


Abstract
Methylage is an epigenetic marker of biological age that exploits the correlation between the methylation state of specific CG dinucleotides (CpGs) and chronological age (in years), gestational age (in weeks), cellular age (in cell cycles or as telomere length, in kilobases). Using DNA methylation data, methylage is measurable via the so called epigenetic clocks. Importantly, alterations of the correlation between methylage and age (age acceleration or deceleration) have been stably associated with pathological states and occur long before clinical signs of diseases become overt, making epigenetic clocks a potentially disruptive tool in preventive, diagnostic and also in forensic applications. Nevertheless, methylage dependency from CpGs selection, mathematical modelling, tissue specificity and age range, still makes the potential of this biomarker limited. In order to enhance model comparisons, interchange, availability, robustness and standardization, we organized a selected set of clocks within a hub webservice, EstimAge (Estimate of methylation Age, http://estimage.iac.rm.cnr.it), which intuitively and informatively enables quick identification, computation and comparison of available clocks, with the support of standard statistics.
Graphical Abstract
Graphical Abstract.
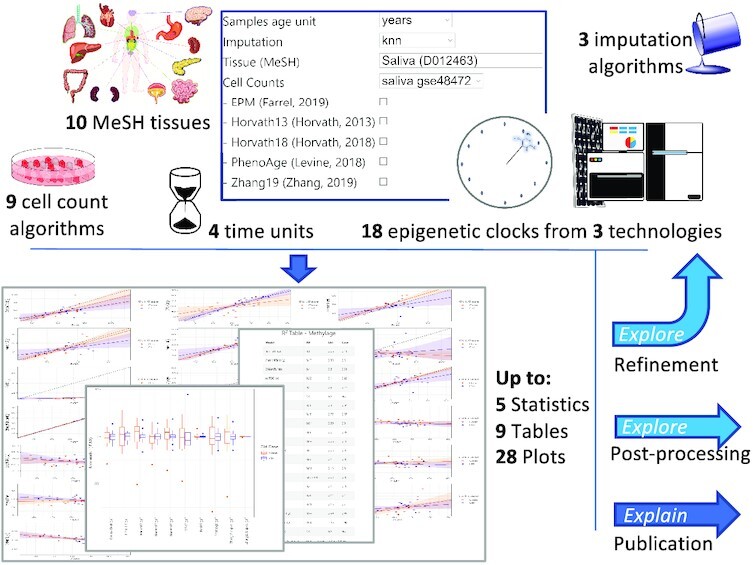
EstimAge enables the computation of methylage via the largest selection of epigenetic clocks, offering multiple tabular and graphical outputs to be readily used for scientific reporting or further processing, including refined/alternative rounds of EstimAge selection.
INTRODUCTION
The discovery of the relevance, ubiquity and, importantly, stability of methylation changes (i.e. reversible and heritable addition of a methyl group at the 5-carbon of the cytosine ring on the DNA chain), has gained popularity in recent years, fostering the systematic study of recurring areas of altered methylation along the genome (CpG islands). Methylation changes, like other epigenetic modifications, can impact on the final quality and yield of proteins translation, in ways that have been recognized crucial in pathological conditions, in physiological embryo development, and as a ubiquitous (by)product of ageing (1–3).
It is particularly in this latter area that epigenomic biomarkers have been under deep and constant investigation. Ageing is (also) the strongest risk factor for multi-morbidities, representing the major economic and social burden in our societies (4). Several biological markers of age have therefore been proposed to track the physiology of ageing, in the hope to better unravel the complexity hidden behind this process and with the aim to identify early divergences (i.e. with an added value in prevention) between chronological and biological ageing, that is age acceleration, a recognized proxy of pathological conditions (5).
Although conceptually not novel, biological estimate of senescence (6) has recently been enriched and quickly dominated by the computation of methylage, via the so called epigenetic clocks (5). In practice, methylage exploits the observed alteration of the methylation state of specific CpGs, that correlate with chronological age in healthy conditions. Regression coefficients that model this relationship can then be reused on the same methylated CpGs, measured across different individuals carrying a variety of pathologies or conditions (7–12). This well conserved mechanism enables also veterinary applications (13–15) with relevance in animal model studies. Similarly, the indication on chronological age that can potentially be extracted from biospecimens such as saliva, blood, or semen, make epigenetic clocks a highly desirable tool in forensic applications (16–20). Finally, the correlation with chronological age that is observable at the systemic level can be identified at the cellular level, with increased cell cycles (i.e. high proliferation, a measure of tumour aggressiveness) being proxied by increased methylage in cells, an issue addressed with mitotic clocks (21,22) and ribosomal clocks, whose conserved structure makes it a trans-species tool for the estimate of age (23).
