

Abstract
Muscle atrophy decreases physical function and overall health. Increased glucocorticoid production and/or use of prescription glucocorticoids can significantly induce muscle atrophy by activating the glucocorticoid receptor, thereby transcribing genes that shift protein balance in favor of net protein degradation. Although mechanical overload can blunt glucocorticoid-induced atrophy in young muscle, those affected by glucocorticoids generally have impaired force generation. It is unknown whether contractile force alters the ability of resistance exercise to mitigate glucocorticoid receptor translocation and induce a desirable shift in protein balance when glucocorticoids are elevated. In the present study, mice were subjected to a single bout of unilateral, electrically induced muscle contractions by stimulating the sciatic nerve at 100 Hz or 50 Hz frequencies to elicit high or moderate force contractions of the tibialis anterior, respectively. Dexamethasone was used to activate the glucocorticoid receptor. Dexamethasone increased glucocorticoid signaling, including nuclear translocation of the receptor, but this was mitigated only by high force contractions. The ability of high force contractions to mitigate glucocorticoid receptor translocation coincided with a contraction-mediated increase in muscle protein synthesis, which did not occur in the dexamethasone-treated mice subjected to moderate force contractions. Though moderate force contractions failed to increase protein synthesis following dexamethasone treatment, both high and moderate force contractions blunted the glucocorticoid-mediated increase in LC3 II:I marker of autophagy. Thus, these data show that force generation is important for the ability of resistance exercise to mitigate glucocorticoid receptor translocation and promote a desirable shift in protein balance when glucocorticoids are elevated.
NEW & NOTEWORTHY Glucocorticoids induce significant skeletal muscle atrophy by activating the glucocorticoid receptor. Our work shows that muscle contractile force dictates glucocorticoid receptor nuclear translocation. We also show that blunting nuclear translocation by high force contractions coincides with the ability of muscle to mount an anabolic response characterized by increased muscle protein synthesis. This work further defines the therapeutic parameters of skeletal muscle contractions to blunt glucocorticoid-induced atrophy.
Keywords: autophagy, dexamethasone, muscle atrophy, protein synthesis
INTRODUCTION
Muscle atrophy is a significant health problem that negatively impacts physical function and increases risk of morbidity and mortality (1). Increased glucocorticoid production and/or use of prescription glucocorticoids can significantly contribute to muscle atrophy in various populations including the elderly and those with cancer, heart disease, obesity, and Alzheimer’s disease (2–8). For instance, endogenous glucocorticoid levels increase up to 50% by the 80th yr of life in humans (9), and it is estimated that 10% of all Americans over 40 yr of age use glucocorticoids therapeutically (10–12). Although both endogenous and pharmacological glucocorticoids can induce muscle atrophy, therapies to prevent or minimize this detrimental side effect are sparse.
Glucocorticoids induce muscle atrophy primarily via activation of the glucocorticoid receptor (13). Receptor activation increases the expression of target genes such as regulated in development and DNA damage 1 (REDD1) that promote muscle atrophy by shifting protein balance in favor of net protein degradation (14–20). Thus, therapies that minimize glucocorticoid receptor activity specifically in the skeletal muscle would effectively maintain a desirable protein balance and presumably blunt the muscle atrophy, while still allowing the desired nonmuscle target tissue to be affected by a glucocorticoid therapy. Resistance exercise would be such a therapy as the high contractile forces generated by synergistic ablation mechanical overload effectively blunt glucocorticoid-induced muscle atrophy in young skeletal muscle (21, 22). Though it is unknown exactly how mechanical overload blunts glucocorticoid-induced muscle atrophy, at least two possibilities exist. First, it has long been speculated that mechanical overload blunts glucocorticoid receptor activation (22–24), though this has yet to be shown. Second, mechanical overload likely counteracts the undesirable shift in protein balance that favors net protein degradation by increasing muscle protein synthesis in part by activating the mechanistic target of rapamycin in complex 1 (mTORC1) (25–28).
Although resistance exercise is promising for those with elevated glucocorticoids, the populations with elevated glucocorticoids generally exhibit some degree of impairment in muscle force-generating capacity (e.g., elderly or critically ill) (29–35). For example, aging has been shown to reduce the force-generating capacity of skeletal muscle by ∼25% (30). The inability to produce high levels of contractile force could alter whether resistance exercise, or a clinical modification of resistance exercise (i.e., electrical stimulation), is effective at modulating the cellular processes that would counteract the glucocorticoid-induced muscle atrophy. Indeed, aged humans with a lower muscle force-generating capacity are unable to increase mTORC1 signaling and muscle protein synthesis to the same magnitude observed in young humans following a bout of resistance exercise (36). Therefore, the purpose of this study was to determine whether a clinically relevant decrease in contractile force differentially regulates muscle glucocorticoid receptor activation and the ability to increase muscle protein synthesis when glucocorticoid levels are elevated. We show that a critical level of muscle contractile force is required to blunt glucocorticoid receptor nuclear translocation and increase muscle protein synthesis in the presence of elevated glucocorticoids.
METHODS
Animals
Male C57BL/6Hsd or C57BL/6J mice were purchased from Envigo (Indianapolis, IN) or Jackson Laboratories (Bar Harbor, ME) at 10–12 wk of age and housed in a temperature- (25°C) and light (12 h/12 h light-dark)-controlled environment within the vivarium at Florida State University. Mice were provided standard 5001 rodent chow (LabDiet, St. Louis, MO) and water ad libitum. All mice were acclimated for at least 7 days before experimentation. The Institutional Animal Care and Use Committee of Florida State University approved the animal facilities and all procedures.
Experimental Design
Experiment 1: Extent to which contractile force modulates glucocorticoid receptor translocation.
The experimental design is shown in Fig. 1A. Resistance exercise was simulated by inducing eccentric muscle contractions via unilateral, electrical stimulation of the sciatic nerve under deep isoflurane anesthesia as previously described (37, 38). The left thigh was shaved, and two bipolar electrodes were inserted subcutaneously in a manner that straddled the sciatic nerve. The nerve was stimulated by a ∼2-mAmp current using a constant current stimulator (Aurora Scientific, ON, Canada) causing all muscles of the lower limb to contract. A stimulation frequency of 100 Hz was used to induce high force muscle contractions of the tibialis anterior (TA) whereas a frequency of 50 Hz was used to induce moderate force muscle contractions (n = 7 male mice per group). Both contraction frequencies resulted in complete plantarflexion of the foot. The high force contractions protocol (100-Hz stimulation) is known to elicit an anabolic response in the TA muscle, which if repeated over time, will induce muscle hypertrophy (25, 39, 40). Preliminary in situ force measurement of the posterior crural muscles showed that summation of a 50-Hz stimulation train reached a peak isometric force that was ∼25% lower than the forces generated by a 100-Hz stimulation. This deficit in force production is clinically relevant as it is similar in magnitude to what has been reported with aging (30). Each stimulus, regardless of frequency, consisted of 300 pulses, each 1 ms in duration. The protocol consisted of 10 sets of 6 stimuli. Each stimulus within a set was separated by a 10-s rest period, and between each of the 10 sets was a 60-s rest period. Following contractions, all mice received a subcutaneous injection of warm saline (500 µL) before being returned to their cages where they had free access to water, but not food. Mice recovered for 2 h before being randomized to receive an intraperitoneal injection of 1 mg/kg dexamethasone (Cat. No. D-2915, Sigma-Aldrich, St. Louis, MO) diluted in saline to activate the glucocorticoid receptor or saline only as a control. This dose of dexamethasone corresponds with a moderately high dose in humans (41). The TA muscles were extracted 2 h after the injection (4 h post contractions) under deep isoflurane anesthesia to allow for full activation of the glucocorticoid receptor (16, 42). Muscles were snap-frozen in liquid nitrogen and stored at −80°C until analysis. Tissue harvest occurred between 1300 and 1500 h, which was during the mouse light cycle.
Figure 1.

Experimental timeline for experiment 1 (A) and experiment 2 (B). DEX, dexamethasone; TA, tibialis anterior.
Experiment 2: Ability to increase muscle protein synthesis when the glucocorticoid receptor is active.
The experimental design is shown in Fig. 1B. Mice were randomized to receive an intraperitoneal injection of 1 mg/kg dexamethasone diluted in saline to activate the glucocorticoid receptor or saline only as a control (n = 8–9 male mice per group). Two hours after the injection, which is the sufficient time to fully activate the glucocorticoid receptor (16, 42), all mice were subjected to the same contraction protocols described in experiment 1. Following contractions, all mice received a subcutaneous injection of warm saline (500 µL) before being returned to their cages where they had free access to water, but not food. Thirty minutes before euthanasia, mice were given an intraperitoneal injection of 0.04 µmol/g body wt of puromycin (Cat. No. P-1033, AG Scientific; San Diego, CA) dissolved in phosphate-buffered saline for measurement of protein synthesis via the SUnSET method (43). Four hours following completion of the contractions protocol (6 h post injection), the TA muscles were extracted, snap-frozen in liquid nitrogen, and stored at −80°C until analysis. Tissue harvest occurred between 1300 and 1500 h, which was during the mouse light cycle. We and others have shown that mTORC1 signaling and protein synthesis are maximized at this postcontraction time point (44–46).
