

Abstract
Chronic hypercapnia (CH) is a hallmark of respiratory diseases such as chronic obstructive pulmonary disease. In such patients, mechanical ventilation is often used to restore normal blood-gas homeostasis. However, little is known regarding physiological changes and neuroplasticity within physiological control networks after termination of CH. Utilizing our goat model of increased inspired CO2-induced CH, we determined whether termination of CH elicits time-dependent physiological and neurochemical changes within brain stem sites of physiological control. Thirty days of CH increased (+15 mmHg) and steady-state ventilation (SS V̇i; 283% of control). Within 24 h after terminating CH, SS V̇i, blood gases, arterial [H+], and most physiological measurements returned to control. However, the acute ventilatory chemoreflex (ΔV̇i/Δ[H+]) was greater than control, and measured SS V̇i exceeded ventilation predicted by arterial [H+] and ΔV̇i/Δ[H+]. Potentially contributing to these differences were increased excitatory neuromodulators serotonin and norepinephrine in the nucleus tractus solitarius, which contrasts with minimal changes observed at 24 h and 30 days of hypercapnia. Similarly, there were minimal changes found in markers of neuroinflammation and glutamate receptor-dependent neuroplasticity upon termination of CH, which were previously increased following 24 h of hypercapnia. Thus, following termination of CH: 1) ventilatory, renal, and other physiological functions rapidly return to control; 2) neuroplasticity within the ventilatory control network may contribute to the difference between measured vs. predicted ventilation and the elevation in the acute ventilatory [H+] chemoreflex; and 3) neuroplasticity is fundamentally distinct from acclimatization to CH.
NEW & NOTEWORTHY In healthy adult goats, steady-state ventilation and most physiological measures return to control within 24 h after termination of chronic hypercapnia (CH). However, the acute [H+] chemoreflex is increased, and measured ventilation exceeds predicted ventilation. At 24 h of recovery, excitatory neuromodulators are above control, but other measured markers of neuroplasticity are unchanged from control. Our data suggest that CH elicits persistent physiological and neurochemical changes for up to 24 h after termination of CH.
Keywords: deacclimatization, hypercapnia, neuromodulation, ventilatory control
INTRODUCTION
Hypercapnia and hypoxemia are hallmarks of respiratory-related diseases such as chronic obstructive pulmonary disease (COPD). Individuals with chronic hypercapnia [partial pressure of arterial CO2 ( > 45 mmHg] have poor prognoses and are at a greater risk for acute “attacks,” known as acute-on-chronic exacerbation (pneumonia, RDS, etc.) that result in life-threatening severe hypercapnia and cardiorespiratory dysfunction (1, 2). During these severe respiratory events, can be dramatically elevated well beyond the already chronically elevated levels of CO2, which further increases the risk for recurrent acute exacerbation of COPD (AECOPD) events, CO2 narcosis, and even death (3, 4). A common therapeutic approach for these individuals is mechanical ventilation to abruptly correct the hypercapnia and hypoxemia. However, it is heavily debated whether hypercapnia should be completely reversed or maintained at low levels that may prevent pulmonary injury (permissive hypercapnia). Some clinical reports suggest abrupt termination of chronically elevated in COPD patients may lead to extreme deviations in acid-base balance, electrolyte homeostasis, and a worsening of the patient’s health (5–7). However, the physiological and neurochemical effects of abrupt termination of hypercapnia on the ventilatory control network are largely understudied.
Limited work on physiological recovery from chronic hypercapnia (CH) (8–10) or chronic hypoxia (11, 12) suggest that, much like the acclimatization process (the process that induces physiological adaptations to chronic changes in and/or ), deacclimatization appears to be a time-dependent process. However, physiological deacclimatization from hypercapnia appears to be a more rapid process than from hypoxia. For example, Jennings and Chen reported a return of ventilation and to baseline within hours following return to normocapnia from CH in dogs exposed to 5% CO2 for 14 days (8). In contrast, the acute ventilatory response to hypoxia in humans remained increased for up to 45 days following a 45-day sojourn at 3,100 m altitude and return to sea level (12). This finding led to the conclusion that adaptations within the ventilatory control network are evident for a prolonged period following restoration of normoxia (12). In addition, available data during acute deacclimatization from CH and hypoxia suggest that the changes observed in arterial and cerebrospinal fluid CO2/[H+] (the major chemical stimuli to breathing) cannot fully explain ventilatory deacclimatization. These data led to the conclusion that correlations between ventilation and arterial/cerebrospinal fluid (CSF) chemical stimuli are “functions rather than determinants of accompanying ventilatory deacclimatization from hypoxia” (11, 13). Studies on the recovery from hypercapnia report a lack of hypoventilation despite arterial alkalosis as observed in both humans (14) and dogs (8), further suggesting that, in the recovery period, levels of arterial or CSF CO2 and/or [H+] do not account for the observed change in ventilation (8, 14). Thus, the data suggest that chronic hypercapnia and/or hypoxia fundamentally alter the neural respiratory control network during the acclimatization and/or deacclimatization process. However, the determinants or factors that contribute to altered ventilatory control mechanisms, particularly during a rapid deacclimatization, have not yet been determined.
We recently established and characterized a large-animal model (adult female goats) of CH induced by 30 days of inhaling 6% inspired CO2 (InCO2) (15). Two of the major findings was that CH led to 1) a sustained, steady-state, minute ventilation (SS V̇i) that exceeded levels predicted based on traditional [H+] stimulus to breathe, and 2) independent adaptation of SS V̇i and the acute ventilatory [H+] chemoreflex (15). To investigate potential substrates that may account for these findings, we studied changes in markers of neuroplasticity. We define neuroplasticity herein as “involvement of time-dependent structural and/or functional changes, arising from cellular and/or molecular mechanisms, that are induced by environmental stimuli and affect physiological responses to the same stimuli” (16). We previously found site- and time-dependent changes in glutamate receptor expression/phosphorylation, dysregulation of tryptophan metabolism, and neuroinflammation within key sites of cardiorespiratory control that may contribute to the multiple physiological adaptations in the early and late exposure of CH (17, 18). However, there were no detectable changes in brain stem neuromodulators, such as serotonin (5-HT) or norepinephrine, within these same brain stem sites. Accordingly, the objective of the present study was to test the hypothesis that abrupt termination of CH leads to time-dependent recovery of ventilatory and integrated physiological control mechanisms and changes in markers of neuroplasticity (glutamate receptor expression/phosphorylation, neuroinflammation, and tryptophan metabolism) without changes in excitatory neuromodulators.
MATERIALS AND METHODS
Ethical Approval
Study protocols were reviewed and approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee, which complies with the Public Health Services Policy on Humane Care and Use of Laboratory Animals (PHS Policy) and by extension all applicable provisions of the Animal Welfare Act and other Federal statutes and regulations relating to animals. The Medical College of Wisconsin has remained continuously accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALACi) since 1968 (AALAC no. 000129). The investigators understand the ethical principles under which this journal operates, and the work herein complies with the journal’s animal ethics checklist.
Study Population and Conditions
Nine female adult goats weighing ∼40–50 kg, independent of our acclimatization study (15), were used in this study. All goats were reared and transported under conditions specified by the US Dept. of Agriculture. Goats were chronically housed in environmental chambers as previously described (15). Temperature and humidity in the chambers were controlled and maintained within normal limits, and photoperiods were fixed between 0600 and 1800 daily. Except during periods of study, food and water was provided ad libitum.
Surgery
The goats underwent surgery to subcutaneously elevate carotid arteries to allow for catherization for serial blood sampling and recording of heart rate and blood pressure. Goats were anesthetized with ketamine (iv), intubated, and mechanically ventilated with 2% isoflurane in 100% O2. The carotid arteries were then isolated from the vagus nerve and superficially elevated above the muscle and sutured in place underneath the skin. Following surgery, for analgesia and to minimize infections, goats were administered flunixin meglumine (2 mg/kg im) once daily for 3 days and ceftiofur sodium (4 mg/kg im) once daily for 7 days. After a 2-wk recovery period, carotid arteries were unilaterally catheterized via an indwelling catheter. Catheters were flushed daily with heparinized saline (0.1% heparin in saline) and pure heparin.
Physiological Measurements
Measurements were adopted as described by Burgraff et al. (15). During physiological studies, a mask was taped to the snout of the goat. A one-way breathing valve was then attached to the mask. The inspiratory port was connected to a pneumotach, and the expiratory port was connected to a Tissot gasometer. Inspiratory flow was measured, recorded, and analyzed digitally with use of LabChart software (AD Instruments, Colorado Springs, CO). Expired air was collected in the Tissot gasometer where mixed expired gas composition was measured via a gas analyzer (OxiGraf, Sunnyvale, CA). The catheter in the carotid artery was used for sampling arterial blood and to measure heart rate and blood pressure throughout the study. Arterial pH and blood gases were measured via a Siemens blood gas analyzer (Rapid Lab 248; Bayer Health Care, Leverkusen, Germany). Arterial blood was also analyzed for arterial electrolytes, which were analyzed on ABL800 FLEX (Radiometer, Copenhagen, Denmark). We emphasize that chronic (hours to days) changes observed in arterial [H+] presumptively reflect changes in cerebrospinal fluid (CSF) at the level of the intracranial chemoreceptors. Studies have suggested that in the chronic steady state (hours to days) changes in arterial [H+] reflect changes in the CSF (11, 19).
Experimental Procedure
This experimental design was adapted from previous design as outlined in detail in Burgraff et al. (15). In the 2-wk surgical recovery period, goats were acclimatized to the environmental chamber while breathing room air. Following the recovery period, control studies were completed while the goats were inhaling room air to obtain baseline physiological variables, including inspiratory minute ventilation (V̇i), breathing frequency (f), tidal volume (Vt), heart rate, and blood pressure, which were measured continuously for 20 min of the control period. Arterial blood was sampled from the carotid artery at 10 and 20 min and placed on ice until assessed for [H+], [], respiratory blood gases ( and ), and electrolytes at the end of the study. At the time of arterial blood sampling, mixed expired air was collected over the course of 5 min. This collection was used for assessment of fractional concentration of expired O2 (FeO2) and CO2 (FeCO2) for use of calculations for oxygen consumption (V̇o2), CO2 excretion (V̇co2), mixed expired oxygen (PeO2), and mixed expired CO2 (PeCO2).
The acute CO2/[H+] chemoreflex was assessed by acutely and incrementally increasing the level of InCO2 at the end of the 20-min control period during room air control, sustained hypercapnia, and at 4 and 24 h of normocapnia. During periods of room air control and 4 and 24 h of room air recovery, the InCO2 was raised to 3, 5, and 7% for 5 min each. During 6% hypercapnic exposure, the InCO2 was raised to 7 and 8% InCO2 to go above the level of sustained InCO2. Arterial blood was sampled during minutes 3–5 during each level of increased FiCO2 to measure changes in arterial CO2 and [H+]. The change in ventilation as a function of changes in arterial CO2 and [H+] were calculated to determine the acute chemoreflex. Using the levels of arterial [H+] and the slopes of the acute [H+] chemoreflex (ΔV̇i/Δ[H+]), we calculated a predicted steady-state ventilation (15).
After control studies, the InCO2 was raised to 6% and remained there for 30 days. During this hypercapnic period, studies were repeated periodically as described for the control period (∼every 2–3 days). At the end of the 30-day exposure the InCO2 was returned to room air levels. Studies were then repeated at 4 and 24 h for all goats and for three additional goats for up to 5 days after the return to room air.
