisms. The server accepts multiple input protein or RNA sequences and computes their catRAPID interaction scores on updated precompiled libraries. Additionally, it is now possible to predict the interactions between a custom protein set and a custom RNA set. Considerable effort has been put into the generation of a new database of RNA-binding motifs that are searched within the predicted RNA targets of proteins. In this update, the sequence fragmentation scheme of the catRAPID fragment module has been included, which allows the server to handle long linear RNAs and to analyse circular RNAs. For the top-scoring protein–RNA pairs, the web server shows the predicted binding sites in both protein and RNA sequences and reports whether the predicted interactions are conserved in orthologous protein–RNA pairs. The catRAPID omics v2.0 web server is a powerful tool for the characterization and classification of RNA-protein interactions and is freely available at http://service.tartaglialab.com/page/catrapid_omics2_group along with documentation and tutorial.
Graphical Abstract
Graphical Abstract.
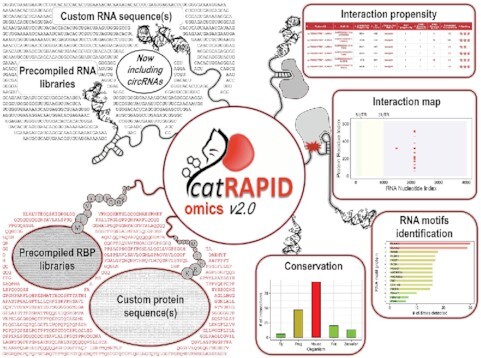
catRAPID omicsv2.0 calculates protein–RNA interaction propensities between user submitted sequences and proteomes/transcriptomes. Transcripts are also scanned for known RNA-binding motifs. For top-scoring pairs, interaction maps and evolutionary conservation analyses are provided.
INTRODUCTION
We previously developed the catRAPID approach to predict protein–RNA interactions (1). Starting from the information contained in both protein and RNA sequences, catRAPID computes secondary structure properties that are combined with physicochemical features, including hydrogen bonding, hydrophobicity and van der Waals contributions, to estimate the binding propensity of a protein–RNA pair (2). We used the method to design experiments aiming to identify the binding partners of non-coding RNAs such as Xist (3), HOTAIR (4), HOTAIRM1 (5) and SAMMSON (6).
To facilitate the calculation of protein–RNA interactions at a high-throughput level, we previously developed the catRAPID omics v1.0 web server (7). catRAPID omics v1.0 exploits precompiled libraries to quickly estimate the interaction propensity of a protein or RNA of interest in different model organisms. For instance, the user could interrogate the human proteome with a non-coding RNA sequence or the mouse transcriptome with a mutant protein in which one region is deleted.
Here, we propose catRAPID omics v2.0, a new version in which several features have been added, including algorithms that we developed and published in the last years. Among the most relevant modifications, we list the integration of the sequence fragmentation approach to identify binding regions in proteins and RNAs (8), the calculation of RNA-binding abilities of proteins (9) and a major update of protein and RNA libraries.
catRAPID omics allows proteome- and transcriptome-wide calculations for the following organisms: Homosapiens, Musmusculus, Rattusnorvegicus, Xenopustropicalis, Daniorerio, Drosophilamelanogaster, Caenorhabditis elegans and Saccharomyces cerevisiae. Importantly, in catRAPID omicsv2.0 we exploit orthology relationships to gain insight into the evolutionary conservation of predicted protein–RNA interactions.
In catRAPID omics v1.0, the precompiled protein set was built by retrieving UniprotKB entries (version 2012_11) (10) annotated as RNA-Binding, DNA-Binding or Nucleic Acid-Binding, plus the addition of RNA-binding Proteins (RBPs) identified through interactome capture experiments (11) and a manually curated database of disordered proteins, for a total of 9269 proteins. RNA motifs recognized by a fraction of these proteins were collected from a small set of sources available at the time and retrieved in RNA sequences by searching for exact matches. In catRAPID omics v2.0, the protein set is mostly composed of experimentally defined RBPs, including non-canonical RNA-binding proteins (12 380 RBPs in total, 2350 of which are in common with the catRAPID omics v1.0 protein library); such set was annotated with an updated database of motifs, many of which have been identified using high-throughput in vivo techniques that were not available at the time of catRAPID omics v1.0 release.
The precompiled RNA library in catRAPID omics v1.0 consisted of 212 773 protein-coding and 46 376 non-coding transcripts from Ensembl 68 (12), with length between 50 and 1200 nucleotides. Transcripts longer than 1200 nt were accepted only as custom input RNA sequences. In catRAPID omics v2.0, we used 141 687 protein-coding and 58 887 non-coding transcripts from Ensembl 101 (13), extending the length limit to 5000 nucleotides and manually including important long non-coding RNAs exceeding this limit. Non-coding RNAs are now divided into long non-coding and small non-coding RNA sets.