Western Blot Analysis
Western blot analysis was conducted as previously described (37). TA muscle protein was extracted by glass-on-glass homogenization in 10 volumes of buffer (10 µL/mg muscle) consisting of 50 mM of HEPES (pH 7.4), 0.1% Triton X-100, 4 mM of EGTA, 10 mM of EDTA, 50 mM of Na4P2O7, 100 mM of β-glycerophosphate, 25 mM of NaF, 5 mM of Na3VO4, and 10 µL/mL of protease inhibitor cocktail (Cat. No. P-8340; Sigma-Aldrich). Muscle extract was centrifuged for 10 min at 10,000 g at 4°C, and the soluble supernatant fraction was quantified via the Bradford method. After quantification, all soluble proteins were diluted to the same concentration in 2× Laemmli buffer. About 20–60 µg of protein was fractionated on 4%–20% Bio-Rad Tris-Glycine Criterion precast gels (Hercules, CA) and transferred to polyvinylidene difluoride membranes. Ponceau-S staining was used to assess the effectiveness of transfer and equal protein loading. Membranes were blocked with 5% nonfat dried milk in Tris-buffered saline + 0.1% Tween 20 (TBST). Membranes were incubated with antibodies diluted in TBST overnight at 4°C. Antibodies against glucocorticoid receptor (Cat. No. 12041), glucocorticoid receptor (Ser211) (Cat. No. 4161), 70 kD ribosomal protein S6 kinase 1 (p70S6K1) Thr389 (Cat. No. 9205), eukaryotic initiation factor 4E binding protein 1 (4EBP1) Ser65 (Cat. No. 9451), microtubule associated protein 1 light chain 3 β (LC3B) (Cat. No. 2775), uncoordinated like kinase 1 (ULK1) Ser757 (Cat. No. 6888), total ULK1 (Cat. No. 8054), and histone H2B (Cat. No. 8135) were obtained from Cell Signaling Technology (Danvers, MA). Antibodies against total p70S6K1 and total 4EBP1 were custom made by Bethyl Laboratories (Montgomery, TX). Antibodies against REDD1 (Cat. No. 10638-1-AP) were obtained from ProteinTech (Chicago, IL). Antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cat. No. sc-32233) were obtained from Santa Cruz Biotechnology (Dallas, TX). Antibodies against puromycin (Cat. No. MABE343) were obtained from Millipore Sigma (Burlington, MA). After incubation with secondary antibodies (Cat. No. A-120-101P or A-90-116P, Bethyl Laboratories), the antigen-antibody complex was visualized by enhanced chemiluminescence using Clarity reagent (Cat. No. 1705061; Bio-Rad) on a Bio-Rad ChemiDoc Touch imaging system. The exposure time for all blots occurred within 10 min. The pixel density for total protein was quantified as the ratio of total protein to a predefined range of the Ponceau-S stain between 25–75 kDa using ImageJ software (National Institutes of Health, Bethesda, MD) or Image Lab Software (Bio-Rad) (47), whereas the pixel density for all other blots was quantified as the phosphorylated-to-total protein ratio using ImageJ software. We have previously shown that our anti-mouse secondary antibody reacts nonspecifically with the presumed endogenous heavy-chain (∼50 kDa) and light-chain (∼25 kDa) IgG within our mouse muscle extracts (48). As such, those bands were excluded from the quantification of puromycin. The antibodies used in this study have been validated by our laboratory (16, 37, 44, 48,49) or through positive control experiments [i.e., increased glucocorticoid receptor (Ser211) phosphorylation following dexamethasone administration].
Cytosolic-Nuclear Fraction Separation
Separation of the nuclear- and cytosolic-enriched fractions was performed as previously described (47, 50). Approximately 20 mg of TA muscle was homogenized using glass on glass in 10 volumes (10 µL/mg tissue) of buffer (hereinafter referred to as buffer A) consisting of 10 mM of NaCl, 1.5 mM of MgCl2, 20 mM of HEPES, 20% glycerol, 0.1% Triton X-100, 1 mM of DTT, and 10 µL/mL of protease inhibitor cocktail (Cat. No. P-8340; Sigma-Aldrich, St. Louis, MO). Samples were centrifuged for 5 min at 2,400 g and 4°C, and the supernatant was collected and saved as the cytosolic-enriched fraction. The cytosolic-enriched fraction was further centrifuged three times, each for 5 min at 3,500 g and 4°C, to pellet and remove any remaining noncytosolic material. The pellet from the initial 2,400 g spin containing the nuclear-enriched fraction was then gently washed three times in 400 µL of buffer A, resuspending the pellet completely during each wash. Between each wash, the nuclear pellet was centrifuged for 5 min at 2,400 g and 4°C. Proteins in the final nuclear pellet were then extracted using glass-on-glass homogenization in 200 µL of the protein extraction buffer described in materials and methods, Western Blot Analysis. The sample was then centrifuged for 10 min at 10,000 g at 4°C. The supernatant was collected and saved as the nuclear-enriched fraction. The protein content of each fraction was quantified by the Bradford method, and equal quantities of protein were diluted into 2× Laemmli buffer for Western blot analysis.
Statistical Analysis
Data are presented as means ± SD. Two-way ANOVA with repeated measures was used to evaluate variables using dexamethasone and contractions as the two factors. Sidak correction for multiple comparisons was used post hoc if an interaction was observed. Because the SUnSET method for protein synthesis does not correct for enrichment of the tracer in the precursor pool (complicating comparison between animals), protein synthesis was presented as a fold change of the contracted muscle relative to the noncontracted muscles within an animal only as previously reported (51–53). Paired Student’s t tests were used to evaluate protein synthesis. All analyses were performed using GraphPad Prism Software (La Jolla, CA). Significance was set at P ≤ 0.05 for all analyses.
RESULTS
Experiment 1: High Force Contractions Mitigate Glucocorticoid Receptor Translocation Whereas Moderate Force Contractions Do Not
Upon activation, the glucocorticoid receptor translocates to the nucleus to alter the transcription of target genes (54, 55). We previously showed that the glucocorticoid-mediated transcription of REDD1 in the skeletal muscle occurred within 2 h of exposure to dexamethasone, and this induction was completely dependent upon a functional glucocorticoid receptor (16). Thus, the glucocorticoid receptor is fully active within 2 h of exposure to the hormone. Accordingly, mice that were previously subjected to a bout of high force muscle contractions (100-Hz stimulation frequency) were injected with vehicle or dexamethasone 2 h before euthanasia to measure glucocorticoid receptor activation. As expected, the quantity of the glucocorticoid receptor in the nuclear-enriched fraction was higher in the noncontracted and contracted muscles in response to dexamethasone (Fig. 2, A–E), but the high force muscle contractions significantly lowered the amount of receptor in the nuclear-enriched fraction (Fig. 2, A–E). Along those lines, the dexamethasone-mediated induction of REDD1, an immediate transcriptional target of the glucocorticoid receptor (16, 42), was lower in the contracted muscle of dexamethasone-treated animals (Fig. 2, F and G). Thus, high force contractions reduce the amount of glucocorticoid receptor in the nuclear fraction when glucocorticoids are elevated.
Figure 2.
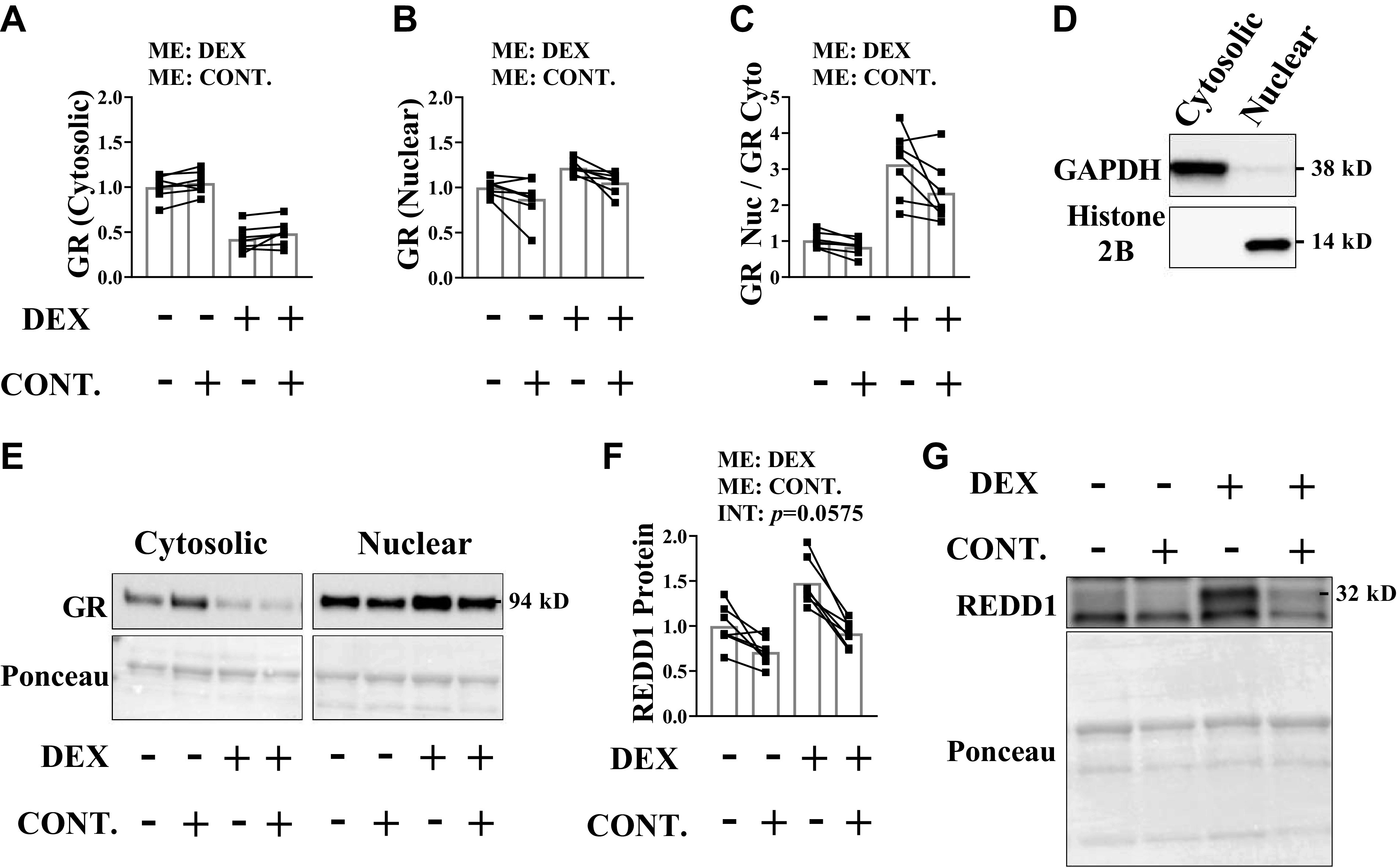
High force contractions mitigate glucocorticoid receptor (GR) translocation. Unilateral, high force muscle contractions were induced in mice before saline or dexamethasone administration. Analysis was conducted on the tibialis anterior (TA) muscle. GR content in the cytosolic-enriched fraction (A), nuclear-enriched fraction (B), and the nuclear to cytosolic ratio (C) was determined by Western blot analysis. D: purity of cytosolic and nuclear fractions. E: representative Western blots. F and G: REDD1 protein content was determined by Western blot analysis. Results are means ± SD. n = 7 male mice per group generated from three experimental replicates. Analyzed by two-way ANOVA with Sidak’s post hoc. Significance at P ≤ 0.05. CONT., contraction; DEX, dexamethasone; INT, interaction; ME, main effect; REDD1, regulated in development and DNA damage 1.