Cognitive function was assessed as previously described (15). Goats were presented with two shapes (X and O) on the outside wall of the Plexiglas environmental chamber for a total of 10 times. For each cognitive function test, one shape was randomly assigned as the correct shape. Upon selection of a correct shape by the goat, a food reward was provided.
After completion of all studies, the goats were euthanized, and brains were extracted for neurochemical analysis. The goats were first sedated with a ketamine-xylazine solution (24:1 iv) and then euthanized by phenobarbital sodium and phenytoin sodium (B-euthanasia, iv). Once euthanized, the entire brain and brain stem were rapidly removed (<10 min) and immediately flash-frozen with dry ice-cooled 2-methylbutane before storage at -80°C prior to sectioning. The brain stem was sectioned into 2-mm coronal slices within a cryostat chamber at −20°C. Bilateral tissue punches (1 mm diameter) were then obtained from nuclei of interest [hypoglossal motor nucleus (XII), nucleus tractus solitarius (NTS)/dorsal motor nucleus of the vagus (DMV), ventral respiratory column (VRC), medullary raphe (MR), ventrolateral medulla (VLM), retrotrapezoid nucleus (RTN), and cuneate nucleus (CN)], as previously published (17, 18). Tissue punches were obtained in homogenizing/loading buffer (Tris·HCl (250 mmol/L, pH 6.8), SDS, glycerol, β-mercaptoethanol, bromophenol, proteinase/phosphatase inhibitor) at 25 mg/mL wt/vol, sonicated on ice, and then stored at −80°C.
Western Blotting
For Western blot analysis, 5 μL of each sample was added to each well of criterion precast gel (10–20% Tris·HCl; Bio-Rad Laboratories Hercules, CA) and was separated at 200 V for ∼30 min. Blots were then transferred onto a PVDF membrane with Bio-Rad Trans-Blot Turbo Transfer (3 min). Membranes were then blocked in 2% BSA in 1× TBST buffer for 90 min. Following the blocking period, membranes were incubated overnight at 4°C in primary antibodies. Western blots were performed with antibodies against NMDA receptor NR1 (1:500; Thermo Fisher Scientific, Waltham, MA), phospho-NR1 S896 (1:500; Millipore-Sigma, Burlington, MA), phospho-NR1 S897 (1:500; Thermo Fisher Scientific, Waltham, MA), AMPA receptor GluR1 (1:500; Millipore-Sigma, Burlington, MA), and GluR2 (1:500; Abcam, Cambridge, UK) and phospho-R2-Y876 (1:500; Thermo Fisher Scientific, Waltham, MA) and phospho-R2 S880 (1:500; Abcam, Cambridge, UK). Protein changes in neuroinflammation were performed with the antibody against interleukin-1β (1:500; Abcam, Cambridge, UK). Antibodies against tryptophan hydroxylase (1:1,000; Millipore-Sigma, Burlington, MA), indolamine 2,3-dioxygenase (1:500; Abcam, Cambridge, UK), dopamine decarboxylase (1:500; Abcam, Cambridge, UK), and acid (1:100; Abcam, Cambridge, UK) were used to measure alterations in tryptophan metabolism. Changes in neuronal count were assessed with antibodies against NeuN (1:500; Millipore-Sigma, Burlington, MA). After overnight incubation, membranes were rinsed three times with 1× TBST, followed by three additional washes in 1× TBST each 5 min. Membranes were then incubated in secondary antibody solution (1:10,000–2 in 2% BSA in 1× TBST) for 60 min. Blot were again rinsed three times with 1× TBST, followed by three additional 5-min washes in 1× TBST. After the third and final wash, blots were developed using either FEMTO developing solution (Millipore-Sigma, Burlington, MA) or Clarity Western ECL substrate (Bio-Rad Laboratories, Hercules, CA). Specific bands were imaged with a ChemiDoc imaging system (Bio-Rad Laboratories, Hercules, CA). Subsequently, blots were stripped with Restore Western blot stripping buffer for 5 min and then were rinsed three times with 1× TBST, followed by three additional 5-min washes in 1× TBST. Blots were blocked in 2% BSA in 1× TBST buffer for 90 min and then reprobed with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primary antibody (1:10,000). Blots were then reimaged and analyzed with Image Lab software. Relative densities of proteins for each individual lane were always normalized to each respective GAPDH on the same blot to correct for protein loading. Six samples of previously collected brain stem tissue from goats exposed to 30 days of room air were run simultaneously with a total of six 24-h recovery samples to allow for equal comparison between room air control goats and 24-h recovery goats. All results are expressed as percent room air control.
High-Performance Liquid Chromatography
HPLC was used to assess monoamine neurotransmitter levels during deacclimatization. For this analysis, tissue punches were thawed in 0.1 M perchloric acid and wet weights determined before sonication and centrifugation at 5,000 rpm (30 min) at 4°C. Supernatant was removed from each sample and subjected to HPLC analysis for norepinephrine (NE), dopamine (DA), 3,4-dihydroxyphenyl acetic acid (DOPAC), and 5-HT, 5-hydroxyindoleacetic acid (5-HIAA). Standards with 3,4-dihydroxybenzylamine (DHBA) (internal standard) and 0.1 M perchloric acid were injected to establish values for quantification. Samples then underwent electrochemical detection [Bioanalytical Systems (BAS) LC4C; 0.65 V, 2 nA, 0.1 Hz filter with Ag/Cl reference electrode] with a Waters Bondapak column (3.9 × 300; Milford, MA) at ambient temperature. Six HPLC tissue samples obtained from brain stem nuclei of 30-day hypercapnic goats from previous studies (18) were compared with samples of the 24-h recovery goats from this study.
Data and Statistical Analysis
Inspiratory flow was quantified breath-by-breath, binned at 5-min intervals and averaged for each 20-min study. Arterial blood gases, electrolytes, metabolic rate values (V̇co2, V̇o2, and RER), mixed expired CO2, heart rate, and blood pressure were averaged across each 20-min study. A one-way repeated-measures ANOVA and Holm–Sidak post hoc test were used to identify differences across condition. Predicted versus actual ventilation data (calculated as described in Ref. 15) was subjected to a two-way ANOVA using time and condition as factors. An analysis of covariance (ANCOVA) was used to assess differences in shifts of the V̇i/[H+] relationships. The slopes of the V̇i/[H+] relationship (acute [H+] chemoreflex; ΔV̇i/Δ[H+]) obtained during room air control and 24-h recovery were subjected to a paired t test. Statistically significant difference was determined as P < 0.05.
For Western blot analysis, expression of target antibodies was normalized to each individual band’s corresponding GAPDH expression. GAPDH-normalized expression values were then averaged across room air control goats within each Western blot to obtain a mean control expression. Each GAPDH-normalized band of every sample was then compared with the mean control expression to obtain expression of the band relative to room air control. All data are presented as percent room air control expression. Statistical differences between 30-day room air control and the 24-h recovery goats were analyzed with a one-way ANOVA, with a Holm–Sidak post hoc test. For HPLC, brain stem tissue levels of 5-HT, catecholamines, and respective metabolites were analyzed through an unpaired t test comparing conditions. Statistically significant difference was determined as P < 0.05.
RESULTS
SS V̇i and Blood Gases during Acclimatization to Chronic Hypercapnia
Ventilatory acclimatization to CH observed in this study was consistent with previous publications (8, 10, 15, 20). Initially, SS V̇i increased from ∼7.16 ± 0.49 L/min to 22.01 ± 2.34 L/min within the first 1 h, and was at 22.99 ± 1.91 L/min and 21.36 ± 2.15 L/min at 24 h and 30 days of hypercapnia. Breathing frequency (f) initially (1 h CO2) increased from 18.99 ± 1.37 breaths per minute (BPM) to 34.90 ± 3.73 BPM. At 24 h and 30 days of hypercapnia, f was 35.90 ± 4.45 BPM and 29.46 ± 2.37 BPM, respectively. Vt increased from 0.39 ± 0.28 to 0.67 ± 0.21 L in the initial hour of 6% InCO2. At 24 h and 30 days of hypercapnia, Vt was 0.69 ± 0.07 L and 0.74 ± 0.06 L, respectively.
Changes in blood gases and acid/base status during acclimatization to CH were also consistent with those previously reported (15). increased from 41.08 ± 0.78 mmHg to 52.2 ± 4.00 mmHg within the first 1 h of hypercapnia. At 24 h, was 53.41 ± 3.27 mmHg and by 30 days reached a level of 54.24 ± 1.12 mmHg. Arterial [H+] initially increased from 34.77 ± 0.45 nmol/L to 40.67 ± 1.16 nmol/L in the first hour of hypercapnia. By 24 h and 30 days of hypercapnia, arterial [H+] was 38.57 ± 0.70 nmol/L and 37.06 ± 0.65 nmol/L, respectively. Arterial [] increased from 28.21 ± 0.57mEq/L to 30.11 ± 0.94mEq/L within the first 1 h of hypercapnia, and by 24 h and 30 days [] was 33.07 ± 0.94mEq/L and 35.00 ± 1.28mEq/L, respectively.
SS V̇i and Blood Gases during Rapid Deacclimatization from Chronic Hypercapnia
At 30 days of 6% inspired CO2-induced hypercapnia, we abruptly returned inspired gases to room air. Four hours after return to room air, V̇i decreased from steady-state levels at day 30 of hypercapnia (P < 0.001), returning to within ∼10% of room air control (P = 0.793). V̇i did not change significantly between 4 and 24 h of return to room air breathing (P = 0.595; Fig. 1A). At 4 h of room air, f had decreased from 30 days values (P < 0.001) and did not significantly differ from control at 24 h of recovery (P = 0.231; Fig. 1B). Similarly, by 4 h of room air breathing Vt decreased from day 30 values (P < 0.001), returning Vt to room air control levels (P = 0.897). Vt remained at that level 24 h later (P = 0.931; Fig. 1C).
Figure 1.

Deacclimatization processes from chronic hypercapnia rapidly occurred for steady-state minute ventilation (V̇i), breathing frequency (f), and tidal volume (Vt). A: 4 h after abrupt termination of chronic hypercapnia, V̇i decreased from ∼283% of control to ∼10% of control and changed minimally thereafter. B and C: during the transition from 30 days of hypercapnia to 4 h of room air recovery, return to control in V̇i was mediated by a decline in both f and Vt. Dotted line provides a reference to control values obtained prior to the hypercapnic period. Ventilatory measurements were obtained from 9 female adult goats. *Significant difference compared with room air control. #Significant difference compared with day 30 of hypercapnia. Significance (P < 0.05) was derived from one-way repeated-measures ANOVA.
In the transition from 30 days of hypercapnia to 4 h of room air breathing, remained increased compared with room air control levels (P = 0.003; Fig. 2C). At 24 h after the return to room air, was further decreased from 4-h levels (P = 0.005), returning to near control levels (P = 0.619). During the transition from 30 days of hypercapnia to 4 h of normocapnia, arterial [H+] significantly decreased from 30-day CO2 values (P = 0.002), returning [H+] to near control levels (P = 0.594). Arterial [H+] did not significantly change thereafter (P = 0.343; Fig. 2A). At 4 h of recovery [] remained increased (∼3 mEq/L) relative to control (P = 0.035; Fig. 2B), but then it fell from 31.08 ± 1.25 mEq/L to 29.89 ± 1.20 mEq/L between 4 and 24 h of deacclimatization, returning [] to near room air control levels (P = 0.208). returned to room air control levels at 4 h (P = 0.827) and did not change thereafter (P = 0.938); Fig. 2D).