In this scenario, the EstimAge (Estimate of methylation Age, http://estimage.iac.rm.cnr.it) web server, offers a uniquely broad and curated repository of heterogenous epigenetic clocks and methylages characterized by the broadest number of variables currently available in literature: tissue, age time unit, cell count correction, imputation pre-processing, and technology (high and low throughput). In particular, we carefully selected from the vast peer-reviewed literature 18 clocks whose source code was available, as proxy of thorough testing and hence robustness (additional clocks not fulfilling these criteria can be found in Supplementary Table S1). We categorized them by tissue, age-span and technology, and we integrated them into EstimAge intuitive web server interface. As an additional feature, tissue-specificity for all clocks is reported according to the National Library of Medicine's (MeSH, (https://meshb-prev.nlm.nih.gov/search) controlled vocabulary thesaurus to avoid ambiguity and enhance standardization. Consequently, EstimAge automatically checks and proposes appropriate clocks for the users’ submitted query, including appropriate reference sets for cells count estimation using standard data sets well known in the community (24,25), although custom computations are also possible. Further, the result page includes publication-quality plots and summary statistics that can be either readily employed for final reports, or used as intermediate exploratory steps towards additional bioinformatic analyses. To the best of our knowledge, this is the most comprehensive effort in this direction.
In fact, we identified only two prior hubs of epigenetic clocks: first the popular, pioneering and prolific work of Horvath's group, that offers a handful of multi- and tissue-specific clocks and variants (1,7–9,26), whose exact algorithms are not fully public (https://dnamage.genetics.ucla.edu, DNAMAGE (1)), and second the recent Bioconductor package methylclock (27), that collects 11 clocks, offline and accessible through R, and hence targets a slightly different type of end-users. Overall EstimAge offers 18 clocks that includes and extends the offer beyond the above mentioned hubs, with the exclusion of two clocks that were either not published in peer-reviewed journals or did not provide the source code (see Supplementary Table S1). Differently from the DNAMAGE and methylclock, EstimAge enables three different types of imputation, manages the presence of missing data if no imputation is selected, and includes clocks whose input comes from low-throughput methylation techniques (see Materials and Methods for details). Further, EstimAge provides four types of clocks, to compute as many types of ages: biological human age (in years, y), gestational age (in weeks, w) and cellular age (cell cycles, cc and telomere length in kilobase, kb). As a direct and intuitive statistic for comparison among clocks, the explicit number of CpGs used by each clock to estimate methylage for the submitted data is also returned.
This comprehensive effort may contribute to overcome four major procedural limitations that hamper the full exploitation of this remarkable biomarker.
First, despite the stability of methylation itself, the identification of the CpGs that present the highest correlation with age remains controversial, as it is highly prone to variations. Indeed, given the cyclic structure of the compacted DNA helix within the cells’ nucleus, accession to the DNA molecule for (de)methylation occurs with remarkable correlation among CpGs (a fact exploited, for example, for efficient imputation of methylation values (28,29)), offering indeed a multiplicity of potentially useful, but non standardized, candidate markers for epigenetic clocks. EstimAge enables a broad spectrum of direct inspection of such clocks, automatically selected by the system once the data tissue is provided, or via custom choices defined by the user, enabling exploratory analyses on the correlation and causation of such divergences.
Second, the reporting for models of epigenetic clocks is not only far from unique, but also in lack of a community consensus. This affects robustness and reproducibility of the results. In particular, clocks are available in three major variety of formats of increasing robustness: (i) the list of CpGs, alone or accompanied by regression coefficients is reported, leading to ambiguous reproducibility; (ii) code and model coefficients are reported within the publication, as online tools or in public repositories; (iii) the code is available in broadly used public repositories (GitHub) or standardized formats (Bioconductor packages (30), Python Package Index (PyPI) packages (https://pypi.org)) granting maximum portability and reproducibility. Only the latter are reported in EstimAge, supporting results robustness, while a list of potentially additional clocks is available in Supplementary Table S1.