In addition to nuclear translocation, full activation of the glucocorticoid receptor requires phosphorylation (55, 56). Of the residues that undergo phosphorylation, Ser211 is most associated with increased transcriptional activity of the receptor (55, 56). Although the nuclear content of glucocorticoid receptor was lower in contracted muscles (Fig. 2) and this coincided with lower expression of the glucocorticoid receptor target gene, REDD1 (Fig. 2), the receptor that entered the nuclear fraction of the contracted muscle was still likely transcriptionally active as the phosphorylated to total protein ratio of the receptor on the Ser211 residue was not affected by contractions (Fig. 3, A and C). However, the absolute quantity of phosphorylated glucocorticoid receptor on Ser211 in the nuclear fraction of the contracted muscle (not corrected for total receptor content) was lower (Fig. 3, B and C), suggesting that high force contractions did not alter the transcriptional activity of the receptor at this time point, but rather, limited the quantity of transcriptionally active receptor in the nuclear fraction.
Figure 3.

High force contractions do not alter relative phosphorylation of the glucocorticoid receptor (GR) on Ser211 in the nuclear enriched fraction. Unilateral, high force muscle contractions were induced in mice before saline or dexamethasone administration. Analysis was conducted on the tibialis anterior (TA) muscle. A: the phosphorylated to total protein ratio of GR (Ser211) in the nuclear-enriched fraction was determined by Western blot analysis. B: the total content of GR (Ser211) was determined by Western blot analysis. C: representative Western blots. Results are means ± SD. n = 7 male mice per group generated from three experimental replicates. *Significantly different at P ≤ 0.05 by paired Student’s t tests. CONT., contraction; DEX, dexamethasone.
To understand whether a threshold of contractile force needs to be met to blunt glucocorticoid receptor translocation, the same experiment was repeated by stimulating the sciatic nerve at 50 Hz to produce contractions that were ∼25% less forceful than those generated by stimulation at 100 Hz. As per the previous experiment, the quantity of glucocorticoid receptor in the nuclear-enriched fraction was higher in the noncontracted and contracted muscles in response to dexamethasone (Fig. 4, A–E). Unlike what was observed with the high force contractions, the moderate force contractions did not alter receptor quantity in the nuclear fraction (Fig. 4, A–E). Consequently, the glucocorticoid-mediated induction of REDD1 was not affected by the moderate force contractions (Fig. 4, F and G), showing that a threshold of force muscle must be produced to mitigate glucocorticoid receptor translocation.
Figure 4.
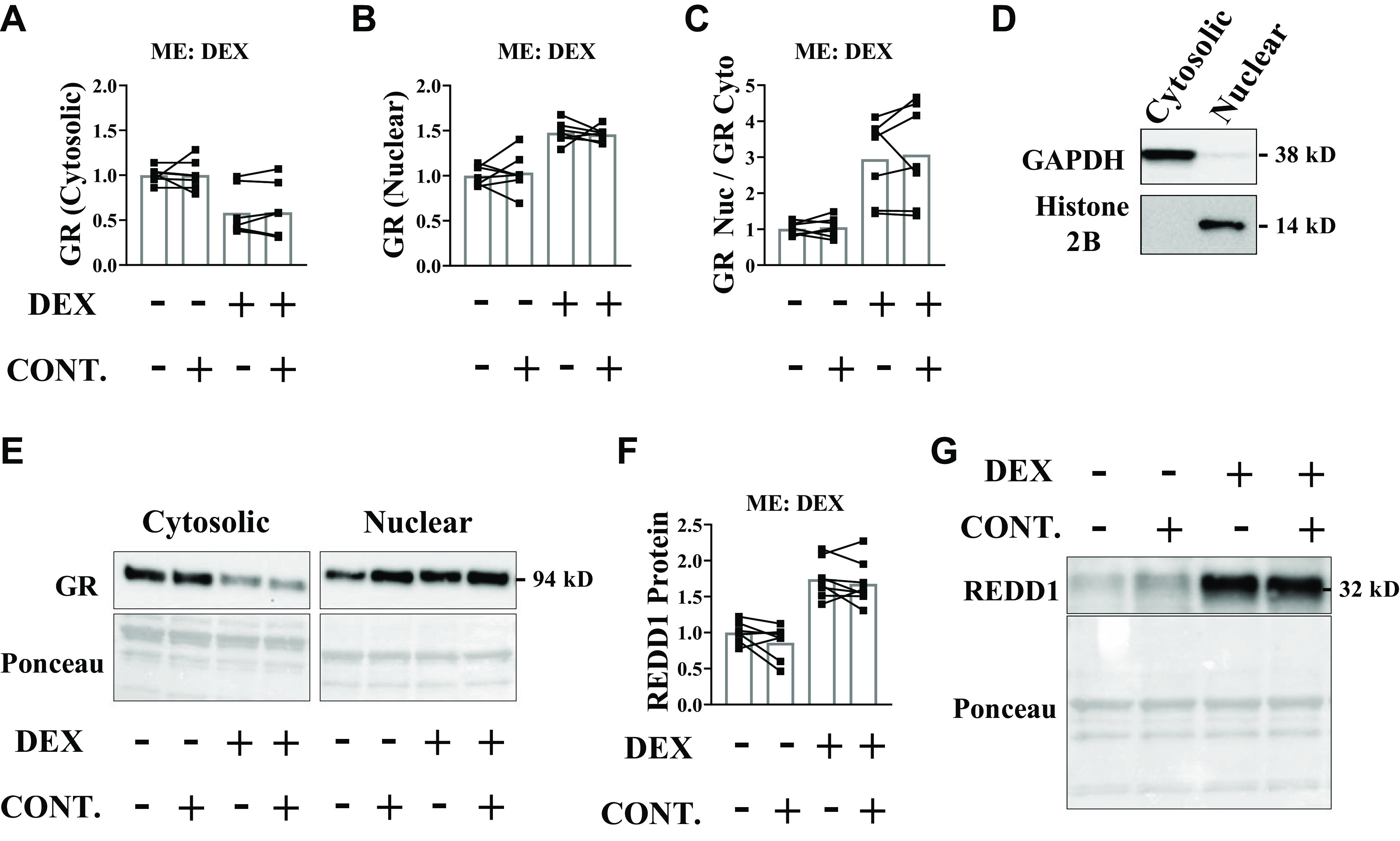
Moderate force contractions fail to mitigate glucocorticoid receptor (GR) translocation. Unilateral, moderate force muscle contractions were induced in mice before saline or dexamethasone administration. Analysis was conducted on the tibialis anterior (TA) muscle. GR content in the cytosolic-enriched fraction (A), nuclear-enriched fraction (B), and the nuclear to cytosolic ratio (C) was determined by Western blot analysis. D: purity of cytosolic and nuclear fractions. E: representative Western blots. F and G: REDD1 protein content was determined by Western blot analysis. Results are means ± SD. n = 7 male mice per group generated from three experimental replicates. Analyzed by two-way ANOVA with Sidak’s post hoc. Significance at P ≤ 0.05. CONT., contraction; DEX, dexamethasone; ME, main effect; REDD1, regulated in development and DNA damage 1.
Experiment 2: The Inability of Moderate Force Muscle Contractions to Mitigate Glucocorticoid Receptor Translocation Coincides with a State of Anabolic Resistance
Resistance exercise shifts protein balance in favor of net protein accretion in large part by increasing rates of muscle protein synthesis (26). The increase in muscle protein synthesis is primarily mediated by activation of mTORC1 (26, 28), which initiates mRNA translation by phosphorylating proteins such as p70S6K1 and 4EBP1. Because glucocorticoids suppress activation of mTORC1 and protein synthesis (18, 19, 57), we tested whether mitigation of glucocorticoid receptor translocation by high force contractions coincided with the ability to increase muscle protein synthesis. Mice were subjected to a bout of muscle contractions 2 h following stimulation of the glucocorticoid receptor, meaning the contractions were initiated when the glucocorticoid receptor was fully active. As expected, REDD1 protein content was increased in response to dexamethasone administration, but high force muscle contractions attenuated this (Fig. 5, A and F), suggesting that glucocorticoid receptor translocation was mitigated. The phosphorylation of both p70S6K1 (Thr389) and 4EBP1 (Ser65) was higher in the contracted muscle of both vehicle- and dexamethasone-treated groups with only a trend (P = 0.09) for dexamethasone to reduce overall 4E-BP1 phosphorylation (Fig. 5, B, C, and F). Consequently, muscle protein synthesis was higher in the contracted muscle of both groups (Fig. 5, D–F).