Figure 2.
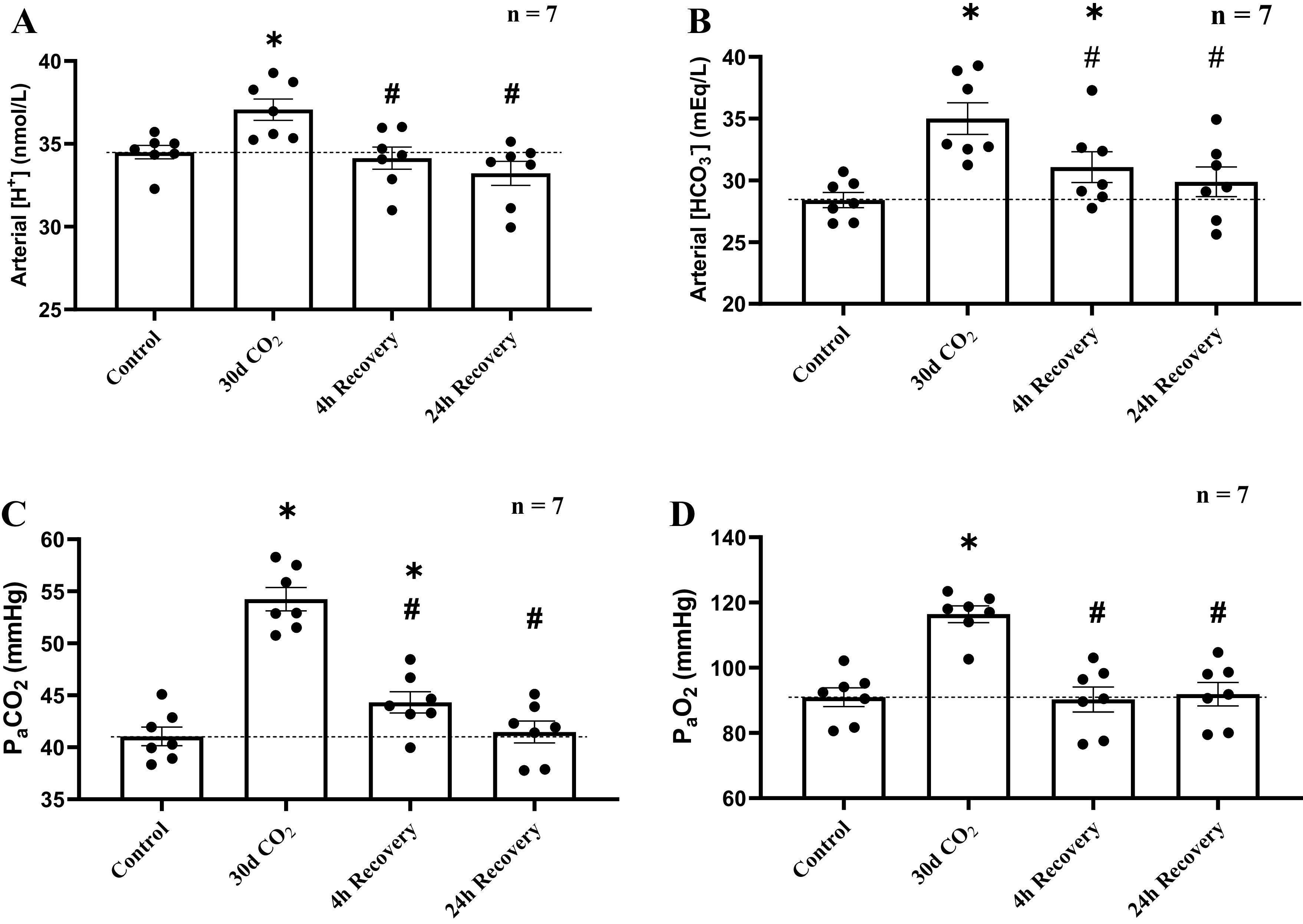
Recovery of arterial blood gases and acid-base status was completed within 24 h of deacclimatization from chronic hypercapnia. Due to a nonfunctioning catheter in 2 goats, blood gases were measured in only 7 of the 9 female adult goats. A: at 4 h of room air recovery, arterial [H+] decreased by 3 nmol/L from chronic hypercapnia, returning values to control values, and did not significantly change thereafter. B: at 4 h of recovery, arterial [] was still significantly elevated compared with room air controls but had returned to room air control levels by 24 h of recovery. C: was elevated relative to room air controls at 4 h of recovery but returned to control levels by 24 h of recovery. D: arterial Po2 immediately returned to control levels 4 h following the return of inspired CO2 (InCO2) to room air and changed minimally thereafter. Dotted line provides a reference to control values obtained prior to the hypercapnic and normocapnic recovery period. *Significant difference compared with room air control. #Significant difference compared with day 30 of hypercapnia. Significance (P < 0.05) was derived from one-way repeated-measures ANOVA.
Ventilation and Blood Gases after 5 Days of Deacclimatization from Chronic Hypercapnia
A subgroup of goats (n = 3) was studied for ∼5 days after the transition from CH to normocapnia (Supplemental Fig. S1 (supplemental materials are found at https://figshare.com/s/1bea57f593b903999cab). In all three animals, V̇i changed minimally throughout the extended recovery period (Supplemental Fig. S1A), However, there were large day-to-day variations in acid/base status within and between the individual goats studied (Supplemental Fig. S1, B and C). Although limited in sample size, the extended recovery data demonstrate that renal compensation was not yet complete, which is consistent with finding of Zouboules et al. that suggests renal adjustments to changes in require at least 5 days (21). However, this finding requires further study.
Ventilatory (V̇i)/Arterial [H+] Relationship and Acute [H+] Sensitivity
In Fig. 3, the first point in each line represents the homeostatic values of SS V̇i and [H+] during the steady state and referred to as the resting “set point.” The slope of each line was obtained by using V̇i and [H+] values that were obtained when acutely increasing InCO2, and thus [H+]. This slope represents the sensitivity of the ventilatory system to acute challenges of [H+] that exceeded the set point levels (acute chemoreflex ΔV̇i/Δ[H+]). Similar to previously reported data (15), we found at 30 days of hypercapnia that there was a leftward-shift in the V̇i/[H+] relationship (P < 0.001). Our data show that at 24 h of normocapnia following hypercapnia, the relationship was insignificantly shifted leftward compared with room air control (P = 0.082). However, in contrast to 30 days of hypercapnia, we found that the slope of the ventilation/arterial [H+] relationship (H+ chemoreflex) at 24 h of recovery was greater than the slope of the relationship at room air control (P = 0.011; Fig. 3, inset). This difference suggests that sensitivity of the ventilatory system to acute changes in [H+] is greater at 24 h of recovery compared with prehypercapnic exposure. The nonsignificant change in the position of the relationship but increase in slope further suggests that steady-state ventilation and the acute chemoreflex can adapt independently of one another.
Figure 3.

Twenty-four hours following termination of hypercapnia, the slope of the ventilatory (V̇i)/arterial [H+] relationship was increased compared with room air control. The first points for each line are steady-state V̇i and arterial [H+] during the chronic steady state. The additional three points are V̇i and [H+], obtained when inspired CO2 (InCO2) was increased above steady-state levels during an acute chemoreflex challenge. The slope of the line represents the sensitivity of the ventilatory system to changes in [H+] (acute chemoreflex; ΔV̇i/Δ[H+]). At 30 days of hypercapnia, the relationship was significantly shifted left compared with room air control without a change in sensitivity. At 24 h of recovery, there was a nonsignificant shift in the relationship between ventilation and arterial [H+] compared with room air control. However, at 24 h of recovery, the slope of the relationship or the sensitivity to acute changes in arterial [H+] was increased compared with room air controls. Inset: slopes (ΔV̇i/Δ[H+]) of goats studied at room air control and 24 h of recovery. Due to a nonfunctioning catheter in 2 goats, ΔV̇i/Δ[H+] was measured in only 7 of the 9 female adult goats. P value for the shift in the relationship was derived from an analysis of covariance (ANCOVA). Significance for ΔV̇i/Δ[H+] (P < 0.05) was derived from a paired t test.
Predicted versus Measured (Actual) Ventilation
As in prior studies during acclimatization to hypercapnia (15) predicted ventilation was calculated as the difference between steady-state arterial [H+] at room air control and arterial [H+] during or after hypercapnia multiplied by the slope of the relationship between V̇i and arterial [H+] during assessment of acute [H+] chemoreflex. This value represents the predicted elevation in V̇i above control room air values and was added to the measured room air control ventilation to derive the predicted ventilation (15). Similar to our previous studies in hypercapnia, actual ventilation was greater than ventilation predicted by the acute [H+] chemoreflex and level of arterial [H+] (P < 0.001). In the present study, we found that actual ventilation exceeded the predicted ventilation at 24 h following the return to normocapnia (P = 0.044; Fig. 4). This finding demonstrates that, despite the return of absolute SS V̇i and arterial [H+] to room air control levels, measured actual ventilation still exceeded what would be predicted based on the level of [H+] and the acute chemoreflex (Fig. 4). Taken together, these data suggest that additional mechanisms beyond the chemical control of breathing are contributing to a greater measured ventilation.
Figure 4.

Actual ventilation significantly exceeded predicted minute ventilation during 30 days of chronic exposure to 6% inspired CO2 (InCO2) and 24 h after return to normocapnia. Predicted ventilation was calculated by use of the steady-state level of arterial [H+] as well as the acute chemoreflex obtained by increasing inspired CO2 above the steady-state level. At 24 h following the transition from hypercapnia to normocapnia, measured ventilation significantly exceeded predicted ventilation, suggesting a disconnect between the steady-state ventilation and the acute chemoreflex and a maintained elevated ventilatory set point, as previously observed in chronic hypercapnia. Due to a nonfunctioning catheter in 2 goats, predicted ventilation was measured in only 7 of the 9 female adult goats. Dotted line provides a reference to control values obtained prior to the hypercapnic and normocapnic recovery period. *Significant difference of actual ventilation compared with predicted ventilation at that time point. P value shown was derived from a two-way ANOVA (predicted vs. actual ventilation and time as factors).
Additional Physiological Measures during Deacclimatization from Chronic Hypercapnia
Plasma electrolytes.
Compared with room air control, there were no significant changes in arterial [Na+] during 30 days of CO2 (P = 0.593) or 4 h of recovery (P = 0.245). However, by 24 h of recovery, [Na+] had decreased below control levels (P = 0.037). Consistent with our previous studies (15), goats subjected to sustained hypercapnia demonstrated a mild hyperkalemia, and reductions in arterial [Cl−] (P = 0.039) and hemoglobin (P = 0.006). During the recovery period, we found that whereas arterial [Cl−] returned to room air control values by 4 h of recovery (P = 0.733), arterial hemoglobin remained decreased at 4 h (P = 0.008) and 24 h (P = 0.002) of room air recovery. There were no significant changes in arterial [K+] (P = 0.935), [Ca2+] (P = 0.963), or glucose (P = 0.740) throughout the entire recovery period (Table 1).
Table 1.