To avoid redundancy and imbalances due to multiple transcript isoforms, we selected only the main isoform of each gene. Furthermore, RNA sequences are divided into overlapping fragments (3,8), which facilitates the handling of large transcripts and the identification of binding regions. Thanks to the fragmentation procedure, catRAPID is also able to deal with circular RNA sequences. Precompiled RNA libraries were supplemented with 28 913 circular RNAs (circRNAs) from the CircAtlas 2.0 database (14), expanding the versatility of the web server. One of the most important features of catRAPID omics v2.0 is that, for the 500 most interacting protein–RNA pairs, the software calculates protein–RNA binding sites. This step, previously validated in our publications (4,8,15,16), significantly increases the power and resolution of our high-throughput method.
RNA-BINDING PROTEIN LIBRARY UPDATE
RNA-binding proteins were gathered from high-throughput detection screens (17) and from EuRBPDB (18), a database of experimentally and computationally identified RBPs. The list of human RBPs was further integrated with a set of manually curated RBPs (19). RBP sequences were obtained from UniprotKB/Swiss-Prot 2020_05 (20).
RNA-binding motifs were collected from several databases, including ATtRACT (21), cisBP-RNA (22), mCrossBase (23), oRNAment (24) and RBPmap (25), and by manual literature search. Further details about motif database construction and motif search are available in Supplementary Methods. RNA-binding proteins with no motifs in the above mentioned resources were assigned those of the most similar RBPs with which they share at least 70% sequence identity, if any (RBPs with this level of sequence identity have been shown to bind similar motifs (22)). MMseqs2 (26) was used to find such similar sequences.
hmmscan tool from the HMMER3 suite (27) was used to scan proteins for Pfam domains annotated with RNA-related terms (28). Orthology-based relationships between RBPs were derived from Ensembl 101 database (13).
The composition of the protein sequence datasets is reported in Table 1.
Table 1.
Precompiled RBP libraries available in catRAPID omics v2.0
| Proteome (UniprotKB 2020_05) | |||||
|---|---|---|---|---|---|
| RBPs | RBPs with motifs | ||||
| Model organism | Source | Total | With orthologs | Original | By similarity |
| Homo sapiens | Hentze et al., Gerstberger et al. | 2064 | 1714 | 275 | 39 |
| Mus musculus | Hentze et al. | 1903 | 1365 | 63 | 188 |
| Drosophila melanogaster | Hentze et al. | 796 | 553 | 56 | 20 |
| Caenorhabditis elegans | Hentze et al. | 491 | 340 | 21 | 6 |
| Saccharomyces cerevisiae | Hentze et al. | 1275 | 604 | 35 | 4 |
| Rattus norvegicus | EuRBPDB | 2174 | 2002 | 7 | 277 |
| Danio rerio | EuRBPDB | 2335 | 1662 | 5 | 159 |
| Xenopus tropicalis | EuRBPDB | 1342 | 1248 | 5 | 79 |
| Total | 12 380 | 9488 | 467 | 772 | |
TRANSCRIPT LIBRARY UPDATE
Ensembl 101 was used for collecting coding and non-coding RNAs. Only transcripts with length between 50 and 5000 nucleotides were allowed. Gene biotype was used to assign transcripts to each class, according to the following criteria:
protein-coding RNAs: protein_coding. For each gene, we selected a single isoform based on (in order of priority) APPRIS score (29), Transcript Support Level and presence in GENCODE Basic set (30). If multiple transcripts had the same flag, we selected the longest one. Orthology relationships were taken directly from Ensembl (31);
long non-coding RNAs: lncRNA, lincRNA, antisense, macro_lncRNA, sense_intronic, sense_overlapping, ncRNA, pseudogene (only for X. Tropicalis). Transcripts shorter than 200 nucleotides were filtered off. Orthology relationships were evaluated transcript-wise: for each transcript, liftOver tool (32) was used to determine the syntenic regions of its exons in other genomes; transcripts whose exons fell in such regions (interspecies overlap) were classified as orthologs. In this way, transcript-level orthology groups were created. From each of such groups, we selected the set of transcripts with the greatest interspecies overlap, allowing one transcript per gene. For each long non-coding RNA gene with no orthologs, we selected a single isoform based on (in order of priority) Transcript Support Level and presence in GENCODE Basic set. If multiple transcripts had the same flag, we selected the longest one;
small non-coding RNAs: ncRNA, miRNA, miscRNA, piRNA, siRNA, snRNA, snoRNA, vaultRNA. Only transcripts shorter than 200 nucleotides were kept. Orthology relationships were retrieved from Ensembl (33).
Full-length circular RNA sequences and their orthology relationships were collected from CircAtlas 2.0 (14). Only circRNAs conserved in at least four organisms (human, mouse, rat and macaque) were kept.
The composition of the RNA sequence datasets is reported in Table 2.
Table 2.