Figure 5.
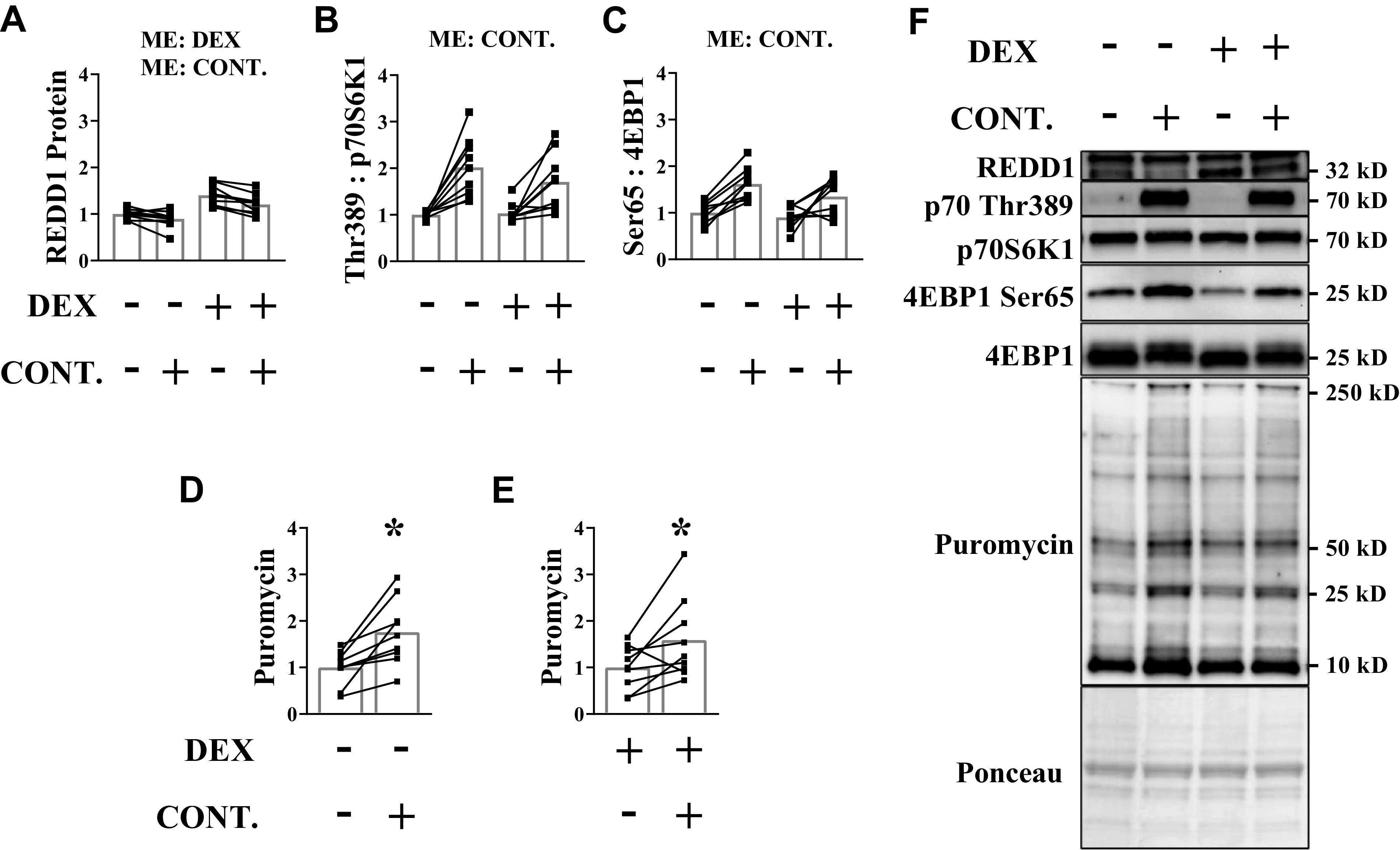
High force contractions can induce an anabolic response when glucocorticoids levels are elevated. Dexamethasone was administered before a bout of unilateral, high force muscle contractions. Analysis was conducted on the tibialis anterior (TA) muscle. A: REDD1 protein content was determined by Western blot analysis. The phosphorylated to total protein ratio of p70S6K1 (Thr389) (B) and 4EBP1 (Ser65) (C) was assessed by Western blot analysis. D and E: puromycin incorporation into muscle protein was assessed in each animal by Western blot analysis. F: representative Western blots. Results are means ± SD. n = 9 male mice per group generated from three experimental replicates. Two-way ANOVA was used to assess REDD1, p70 (Thr389), and 4EBP1 (Ser65). Paired Student’s t test was used to assess puromycin within each condition. *Significantly different than noncontracted muscle within a treatment. CONT., contraction; DEX, dexamethasone; ME, main effect; p70S6K1, 70 kD ribosomal protein S6 kinase 1; REDD1, regulated in development and DNA damage 1eukaryotic initiation factor; 4EBP1, 4E binding protein 1.
Next, we tested whether the inability of moderate force contractions to mitigate glucocorticoid receptor translocation coincided with a change in the contraction-induced anabolic response. Glucocorticoid signaling was effectively increased by dexamethasone as REDD1 protein content was higher in the noncontracted and contracted muscles of treated mice, but this induction was not affected by the moderate force contractions (Fig. 6 A and F). The phosphorylation of both p70S6K1 (Thr389) and 4EBP1 (Ser65) was increased in the contracted muscle of control mice, but this was either absent (p70S6K1) or significantly blunted (4EBP1) in the dexamethasone-treated mice (Fig. 6, B, C, and F). The expected contraction-mediated increase in synthesis was observed in the absence of dexamethasone, but attenuation of contraction-induced mTORC1 signaling following dexamethasone treatment coincided with the failure to significantly increase muscle protein synthesis (Fig. 6, D–F).
Figure 6.
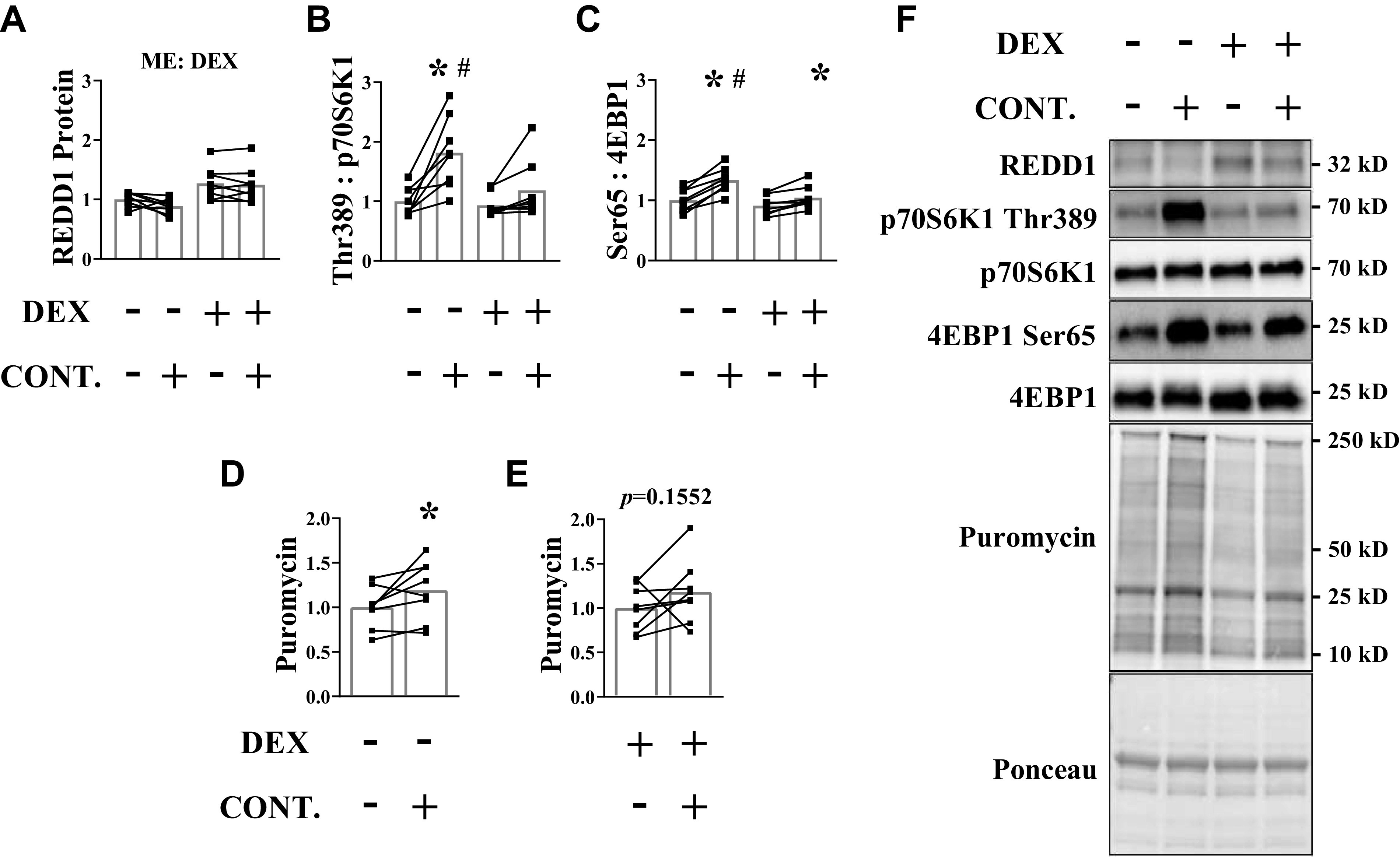
Moderate force contractions do not induce an anabolic response when glucocorticoid levels are elevated. Dexamethasone was administered before a bout of unilateral, moderate force muscle contractions. Analysis was conducted on the tibialis anterior (TA) muscle. A: REDD1 protein content was determined by Western blot analysis. The phosphorylated to total protein ratio of p70 (Thr389) (B) and 4EBP1 (Ser65) (C) was assessed by Western blot analysis. D and E: puromycin incorporation into muscle protein was assessed in each animal by Western blot analysis. F: representative Western blots. Results are means ± SD. n = 8 male mice per group generated from three experimental replicates. Two-way ANOVA was used to assess REDD1, p70S6K1 (Thr389), and 4EBP1 (Ser65). Sidak correction was used post hoc. Paired Student’s t test was used to assess puromycin incorporation in each condition. *Significantly different than noncontracted muscle within a condition. #Significantly different than contracted muscle in the opposite condition. Significant at P ≤ 0.05. CONT., contraction; DEX, dexamethasone; ME, main effect; p70S6K1, 70 kD ribosomal protein S6 kinase 1; REDD1, regulated in development and DNA damage 1eukaryotic initiation factor; 4EBP1, 4E binding protein 1.