Integrated physiological responses to deacclimatization from chronic hypercapnia
| Control | 30-day CO2 | 4-h Recovery | 24-h Recovery | |
|---|---|---|---|---|
| Arterial [Na+] | 145.67 ± 1.01 | 144.43 ± 0.71 | 143.57 ± 0.44 | 142.57 ± 0.43* |
| Arterial [Cl−] | 107.09 ± 0.73 | 99.42 ± 3.15* | 106.29 ± 1.45 | 104.00 ± 0.96 |
| Arterial hemoglobin [Hb] | 8.77 ± 0.44 | 7.57 ± 0.43* | 7.79 ± 0.46* | 7.40 ± 0.43* |
| Arterial potassium [K+] | 4.05 ± 0.07 | 4.18 ± 0.04 | 4.15 ± 0.05 | 4.10 ± 0.06 |
| Arterial calcium [Ca2+] | 1.09 ± 0.03 | 1.07 ± 0.02 | 1.05 ± 0.03 | 1.08 ± 0.02 |
| Arterial glucose | 58.70 ± 1.20 | 57.86 ± 2.04 | 61.93 ± 2.67 | 56.07 ± 1.39 |
| -PeCO2 | 16.74 ± 0.52 | 1.38 ± 1.21* | 18.80 ± 0.71 | 16.74 ± 1.21 |
| Oxygen cConsumption (V̇o2) | 0.23 ± 0.02 | 0.30 ± 0.02* | 0.26 ± 0.02 | 0.21 ± 0.01 |
| CO2 excretion (V̇co2) | 0.17 ± 0.02 | 0.20 ± 0.03 | 0.22 ± 0.01 | 0.17 ± 0.01 |
| Respiratory exchange ratio (RER) | 0.77 ± 0.02 | 0.67 ± 0.10 | 0.87 ± 0.01* | 0.81 ± 0.01 |
| Heart rate | 61.91 ± 4.86 | 66.47 ± 4.60 | 71.05 ± 3.90* | 63.04 ± 4.92 |
| arterial blood pressure | 74.26 ± 3.41 | 70.28 ± 3.35 | 72.54 ± 3.35 | 67.01 ± 4.26 |
| Cognitive function | 8 ± 0.3 | 5 ± 0.4* | 8 ± 0.4 | 9 ± 0.2 |
Values are means ± SE for several physiological variables of adult goats prior to chronic hypercapnia (CH; control), at 30 days of CH, and after 4 and 24 h of recovery from CH. As indicated by boldface values, at 30 days of hypercapnia there were significant decreases in arterial [Cl−], hemoglobin, and the arterial-mixed expired CO2 difference and a significant increase in V̇o2. Four hours after abrupt termination of hypercapnia, there was a significant decrease in arterial hemoglobin and significant (P < 0.05) increases in heart rate and RER. Twenty-four hours following termination of hypercapnia, there were significant reductions in arterial [Na+] and hemoglobin relative to control. *Boldface values indicate P < 0.05 derived from a one-way repeated-measures ANOVA.
Arterial–mixed expired Pco2 difference.
The data demonstrated here and previously (15) show that exposure to 30 days of CH results in a decrease of the arterial-mixed expired Pco2 difference (P < 0.001). Four hours after the transition from CH to normocapnia the arterial-mixed expired Pco2 difference returned to room air control levels (P = 0.159) and changed minimally thereafter (P = 0.109). The return in the -PeCO2 difference was due to a decrease in PeCO2 in the transition from 30 days of hypercapnia to 4 h of recovery (P < 0.001). As discussed in Burgraff et al. (15), during hypercapnia an additional buffering mechanism in our model may be gastric excretion of CO2, thus causing an increase in PeCO2 at 30 days of hypercapnia [see Dean et al. 2011 for review (22)]. Therefore, it is likely that the return of the arterial-mixed expired Pco2 difference during deacclimatization is partially driven by the decrease in PeCO2 which is a result of decreased contribution of gastric CO2 excretion. Overall, the data suggest that, like acclimatization, deacclimatization results in rapid adaptations to whole body CO2 buffering mechanisms (Table 1).
Metabolic rate.
Consistent with previous studies (17), 30 days of CH resulted in an increased V̇o2 relative to control (P = 0.005). Conversely, V̇co2 remained near control levels throughout the entire hypercapnic period (P = 0.7891). These data likely reflect tissue storage of CO2 (15) (Table 1). Four hours after the transition from hypercapnia to normocapnia, V̇o2 returned to room air control values (P = 0.118). We believe this could be attributed to the decline in the O2 cost of breathing due to the reduction in ventilation. Although insignificant (P = 0.305), there was a +0.05-mmHg increase (relative to control) in V̇co2, which likely reflects tissue release of excess CO2 stores (15, 23, 24). The RER was elevated (P = 0.032) relative to controls at 4 h of recovery but returned to room air control levels by 24 h of recovery (Table 1). Taken together, these data suggest that the metabolic rate in the off-transition rapidly reverts to prehypercapnic values and there is a time-dependent release of tissues CO2 stores.
Heart rate and blood pressure.
Our laboratory has previously reported that CH results in both an elevated heart rate and hypertension (15). Our data show that heart rate was elevated 4 h after termination of hypercapnia (P = 0.007) but returned to control levels by 24 h of deacclimatization (P = 0.266). Blood pressure, however, was not significantly different from control levels at 4 h (P = 0.933) and 24 h of recovery (P = 0.266; Table 1). Thus, cardiovascular deacclimatization was a relatively rapid process.
Cognitive function.
Our laboratory and others have reported the negative impact hypercapnia has on cognitive functioning abilities (15, 25, 26). Herein, we tested whether the sudden termination of hypercapnia affected cognitive function. Consistent with our acclimatization studies, 30 days of 6% InCO2 resulted in a reduction of the number of correct choices (P < 0.003; Table 1), suggesting that hypercapnia resulted in a decline of cognitive function. Within 4 h of the abrupt termination of CH, the number of correct choices had returned to prehypercapnic values (P = 0.237) and remained at that level throughout the remaining 24-h period (P = 0.176; Table 1). These data strongly suggest that consequences of hypercapnia on cognitive function are rapidly reversed once CO2 levels are restored. However, the underlying mechanisms driving the cognitive decline induced by chronic hypercapnic, and those that govern the reversal of this decline remain unknown.
Neurochemical Adaptations 24 h after Abrupt Termination of Chronic Hypercapnia
Neuromodulator levels and metabolism.
To determine whether the transition from CH to normocapnia resulted in changes in monoamine neurotransmitters, we utilized HPLC to measure changes in serotonin, norepinephrine, and dopamine within important brain stem respiratory nuclei including the VLM, MR, VRC, NTS, and RTN.
Within the NTS there was a significant increase in norepinephrine (P < 0.001), serotonin (P = 0.007), dopamine (P < 0.001), and DOPAC (P = 0.025) between 30-day hypercapnia goats and 24-h recovery goats (Fig. 5, A–E). There were no significant differences in norepinephrine between the 30-day hypercapnic goats and 24-h recovery goats within the VRC (1380.18 ± 569.27 vs. 448.34 ± 138.75; P = 0.143), MR (315.65 ± 85.25 vs. 305.00 ± 62.01; P = 0.938), VLM (377.38 ± 73.44 vs. 516.44 ± 80.57; P = 0.235), and RTN (439.63 ± 44.62 vs. 515.28 ± 95.86; P = 0.445). Similarly, there were no significant differences in serotonin (5-HT) or its metabolite HIAA between the 30-day hypercapnic goats and the recovery goats within the VRC (5-HT: 1,968.60 ± 997.86 vs. 675.23 ± 196.37; P = 0.232; HIAA: 3,377.73 ± 1,475.38 vs. 1,886.81 ± 313.7; P = 0.346), RTN (5-HT: 573.82 ± 73.62 vs. 589.99 ± 94.81; P = 0.895; HIAA: 700.85 ± 158.85 vs. 872.18 ± 258.37; P = 0.564), and the MR (5-HT: 467.65 ± 111.71 vs.765.43 ± 35.04; P = 0.114; HIAA: 1570.95 ± 304.75 vs. 777.56 ± 62.01; P = 0.120). Within the VLM there was slight but nonsignificant increase in 5-HT (488.28 ± 118.25 vs.786.68 ± 90.87; P = 0.076), but not HIAA (634.50 ± 97.99 vs.776.19 ± 76.28; P = 0.272). The HIAA/5-HT ratio, an index of 5-HT turnover significantly decreased within the MR (P < 0.001) but not the VLM (P = 0.064) (Fig. 6).
Figure 5.
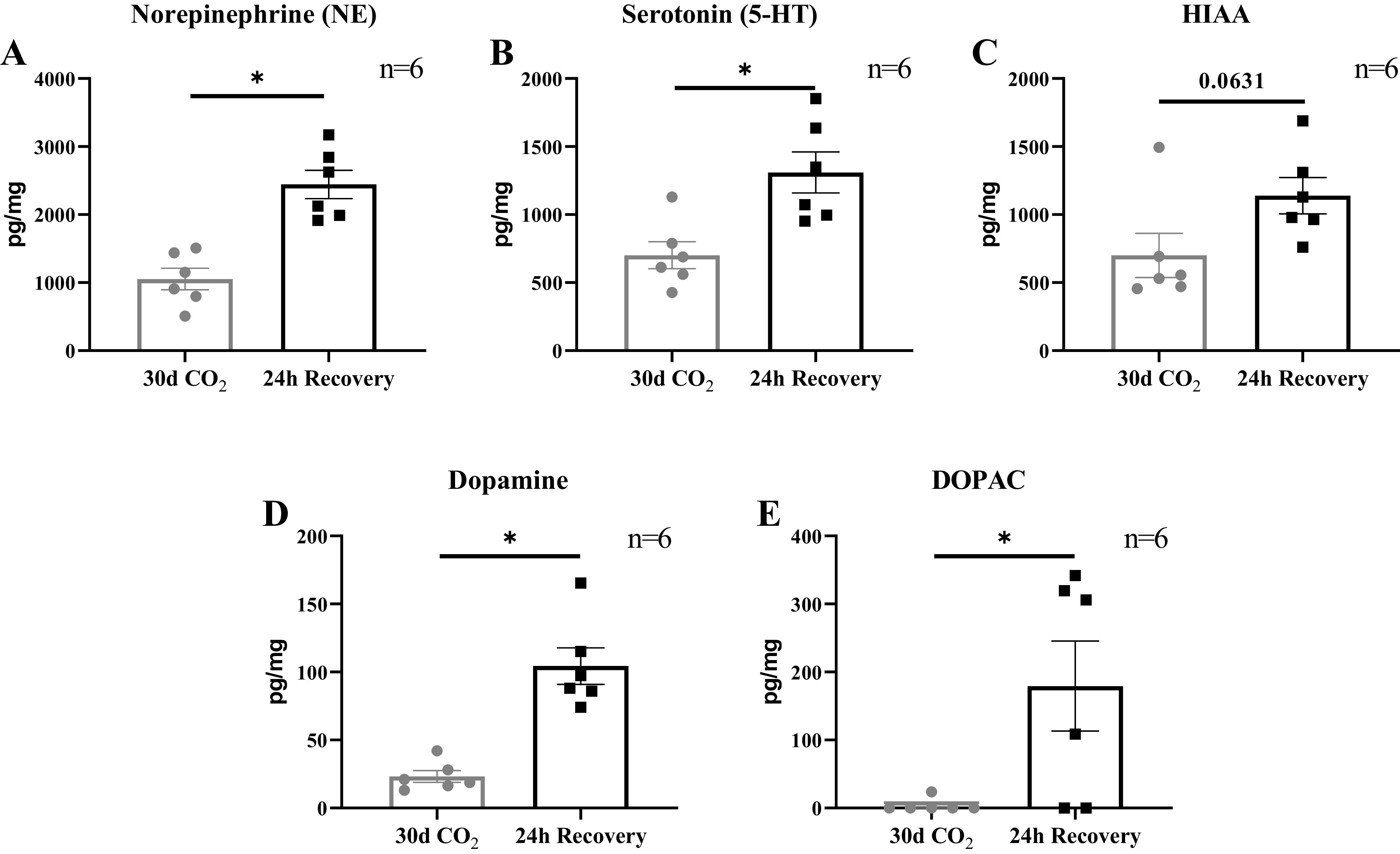
Excitatory neuromodulators and their metabolites in the nucleus tractus solitarius (NTS) were greater after 24 h of recovery compared with 30 days of chronic hypercapnia. Bulk tissue HPLC analysis within the NTS showed a significant increase between 24-h recovery female adult goats (n = 6) compared with 30-day hypercapnic female adult goats (n = 6) in norepinephrine (A), serotonin (B), dopamine (D), and 3,4-dihydroxyphenyl acetic acid (DOPAC; E). There was a slight but nonsignificant increase in 5-hydroxyindoleacetic acid (HIAA; C) within the NTS (P = 0.063). P value represents the significance term from an unpaired t test. *P < 0.05.