Third, the data hungry mathematical models adopted in epigenetic clocks can be applied with limited effectiveness to public data, offering a large predominance of blood samples on other tissues. It is therefore still matter of debate whether multi-tissue clocks are sufficient to explain methylage, whether they are indeed blood clocks and whether tissue-specific clocks must be designed in order to address differential ageing organ-wise. Fair comparisons require therefore to assess side-by-side tissue-specific clocks that have been generated by project-specific data sets. EstimAge collection provides a natural place to showcase all existing robust clocks controlling the potentially redundant proliferation of clocks, and enhancing cross fertilization among neighbouring areas. A definitely needed approach as it is, for example, easily observable from the citation patterns between forensic and biomedical published clocks, or mathematical modelling with the non-linear EPM model (31) virtually not cited, i.e. unexplored despite its inception in 2016 within the biomedical area.
Fourth, the theoretical exploration of mathematical models that best describe the relationship between ageing and methylation is to date very limited. Indeed, from its inception in 2013, when the pioneering publications of Horvath (1), Hannum (26) and Wiedner (32) described the concept of epigenomic clocks, methylage computation has been dominated by the elastic net regression model that has become a de facto standard, with very few examples of non-linear mathematical modelling of the phenomenon (33). EstimAge is currently not only the broadest, but also the only repository including also non-linear modelling of methylage.
We expect this service to be further expanded: in addition to continuing patrolling the upcoming literature, Supplementary Table S1 lists several potential additional clocks currently only lacking of public source code or peer-reviewed publication to be added.
MATERIALS AND METHODS
Clock selection
In addition to the work known by our team, a systematic search of the terms ‘methylage’, ‘epigenetic clock’, ‘methylation clock’ was run on Google and Pubmed, backed by cross referencing. Curation with respect to the form in which the clock was provided was done manually (Supplementary Table S1). In order to provide validated models and reproducible results, only clocks published in peer-reviewed articles and whose source code was available were retained in EstimAge. Among the clocks that did not provide the code within their publication, five (8,34–37) were implemented within the methylclock R package (27) and were therefore included in EstimAge as shown in Table 1.
Table 1.
List of EstimAge available clocks characterized by the original publication Name when available, or FirstAuthorLastName followed by PublicationYear; Tissue characterized by MeSH heading; Unique ID, enabling automatic filtering of appropriate clocks; Life phase, i.e. training age span, where Pediatric corresponds to 0–18 years and Adult to 18–80 years; Technology it can compute data from; Cit. reporting the reference. All clocks are loosely grouped by application: multi-tissue for general purpose investigation, tissue-specific for more focused applications; gestational (which output results in weeks, w), forensic, as well as miscellaneous clocks computing acceleration in cell cycles (cc) or kilobases (kb).
| Name | Tissue | Life phase | Technology | Cit. |
|---|---|---|---|---|
| Multi-tissue (years) | ||||
| Zhang19.enpred Zhang19.blupred | Tissues; D014024 | lifespan | HM450, HMEPIC | (13) |
| Horvath13 | Tissues; D014024 | lifespan | HM27, HM450 | (1) |
| EPM | Tissues; D014024 | lifespan | HM450 | (33) |
| PhenoAge | Tissues; D014024 | adult | HM450, HMEPIC | (8) |
| Tissue-specific (years) | ||||
| Hannum13 | Blood; D001769 | adult | HM450 | (26) |
| CorticalClock | Cerebral Cortex; D002540 | lifespan | HM450, HMEPIC | (38) |
| MEAT | Muscle, Skeletal; D018482 | adult | HM27, HM450, HMEPIC | (11) |
| PedBE | Mouth Mucosa; D009061 | pediatric | HM450, HMEPIC | (10) |
| Horvath18 | Mouth Mucosa;D009061 Blood;D001769 Saliva; D012463 Skin; D012867 Endothelial cells; D042783 | lifespan | HM450, HMEPIC | (7) |
| Gestational* (weeks) | ||||
| Knight16 | Fetal Blood; D005312 | foetal | HM27, HM450 | (39) |
| Bohlin16 | Fetal Blood; D005312 | foetal | HM450 | (34) |
| Mayne17 | Placenta; A16.710; D010920 | foetal | HM27, HM450 | (37) |
| Lee19.RPC Lee19.CPC Lee19.rRPC | Placenta; D010920 | foetal | HM450 | (35) |
| Forensic applications (years) | ||||
| ZPiekarska15 | Blood; D001769 | lifespan | Bisulfite conversion+ pyrosequencing assays | (20) |
| FAradas16 | Blood; D001769 | adult | HM450k EpiTYPER | (17) |
| Miscellaneous clocks (cell cycles, kilobases) | ||||
| EPIToc (mitotic clock**) | Neoplasms; D009369 | adult | HM450 | (21) |
| MiAge (mitotic clock**) | Neoplasms; D009369 | adult | HM450 | (22) |
| DNAmTL (telomere Length estimator***) | Blood; D001769 | adult | HM450, HMEPIC | (36) |
Input data
EstimAge requires two comma separated csv files: one containing the DNA methylation beta-values, and the other containing associated metadata, as described below (see also Tutorial as well as examples on the webservice main page).