Precompiled RNA libraries available in catRAPID omics v2.0
| Transcriptome (Ensembl 101) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Protein-coding | Long non-coding | Small non-coding | Circular RNAs (CircAtlas 2.0) | |||||
| Model organism | Total | With orthologs | Total | With orthologs | Total | With orthologs | Total | With orthologs |
| Homo sapiens | 19 175 | 17 684 | 16 523 | 1001 | 4912 | 1785 | 9997 | 6678 |
| Mus musculus | 20 823 | 20 053 | 8779 | 999 | 5162 | 2535 | 10714 | 9452 |
| Drosophila melanogaster | 13 286 | 7452 | 2445 | - | 601 | 18 | - | - |
| Caenorhabditis elegans | 19 929 | 7270 | 1583 | - | 6933 | 6 | - | - |
| Saccharomyces cerevisiae | 6523 | 2819 | 16 | - | 61 | 3 | - | - |
| Rattus norvegicus | 20 894 | 19 758 | 2828 | 281 | 4662 | 2829 | 8202 | 7735 |
| Danio rerio | 24 628 | 18 383 | 2164 | 50 | 1169 | 438 | - | - |
| Xenopus tropicalis | 16 429 | 14 158 | 76 | 5 | 973 | 845 | - | - |
| Total | 141 687 | 107 577 | 34 414 | 2336 | 24 473 | 8459 | 28913 | 23 865 |
IMPROVEMENTS ON LARGE-SCALE COMPUTATION OF PROTEIN-RNA INTERACTIONS
In case the user submits only RNA or protein sequences, interactions are evaluated against a precompiled RBP or RNA library, respectively. Differently from catRAPID omics v1.0, the web server accepts multiple (up to 10) query sequences. Furthermore, it is now possible to compute all the possible pairwise interactions between a custom set of proteins and a custom set of transcripts (each composed of 500 sequences maximum). While in catRAPID omics v1.0 fragmentation occurred only for query transcripts longer than 1200 nt, it is now applied to all transcripts longer than 51 nt, whether they are submitted by the user or belonging to precompiled libraries.
Input sequences are compared to the precompiled RNA-binding proteins and RNA libraries using MMseqs2 (26). Each sequence is assigned the orthology-based relationships and the RNA-binding motifs (in case of proteins) of the best match, provided that sequence identity is higher than 70%. Submitted proteins also undergo a catRAPID signature run (9) to calculate their overall RNA-binding propensity, and an hmmscan run (27) to identify RNA-binding domains. If a submitted protein sequence is not similar to any RBP from the precompiled libraries and its catRAPID signature score is lower than 0.5, the web server warns the user that the protein is unlikely to bind RNA, but it still shows the interactions predicted by the catRAPID algorithm.
The main result of any catRAPID omics run consists of a list of interaction propensity values and corresponding z-scores calculated for each possible protein–RNA pair. When transcripts are fragmented, the values reported in the main tables are those relative to the top-scoring RNA fragment (a full table with the interaction propensity values for all RNA fragments is also available). If a fragment is produced from an mRNA or a circRNA, an annotation is provided, specifying whether it falls in a translated and/or in an untranslated region or if it overlaps with the back-splicing junction, respectively.
To help the user to rank the results, a star rating system is provided. Although conceptually similar to the catRAPID omics v1.0 star rating system, the new one is calculated in a slightly different way, being the sum of:
catRAPID normalized propensity: z-score values between -4 and 4 are mapped to [0,1] range. z-score values under -4 are assigned 0, those above 4 are assigned 1;
RBP propensity: a measure of the propensity of the protein to bind RNA. It equals 1 if the protein is in the precompiled RBP library or it is similar to one of such RBPs. Otherwise, it is set to catRAPID signature overall score;
known RNA-binding motifs: 0 if no RBP-specific RNA motif is found on the RNA sequence, 0.5 if only one of such motif occurrences is found, 1 if multiple motif occurrences are found. See Supplementary Methods for a description of how motif presence is evaluated.
After summing these values, the ranking score is scaled to [0,1] range.
Another new feature available in the main results page is a panel of plots (Figure 1) showing the number and identity of RNA-binding domains and RNA-binding motifs identified, as well as the number of conserved interactions (see next paragraph).
Figure 1.

Summary displayed in the main output page. Top-left: number of proteins having one or more RNA-binding domains (RBDs). Top-right: number of proteins in which the most detected RBDs were found. Bottom-left: number of RNA-binding motifs occurrences found in the analysed transcripts; only the most represented RBPs are displayed. Bottom-right: number of interactions predicted to be conserved in other organisms.
ANNOTATION OF TOP-SCORING INTERACTIONS
Top-scoring pairs are directly shown in a table in the main result page (Figure 2). Such protein–RNA couples are selected by taking the 500 interactions with the highest interaction propensity value. If N query sequences are submitted for analysis versus a precompiled library, the top 500/N interactions are reported for each query. In the new catRAPID omics the top-scoring pairs undergo two further analyses, allowing a more complete characterization of their interactions:
Figure 2.