Figure 6.

Serotonin (5-HT) metabolism is decreased within the medullary raphe (MR) at 24 h of recovery. Within the MR (a primary site of 5-HT synthesis) in the 24-h recovery goats, there was a significant decrease in the 5-hydroxyindoleacetic acid (HIAA)/5-HT ratio (index of 5-HT metabolism) compared with goats exposed to 30 days of hypercapnia. Within the ventrolateral medulla (VLM), there was a nonsignificant change in this ratio between the 2 groups of animals. Changes in the HIAA/5-HT ratio may contribute to elevated levels of 5-HT. P value represents the significance term from an unpaired t test. *P < 0.05.
NMDA and AMPA receptor subunit expression and phosphorylation.
Adaptations in glutamate receptor plasticity have been previously observed during the acclimatization to hypercapnia (15). To determine whether similar adaptations of glutamate plasticity occurred during deacclimatization, we studied specific markers of NMDA and AMPA receptor subunit expression and activation within individual regions of brain stem respiratory nuclei that were shown to be significantly altered during either 24 h or 30 days of hypercapnia. There were no significant differences in NMDA (GluN1) or AMPA (GluR1, GluR2) receptor subunit expression or phosphorylation sites between 24-h recovery goats and 30-day room air control (Table 2). These data suggest that, in contrast to the acclimatization period, glutamate receptor-dependent neuroplasticity is likely not occurring during deacclimatization from CH.
Table 2.
GAPDH normalized glutamate receptor expression/phosphorylation expression relative to control 24 h after abrupt termination of chronic hypercapnia
| Target | Nuclei | 24-h Recovery (%) | 30-daysControl (%) | P Value |
|---|---|---|---|---|
| GluN1 | Rostral solitary complex | 115 ± 10 | 100 ± 18 | 0.483 |
| Rostral VLM | 120 ± 4 | 100 ± 19 | 0.327 | |
| Caudal MR | 160 ± 31 | 100 ± 19 | 0.128 | |
| GluN1 S896 | Rostral VLM | 146 ± 22 | 100 ± 18 | 0.137 |
| Rostral XII | 112 ± 19 | 100 ± 12 | 0.589 | |
| GluN1 S897 | Rostral VLM | 130 ± 25 | 100 ± 29 | 0.452 |
| Caudal VRC | 80 ± 7 | 100 ± 7 | 0.075 | |
| GluR1 | Caudal RTN | 132 ± 30 | 100 ± 14 | 0.354 |
| GluR2 | Rostral solitary complex | 111 ± 9 | 100 ± 14 | 0.495 |
| Caudal VLM | 121 ± 10 | 100 ± 4 | 0.089 | |
| GluR2 S880 | Caudal VLM | 99 ± 10 | 100 ± 11 | 0.990 |
| Rostral VLM | 135 ± 14 | 100 ± 21 | 0.193 | |
| Rostral VRC | 118 ± 16 | 100 ± 6 | 0.325 | |
| GluR2 Y876 | Caudal XII | 79 ± 19 | 100 ± 10 | 0.372 |
Twenty-four hours after abrupt termination of 6% inspired CO2 (InCO2), there were no significant (P > 0.05) differences from control goats in protein expression or phosphorylation of glutamate receptors (GluR) within nuclei that had previously been altered from control during either acute (24-h) or chronic (30-day) hypercapnia. For each glutamate receptor, the 24-recovery values are expressed as percentage of the 30-day room air control goats ± SE. Values of the 30-day control goats were averaged and are presented as 100% ± SE to reflect the variation within these goats. MR, medullary raphe; RTN, retrotrapezoid nucleus; VLM, ventrolateral medulla; VRC, ventral respiratory column; XII, hypoglossal motor nucleus.
Neuroinflammation.
The inflammatory cytokine IL-1β as a marker of neuroinflammation in the VRC, MR, and cuneate nucleus (CN) was greater than control at 24 h of CH (18). Across these sites, there were no differences between the 24-h recovery goats and the 30-day room air control goats. These data suggest that, unlike the acclimatization, neuroinflammation is likely not occurring in the transition from hypercapnia to normocapnia (Table 3).
Table 3.
GAPDH normalized IL-1β and tryptophan metabolism enzyme expression relative to control 24 h after abrupt termination of chronic hypercapnia
| Target | Nuclei | 24-h Recovery (%) | 30-days Control (%) | P Value |
|---|---|---|---|---|
| IL-1β | Caudal cuneate | 132 ± 17 | 100 ± 11 | 0.148 |
| Rostral cuneate | 80 ± 13 | 100 ± 24 | 0.490 | |
| Caudal MR | 118 ± 21 | 100 ± 19 | 0.533 | |
| Rostral MR | 93 ± 14 | 100 ± 14 | 0.741 | |
| Caudal VRC | 103 ± 16 | 100 ± 22 | 0.914 | |
| Rostral VRC | 87 ± 12 | 100 ± 15 | 0.518 | |
| TPH | Rostral VLM | 61 ± 9* | 100 ± 10 | 0.018 |
| Caudal MR | 116 ± 15 | 100 ± 17 | 0.492 | |
| Rostral MR | 50 ± 4* | 100 ± 10 | <0.001 | |
| IDO | Caudal VLM | 123 ± 33 | 100 ± 15 | 0.539 |
| Rostral VLM | 52 ± 15* | 100 ± 10 | 0.024 | |
| Caudal MR | 81 + 21 | 100 ± 26 | 0.602 | |
| Rostral MR | 73 ± 11* | 100 ± 5 | 0.040 | |
| DDC | Rostral VLM | 72 ± 10 | 100 ± 13 | 0.127 |
| Caudal MR | 153 ± 47 | 100 ± 8 | 0.292 | |
| QA | Rostral VLM | 74 ± 12 | 100 ± 9 | 0.321 |
Twenty-four hours after abrupt termination of 6% inspired CO2 (InCO2), IDO, and TPH in the rostral regions of the VLM and MR were significantly (*P < 0.05) lower compared with 30-day control goats. However, 24 h after abrupt termination of 6% InCO2, there were no differences in IL-1β or downstream products of tryptophan metabolism (DDC and QA) within nuclei that had previously been altered during either acute (24-h) or chronic (30-day) hypercapnia. There were no differences in TPH or IDO expression in the caudal regions of the VLM and MR. For both IDO and TPH, the 24-hrecovery values are expressed as percentage of the 30-day control goats ± SE. Values of the 30-day control goats were averaged and are presented as 100% ± SE to reflect the variation within these goats. MR, medullary raphe; RTN, retrotrapezoid nucleus; VLM, ventrolateral medulla; VRC, ventral respiratory column; XII, hypoglossal motor nucleus; DDC, dopamine decarboxylase; QA, quinolinic acid; IDO, indolamine 2,3-dioxygenase; TPH, tryptophan hydroxylase.
Tryptophan metabolism.
Since key enzymes of tryptophan metabolism were reduced from control after 30 days of CH, we determined whether the abrupt termination of hypercapnia restored the levels of TPH and IDO to control. Within the rostral (+2–4 mm to obex) region of the MR, IDO (P = 0.040) and TPH (P < 0.001) were decreased by ∼28 and ∼50%, respectively, relative to 30-day room air controls (Fig. 7A). Similar changes were observed within the rostral (+2–4 mm to obex) region of the VLM, where IDO was 48% decreased relative to room air controls (P = 0.024), and TPH was 39% decreased compared with 30-day room air controls (P = 0.018; Fig. 7B). In addition, neuronal marker NeuN was decreased by ∼59% in the rostral VLM (P = 0.017) and ∼62% in rostral MR (P = 0.037) relative to 30-day room air controls (Fig. 7, A and B). We suspect that the reduction in NeuN in the recovery goats was likely a sustained effect of CH, which has been shown to reduce NeuN expression in these regions (18).
Figure 7.
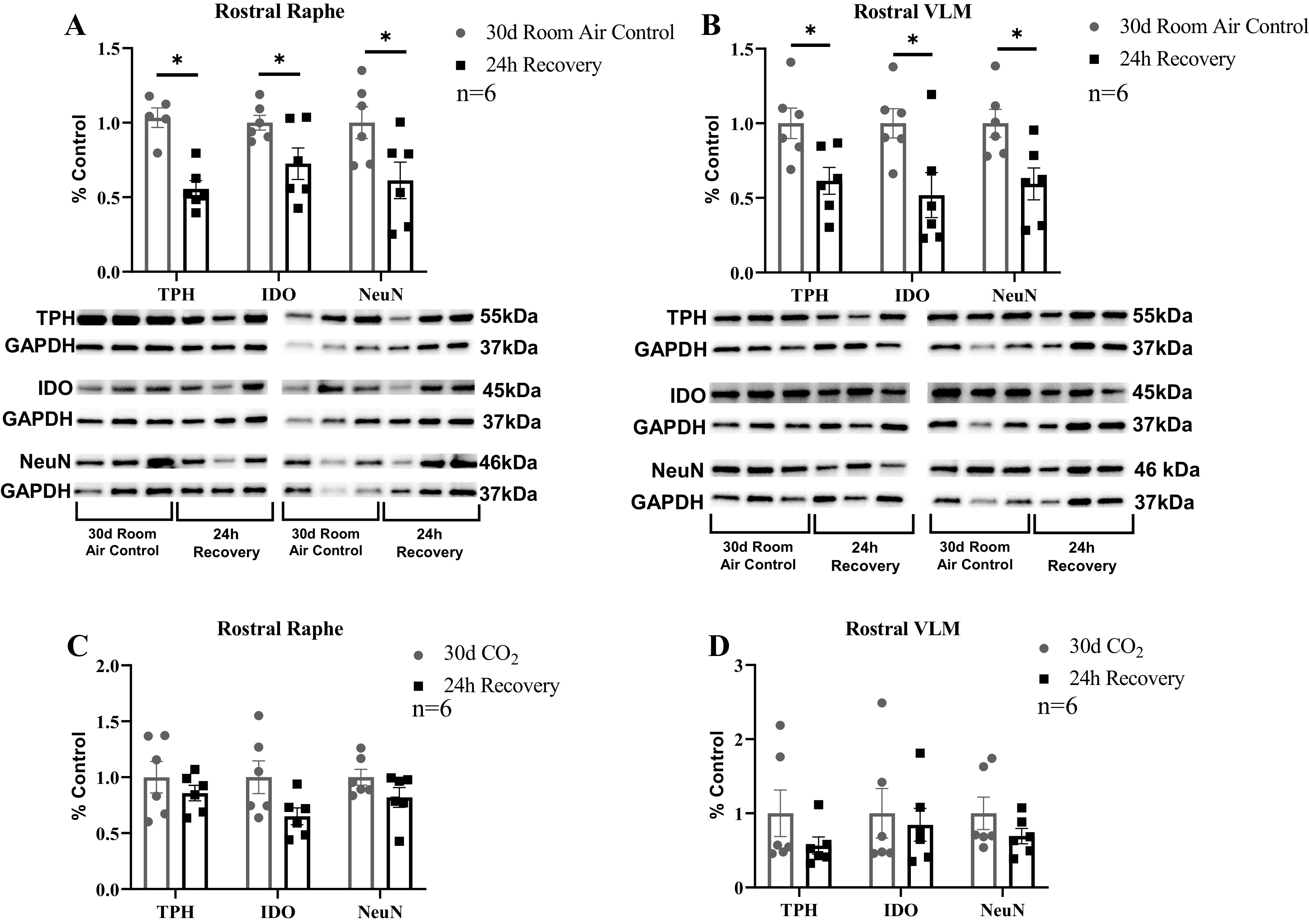
Selected enzymes of tryptophan metabolism and total number of neurons remained decreased 24 h after termination of chronic hypercapnia. Within the rostral (+2–4 mm from obex) regions of the medullary raphe (MR; A) and ventrolateral medulla (VLM; B), enzymes tryptophan hydroxylase (TPH), indolamine 2,3-dioxygenase (IDO), and total neuron number (NeuN) were significantly reduced in 24-h recovery goats compared with 30-day room air control goats. However, in both the rostral MR (C) and VLM (D), there was no significant difference between 30-day hypercapnia adult female goats (n = 6) and 24-h (n = 6) adult female recovery goats. Significance was derived from one-way ANOVA with *P < 0.05.