The beta-values table should contain CpGs on the rows and samples on the columns and its first column, named ‘ID_REF’, should contain the CpGs IDs.
The associated metadata table should have the samples on the rows and contain the following columns. (i) ‘SampleID’, that are the same IDs used in the beta-values table columns; (ii) ‘Age’, containing the samples age (in years, weeks, cell counts or kilobases); (iii) (optional) ‘Ctrl.Case’, specifying if the sample is in the control group (‘Ctrl’) or not (‘Case’). This column is used by EstimAge to generate the outputs discussed in Results. When this information is missing, all samples are considered to be controls; (iv) (optional, only used in EPM) ‘Train.Test’, specifying whether the sample should be in the training or in the test set. This information is required and used only when running EPM. In fact, while all other clocks provide the selected sets of CpGs and the respective model coefficients in the original publications, EPM needs to be trained data set-specifically. It is hence up to the user to group the samples effectively into the training set (which EPM will be trained on) and test set (whose methylage will be predicted based on the trained model). If the ‘Train.Test’ column is missing from the metadata, all samples will be included in the training set when running EPM.
All clocks can be run in EstimAge if the two input tables are available. However, since in the original implementations the required format is not unique (e.g. some clocks expect the presence of the CpGs identifiers column in the beta-values table, others do not), we added, where needed, a formatting step before the computation of the clocks, thus removing the need of ad hoc data formatting from the user.
Data imputation
EstimAge provides three possible algorithms for data imputation: k-nearest neighbours (knn), the mean or methyLImp (28). However, in the absence of users’ preference (i.e. no selection) EstimAge follows the original publication clock's solution to guarantee maximum adherence to the original intent of the authors. It has to be noted that imputation algorithms fail when the values of a CpG are missing in all the samples, therefore NA value are still possible after the imputation step. Additional details are given in the Supplementary Data.
Additional notes on implementation
In addition to the standard outputs statistics and figures (R2, regression and box plots) EstimAge returns the number of clock CpGs (after imputation) that are in fact available in the data set, over the number of CpGs that model the clock (i.e. CpGs available versus total CpGs needed, parameter Used/Total). Since EPM computes a new set of CpGs (whose size is CpGtraining) for each new data set, the ratio will always be 1, i.e. CpGtraining/CpGtraining. For Horvath13 and Knight16, the clock is computed only if all CpGs are present in the data set, therefore the ratio will again always be 1. Shall the clock computation fail, the result will return NA (see Supplementary Data for further details).
Webserver implementation
The front-end for EstimAge follows the Model-View-Controller (MVC) paradigm, thanks to the web2py framework (http://www.web2py.com/). Technical strategies for safety and ease of use include: (i) rename safely the uploaded files to ensure anonymity and prevent directory traversal attacks; (ii) use a local scheduler that runs the jobs sequentially in order to prevent system overload; (iii) keep the uploaded data until the job is completed and then erase them; (iv) allow access to the computation results through an html page. The EstimAge webserver has been tested with Chrome and Firefox (on Linux, MacOS, Window), Edge and Safari (Windows and MacOS).