An example of the table displayed in the main output page. For each protein–RNA pair, the interaction propensity and z-score are shown. Values in the Protein ID and RNA ID columns link to the Uniprot, Ensembl or CircAtlas v2.0 entries, or to the custom sequence. A click on the text within the Interaction Matrix column gives access to the interaction matrix in tabular or graphic format, as produced by a parallel catRAPID run upon protein fragmentation. RBP propensity equals 1 if the protein is in the RBP precompiled library or it is similar to one of such RBPs; otherwise, it is set to catRAPID signature overall score. By clicking on the number of RNA-binding domain and motif instances, a page is displayed showing their position within the protein and RNA sequence, respectively. The Conserved Interactions column reports the number of organisms in which the interaction is conserved out of those in which an orthologous pair is found; by clicking on the text, it is possible to access a new page with the orthologous pairs that are predicted to interact. Star rating system is displayed in the Ranking column.
a second catRAPID run is performed, in which also the protein undergoes sequence fragmentation (3,8). This allows to build a protein–RNA interaction matrix, in which an interaction propensity score is assigned to each protein–RNA fragment pair. Such matrix, which allows to find the protein and RNA regions that are more likely to interact with each other, is available both in tabular form and as a graphical representation (interaction map), both annotated with the localization of UTRs and CDS (in case of mRNA; Figure 3) or of the back-splicing junction (in case of circRNA);
an evolutionary conservation analysis across all the species available in the web server is performed, in which the orthologous RBP-RNA pairs (if any) undergo a parallel catRAPID analysis. The result of such analysis is the number of orthologous pairs in which the interaction is putatively conserved out of the total number of orthologous pairs. For an interaction to be classified as conserved, the z-score for the ortholog pair must be higher than the z-score of the pair under analysis minus 0.5, which is an arbitrary cutoff for detecting similar interactions.
Figure 3.
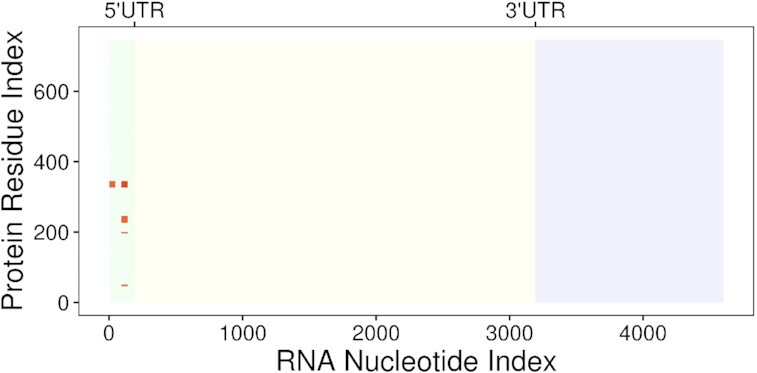
Example of interaction map describing the binding between a generic RBP and an mRNA. Red marks correspond to the predicted binding regions, the intensity being proportional to the interaction propensity (8). 5′UTR label indicates the end of the 5′ Untranslated Region, while 3’UTR marks the start of the 3’ Untranslated Region.
CALCULATION OF RBP–circRNA INTERACTIONS
The fragmentation procedure applied to circRNA sequences is shown in Figure 4. This approach allows to evenly cover the region around the back-splicing junction, an area that is particularly important since it contains the sequence determinants that distinguish the circRNA from its linear counterpart. Such fragmentation is applied not only to the circRNAs belonging to the precompiled library, but also to the circular RNA sequences submitted by the user. Following this approach we found a case of predicted protein–circRNA interaction supported by experimental evidence. We submitted a panel of six human RBPs, including the three FraX proteins (FMRP, FXR1 and FXR2), against the human precompiled circRNA library (this run is also available in the web server as a sample analysis). FraX proteins form a set of homologous RBPs (34) whose most known member is FMRP, a protein linked to Fragile X syndrome and autism (35–37). FraX proteins are highly expressed in neurons (38), in which they also have been shown to localize to neuronal projections, where they could have a role in presynaptic mRNA transport and translation (39–43). PAR-CLIP showed that most of the RNA targets of the three FraX proteins overlap, and that they have identical sequence binding preferences, consisting in ACUK and WGGA motifs (44). FMRP has also been shown to bind circRNAs (45,46).
Figure 4.