We also compared protein levels of these targets in 30-day hypercapnic goats with the 24-h recovery goats. Within the MR (Fig. 7C), there were no significant differences between 30-day hypercapnic and 24-h recovery goats in TPH (P = 0.386), IDO (P = 0.088), or NeuN (P = 0.142). Similarly, within the VLM (Fig. 7D) there were no significant differences between 30-day hypercapnic and 24-h recovery goats in TPH (P = 0.222), IDO (P = 0.707), or NeuN (P = 0.233). In sum, these data suggest that termination of CH does not restore IDO and TPH levels to control.
In the caudal regions (+0–2 mm to obex) of the MR, there were no significant differences in expression of TPH or IDO between 24-h recovery goats and 30-day room air control goats (Table 3). Similarly, in the caudal VLM, no differences were found between 24-h recovery goats and 30-day room air goats in expression levels of IDO (Table 3). We have previously shown that dopamine decarboxylase (DDC) in the caudal MR and rostral VLM, and quinolinic acid (QA) in the rostral VLM were found to be altered at either 24 h or 30 days of hypercapnia compared with room air controls (18). However, in 24-h recovery goats, there was no difference in DDC expression in the caudal MR or rostral VLM compared with 30-day room air controls (Table 3). Similarly, QA did not differ between 24-h recovery goats and 30-day room air control goats within the rostral VLM (Table 3).
DISCUSSION
The physiological consequences of rapid normalization of in patients with diseases marked or defined by CH are not well understood. In the current study, we address this knowledge gap by characterizing the integrated physiological and neurochemical adaptations that occur after abrupt termination of 30 days of increased InCO2-induced CH in adult goats. We tested the hypothesis that a rapid transition from CH to room air leads to a time-dependent restoration of physiological functions. Our physiological data support this hypothesis, as restoration of steady-state ventilatory, acid-base balance, and electrolyte control mechanisms were partially complete by 4 h and complete by 24 h of normocapnia. However, we also found an elevated acute [H+] chemoreflex, along with a measured ventilation that exceeded predicted at 24 h of normocapnia, an important and notable exception to complete and rapid normalization after termination of CH.
On the basis of previous data (17, 18) during acclimatization to hypercapnia, we also tested the hypothesis that abrupt termination of a 30-day period of CH would significantly change markers of neuroplasticity, including glutamate receptor expression/phosphorylation state, neuroinflammation, and tryptophan metabolism, without changes in excitatory neuromodulators. Our data do not fully support this hypothesis, as abrupt termination of CH elicited increases in excitatory neuromodulators but did not change markers of neuroplasticity. Collectively, our physiological and neurochemical data suggest that 24 h following the termination of hypercapnia, 1) most integrated physiological systems demonstrate a rapid return to control despite persistent increases in the acute [H+] chemoreflex and the difference between actual versus predicted ventilation after 24 h; 2) increases in excitatory neuromodulators at 24 h of recovery may contribute to the reported elevated chemoreflex and steady-state ventilation exceeding predicted ventilation; and 3) the mechanisms of neuroplasticity occurring throughout acclimatization to and deacclimatization from CH are fundamentally different.
Physiological Deacclimatization from Chronic Hypercapnia
Ventilation and acute chemoreflex.
Data presented in this study are consistent with previous data from other species, which suggests that within 24 h of termination of hypercapnia, steady-state ventilation, acid-base status, and arterial blood gases have returned to prehypercapnic levels (8, 10, 27). However, we also found that termination of CH resulted in an increase in the acute ventilatory [H+] chemoreflex and a greater than predicted ventilation at 24 h of recovery. We have previously shown that CH elicits a sustained increase in steady-state ventilation and an initial suppression of the acute ventilatory [H+] chemoreflex that is returned to control within 7 days of CH. Taken together, our previous and current data support the conclusion that steady-state ventilation and the acute chemoreflex can adapt independently of each other (15). However, the data in this study show that the adaptation of the acute chemoreflex during deacclimatization is opposite the changes observed during the initial 24-h acclimatization period when the chemoreflex is significantly suppressed (15). The differences among these adaptations between acclimatization to and deacclimatization from CH suggest that these processes are mediated by different mechanisms of neuroplasticity. Herein, we report both sustained and novel neural adaptations during recovery (discussed below) that likely play a role in the increased sensitivity to acute changes in [H+] and the difference between actual versus predicted ventilation during deacclimatization.
Integrated physiological responses.
Overall, our findings presented in this study suggest a rapid return of most responses including metabolic rate, mixed arterial-expired Pco2 difference, most electrolytes, and cognitive function. However, the most significant change in physiological responses was hyponatremia at 24 h of recovery. This finding is consistent with reports of patients who experience acute exacerbation of COPD (AECOPD) and have hypercapnia reversed too quickly during mechanical ventilation. Hypercapnic COPD patients have been reported to be at a higher risk of a syndrome of inappropriate secretion of antidiuretic hormone (SIADH) (28). Whether the hyponatremia reported in this study is an effect of a compensatory mechanisms to maintain electrical neutrality, a consequence of SIADH, or both is unknown and extends beyond the scope of this study.
Brain Stem Site-Dependent Changes in Neuromodulation at 24 Hours of Recovery
Here, we found that the transition from hypercapnia to normocapnia resulted in an increase in excitatory neuromodulators (5-HT, NE, and DA) within the NTS but no other brain stem respiratory nuclei. Although our previous studies have shown the NTS has no clear role in the mechanisms of physiological adaptation to CH (17, 18), there is considerable evidence implicating mechanisms of ventilatory acclimatization to hypoxia involving neuromodulators and neurotransmitters within the NTS (29–31). The increases in neuromodulators also contrasts with our previous findings where no changes in neuromodulators were reported during either the acclimatization to or sustained periods of CH (18). The differences in changes within the NTS and levels of excitatory neuromodulators during acclimatization to and deacclimatization from hypercapnia further suggest that these processes depend on distinct neural mechanisms.
We speculate that the shift in the neuromodulatory “tone” within the central ventilatory control network to favor excitation may be a mechanism underlying the increased acute ventilatory [H+] chemoreflex and/or the difference between actual and predicted ventilation observed at 24 h of recovery. It has been well established that within the respiratory control network neuromodulators play an important role in determining the overall state of the respiratory network and are able to compensate for shifts in ventilatory stimuli, as previously noted in a phenomenon known as “compensatory neuromodulation” (32–35). As such, alterations in key neuromodulators such as 5-HT (36, 37) and NE (38–40) throughout respiratory control nuclei may contribute to determining ventilation and the ventilatory response to acute changes in respiratory stimuli, such as [H+]. Specifically, the NTS and associated excitatory neuromodulators may play a role in the increased acute ventilatory [H+] chemoreflex as well as the difference between actual versus predicted ventilation that we observed at 24 h of recovery. However, the exact mechanistic significance of NTS-specific neuromodulators has yet to be elucidated. Finally, the elevated levels of NE within the NTS may also contribute to the observed significant elevation in heart rate at 4 h of recovery, whereby increased catecholamine release within the NTS could result in an increased sympathetic tone within cardiovascular centers, resulting in elevated contractility and/or increased pacemaker firing at the level of the sinoatrial node.
Specific Markers of Neuroplasticity Were Unchanged during Deacclimatization
We have previously demonstrated that correlative changes in glutamate receptor expression/phosphorylation state, increased markers of neuroinflammation, and decreased markers of tryptophan metabolism within brain stem respiratory nuclei may contribute to the temporal pattern of ventilation during acclimatization to CH (17, 18). In the present study, we have shown that, unlike the acclimatization to hypercapnia (17, 18), markers of glutamate receptor activity and neuroinflammation remain unchanged from room air control during deacclimatization from hypercapnia, suggesting that these mechanisms likely do not contribute to the restoration of ventilation or other physiological systems to return to a prehypercapnic state. Moreover, the contrast in changes of markers of neuroplasticity during acclimatization to and deacclimatization from hypercapnia may contribute the differences in acute ventilatory chemoreflex adaptations during these time points and further supports our conclusion that these processes rely on distinct neural mechanisms. However, it should be noted that TPH and IDO do not significantly differ between 30 days of hypercapnia and 24 h of recovery. These results suggest that, indeed, some changes that occurred during CH were sustained in the recovery period. Consistent with our previous conclusions during CH, the reduction in TPH and IDO may be the result of the relatively selective reduction of serotonergic neurons within the brain stem.
An alternative hypothesis that could underlie the observed changes in ventilatory control, which were not studied herein, is redox and nitrosative signaling mechanisms. Others have demonstrated that molecular CO2 produces oxidative and nitrosative intermediates via reactions with peroxynitraite (ONOO−, O2-induced reactive species (41). During times of hypercapnic acidosis, the reaction between molecular CO2 and ONOO− has been implicated in the oxidative and nitrosative modulation of brain stem neurons including chemosensitive neurons. There is growing evidence that chemosensitive neurons, notably within the solitary complex (combined NTS and DMV) are stimulated by increased CO2 and/or O2-induced redox and nitrosative signaling mechanisms (41, 42). This change is of particular interest, given the hypercapnia and (very mild) hyperoxia that was experienced by our goat model during 6% InCO2. Future studies should address measurement of reactive species during both acclimatization and deacclimatization to determine the role of reactive species in these processes.