High and Moderate Force Contractions Reduce the LC3 II:I Marker of Autophagy following Glucocorticoid Administration
Autophagy is the process of bulk clearance of misfolded/aggregate proteins and dysfunctional/damaged organelles (e.g., mitochondria via mitophagy) via the lysosome (58). Various cellular signals, including mTORC1, regulate the initiation and progression of autophagy in part by phosphorylating ULK1 (58, 59). ULK1 induces autophagy by initiating formation and maturation of a double membrane structure called a phagophore (immature autophagosome) (58). When the phagophore nears maturity, LC3B is lipidated to allow closure of the autophagosome and subsequent delivery of the cargo to the lysosome (58). Thus, the ratio of the lipidated to nonlipidated form of LC3 is used as a surrogate marker of autophagy activation (60). We and others have shown that resistance exercise in humans or mechanical overload in rodents decreases the LC3 II:I ratio in skeletal muscle (37, 38, 48, 61), and this coincides with increased phosphorylation of ULK1 on inhibitory Ser757 residue by mTORC1. Because previous work showed that glucocorticoids increase the LC3 II:I ratio (62), we tested whether contractile force differentially affected the ability of contractions to suppress this marker of autophagy. The LC3 II:I ratio was higher in the muscle following dexamethasone treatment, but this increase was attenuated following high force contractions (Fig. 7, A and C) and modestly attenuated following moderate force contractions (Fig. 7, D and F). Interestingly, the phosphorylation of ULK1 (Ser757) was increased following high and moderate force contractions in the saline groups, but dexamethasone prevented the increase in ULK (Ser757) phosphorylation only in the mice subjected to moderate force contractions (Fig. 7, B, C, E, and F). Our previous work showed that contractions decrease the LC3 II:I ratio even when mTORC1 was inhibited by rapamycin (37), suggesting that the decrease in the LC3 II:I ratio following mechanical stimuli is likely mediated by factors other than mTORC1. These findings suggest that both high and moderate force contractions can blunt the glucocorticoid-mediated increase in the LC3 II:I marker of autophagy.
Figure 7.
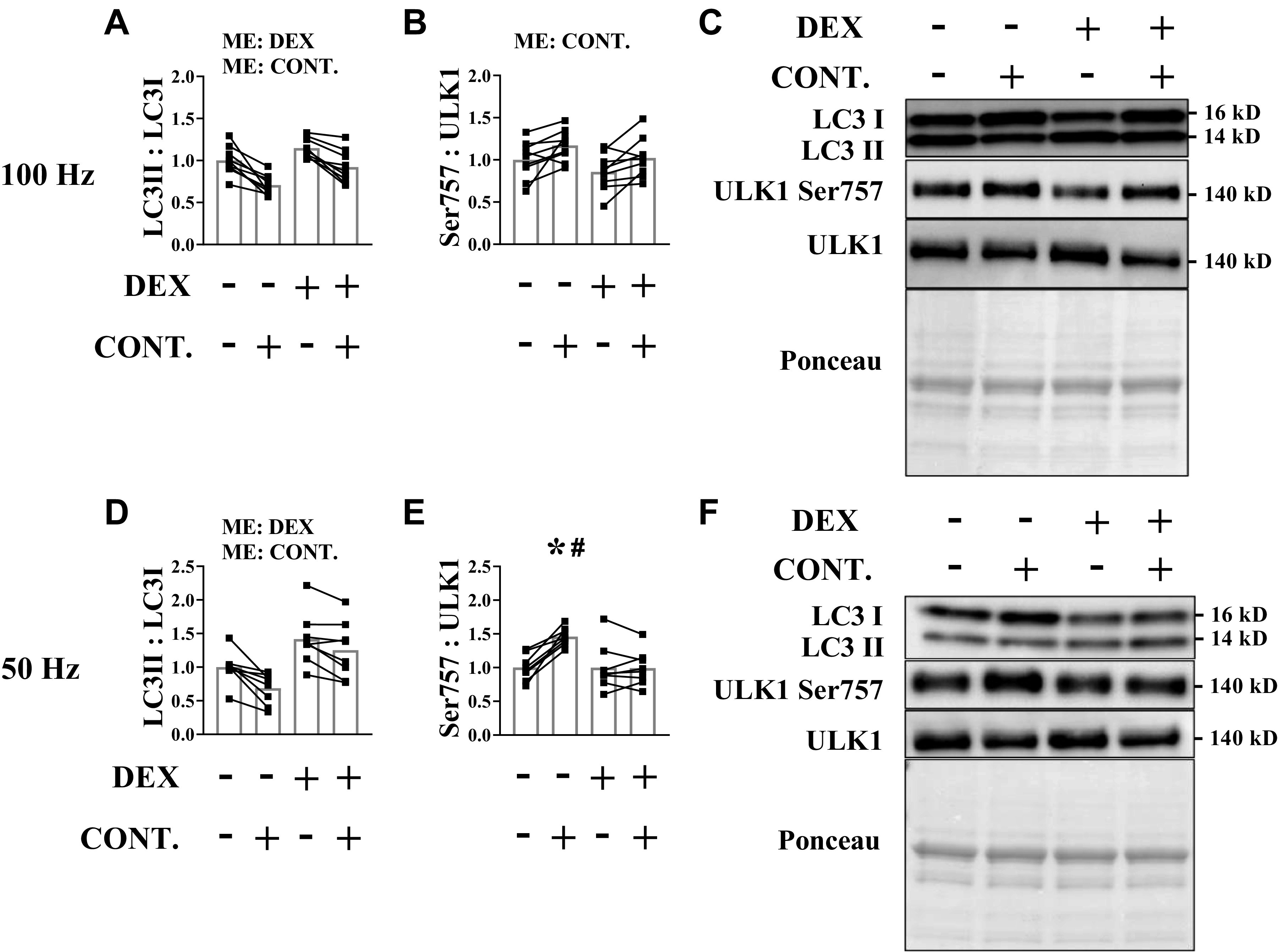
High and moderate force contractions reduce the light chain 3 (LC3) II:I marker of autophagy following glucocorticoid administration. Dexamethasone was administered before unilateral muscle contractions. Analysis was conducted on the tibialis anterior (TA) muscle. A and D: LC3II:I ratio was determined by Western blot analysis. B and E: the phosphorylated to total protein ratio of uncoordinated like kinase 1 (ULK1) (Ser757) was assessed by Western blot analysis. C and F: representative Western blots. Results are means ± SD. n = 8–9 male mice per group generated from three experimental replicates. Two-way ANOVA was used to assess LC3 II:I ratio and ULK1 (Ser757). Sidak correction was used post hoc. Paired Student’s t test used to assess puromycin within each condition*Significantly different than noncontracted muscle within a treatment. #Significantly different than contracted muscle in the opposite condition. Significant at P ≤ 0.05.CONT., contraction; DEX, dexamethasone; ME, main effect.