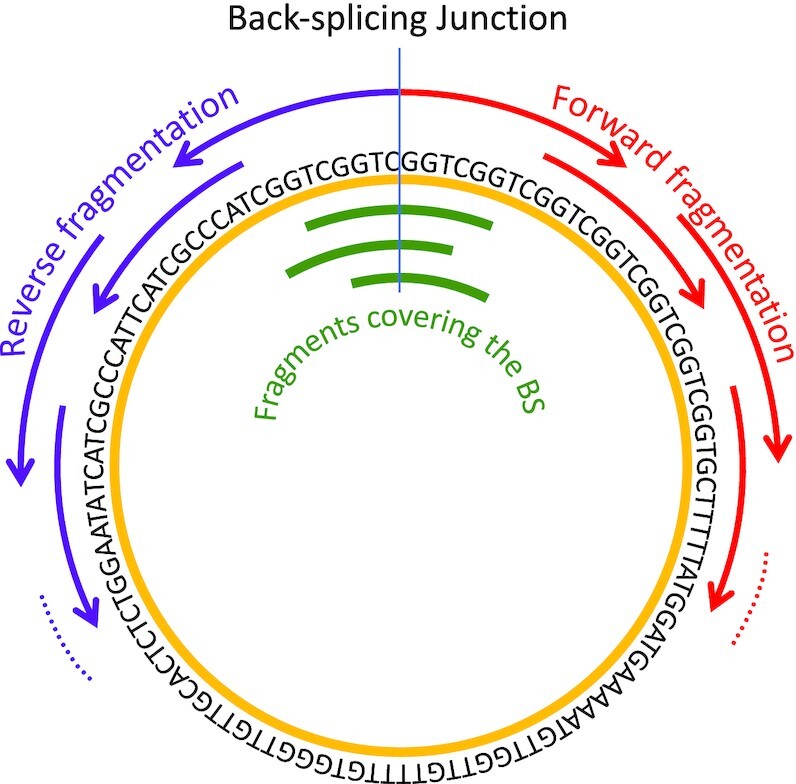
Schematic representation of the fragmentation procedure applied to circular RNA sequences. The sequence fragmentation approach applied to any linear RNA is based on the division of RNA sequences with length 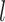 into overlapping fragments with length
into overlapping fragments with length 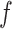 . These fragments expand from the 5' to the 3' end of sequence with a step equal to the half of the fragment length (
. These fragments expand from the 5' to the 3' end of sequence with a step equal to the half of the fragment length (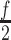 ). Similarly, in order to avoid uncovered parts in the 3'end, the fragmentation procedure is also applied in reverse, expanding from the 3' to the 5' end of the sequence. The same process is applied to circRNAs, but with three additional fragments that cover the back-splicing junction (BS), one that is centered exactly on the BS and two more fragments starting at positions
). Similarly, in order to avoid uncovered parts in the 3'end, the fragmentation procedure is also applied in reverse, expanding from the 3' to the 5' end of the sequence. The same process is applied to circRNAs, but with three additional fragments that cover the back-splicing junction (BS), one that is centered exactly on the BS and two more fragments starting at positions 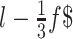 and
and 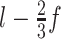 , respectively.
, respectively.
Ranking the top results of the above mentioned catRAPID omics run by star score, we found that one of the best FXR2 interactors was hsa-CHD7_0003 (21st out of 9997 circRNAs), a circRNA arising from four exons of the CHD7 gene, which encodes for a chromatin remodeling factor implicated in CHARGE syndrome (47). According to CircAtlas 2.0, hsa-CHD7_0003 is mainly expressed in the brain, where the reads mapping on its back-splicing junction account for about 10% of the expression of all the RNAs produced using the splice junctions flanking the circRNA. We found such interaction particularly compelling since it was supported by the presence of the WGGA motif and predicted to be conserved in Rattus norvegicus; most interestingly, the RNA binding site was predicted to be located across the back-splicing junction (Figure 5). While the z-score supporting this interaction is high (1.03; z-scores obtained for all the 57450 protein–RNA pairs range from –1.59 to 1.60), those calculated for the interaction with FMRP and FXR1 are rather low (0.11 and 0.07, respectively), suggesting that, even if the common RNA recognition element WGGA is present, hsa-CHD7_0003 is able to interact only with FXR2. Strikingly, according to the CLIP-Seq annotation available in CircAtlas 2.0, hsa-CHD7_0003 exonic sequence contains a binding site for FXR2, but not for the other two FraX proteins, supporting the fact that hsa-CHD7_0003 is specifically recognized by the former protein. This example shows how, with the new ability to submit multiple queries, catRAPID omics can be used to detect different binding preferences among proteins that recognize the same primary structure elements, as well as its capacity to handle circular RNA molecules thanks to the sequence fragmentation approach.
Figure 5.
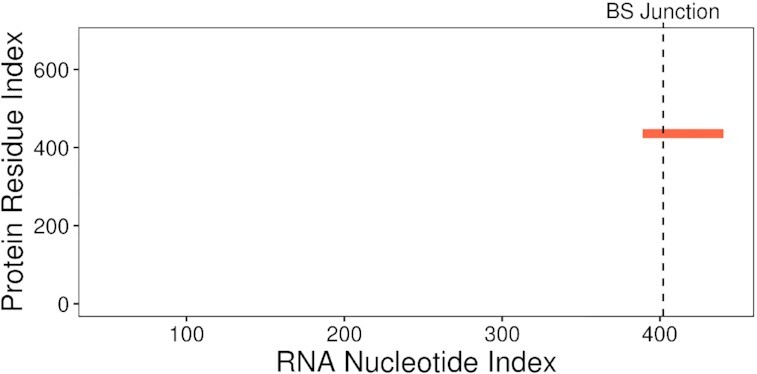
Interaction map of FXR2 binding to the circular RNA hsa-CHD7_0003. The red rectangle corresponds to the predicted binding region (8). BS Junction label indicates the position of the back-splicing junction.