Tryptophan and 5-HT Metabolism
We previously reported that 30 days of hypercapnia results in a significant reduction in tryptophan enzymes tryptophan hydroxylase (TPH) and indolamine 2,3-dioxygenase (IDO) and neuronal marker NeuN (18) within rostral regions of the VLM and MR, which are primary sites of 5-HT-producing neurons. However, despite the putative loss of 5-HT neurons, there were no changes from control during CH in 5-HT levels within the VLM and MR or nuclei that are a target of direct serotonergic input (18). Our current data demonstrate that the transition from CH to acute normocapnia does not reverse the aforementioned changes in tryptophan metabolism but results in an increase in 5-HT within a target site (NTS) and a trending increase within a site of 5-HT synthesis (VLM). The elevation in 5-HT despite the sustained reduction of TPH relative to room air control in the acute recovery likely reflects alterations in 5-HT breakdown/metabolism, which requires uptake by the serotonin transporter (SERT) and enzymatic breakdown by monoamine oxidases (MAOs). Within the MR the ratio of HIAA to 5-HT is decreased, suggesting that the turnover or breakdown of 5-HT is reduced compared with previous time points and could be a contributor to the elevated levels of 5-HT within these sites in the absence of an increase in TPH. However, it remains unclear whether the reduced 5-HT turnover results from decreased enzymatic activity of MAOs or through decreased SERT, which were not measured herein but could explain these results.
Differences between Hypercapnic and Hypoxic Deacclimatization
Previous data (8, 14, 43) and those herein suggest that hypercapnic ventilatory deacclimatization occurs more rapidly than that for hypoxia (11, 12), suggesting different mechanisms of neuroplasticity may also mediate the deacclimatization from chronic hypoxia and hypercapnia. One potential difference in underlying mechanisms is the role of carotid chemoreceptor activity in these processes. Hypoxic stimulation of carotid chemoreceptors results in an acute and a time-dependent increase in carotid afferent fiber output and subsequently an acute and a time-dependent secondary progressive increase in ventilation (44, 45). In contrast, hypercapnia acutely increases carotid chemoreceptor activity and ventilation, but there is no time-dependent progressive increase in carotid chemoreceptor activity and ventilation with sustained hypercapnia (44). Indeed, mammals with denervated carotid bodies (CBD) exhibit an attenuated time-dependent ventilatory acclimatization to chronic hypoxia (VAH) (46, 47). Conversely, our unpublished data show that bilaterally carotid denervated goats had the same temporal adaptation of steady-state ventilation during hypercapnia as carotid-intact goats. Accordingly, we speculate that the carotid bodies likely play a minor role in not only the acclimatization to but also the deacclimatization from CH. In addition, there are also differences in monoamine neurotransmitter metabolism during acclimatization to and deacclimatization from CH (Figs. 5 and 6) and hypoxia (48). Overall, these data indicate that changes in excitatory neuromodulators need to be considered in attempting to account for physiological acclimatization to and deacclimatization from chronic hypoxia and hypercapnia.
We recognize that our data are drawn from exposure to one level of (mild) CH over a relatively short 30-day time-period, whereby levels were increased through increases in inspired CO2. Our findings are likely translatable to healthy individuals enclosed in a poorly ventilated space where there is an increase in CO2 in the inspired air but may also reasonably translate to patients with chronic hypoventilation. The effects of CH might be both time and dose dependent, as suggested by effects of CH in COPD and in other patients with more severe and more prolonged hypercapnia who have an attenuated ventilatory CO2/H+ chemoreflex (49, 50). Similarly, the effects of chronic hypoxia are also dose and time dependent (12, 51–53). High-altitude natives and lowlanders who moved and remained at high altitude before puberty also have an attenuated response to hypoxia (12, 54). Moreover, chronic disease-induced hypoxemia also decreases the ventilatory response to hypoxia (55). Finally, the effects of long-term hypoxia are not reversed upon normalization of arterial blood gases, indicating that permanent changes have occurred in the control of the chemoreflexes (51, 55, 56)
Summary and Importance of Present Findings
The data presented herein provide a comprehensive assessment of the physiological and neurochemical changes that occur in the abrupt termination of CH. First, our data suggest that abrupt termination of CH results in a rapid return of ventilation, blood gases, acid-base status, and most integrated physiological responses to prehypercapnic levels. However, there are indications of persistent changes in ventilatory control at 24 h of recovery, indicated by the significant increase in the acute ventilatory [H+] chemoreflex and steady-state V̇i significantly exceeding predicted ventilation. These findings also support previous observations that steady-state ventilation and the acute chemoreflex can adapt independently (15). Second, our data suggest that acclimatization to and deacclimatization from CH appear to rely on distinct mechanisms of neuroplasticity. Third, we have identified decreases in 5-HT neurons in the MR and VLM in two different experimental models of CH (18, 57), but herein we found that at 24 h of recovery from hypercapnia there was an increased 5-HT level within the NTS. If 5-HT is also increased in patients mechanically ventilated to alleviate CH, our findings have important implications in pharmacotherapies targeting the 5-HT system (i.e., Prozac), as reversing hypercapnia in these patients may further increase 5-HT to pathological levels, which may result in life-threatening consequences, such as serotonin syndrome. Finally, after 24 h of return to normocapnia, the increase in 5-HT occurs despite lower expression of TPH, which may suggest a decreased breakdown or reuptake of 5-HT. These decreases were evident at 30 days of hypercapnia and were sustained to 24 h of return to normocapnia, suggesting that CH may result in sustained alterations in 5-HT metabolism.
Overall, this study demonstrates that the abrupt termination of CH results in a time-dependent reestablishment of physiological functions. These data provide insights into the adaptive and/or maladaptive processes that may occur during CO2 weaning in mechanically ventilated-COPD patients following acute exacerbations. Furthermore, the data presented in the present study provide insights into changes that may occur in a wide range of clinical conditions (respiratory diseases, mechanical ventilation) and occupational settings (sealed or poorly ventilated environments) that vary in severity and duration of hypercapnia. Although the abrupt restoration of mild experimental hypercapnia was followed by a rapid physiological and cognitive recovery without major compromise or permanent negative effects, additional physiological and molecular studies characterizing the (mal)adaptive responses to more severe levels of CH are needed to better understand compensatory responses to exacerbations of CH in patients with respiratory disease.
GRANTS
Funding for this work was provided by the National Heart, Lung, and Blood Institute Grant HL-007852 and Department of Veterans Affairs Grant BX003284.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.J.B., N.J.B., M.R.H., L.G.P., and H.V.F. conceived and designed research; K.J.B., N.J.B., S.E.N., and L.G.P. performed experiments; K.J.B. analyzed data; K.J.B., N.J.B., M.R.H., and H.V.F. interpreted results of experiments; K.J.B. prepared figures; K.J.B. drafted manuscript; K.J.B., N.J.B., S.E.N., M.R.H., and H.V.F. edited and revised manuscript; K.J.B., N.J.B., S.E.N., M.R.H., L.G.P., and H.V.F. approved final version of manuscript.
REFERENCES
- 1.Groenewegen KH, Schols AM, Wouters EF. Mortality and mortality-related factors after hospitalization for acute exacerbation of COPD. Chest 124: 459–467, 2003. doi: 10.1378/chest.124.2.459. [DOI] [PubMed] [Google Scholar]
- 2.Kessler R, Faller M, Fourgaut G, Mennecier B, Weitzenblum E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 159: 158–164, 1999. doi: 10.1164/ajrccm.159.1.9803117. [DOI] [PubMed] [Google Scholar]
- 3.Costello R, Deegan P, Fitzpatrick M, McNicholas WT. Reversible hypercapnia in chronic obstructive pulmonary disease: a distinct pattern of respiratory failure with a favorable prognosis. Am J Med 102: 239–244, 1997. doi: 10.1016/S0002-9343(97)00017-X. [DOI] [PubMed] [Google Scholar]
- 4.Slenter RH, Sprooten RT, Kotz D, Wesseling G, Wouters EF, Rohde GG. Predictors of 1-year mortality at hospital admission for acute exacerbations of chronic obstructive pulmonary disease. Respiration 85: 15–26, 2013. doi: 10.1159/000342036. [DOI] [PubMed] [Google Scholar]
- 5.Bruno CM, Valenti M. Acid-base disorders in patients with chronic obstructive pulmonary disease: a pathophysiological review. J Biomed Biotechnol 2012: 915150, 2012., doi: 10.1155/2012/915150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das P, Bandyopadhay M, Baral K, Paul R, Banerjee AK. Dyselectrolytemia in chronic obstructive pulmonary diseases with acute exacerbation. Niger J Physiol Sci 25: 25–27, 2010. [PubMed] [Google Scholar]
- 7.Webster NR, Kulkarni V. Metabolic alkalosis in the critically ill. Crit Rev Clin Lab Sci 36: 497–510, 1999. doi: 10.1080/10408369991239286. [DOI] [PubMed] [Google Scholar]
- 8.Jennings DB, Chen CC. Ventilation in conscious dogs during acute and chronic hypercapnia. J Appl Physiol 41: 839–847, 1976. doi: 10.1152/jappl.1976.41.6.839. [DOI] [PubMed] [Google Scholar]
- 9.Jennings DB, Davidson JS. Acid-base and ventilatory adaptation in conscious dogs during chronic hypercapnia. Respir Physiol 58: 377–393, 1984. doi: 10.1016/0034-5687(84)90013-6. [DOI] [PubMed] [Google Scholar]
- 10.Schaefer KE, Hastings BJ, Carey CR, Nichols G Jr.. Respiratory Acclimatization to Carbon Dioxide. J Appl Physiol 18: 1071–1078, 1963. doi: 10.1152/jappl.1963.18.6.1071. [DOI] [PubMed] [Google Scholar]
- 11.Dempsey JA, Forster HV, Bisgard GE, Chosy LW, Hanson PG, Kiorpes AL, Pelligrino DA. Role of cerebrospinal fluid [H+] in ventilatory deacclimatization from chronic hypoxia. J Clin Invest 64: 199–205, 1979. doi: 10.1172/JCI109440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forster HV, Dempsey JA, Birnbaum ML, Reddan WG, Thoden J, Grover RF, Rankin J. Effect of chronic exposure to hypoxia on ventilatory response to CO 2 and hypoxia. J Appl Physiol 31: 586–592, 1971. doi: 10.1152/jappl.1971.31.4.586. [DOI] [PubMed] [Google Scholar]
- 13.Dempsey JA, Forster HV. Mediation of ventilatory adaptations. Physiol Rev 62: 262–346, 1982. doi: 10.1152/physrev.1982.62.1.262. [DOI] [PubMed] [Google Scholar]