DISCUSSION
Excess glucocorticoid production and/or therapeutic use of glucocorticoids can promote skeletal muscle atrophy in what has been proposed to be a glucocorticoid receptor dependent manner (13). Although this is known, therapies that mitigate the atrophy by blunting glucocorticoid receptor activation specifically in the muscle are limiting. Previous work has demonstrated that mechanical overload (i.e., synergistic ablation) attenuates glucocorticoid-induced atrophy in young skeletal muscle in what has been proposed to be through mitigation of the glucocorticoid receptor activation (22, 63). Our current data show that high force contractions mitigate glucocorticoid receptor translocation, and therefore, resistance exercise could serve as an immediate way to blunt the undesired effects of glucocorticoid receptor signaling in multiple muscle groups, especially the muscles composed of type II fibers as they are most susceptible to glucocorticoid-induced atrophy (63). This would also benefit those taking therapeutic glucocorticoids as it would blunt receptor activation in the exercised muscle while likely still allowing the glucocorticoid to exert beneficial effects in nonmuscle tissue (e.g., cancerous tumor). However, force-generating capacity is typically compromised in the populations with elevated glucocorticoids (i.e., elderly, diseased) (29–32), and the results of the current study show that a certain threshold of muscle contractile force must be met to blunt receptor translocation in the muscle. The failure to blunt receptor translocation coincided with a state of anabolic resistance, which by extension, would limit the therapeutic efficacy of resistance exercise against glucocorticoid-induced atrophy. Despite this, previous work showed that resistance exercise partially blunted glucocorticoid-induced atrophy in patients with heart transplant (64), which is a population that exhibits a loss of strength (65). This suggests that the benefits of resistance exercise against glucocorticoids may extend beyond simply limiting glucocorticoid receptor translocation. Indeed, our data show that moderate force contractions attenuated the dexamethasone-mediated increase in the LC3 II:I ratio marker of autophagy independent of contractile force. Therefore, resistance exercise may protect against some atrophy regardless of a person’s force-generating capacity in part by maintaining a desirable protein balance through mitigation of autophagy, which likely occurs via nongenomic actions of the hormone (66). In addition, previous work showed that mechanical overload via synergist ablation reduced 20S proteasome activity in skeletal muscle (67). Though our laboratory did not detect any differences in ubiquitylated proteins in our samples (data not shown), it is unknown whether contractions would attenuate a glucocorticoid-mediated increase in ubiquitin proteasome activity at later time points post contraction or following repeated bouts of contractions. Regardless, the greatest preservation of protein balance would likely occur in those that can generate enough force to also limit receptor translocation and mount a subsequent anabolic response (i.e., increase protein synthesis). These results also highlight a potential limitation to the therapeutic ability of electrically induced muscle contractions (68) in those that are either too sick or frail to partake in traditional resistance exercise. In those individuals, a high degree of muscle force would need to be produced by the electrically induced contractions to mitigate glucocorticoid receptor translocation and achieve the greatest prevention of muscle atrophy.
Though high force contractions decreased nuclear translocation of the glucocorticoid receptor, the underlying mechanism(s) remain(s) unknown. The putative glucocorticoid receptor signaling cascade suggests that in the absence of ligand, the glucocorticoid receptor resides in the cytosol, bound to a chaperone heterocomplex of proteins that includes heat shock proteins (69). Upon glucocorticoid binding to the receptor, the glucocorticoid receptor is phosphorylated before translocation to the nucleus where it alters the gene expression signature by interacting with transcriptional machinery (54–56). Our data suggest that the initial events in the cascade following contractions are likely intact up through at least the step of glucocorticoid receptor phosphorylation as phosphorylation of the glucocorticoid receptor (Ser211) in the nuclear-enriched fraction was not affected by high force contractions. Rather, the absolute content of the glucocorticoid receptor was reduced in the nuclear fraction in response to contractions. Therefore, we speculate that the muscle contractions reduce the amount of receptor in the nuclear fraction by acting downstream of the initiating steps of receptor activation by either limiting nuclear receptor import, increasing nuclear receptor export, increasing degradation of the receptor, or a combination of those events. Accordingly, there is evidence that the nucleus is a mechanosensitive organelle that responds to stretch by altering nuclear pore complex permeability, the availability of chromatin, and exportation of transcription factors (70, 71). Our data would seem to contradict previous work which showed that the phosphorylation of the glucocorticoid receptor on Ser211 was decreased in the muscle of humans 10 min after an acute bout of resistance exercise (23), whereas a phosphoproteome analysis showed that phosphorylation of the glucocorticoid receptor on Ser229 was decreased in the TA muscle of mice 30 min following the high force contractions protocol used in the present study (24). Those data imply that the receptor’s transcriptional activity is blunted immediately following contractions, but that this effect wanes in the post exercise recovery. Then, at later time points in the recovery, the transcriptional activity remains blunted by limiting the amount of receptor in the nucleus. It is unknown whether the transcriptional activity of the glucocorticoid receptor is modulated by accumulation of multiple bouts of exercise, which if it is, could also explain why long-term resistance exercise training was able to blunt muscle atrophy in patients with heart failure that exhibit a loss of strength (64, 65). The degree of glucocorticoid receptor transcriptional activity throughout the post exercise recovery period, as well as the effect of multiple exercise bouts (i.e., training duration) on receptor activation, will require additional investigation.
We show here that the inability of moderate force contractions to blunt glucocorticoid receptor activation induces a state of anabolic resistance characterized by the inability to stimulate mTORC1 signaling and muscle protein synthesis. The current model for mTORC1 activation suggests that mTORC1 is localized at the lysosome through the GATOR1/2-RAGULATOR-RAG GTPase protein complex in response to factors such as amino acids (72–75), where it can then be activated by factors such as Ras homolog enriched in brain (Rheb) and/or phosphatidic acid (PA) (45, 76). Mechanical overload is currently thought to induce mTORC1 signaling through activation of Rheb and the generation of PA (77, 78). Though REDD1 suppresses mTORC1 by limiting Rheb activation (79), we have shown that REDD1 blunts, but does not prevent, mechanical overload-induced activation of mTORC1 (44, 48, 49). Thus, the failure of moderate force contractions to activate mTORC1 following dexamethasone administration is likely due to changes in various mTORC1 regulatory factors beyond just a sustained increased in expression of REDD1. Like REDD1, SESTRIN1 is also an immediate transcriptional target of the glucocorticoid receptor in skeletal muscle (42), and recent work suggests that SESTRIN1 limits mTORC1 via regulation of the GATOR1/2 complex and subsequent localization of mTORC1 at the lysosome (80, 81). Induction of Kruppel-like factor 15 (KLF15) by glucocorticoids could also contribute to anabolic resistance by promoting the degradation of branched chain amino acids via induction of the Branched Chain Amino Transferase 2 (BCAT2) (20), which would also limit mTORC1 localization at the lysosome. The role of KLF15 would likely occur at later time points (≥6 h post dexamethasone) as the induction of Bcat2 mRNA in skeletal muscle of dexamethasone-treated mice did not occur until 6 h post treatment (20). Therefore, an early increase in both SESTRIN1 and REDD1 by dexamethasone would synergistically limit activation of mTORC1 and promote anabolic resistance to moderate force contractions by inhibiting both mTORC1 lysosomal localization (i.e., SESTRIN1) and activation of mTORC1 via Rheb (i.e., REDD1). Then, the anabolic resistance observed following moderate force contractions via mTORC1 suppression would likely continue, or be enhanced, at later time points by the induction of genes such as Klf15 and Bcat2.
In conclusion, we show that a threshold of muscle force must be generated to acutely mitigate glucocorticoid receptor nuclear translocation. The failure to mitigate nuclear localization when this threshold of force is not met coincided with a state of anabolic resistance characterized by the inability to stimulate mTORC1 signaling and increase muscle protein synthesis. Such an effect would limit the ability of this exercise modality to maintain a desirable protein balance in the muscle. As the ability to mount an anabolic response is a major factor underlying a modality’s potential to limit muscle atrophy during catabolic states, the capacity of the skeletal muscle to generate force and mitigate glucocorticoid receptor translocation could dictate the extent by which resistance exercise would blunt glucocorticoid-induced atrophy. Future work will need to define the threshold of force necessary to blunt glucocorticoid receptor activation and whether lower doses of glucocorticoids or even different glucocorticoids (i.e., prednisone) also induce anabolic resistance when force generating capacity is limited. It will also need to identify the specific glucocorticoid genes/gene programs that can be altered by contractions and whether a prolonged electrical stimulation protocol is capable of preserving skeletal muscle mass despite the chronic administration of glucocorticoids.
GRANTS
The study is supported by National Institutes of Health (NIH) Grant DK15658 (to S.R.K.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.R.D. and B.S.G. conceived and designed research; K.R.D., M.L.R., and B.S.G. performed experiments; K.R.D., J.L.S., M.L.R., S.R.K., and B.S.G. analyzed data; K.R.D., J.L.S., S.R.K., and B.S.G. interpreted results of experiments; K.R.D. and B.S.G. prepared figures; K.R.D. and B.S.G. drafted manuscript; K.R.D., J.L.S., S.R.K., and B.S.G. edited and revised manuscript; K.R.D., J.L.S., M.L.R., S.R.K., and B.S.G. approved final version of manuscript.