CONCLUSIONS AND FUTURE PERSPECTIVES
With catRAPID omics v2.0 we expanded the application of the catRAPID algorithm to large datasets, both by updating the catRAPID omicsv1.0 RBP, mRNA and non-coding RNA libraries, and by introducing circRNAs, a novel class of RNAs whose functional characterization is a hot topic in RNA biology (48,49). The ability to handle circular RNA molecules was made possible thanks to the RNA sequence fragmentation (8) that is now applied to all the transcripts. Fragmentation occurs also at the protein level for the top-scoring interactions, allowing to better rationalize the elements that contribute to molecular recognition (3,8). The parallel calculation of orthologous binding pairs and the employment of catRAPID signature (9) to predict the RNA-binding propensity provide an extra confidence layer to our predictions. We also improved the flexibility of our method by allowing multiple input sequences and custom proteins versus custom RNAs predictions.
The downside of the fragmentation approach is that it increases the amount of calculations required to predict the interaction. Thanks to efficient parallelization and to the employment of only the main isoforms in the RNA precompiled libraries preparation, we managed to obtain execution times that are similar to those of catRAPID omics v1.0 (see catRAPID omics v2.0 online documentation), while performing further annotation steps that require additional catRAPID runs. Since the choice of the main isoform is based on the APPRIS score (29) (when available), which favours the most evolutionary conserved isoforms, the conservation analysis of the interaction is likely performed on orthologous transcripts. We are planning to implement a gene-level fragmentation scheme, which will allow us to capture all the transcript isoforms of a gene with a minimal set of fragments. Available transcript-level expression data will be integrated in order to identify co-expressed protein–RNA pairs among those which are predicted to interact, following a scheme we previously implemented (50). One of the main limitations of catRAPID omics is that the precompiled libraries only contain mature transcripts, while many protein–RNA interactions occur in the introns (51). To study the interactions involving intronic regions, users must submit them as custom sequences, with the limit of 500 RNA sequences per run. To improve the throughput of protein-intron interaction prediction we will introduce precompiled libraries of intronic sequences. These improvements, together with a major revamping of the catRAPID core algorithm, will form the basis for a future update of the catRAPID omics web server.
DATA AVAILABILITY
catRAPID omics v2.0 can be freely accessed at http://service.tartaglialab.com/page/catrapid_omics2_group.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the members of the Tartaglia and Gustincich laboratories.
Authors contribution: A.A. and A.C. developed the web server with the aid of G.P., J.R. and G.G.T. G.P. and A.C. built the protein and RNA libraries and collected motif databases. A.A. built the computational framework. A.C. and G.G.T. wrote the manuscript.
Notes
Present address: Alessio Colantoni, Center for Life Nano- & Neuro-Science, Fondazione Istituto Italiano di Tecnologia (IIT), Rome 00161, Italy.
Contributor Information
Alexandros Armaos, Center for Human Technology, Fondazione Istituto Italiano di Tecnologia (IIT), Genoa 16152, Italy.
Alessio Colantoni, Department of Biology and Biotechnology Charles Darwin, Sapienza University of Rome, Rome 00185, Italy.
Gabriele Proietti, Center for Human Technology, Fondazione Istituto Italiano di Tecnologia (IIT), Genoa 16152, Italy; Dipartimento di Neuroscienze, University of Genova, Genoa 16126, Italy.
Jakob Rupert, Center for Human Technology, Fondazione Istituto Italiano di Tecnologia (IIT), Genoa 16152, Italy; Department of Biology and Biotechnology Charles Darwin, Sapienza University of Rome, Rome 00185, Italy.
Gian Gaetano Tartaglia, Center for Human Technology, Fondazione Istituto Italiano di Tecnologia (IIT), Genoa 16152, Italy; Department of Biology and Biotechnology Charles Darwin, Sapienza University of Rome, Rome 00185, Italy; Center for Life Nano- & Neuro-Science, Fondazione Istituto Italiano di Tecnologia (IIT), Rome 00161, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council [RIBOMYLOME_309545, ASTRA_855923]; H2020 projects [IASIS_727658, INFORE_825080]. Funding for open access charge: ERC [ASTRA_855923].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bellucci M., Agostini F., Masin M., Tartaglia G.G.. Predicting protein associations with long noncoding RNAs. Nat. Methods. 2011; 8:444–445. [DOI] [PubMed] [Google Scholar]
- 2. Cid-Samper F., Gelabert-Baldrich M., Lang B., Lorenzo-Gotor N., Ponti R.D., Severijnen L.A.W.F.M., Bolognesi B., Gelpi E., Hukema R.K., Botta-Orfila T.et al.. An integrative study of protein-RNA condensates identifies scaffolding RNAs and reveals players in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2018; 25:3422–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cirillo D., Blanco M., Armaos A., Buness A., Avner P., Guttman M., Cerase A., Tartaglia G.G.. Quantitative predictions of protein interactions with long noncoding RNAs. Nat. Methods. 2016; 14:5–6. [DOI] [PubMed] [Google Scholar]