RESULTS
EstimAge requires: two input tables with CpGs methylation values and matching samples meta information; samples tissue to be selected among MeSH terms; age units (years for human tissues, weeks for gestational clocks, cell cycles for mitoclocks and kilobases for telomere clocks); it offers (methylation-specific) imputation options and automatically recommends the appropriate tissue-specific clocks and reference data sets for cell count estimation (Supplementary Table S2). All options can be skipped (no selection) or overruled (options ‘other’ or ‘none’). EstimAge computes the samples methylages and age accelerations using the selected clocks and options. The results are reported in intuitive tabular formats and plots. In the following, the results obtained from the example provided on the webpage is presented for clarity. The input data and metadata used as example are a subset of GEO Series GSE72776 (see Supplementary Data for details), containing DNA methylation profiles of human blood samples from healthy subjects and subjects with Parkinson's disease (PD). The example includes 40 of the original 84 samples: 20 healthy and 20 PD subjects. Moreover, while in the tutorial all CpG sites are preserved (485 512), the example in this article was further pruned and only the first 299 999 rows were kept, enabling observation of additional variations in the parameters (namely, Used/Total CpGs). In this example no imputation was explicitly run by EstimAge.
First, given the relatively large number of parameters, a summary table containing the clocks selected by the user and all input specifications is provided. It is here that the user can check at a glance also the compliance of her dataset with the models: column ‘Used/Total CpGs’ returns for each computed clock the ratio of CpGs in the data set (following imputation, if any) over the total number of CpGs that model the clock (see Figure 1). The presence of missing data can affect the computation of all the clocks except EPM, which computes on-the-fly the reference CpGs to be used. In particular, missing data may cause a clock to fail in the computation of methylage. This is the case of Horvath13 and ZPierkarska15 in the example in Figure 1, which failed to produce an output due to the high number of missing values in the input data.
Figure 1.

Summary table including all features and appropriateness of the selected clocks based on the number of available CpGs in the data set, over the number of CpGs that model the clock. The high variability of the total number of CpGs included in the clocks (ranging from 3 to 513) and of the number (ranging from 0 to 210) and percentage (ranging from 0 to 100%) of CpGs actually used to compute methylage, is clear in the ‘Used/Total CpGs’ column. ‘TotalCpGs’ is the number of reference CpGs used by the clock, while ‘Used’ is the number of reference CpGs actually available in the input data. As shown in the following results, when no CpG of the clock is present in the data (e.g. in Zpiekarska15), the clock is not usable. The ‘Used/Total CpGs’ of Horvath13 is NA because, besides the availability of some of the CpGs of the clock, the computation of methylage failed (due to the presence of too many missing data, in this example, see Supplementary Information for details.
Follow two tables with computation of methylage, R2 of the regression model of methylage versus age, and clock-specific regression plots (see Figure 2). For all clocks but EPM, the regression model used to relate methylage and age is a linear regression. In EPM, instead, non-linear relationships between methylage and age are allowed and the regression model is of the form methylage = a + b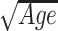 as in (33).
as in (33).
Figure 2.

On the left, table of R2 of the regression of methylage versus age obtained for each selected clock considering all samples (All), only controls (Ctrl), or only cases (Case). DNAmTL outputs results in kb, ZPiekarska15 uses CpGs from low-throughput technologies and therefore returns NA, the current input being from HM450. EPM has used for training the CpGs correlating at 80% with age (see Materials and Methods for details). The model performances are highly variable and depend on both the selected clock and the selected samples (Ctrl, Case or All). For example, EPM fits the Ctrl group (R2 = 0.73) better than the Case group (R2 = 0.22) and, besides using only three CpGs (see Figure 1), its performances on the Ctrl group are better than those of PhenoAge (R2 = 0.32), that was computed with 210 CpGs. The user could for example choose to attempt imputation to improve the performances, or decide to adopt for this specific data set Hannum13, whose R2 is the highest when considering all samples (All). On the right, example of regression plot for the Hannum13 clock, showing age (y) on the x axis and methlyage (y) on the y axis.
In the regression plots, samples are color-coded according to input metadata (Ctrl.Case column, see Methods for details). If control samples (Ctrl) are available, a regression line for such samples is also shown, together with its confidence interval. When the samples age and the methylage units are the same, the scale of the x and y axes are forced to be the same and the bisector is also plotted (black line).