- 14.Lambertson CJ, Gelfand R, Kemp RA. Dynamic Response Characteristics of Several CO2-Reactive Components of the Respiratory Control System. Oxford, UK: Blackwell, 1965. [Google Scholar]
- 15.Burgraff NJ, Neumueller SE, Buchholz K, Langer TM 3rd, Hodges MR, Pan L, Forster HV. Ventilatory and integrated physiological responses to chronic hypercapnia in goats. J Physiol 596: 5343–5363, 2018. doi: 10.1113/JP276666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol (1985) 94: 358–374, 2003. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- 17.Burgraff NJ, Neumueller SE, Buchholz KJ, Hodges MR, Pan L, Forster HV. Glutamate receptor plasticity in brain stem respiratory nuclei following chronic hypercapnia in goats. Physiol Rep 7: e14035, 2019. doi: 10.14814/phy2.14035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgraff NJ, Neumueller SE, Buchholz KJ, LeClaire J, Hodges MR, Pan L, Forster HV. Brain stem serotonergic, catecholaminergic, and inflammatory adaptations during chronic hypercapnia in goats. FASEB J 33: 14491–14505, 2019. doi: 10.1096/fj.201901288RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siesjo BK. Symposium on acid-base homeostasis. The regulation of cerebrospinal fluid pH. Kidney Int 1: 360–374, 1972. doi: 10.1038/ki.1972.47. [DOI] [PubMed] [Google Scholar]
- 20.Kondo T, Kumagai M, Ohta Y, Bishop B. Ventilatory responses to hypercapnia and hypoxia following chronic hypercapnia in the rat. Respir Physiol 122: 35–43, 2000. doi: 10.1016/s0034-5687(00)00134-1. [DOI] [PubMed] [Google Scholar]
- 21.Zouboules SM, Lafave HC, O'Halloran KD, Brutsaert TD, Nysten HE, Nysten CE, Steinback CD, Sherpa MT, Day TA. Renal reactivity: acid-base compensation during incremental ascent to high altitude. J Physiol 596: 6191–6203, 2018. doi: 10.1113/JP276973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dean JB. Theory of gastric CO2 ventilation and its control during respiratory acidosis: implications for central chemosensitivity, pH regulation, and diseases causing chronic CO2 retention. Respir Physiol Neurobiol 175: 189–209, 2011. doi: 10.1016/j.resp.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Reichart E, Claudon F, Sabliere S. [CO2 storage in various organs during chronic experimental hypercapnia (author's transl)]. Bull Eur Physiopathol Respir 12: 19–32, 1976. [PubMed] [Google Scholar]
- 24.Schaefer KE. Effects of increased ambient CO2 levels on human and animal health. Experientia 38: 1163–1168, 1982. doi: 10.1007/BF01959726. [DOI] [PubMed] [Google Scholar]
- 25.Incalzi RA, Gemma A, Marra C, Capparella O, Fuso L, Carbonin P. Verbal memory impairment in COPD: its mechanisms and clinical relevance. Chest 112: 1506–1513, 1997. doi: 10.1378/chest.112.6.1506. [DOI] [PubMed] [Google Scholar]
- 26.Schou L, Østergaard B, Rasmussen LS, Rydahl-Hansen S, Phanareth K. Cognitive dysfunction in patients with chronic obstructive pulmonary disease–a systematic review. Respir Med 106: 1071–1081, 2012. doi: 10.1016/j.rmed.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Davidson JS, Jennings DB. Measurement of total ammonia levels in plasma, whole blood, and cerebrospinal fluid of dogs using an ion-specific electrode. Can J Physiol Pharmacol 58: 550–556, 1980. doi: 10.1139/y80-090. [DOI] [PubMed] [Google Scholar]
- 28.Chalela R, González-García JG, Chillarón JJ, Valera-Hernández L, Montoya-Rangel C, Badenes D, Mojal S, Gea J. Impact of hyponatremia on mortality and morbidity in patients with COPD exacerbations. Respir Med 117: 237–242, 2016. doi: 10.1016/j.rmed.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Goiny M, Lagercrantz H, Srinivasan M, Ungerstedt U, Yamamoto Y. Hypoxia-mediated in vivo release of dopamine in nucleus tractus solitarii of rabbits. J Appl Physiol (1985) 70: 2395–2400, 1991. doi: 10.1152/jappl.1991.70.6.2395. [DOI] [PubMed] [Google Scholar]
- 30.Pamenter ME, Carr JA, Go A, Fu Z, Reid SG, Powell FL. Glutamate receptors in the nucleus tractus solitarius contribute to ventilatory acclimatization to hypoxia in rat. J Physiol 592: 1839–1856, 2014. doi: 10.1113/jphysiol.2013.268706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pamenter ME, Nguyen J, Carr JA, Powell FL. The effect of combined glutamate receptor blockade in the NTS on the hypoxic ventilatory response in awake rats differs from the effect of individual glutamate receptor blockade. Physiol Rep 2: e12092, 2014. doi: 10.14814/phy2.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doi A, Ramirez J-M. Neuromodulation and the orchestration of the respiratory rhythm. Respir Physiol Neurobiol 164: 96–104, 2008. doi: 10.1016/j.resp.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doi A, Ramirez J-M. State-dependent interactions between excitatory neuromodulators in the neuronal control of breathing. J Neurosci 30: 8251–8262, 2010. doi: 10.1523/JNEUROSCI.5361-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forster HV, Julius H. Comroe Distinguished Lecture: Interdependence of neuromodulators in the control of breathing. J Appl Physiol (1985) 125: 1511–1525, 2018. doi: 10.1152/japplphysiol.00477.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langer TM 3rd, Neumueller SE, Crumley E, Burgraff NJ, Talwar S, Hodges MR, Pan L, Forster HV. State-dependent and -independent effects of dialyzing excitatory neuromodulator receptor antagonists into the ventral respiratory column. J Appl Physiol (1985) 122: 327–338, 2017. doi: 10.1152/japplphysiol.00619.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodges MR, Richerson GB. Medullary serotonin neurons and their roles in central respiratory chemoreception. Respir Physiol Neurobiol 173: 256–263, 2010. doi: 10.1016/j.resp.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hodges MR, Richerson GB. The role of medullary serotonin (5-HT) neurons in respiratory control: contributions to eupneic ventilation, CO2 chemoreception, and thermoregulation. J Appl Physiol (1985) 108: 1425–1432, 2010. doi: 10.1152/japplphysiol.01270.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viemari JC. Noradrenergic modulation of the respiratory neural network. Respir Physiol Neurobiol 164: 123–130, 2008. doi: 10.1016/j.resp.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 39.Viemari JC, Ramirez JM. Norepinephrine differentially modulates different types of respiratory pacemaker and nonpacemaker neurons. J Neurophysiol 95: 2070–2082, 2006. doi: 10.1152/jn.01308.2005. [DOI] [PubMed] [Google Scholar]
- 40.Zanella S, Doi A, Garcia AJ, Elsen F, Kirsch S, Wei AD, Ramirez J-M. When norepinephrine becomes a driver of breathing irregularities: how intermittent hypoxia fundamentally alters the modulatory response of the respiratory network. J Neurosci 34: 36–50, 2014. 10.1523/JNEUROSCI.3644-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dean JB. Hypercapnia causes cellular oxidation and nitrosation in addition to acidosis: implications for CO2 chemoreceptor function and dysfunction. J Appl Physiol 108: 1786–1795, 2010. doi: 10.1152/japplphysiol.01337.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciarlone GE, Hinojo CM, Stavitzski NM, Dean JB. CNS function and dysfunction during exposure to hyperbaric oxygen in operational and clinical settings. Redox Biol 27: 101159, 2019. doi: 10.1016/j.redox.2019.101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clark JM, Sinclair RD, Welch RE. Rate of acclimitization to chronic hypercapnia in man. In: Underwater Physiology. Proceedings of the Fourth Symposium on Underwater Physiology, edited by Lambertson CJ. New York: Academic, 1971, p. 399–408. [Google Scholar]
- 44.Bisgard GE, Busch MA, Daristotle L, Berssenbrugge AD, Forster HV. Carotid body hypercapnia does not elicit ventilatory acclimatization in goats. Respir Physiol 65: 113–125, 1986. doi: 10.1016/0034-5687(86)90010-1. [DOI] [PubMed] [Google Scholar]
- 45.Dwinell MR, Janssen PL, Pizarro J, Bisgard GE. Effects of carotid body hypocapnia during ventilatory acclimatization to hypoxia. J Appl Physiol (1985) 82: 118–124, 1997. doi: 10.1152/jappl.1997.82.1.118. [DOI] [PubMed] [Google Scholar]
- 46.Forster HV, Bisgard GE, Klein JP. Effect of peripheral chemoreceptor denervation on acclimatization of goats during hypoxia. J Appl Physiol Respir Environ Exerc Physiol 50: 392–398, 1981. doi: 10.1152/jappl.1981.50.2.392. [DOI] [PubMed] [Google Scholar]
- 47.Forster HV, Bisgard GE, Rasmussen B, Orr JA, Buss DD, Manohar M. Ventilatory control in peripheral chemoreceptor-denervated ponies during chronic hypoxemia. J Appl Physiol 41: 878–885, 1976. doi: 10.1152/jappl.1976.41.6.878. [DOI] [PubMed] [Google Scholar]
- 48.Olson EB Jr, Vidruk EH, McCrimmon DR, Dempsey JA. Monoamine neurotransmitter metabolism during acclimatization to hypoxia in rats. Respir Physiol 54: 79–96, 1983. doi: 10.1016/0034-5687(83)90115-9. [DOI] [PubMed] [Google Scholar]
- 49.Gelb AF, Klein E, Schiffman P, Lugliani R, Aronstam P. Ventilatory response and drive in acute and chronic obstructive pulmonary disease. Am Rev Respir Dis 116: 9–16, 1977. doi: 10.1164/arrd.1977.116.1.9. [DOI] [PubMed] [Google Scholar]
- 50.Kepron W, Cherniack RM. The ventilatory response to hypercapnia and to hypoxemia in chronic obstructive lung disease. Am Rev Respir Dis 108: 843–850, 1973. doi: 10.1164/arrd.1973.108.4.843. [DOI] [PubMed] [Google Scholar]
- 51.Lahiri S, Kao FF, Velásquez T, Martinez C, Pezzia W. Irreversible blunted respiratory sensitivity to hypoxia in high altitude natives. Respir Physiol 6: 360–374, 1969. doi: 10.1016/0034-5687(69)90034-6. [DOI] [PubMed] [Google Scholar]
- 52.Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol 112: 123–134, 1998. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- 53.Zhuang J, Droma T, Sun S, Janes C, McCullough RE, McCullough RG, Cymerman A, Huang SY, Reeves JT, Moore LG. Hypoxic ventilatory responsiveness in Tibetan compared with Han residents of 3,658 m. J Appl Physiol (1985) 74: 303–311, 1993. doi: 10.1152/jappl.1993.74.1.303. [DOI] [PubMed] [Google Scholar]
- 54.Weil JV, Byrne-Quinn E, Sodal IE, Filley GF, Grover RF. Acquired attenuation of chemoreceptor function in chronically hypoxic man at high altitude. J Clin Invest 50: 186–195, 1971. doi: 10.1172/JCI106472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sorensen SC, Severinghaus JW. Respiratory insensitivity to acute hypoxia persisting after correction of tetralogy of Fallot. J Appl Physiol 25: 221–223, 1968. doi: 10.1152/jappl.1968.25.3.221. [DOI] [PubMed] [Google Scholar]
- 56.Vargas M, León-Velarde F, Monge CC, Palacios JA, Robbins PA. Similar hypoxic ventilatory responses in sea-level natives and high-altitude Andean natives living at sea level. J Appl Physiol (1985) 84: 1024–1029, 1998. doi: 10.1152/jappl.1998.84.3.1024. [DOI] [PubMed] [Google Scholar]
- 57.Miller JR, Neumueller S, Muere C, Olesiak S, Pan L, Hodges MR, Forster HV. Changes in neurochemicals within the ventrolateral medullary respiratory column in awake goats after carotid body denervation. J Appl Physiol (1985) 115: 1088–1098, 2013. doi: 10.1152/japplphysiol.00293.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]