- 4. Battistelli C., Garbo S., Riccioni V., Montaldo C., Santangelo L., Vandelli A., Strippoli R., Tartaglia G.G., Tripodi M., Cicchini C.. Design and functional validation of a mutant variant of the LncRNA HOTAIR to counteract snail function in epithelial-to-mesenchymal transition. Cancer Res. 2021; 81:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rea J., Menci V., Tollis P., Santini T., Armaos A., Garone M.G., Iberite F., Cipriano A., Tartaglia G.G., Rosa A.et al.. HOTAIRM1 regulates neuronal differentiation by modulating NEUROGENIN 2 and the downstream neurogenic cascade. Cell Death. Dis. 2020; 11:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vendramin R., Verheyden Y., Ishikawa H., Goedert L., Nicolas E., Saraf K., Armaos A., Delli Ponti R., Izumikawa K., Mestdagh P.et al.. SAMMSON fosters cancer cell fitness by concertedly enhancing mitochondrial and cytosolic translation. Nat. Struct. Mol. Biol. 2018; 25:1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agostini F., Zanzoni A., Klus P., Marchese D., Cirillo D., Tartaglia G.G.. CatRAPID omics: a web server for large-scale prediction of protein–RNA interactions. Bioinformatics. 2013; 29:2928–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cirillo D., Agostini F., Klus P., Marchese D., Rodriguez S., Bolognesi B., Tartaglia G.G.. Neurodegenerative diseases: quantitative predictions of protein–RNA interactions. RNA. 2013; 19:129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Livi C.M., Klus P., Delli Ponti R., Tartaglia G.G.. catRAPID signature: identification of ribonucleoproteins and RNA-binding regions. Bioinformatics. 2016; 32:773–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Apweiler R., Martin M.J., O’Donovan C., Magrane M., Alam-Faruque Y., Alpi E., Antunes R., Arganiska J., Casanova E.B., Bely B.et al.. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic. Acids. Res. 2013; 41:D43–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castello A., Horos R., Strein C., Fischer B., Eichelbaum K., Steinmetz L.M., Krijgsveld J., Hentze M.W.. System-wide identification of RNA-binding proteins by interactome capture. Nat. Protoc. 2013; 8:491–500. [DOI] [PubMed] [Google Scholar]
- 12. Flicek P., Ahmed I., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S.et al.. Ensembl 2013. Nucleic Acids Res. 2013; 41:D48–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R.et al.. Ensembl 2020. Nucleic Acids Res. 2020; 48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu W., Ji P., Zhao F.. CircAtlas: an integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020; 21:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agostini F., Cirillo D., Bolognesi B., Tartaglia G.G.. X-inactivation: quantitative predictions of protein interactions in the Xist network. Nucleic Acids Res. 2013; 41:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lang B., Armaos A., Tartaglia G.G.. RNAct: protein-RNA interaction predictions for model organisms with supporting experimental data. Nucleic Acids Res. 2019; 47:D601–D606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
- 18. Liao J.-Y.Y., Yang B., Zhang Y.-C.C.Y., Wang X.-J.J., Ye Y., Peng J.-W.W., Yang Z.-Z.Z., He J.-H.H., Zhang Y.-C.C.Y., Hu K.S.et al.. EuRBPDB: a comprehensive resource for annotation, functional and oncological investigation of eukaryotic RNA binding proteins (RBPs). Nucleic Acids Res. 2020; 48:307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gerstberger S., Hafner M., Tuschl T.. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bateman A., Martin M.J., Orchard S., Magrane M., Agivetova R., Ahmad S., Alpi E., Bowler-Barnett E.H., Britto R., Bursteinas B.et al.. UniProt: the universal protein knowledgebase in 2021. Nucleic. Acids. Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giudice G., Sánchez-Cabo F., Torroja C., Lara-Pezzi E.. ATtRACT – a database of RNA-binding proteins and associated motifs. Database. 2016; 2016:baw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A.et al.. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013; 499:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng H., Bao S., Rahman M.A., Weyn-Vanhentenryck S.M., Khan A., Wong J., Shah A., Flynn E.D., Krainer A.R., Zhang C.. Modeling RNA-binding protein specificity in vivo by precisely registering protein-RNA crosslink sites. Mol. Cell. 2019; 74:1189–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Benoit Bouvrette L.P., Bovaird S., Blanchette M., Lécuyer E.. ORNAment: s database of putative RNA binding protein target sites in the transcriptomes of model species. Nucleic Acids Res. 2020; 48:D166–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paz I., Kosti I., Ares M., Cline M., Mandel-Gutfreund Y.. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014; 42:W361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steinegger M., Söding J.. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:1026–1028. [DOI] [PubMed] [Google Scholar]
- 27. Finn R.D., Clements J., Eddy S.R.. HMMER web server: interactive sequence similarity searching. Nucleic. Acids. Res. 2011; 39:W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finn R.D., Mistry J., Tate J., Coggill P., Heger A., Pollington J.E., Gavin O.L., Gunasekaran P., Ceric G., Forslund K.et al.. The Pfam protein families database. NucleicAcids Res. 2009; 38:D290–D301. [Google Scholar]