Three standard types of age acceleration are also provided in the form of (i) tables reporting the acceleration of each sample and (ii) box plots, in which, for each clock, age accelerations are drawn separately for cases and controls.
The first age acceleration estimate is obtained as the difference between methylage and age. In this case, age acceleration is meaningful only when the samples age is compatible with the clock's methylage units (y/w/cc/kb). Therefore, EstimAge reports only the clocks for which this condition is true.
The second estimate, Extrinsic Epigenetic Age Acceleration (EEAA), is computed as the residuals from a regression (non-linear for EPM, linear for all other clocks) of methylage on the samples age (40,41). Regression is computed in priority on control samples, but if less than three are available, then all samples are considered. If the total number of samples is less than three, EEAA cannot be computed. The R2 coefficients of each regression model are those reported in the R2 table previously described (Figure 2).
The third estimate, Intrinsic Epigenetic AgeAacceleration (IEAA), is computed as the residuals from a regression (non-linear for EPM, linear for all other clocks) of methylage on the samples age, adjusted for the estimated cell counts (40,41). In this case, the table of the estimated cell counts is also provided (only for cell types whose maximum relative abundance is higher than 10−5). IEAA is computed in priority on control samples. If the number of control samples is equal or less than number of cell types included in the model, then all samples are considered. If the total number of samples is less than the number of cell types, IEAA cannot be computed. A table including the R2 of each regression model is also returned.
Overall, the output tables and plots of EstimAge offer the possibility to qualitatively see and quantitatively inspect and compare the results of different methylage clocks. Starting from the immediate feedback on the appropriateness of the clock to the data set (number of CpGs available versus number of CpGs needed, Figure 1), the R2 tables and the regression plots (Figure 2) highlight the goodness of fit of the models; Finally the age acceleration tables and box plots (Figure 3) allow to verify whether, for example, the case samples are biologically older than the control samples, and to determine which clock captures which amount of such age acceleration; the cell-adjusted age acceleration tables and box-plots (not shown) reveal how age acceleration is different between samples and between clocks, when the complex tissue composition is taken into account (Figure 4). The variability observed in the proposed example (real data) shows how relevant it is to assess this parameter with a variety of approaches and enable easy refinement of the clocks choice.
Figure 3.

EEAA acceleration table, EEAA box plots for clocks in years, and EEAA box plot for telomere length clock, in kb.
Figure 4.

Cells count table for the calculation of IEAA.
DATA AVAILABILITY
EstimAge is an open source webserver available at: http://estimage.iac.rm.cnr.it.
A portion of the data set GSE72776 on GEO has been elaborated and further used as example as described in the Results or on the Website.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Ilaria Gonnella for her prompt technical support and Filippo Castiglione for his generous material support. The authors acknowledge the use of computational resources from the parallel computing cluster of the Open Physics Hub (https://site.unibo.it/openphysicshub/en) at the Physics and Astronomy Department in Bologna.
Contributor Information
Pietro Di Lena, Department of Computer Science and Engineering - DISI, University of Bologna, Bologna 40100, Italy.
Claudia Sala, Department of Physics and Astronomy, University of Bologna, Bologna 40100, Italy.
Christine Nardini, CNR IAC “Mauro Picone”, Rome, Rome 00185, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
iPC Individualized Paediatric Cure European Union's Horizon 2020 research and innovation programme [826121]; H2020-MSCA-ITN [721815 to C.S.]. Funding for open access charge: iPC Individualized Paediatric Cure European Union's Horizon 2020 Research and Innovation Programme.
Conflict of interest statement. None declared.