REFERENCES
- 1.Powers SK, Lynch GS, Murphy KT, Reid MB, Zijdewind I. Disease-induced skeletal muscle atrophy and fatigue. Med Sci Sports Exerc 48: 2307–2319, 2016. doi: 10.1249/MSS.0000000000000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burns JM, Johnson DK, Watts A, Swerdlow RH, Brooks WM. Reduced lean mass in early Alzheimer disease and its association with brain atrophy. Arch Neurol 67: 428–433, 2010. doi: 10.1001/archneurol.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry BD, Caldow MK, Brennan-Speranza TC, Sbaraglia M, Jerums G, Garnham A, Wong C, Levinger P, Asrar Ul Haq M, Hare DL, Price SR, Levinger I. Muscle atrophy in patients with Type 2 Diabetes Mellitus: roles of inflammatory pathways, physical activity and exercise. Exerc Immunol Rev 22: 94–109, 2016. [PMC free article] [PubMed] [Google Scholar]
- 4.Roy B, Curtis ME, Fears LS, Nahashon SN, Fentress HM. Molecular mechanisms of obesity-induced osteoporosis and muscle atrophy. Front Physiol 7: 439, 2016. doi: 10.3389/fphys.2016.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki T, Palus S, Springer J. Skeletal muscle wasting in chronic heart failure. ESC Heart Fail 5: 1099–1107, 2018. doi: 10.1002/ehf2.12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vinciguerra M, Musaro A, Rosenthal N. Regulation of muscle atrophy in aging and disease. Adv Exp Med Biol 694: 211–233, 2010. doi: 10.1007/978-1-4419-7002-2_15. [DOI] [PubMed] [Google Scholar]
- 7.White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, Carson JA. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the ApcMin/+ mouse. PLoS One 6: e24650, 2011. doi: 10.1371/journal.pone.0024650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White JP, Puppa MJ, Narsale A, Carson JA. Characterization of the male ApcMin/+ mouse as a hypogonadism model related to cancer cachexia. Biol Open 2: 1346–1353, 2013. doi: 10.1242/bio.20136544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Cauter E, Leproult R, Kupfer DJ. Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. J Clin Endocrinol Metab 81: 2468–2473, 1996. doi: 10.1210/jcem.81.7.8675562. [DOI] [PubMed] [Google Scholar]
- 10.Kavanaugh A, Wells AF. Benefits and risks of low-dose glucocorticoid treatment in the patient with rheumatoid arthritis. Rheumatology (Oxford) 53: 1742–1751, 2014. doi: 10.1093/rheumatology/keu135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lossignol D. A little help from steroids in oncology. J Transl Int Med 4: 52–54, 2016. doi: 10.1515/jtim-2016-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overman RA, Yeh J-Y, Deal CL. Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res (Hoboken) 65: 294–298, 2013. doi: 10.1002/acr.21796. [DOI] [PubMed] [Google Scholar]
- 13.Watson ML, Baehr LM, Reichardt HM, Tuckermann JP, Bodine SC, Furlow JD. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am J Physiol Endocrinol Metab 302: E1210–E1220, 2012. doi: 10.1152/ajpendo.00512.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Britto FA, Begue G, Rossano B, Docquier A, Vernus B, Sar C, Ferry A, Bonnieu A, Ollendorff V, Favier FB. REDD1 deletion prevents dexamethasone-induced skeletal muscle atrophy. Am J Physiol Endocrinol Metab 307: E983–E993, 2014. doi: 10.1152/ajpendo.00234.2014. [DOI] [PubMed] [Google Scholar]
- 15.Favier FB, Costes F, Defour A, Bonnefoy R, Lefai E, Baugé S, Peinnequin A, Benoit H, Freyssenet D. Downregulation of Akt/mammalian target of rapamycin pathway in skeletal muscle is associated with increased REDD1 expression in response to chronic hypoxia. Am J Physiol Regul Integr Comp Physiol 298: R1659–R1666, 2010. doi: 10.1152/ajpregu.00550.2009. [DOI] [PubMed] [Google Scholar]
- 16.Gordon BS, Steiner JL, Rossetti ML, Qiao S, Ellisen LW, Govindarajan SS, Eroshkin AM, Williamson DL, Coen PM. REDD1 induction regulates the skeletal muscle gene expression signature following acute aerobic exercise. Am J Physiol Endocrinol Metab 313: E737–E747, 2017. [Erratum in Am J Physiol Endocrinol Metab 319: E1121, 2020]. doi: 10.1152/ajpendo.00120.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumari R, Willing LB, Jefferson LS, Simpson IA, Kimball SR. REDD1 (regulated in development and DNA damage response 1) expression in skeletal muscle as a surrogate biomarker of the efficiency of glucocorticoid receptor blockade. Biochem Biophys Res Commun 412: 644–647, 2011. doi: 10.1016/j.bbrc.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah OJ, Anthony JC, Kimball SR, Jefferson LS. Glucocorticoids oppose translational control by leucine in skeletal muscle. Am J Physiol Endocrinol Metab 279: E1185–E1190, 2000. doi: 10.1152/ajpendo.2000.279.5.E1185. [DOI] [PubMed] [Google Scholar]
- 19.Shah OJ, Iniguez-Lluhi JA, Romanelli A, Kimball SR, Jefferson LS. The activated glucocorticoid receptor modulates presumptive autoregulation of ribosomal protein S6 protein kinase, p70 S6K. J Biol Chem 277: 2525–2533, 2002. doi: 10.1074/jbc.M105935200. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S, Nakae J, Tagata Y, Nishitani S, Takehana K, Sano M, Fukuda K, Suematsu M, Morimoto C, Tanaka H. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 13: 170–182, 2011. doi: 10.1016/j.cmet.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Gardiner PF, Hibl B, Simpson DR, Roy R, Edgerton VR. Effects of a mild weight-lifting program on the progress of glucocorticoid-induced atrophy in rat hindlimb muscles. Pflugers Arch 385: 147–153, 1980. doi: 10.1007/BF00588695. [DOI] [PubMed] [Google Scholar]
- 22.Kurowski TT, Chatterton RT Jr, Hickson RC. Countereffects of compensatory overload and glucocorticoids in skeletal muscle: androgen and glucocorticoid cytosol receptor binding. J Steroid Biochem 21: 137–145, 1984. doi: 10.1016/0022-4731(84)90374-1. [DOI] [PubMed] [Google Scholar]
- 23.Nicoll JX, Fry AC, Mosier EM, Olsen LA, Sontag SA. MAPK, androgen, and glucocorticoid receptor phosphorylation following high-frequency resistance exercise non-functional overreaching. Eur J Appl Physiol 119: 2237–2253, 2019. doi: 10.1007/s00421-019-04200-y. [DOI] [PubMed] [Google Scholar]
- 24.Potts GK, McNally RM, Blanco R, You J-S, Hebert AS, Westphall MS, Coon JJ, Hornberger TA. A map of the phosphoproteomic alterations that occur after a bout of maximal-intensity contractions. J Physiol 595: 5209–5226, 2017. doi: 10.1113/JP273904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baar K, Esser K. Phosphorylation of p70S6k correlates with increased skeletal muscle mass following resistance exercise. Am J Physiol 276: C120–C127, 1999. doi: 10.1152/ajpcell.1999.276.1.C120. [DOI] [PubMed] [Google Scholar]
- 26.Drummond MJ, Fry CS, Glynn EL, Dreyer HC, Dhanani S, Timmerman KL, Volpi E, Rasmussen BB. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. J Physiol 587: 1535–1546, 2009. doi: 10.1113/jphysiol.2008.163816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodman CA, Frey JW, Mabrey DM, Jacobs BL, Lincoln HC, You JS, Hornberger TA. The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J Physiol 589: 5485–5501, 2011. doi: 10.1113/jphysiol.2011.218255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogasawara R, Fujita S, Hornberger TA, Kitaoka Y, Makanae Y, Nakazato K, Naokata I. The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Sci Rep 6: 31142, 2016. doi: 10.1038/srep31142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alamdari N, Toraldo G, Aversa Z, Smith I, Castillero E, Renaud G, Qaisar R, Larsson L, Jasuja R, Hasselgren PO. Loss of muscle strength during sepsis is in part regulated by glucocorticoids and is associated with reduced muscle fiber stiffness. Am J Physiol Regul Integr Comp Physiol 303: R1090–R1099, 2012. doi: 10.1152/ajpregu.00636.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 404: 71–82, 1988. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faulkner JA, Brooks SV, Zerba E. Muscle atrophy and weakness with aging: contraction-induced injury as an underlying mechanism. J Gerontol A Biol Sci Med Sci 50: 124–129, 1995. doi: 10.1093/gerona/50a.special_issue.124. [DOI] [PubMed] [Google Scholar]
- 32.Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, Simonsick EM, Tylavsky FA, Visser M, Newman AB. The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 61: 1059–1064, 2006. doi: 10.1093/gerona/61.10.1059. [DOI] [PubMed] [Google Scholar]
- 33.Moran AL, Nelson SA, Landisch RM, Warren GL, Lowe DA. Estradiol replacement reverses ovariectomy-induced muscle contractile and myosin dysfunction in mature female mice. J Appl Physiol (1985) 102: 1387–1393, 2007. doi: 10.1152/japplphysiol.01305.2006. [DOI] [PubMed] [Google Scholar]
- 34.Roberts BM, Ahn B, Smuder AJ, Al-Rajhi M, Gill LC, Beharry AW, Powers SK, Fuller DD, Ferreira LF, Judge AR. Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J 27: 2600–2610, 2013. doi: 10.1096/fj.12-222844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.VanderVeen BN, Hardee JP, Fix DK, Carson JA. Skeletal muscle function during the progression of cancer cachexia in the male ApcMin/+ mice. J Appl Physiol (1985) 124: 684–695, 2018. doi: 10.1152/japplphysiol.00897.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fry CS, Drummond MJ, Glynn EL, Dickinson JM, Gundermann DM, Timmerman KL, Walker DK, Dhanani S, Volpi E, Rasmussen BB. Aging impairs contraction-induced human skeletal muscle mTORC1 signaling and protein synthesis. Skelet Muscle 1: 11, 2011. doi: 10.1186/2044-5040-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gordon BS, Rossetti ML, Eroshkin AM. Arrdc2 and Arrdc3 elicit divergent changes in gene expression in skeletal muscle following anabolic and catabolic stimuli. Physiol Genomics 51: 208–217, 2019. doi: 10.1152/physiolgenomics.00007.2019. [DOI] [PubMed] [Google Scholar]