- 29. Rodriguez J.M., Maietta P., Ezkurdia I., Pietrelli A., Wesselink J.J., Lopez G., Valencia A., Tress M.L.. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 2013; 41:D110–D117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Herrero J., Muffato M., Beal K., Fitzgerald S., Gordon L., Pignatelli M., Vilella A.J., Searle S.M.J., Amode R., Brent S.et al.. Ensembl comparative genomics resources. Database. 2016; 2016:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hinrichs A.S., Karolchik D., Baertsch R., Barber G.P., Bejerano G., Clawson H., Diekhans M., Furey T.S., Harte R.A., Hsu F.et al.. The UCSC Genome Browser Database: update 2006. NucleicAcids Res. 2006; 34:D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pignatelli M., Vilella A.J., Muffato M., Gordon L., White S., Flicek P., Herrero J.. NcRNA orthologies in the vertebrate lineage. Database. 2016; 2016:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kirkpatrick L.L., McIlwain K.A., Nelson D.L.. Comparative genomic sequence analysis of the FXR gene family: FMR1, FXR1, and FXR2. Genomics. 2001; 78:169–177. [DOI] [PubMed] [Google Scholar]
- 35. Hoogeveen A.T., Willemsen R., Oostra B.A.. Fragile X syndrome, the fragile X related proteins, and animal models. Microsc. Res. Tech. 2002; 57:148–155. [DOI] [PubMed] [Google Scholar]
- 36. Farzin F., Perry H., Hessl D., Loesch D., Cohen J., Bacalman S., Gane L., Tassone F., Hagerman P., Hagerman R.. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J. Dev. Behav. Pediatr. 2006; 27:S137–S144. [DOI] [PubMed] [Google Scholar]
- 37. Hagerman R., Hoem G., Hagerman P.. Fragile X and autism: intertwined at the molecular level leading to targeted treatments. Mol. Autism. 2010; 1:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tamanini F., Willemsen R., Van Unen L., Bontekoe C., Galjaard H., Oostra B.A., Hoogeveen A.T.. Differential expression of FMR1, FXR1 and FXR2 proteins in human brain and testis. Hum. Mol. Genet. 1997; 6:1315–1322. [DOI] [PubMed] [Google Scholar]
- 39. Kanai Y., Dohmae N., Hirokawa N.. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004; 43:513–525. [DOI] [PubMed] [Google Scholar]
- 40. Dictenberg J.B., Swanger S.A., Antar L.N., Singer R.H., Bassell G.J.. A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell. 2008; 14:926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Akins M.R., Berk-Rauch H.E., Kwan K.Y., Mitchell M.E., Shepard K.A., Korsak L.I.T., Stackpole E.E., Warner-Schmidt J.L., Sestan N., Cameron H.A.et al.. Axonal ribosomes and mRNAs associate with fragile X granules in adult rodent and human brains. Hum. Mol. Genet. 2017; 26:192–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shepard K.A., Korsak L.I.T., DeBartolo D., Akins M.R. Axonal localization of the fragile X family of RNA binding proteins is conserved across mammals. J. Comp. Neurol. 2020; 528:502–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goering R., Hudish L.I., Guzman B.B., Raj N., Bassell G.J., Russ H.A., Dominguez D., Taliaferro J.M.. FMRP promotes RNA localization to neuronal projections through interactions between its RGG domain and g-quadruplex RNA sequences. eLife. 2020; 9:e52621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ascano M., Mukherjee N., Bandaru P., Miller J.B., Nusbaum J.D., Corcoran D.L., Langlois C., Munschauer M., Dewell S., Hafner M.et al.. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012; 492:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu Y.J., Zheng B., Luo G.J., Ma X.K., Lu X.Y., Lin X.M., Yang S., Zhao Q., Wu T., Li Z.X.et al.. Circular RNAs negatively regulate cancer stem cells by physically binding FMRP against CCAR1 complex in hepatocellular carcinoma. Theranostics. 2019; 9:3526–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu J., Ji L., Liang Y., Wan Z., Zheng W., Song X., Gorshkov K., Sun Q., Lin H., Zheng X.et al.. CircRNA-SORE mediates sorafenib resistance in hepatocellular carcinoma by stabilizing YBX1. Signal Transduct. Target. Ther. 2020; 5:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Basson M.A., van Ravenswaaij-Arts C.. Functional insights into chromatin remodelling from studies on CHARGE syndrome. Trends Genet. 2015; 31:600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu C.Y., Kuo H.C.. The emerging roles and functions of circular RNAs and their generation. J. Biomed. Sci. 2019; 26:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang A., Zheng H., Wu Z., Chen M., Huang Y.. Circular RNA-protein interactions: functions, mechanisms, and identification. Theranostics. 2020; 10:3506–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cirillo D., Marchese D., Agostini F., Livi C.M., Botta-Orfila T., Tartaglia G.G.. Constitutive patterns of gene expression regulated by RNA-binding proteins. Genome Biol. 2014; 15:R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Van Nostrand E.L., Freese P., Pratt G.A., Wang X., Wei X., Xiao R., Blue S.M., Chen J.-Y., Cody N.A.L., Dominguez D.et al.. A large-scale binding and functional map of human RNA-binding proteins. Nature. 2020; 583:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
catRAPID omics v2.0 can be freely accessed at http://service.tartaglialab.com/page/catrapid_omics2_group.