REFERENCES
- 1. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Greenberg M.V.C., Bourc’his D.. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019; 20:590–607. [DOI] [PubMed] [Google Scholar]
- 3. Sandoval J., Esteller M.. Cancer epigenomics: beyond genomics. Curr. Opin. Genet. Dev. 2012; 22:50–55. [DOI] [PubMed] [Google Scholar]
- 4. Calderón-Larrañaga A., Santoni G., Wang H.X., Welmer A.K., Rizzuto D., Vetrano D.L., Marengoni A., Fratiglioni L.. Rapidly developing multimorbidity and disability in older adults: does social background matter?. J. Intern. Med. 2018; 283:489–499. [DOI] [PubMed] [Google Scholar]
- 5. Horvath S., Raj K.. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018; 19:371–384. [DOI] [PubMed] [Google Scholar]
- 6. Nardini C., Moreau J.-F., Gensous N., Ravaioli F., Garagnani P., Bacalini M.G.. The epigenetics of inflammaging: the contribution of age-related heterochromatin loss and locus-specific remodelling and the modulation by environmental stimuli. Semin. Immunol. 2018; 40:49–60. [DOI] [PubMed] [Google Scholar]
- 7. Horvath S., Oshima J., Martin G.M., Lu A.T., Quach A., Cohen H., Felton S., Matsuyama M., Lowe D., Kabacik S.et al.. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY). 2018; 10:1758–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levine M.E., Lu A.T., Quach A., Chen B.H., Assimes T.L., Bandinelli S., Hou L., Baccarelli A.A., Stewart J.D., Li Y.et al.. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018; 10:573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu A.T., Quach A., Wilson J.G., Reiner A.P., Aviv A., Raj K., Hou L., Baccarelli A.A., Li Y., Stewart J.D.et al.. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY). 2019; 11:303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McEwen L.M., O’Donnell K.J., McGill M.G., Edgar R.D., Jones M.J., MacIsaac J.L., Lin D.T.S., Ramadori K., Morin A., Gladish N.et al.. The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:23329–23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Voisin S., Harvey N.R., Haupt L.M., Griffiths L.R., Ashton K.J., Coffey V.G., Doering T.M., Thompson J.-L.M., Benedict C., Cedernaes J.et al.. An epigenetic clock for human skeletal muscle. J. Cachexia Sarcopenia Muscle. 2020; 11:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Q., Vallerga C.L., Walker R.M., Lin T., Henders A.K., Montgomery G.W., He J., Fan D., Fowdar J., Kennedy M.et al.. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 2019; 11:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stubbs T.M., Bonder M.J., Stark A.-K., Krueger F., Bolland D., Butcher G., Chandra T., Clark S.J., Corcoran A., Eckersley-Maslin M.et al.. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017; 18:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thompson M.J., Chwiałkowska K., Rubbi L., Lusis A.J., Davis R.C., Srivastava A., Korstanje R., Churchill G.A., Horvath S., Pellegrini M.. A multi-tissue full lifespan epigenetic clock for mice. Aging (Albany NY). 2018; 10:2832–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thompson M.J., vonHoldt B., Horvath S., Pellegrini M.. An epigenetic aging clock for dogs and wolves. Aging (Albany NY). 2017; 9:1055–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dias H.C., Cordeiro C., Real F.C., Cunha E., Manco L.. Age estimation based on DNA methylation using blood samples from deceased individuals. J. Forensic Sci. 2020; 65:465–470. [DOI] [PubMed] [Google Scholar]
- 17. Freire-Aradas A., Mosquera-Miguel A., Girón-Santamaría L., Gómez-Tato A., Casares de Cal M., Álvarez-Dios J., Ansede-Bermejoc J., Torres-Español M., Schneiderd P.M., Pospiech E.et al.. Development of a methylation marker set for forensic age estimation using analysis of public methylation data and the Agena Bioscience EpiTYPER system. Forensic Sci. Int. Genet. 2016; 24:65–74. [DOI] [PubMed] [Google Scholar]
- 18. Hong S.R., Jung S.-E., Lee E.H., Shin K.-J., Yang W.I., Lee H.Y.. DNA methylation-based age prediction from saliva: High age predictability by combination of 7 CpG markers. Forensic Sci. Int. Genet. 2017; 29:118–125. [DOI] [PubMed] [Google Scholar]
- 19. Lee H.Y., Jung S.-.E., Oh YuNa, Choi A., Yang W.I., Shin K.-.J.. Epigenetic age signatures in the forensically relevant body fluid of semen: a preliminary study. Forensic Sci. Int. Genet. 2015; 19:28–34. [DOI] [PubMed] [Google Scholar]
- 20. Zbieć-Piekarska R., Spólnicka M., Kupiec T., Parys-Proszek A., Makowska Ż., Pałeczka A., Kucharczyk K., Płoski R., Branicki W.. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015; 17:173–179. [DOI] [PubMed] [Google Scholar]