- 38.Steiner JL, Fukuda DH, Rossetti ML, Hoffman JR, Gordon BS. Castration alters protein balance after high-frequency muscle contraction. J Appl Physiol (1985) 122: 264–272, 2017. doi: 10.1152/japplphysiol.00740.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hardee JP, Fix DK, Koh H-J, Wang X, Goldsmith EC, Carson JA. Repeated eccentric contractions positively regulate muscle oxidative metabolism and protein synthesis during cancer cachexia in mice. J Appl Physiol (1985) 128: 1666–1676, 2020. doi: 10.1152/japplphysiol.00908.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardee JP, Mangum JE, Gao S, Sato S, Hetzler KL, Puppa MJ, Fix DK, Carson JA. Eccentric contraction-induced myofiber growth in tumor-bearing mice. J Appl Physiol (1985) 120: 29–37, 2016. doi: 10.1152/japplphysiol.00416.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gensler LS. Glucocorticoids: complications to anticipate and prevent. Neurohospitalist 3: 92–97, 2013. doi: 10.1177/1941874412458678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang J-C. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci USA 109: 11160–11165, 2012. doi: 10.1073/pnas.1111334109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, Hornberger TA. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J 25: 1028–1039, 2011. doi: 10.1096/fj.10-168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gordon BS, Steiner JL, Lang CH, Jefferson LS, Kimball SR. Reduced REDD1 expression contributes to activation of mTORC1 following electrically induced muscle contraction. Am J Physiol Endocrinol Metab 307: E703–E711, 2014. doi: 10.1152/ajpendo.00250.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Neil TK, Duffy LR, Frey JW, Hornberger TA. The role of phosphoinositide 3-kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J Physiol 587: 3691–3701, 2009. doi: 10.1113/jphysiol.2009.173609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steiner JL, Lang CH. Alcohol impairs skeletal muscle protein synthesis and mTOR signaling in a time-dependent manner following electrically stimulated muscle contraction. J Appl Physiol (1985) 117: 1170–1179, 2014. doi: 10.1152/japplphysiol.00180.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossetti ML, Tomko RJ Jr, Gordon BS. Androgen depletion alters the diurnal patterns to signals that regulate autophagy in the limb skeletal muscle. Mol Cell Biochem 476: 959–969, 2021. doi: 10.1007/s11010-020-03963-9. [DOI] [PubMed] [Google Scholar]
- 48.Gordon BS, Liu C, Steiner JL, Nader GA, Jefferson LS, Kimball SR. Loss of REDD1 augments the rate of the overload-induced increase in muscle mass. Am J Physiol Regul Integr Comp Physiol 311: R545–R557, 2016. doi: 10.1152/ajpregu.00159.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon BS, Williamson DL, Lang CH, Jefferson LS, Kimball SR. Nutrient-induced stimulation of protein synthesis in mouse skeletal muscle is limited by the mTORC1 repressor REDD1. J Nutr 145: 708–713, 2015. doi: 10.3945/jn.114.207621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rossetti ML, Esser KA, Lee C, Tomko RJ Jr, Eroshkin AM, Gordon BS. Disruptions to the limb muscle core molecular clock coincide with changes in mitochondrial quality control following androgen depletion. Am J Physiol Endocrinol Metab 317: E631–E645, 2019. doi: 10.1152/ajpendo.00177.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baehr LM, West DWD, Marshall AG, Marcotte GR, Baar K, Bodine SC. Muscle-specific and age-related changes in protein synthesis and protein degradation in response to hindlimb unloading in rats. J Appl Physiol (1985) 122: 1336–1350, 2017. doi: 10.1152/japplphysiol.00703.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.West DW, Baehr LM, Marcotte GR, Chason CM, Tolento L, Gomes AV, Bodine SC, Baar K. Acute resistance exercise activates rapamycin-sensitive and -insensitive mechanisms that control translational activity and capacity in skeletal muscle. J Physiol 594: 453–468, 2016. doi: 10.1113/JP271365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.West DWD, Marcotte GR, Chason CM, Juo N, Baehr LM, Bodine SC, Baar K. Normal ribosomal biogenesis but shortened protein synthetic response to acute eccentric resistance exercise in old skeletal muscle. Front Physiol 9: 1915, 2018. doi: 10.3389/fphys.2018.01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Echeverría PC, Mazaira G, Erlejman A, Gomez-Sanchez C, Piwien Pilipuk G, Galigniana MD. Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin beta. Mol Cell Biol 29: 4788–4797, 2009. doi: 10.1128/MCB.00649-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vandevyver S, Dejager L, Libert C. On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic 13: 364–374, 2012. doi: 10.1111/j.1600-0854.2011.01288.x. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation "code" of the glucocorticoid receptor in vivo. J Biol Chem 277: 26573–26580, 2002. doi: 10.1074/jbc.M110530200. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem 281: 39128–39134, 2006. doi: 10.1074/jbc.M610023200. [DOI] [PubMed] [Google Scholar]
- 58.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364, 2018. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 59.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141, 2011. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ju J-S, Varadhachary AS, Miller SE, Weihl CC. Quantitation of "autophagic flux" in mature skeletal muscle. Autophagy 6: 929–935, 2010. doi: 10.4161/auto.6.7.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fry CS, Drummond MJ, Glynn EL, Dickinson JM, Gundermann DM, Timmerman KL, Walker DK, Volpi E, Rasmussen BB. Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults. J Gerontol A Biol Sci Med Sci 68: 599–607, 2013. doi: 10.1093/gerona/gls209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Troncoso R, Paredes F, Parra V, Gatica D, Vásquez-Trincado C, Quiroga C, Bravo-Sagua R, López-Crisosto C, Rodriguez AE, Oyarzún AP, Kroemer G, Lavandero S. Dexamethasone-induced autophagy mediates muscle atrophy through mitochondrial clearance. Cell Cycle 13: 2281–2295, 2014. doi: 10.4161/cc.29272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goldberg AL, Goodman HM. Relationship between cortisone and muscle work in determining muscle size. J Physiol 200: 667–675, 1969. doi: 10.1113/jphysiol.1969.sp008715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Braith RW, Welsch MA, Mills RM Jr, Keller JW, Pollock ML. Resistance exercise prevents glucocorticoid-induced myopathy in heart transplant recipients. Med Sci Sports Exerc 30: 483–489, 1998. doi: 10.1097/00005768-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 65.Braith RW, Limacher MC, Leggett SH, Pollock ML. Skeletal muscle strength in heart transplant recipients. J Heart Lung Transplant 12: 1018–1023, 1993. [PubMed] [Google Scholar]
- 66.Ramamoorthy S, Cidlowski JA. Corticosteroids: mechanisms of action in health and disease. Rheum Dis Clin North Am 42: 15–31, 2016. doi: 10.1016/j.rdc.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roberts MD, Mobley CB, Vann CG, Haun CT, Schoenfeld BJ, Young KC, Kavazis AN. Synergist ablation-induced hypertrophy occurs more rapidly in the plantaris than soleus muscle in rats due to different molecular mechanisms. Am J Physiol Regul Integr Comp Physiol 318: R360–R368, 2020. doi: 10.1152/ajpregu.00304.2019. [DOI] [PubMed] [Google Scholar]
- 68.Nussbaum EL, Houghton P, Anthony J, Rennie S, Shay BL, Hoens AM. Neuromuscular electrical stimulation for treatment of muscle impairment: critical review and recommendations for clinical practice. Physiother Can 69: 1–76, 2017. doi: 10.3138/ptc.2015-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bresnick EH, Dalman FC, Sanchez ER, Pratt WB. Evidence that the 90-kDa heat shock protein is necessary for the steroid binding conformation of the L cell glucocorticoid receptor. J Biol Chem 264: 4992–4997, 1989. doi: 10.1016/S0021-9258(18)83689-4. [DOI] [PubMed] [Google Scholar]
- 70.Burkholder TJ. Mechanotransduction in skeletal muscle. Front Biosci 12: 174–191, 2007. doi: 10.2741/2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Donnaloja F, Jacchetti E, Soncini M, Raimondi MT. Mechanosensing at the nuclear envelope by nuclear pore complex stretch activation and its effect in physiology and pathology. Front Physiol 10: 896, 2019. doi: 10.3389/fphys.2019.00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340: 1100–1106, 2013. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150: 1196–1208, 2012. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan K-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10: 935–945, 2008. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501, 2008. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dennis MD, Baum JI, Kimball SR, Jefferson LS. Mechanisms involved in the coordinate regulation of mTORC1 by insulin and amino acids. J Biol Chem 286: 8287–8296, 2011. doi: 10.1074/jbc.M110.209171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jacobs BL, McNally RM, Kim K-J, Blanco R, Privett RE, You J-S, Hornberger TA. Identification of mechanically regulated phosphorylation sites on tuberin (TSC2) that control mechanistic target of rapamycin (mTOR) signaling. J Biol Chem 292: 6987–6997, 2017. doi: 10.1074/jbc.M117.777805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.You JS, Frey JW, Hornberger TA. Mechanical stimulation induces mTOR signaling via an ERK-independent mechanism: implications for a direct activation of mTOR by phosphatidic acid. PLoS One 7: e47258, 2012. doi: 10.1371/journal.pone.0047258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dennis MD, Coleman CS, Berg A, Jefferson LS, Kimball SR. REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci Signal 7: ra68, 2014. [Erratum in Sci Signal 8: er4, 2015]. doi: 10.1126/scisignal.2005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep 9: 1–8, 2014. doi: 10.1016/j.celrep.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu D, Shimkus KL, Lacko HA, Kutzler L, Jefferson LS, Kimball SR. Evidence for a role for Sestrin1 in mediating leucine-induced activation of mTORC1 in skeletal muscle. Am J Physiol Endocrinol Metab 316: E817–E828, 2019. doi: 10.1152/ajpendo.00522.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]