- 21. Yang Z., Wong A., Kuh D., Paul D.S., Rakyan V.K., Leslie R.D., Zheng S.C., Widschwendter M., Beck S., Teschendorff A.E.. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016; 17:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Youn A., Wang S.. The MiAge calculator: a DNA methylation-based mitotic age calculator of human tissue types. Epigenetics. 2018; 13:192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang M., Lemos B.. Ribosomal DNA harbors an evolutionarily conserved clock of biological aging. Genome Res. 2019; 29:325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aryee M.J., Jaffe A.E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A.P., Hansen K.D., Irizarry R.A.. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014; 30:1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Min J.L., Hemani G., Davey Smith G., Relton C., Suderman M.. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018; 34:3983–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hannum G., Guinney J., Zhao L., Zhang L., Hughes G., Sadda S., Klotzle B., Bibikova M., Fan J.B., Gao Y.et al.. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013; 49:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pelegí-Sisó D., de Prado P., Ronkainen J., Bustamante M., González J.R.. methylclock: a Bioconductor package to estimate DNA methylation age. Bioinformatics. 2020; doi:10.1093/bioinformatics/btaa825. [DOI] [PubMed] [Google Scholar]
- 28. Di Lena P., Sala C., Prodi A., Nardini C.. Missing value estimation methods for DNA methylation data. Bioinformatics. 2019; 35:3786–3793. [DOI] [PubMed] [Google Scholar]
- 29. Lena P.D., Sala C., Prodi A., Nardini C.. Methylation data imputation performances under different representations and missingness patterns. BMC Bioinformatics. 2020; 21:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J.et al.. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Snir S., vonHoldt B.M., Pellegrini M.. A statistical framework to identify deviation from time linearity in epigenetic aging. PLoS Comput. Biol. 2016; 12:e1005183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weidner C.I., Lin Q., Koch C.M., Eisele L., Beier F., Ziegler P., Bauerschlag D.O., Jockel K.H., Erbel R., Muhleisen T.W.et al.. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014; 15:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Farrell C., Snir S., Pellegrini M.. The epigenetic pacemaker - modeling epigenetic states under an evolutionary framework. Bioinformatics. 2020; 36:4662–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bohlin J., Håberg S.E., Magnus P., Reese S.E., Gjessing H.K., Magnus M.C., Parr C.L., Page C.M., London S.J., Nystad W.. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 2016; 17:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee Y., Choufani S., Weksberg R., Wilson S.L., Yuan V., Burt A., Marsit C., Lu A.T., Ritz B., Bohlin J.et al.. Placental epigenetic clocks: estimating gestational age using placental DNA methylation levels. Aging (Albany NY). 2019; 11:4238–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu A.T., Seeboth A., Tsai P.-C., Sun D., Quach A., Reiner A.P., Kooperberg C., Ferrucci L., Hou L., Baccarelli A.A.et al.. DNA methylation-based estimator of telomere length. Aging (Albany NY). 2019; 11:5895–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mayne B.T., Leemaqz S.Y., Smith A.K., Breen J., Roberts C.T., Bianco-Miotto T.. Accelerated placental aging in early onset preeclampsia pregnancies identified by DNA methylation. Epigenomics. 2017; 9:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shireby G.L., Davies J.P., Francis P.T., Burrage J., Walker E.M., Neilson G.W.A., Dahir A., Thomas A.J., Love S., Smith R.G.et al.. Recalibrating the epigenetic clock: implications for assessing biological age in the human cortex. Brain. 2020; 143:3763–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Knight A.K., Craig J.M., Theda C., Bækvad-Hansen M., Bybjerg-Grauholm J., Hansen C.S., Hollegaard M.V., Hougaard D.M., Mortensen P.B., Weinsheimer S.M.et al.. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 2016; 17:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Horvath S., Ritz B.R.. Increased epigenetic age and granulocyte counts in the blood of Parkinson's disease patients. Aging (Albany NY). 2015; 7:1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith J.A., Raisky J., Ratliff S.M., Liu J., Kardia S.L.R., Turner S.T., Mosley T.H., Zhao W.. Intrinsic and extrinsic epigenetic age acceleration are associated with hypertensive target organ damage in older African Americans. BMC Med. Genomics. 2019; 12:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
EstimAge is an open source webserver available at: http://estimage.iac.rm.cnr.it.
A portion of the data set GSE72776 on GEO has been elaborated and further used as example as described in the Results or on the Website.