

Optical barcoding reveals enhanced TNFα activation and polyclonality in lung, but not liver, metastases from breast cancer.
Abstract
Intratumoral heterogeneity is a driver of breast cancer progression, but the nature of the clonal interactive network involved in this process remains unclear. Here, we optimized the use of optical barcoding to visualize and characterize 31 cancer subclones in vivo. By mapping the clonal composition of thousands of metastases in two clinically relevant sites, the lungs and liver, we found that metastases were highly polyclonal in lungs but not in the liver. Furthermore, the transcriptome of the subclones varied according to their metastatic niche. We also identified a reversible niche-driven signature that was conserved in lung and liver metastases collected during patient autopsies. Among this signature, we found that the tumor necrosis factor–α pathway was up-regulated in lung compared to liver metastases, and inhibition of this pathway affected metastasis diversity. These results highlight that the cellular and molecular heterogeneity observed in metastases is largely dictated by the tumor microenvironment.
INTRODUCTION
Breast cancer is a heterogeneous disease, associated with a large range of clinical outcomes within and across molecular subtypes. These disparities are mostly dependent on the distinct ability of individual cancer cells to metastasize to distant organs and to escape standard therapies (1). Recent studies based on single-cell sequencing have shown that patient samples displayed varying degrees of heterogeneity (2–4) and that tumors with a high degree of intratumoral heterogeneity are often associated with poor outcomes because of their likelihood of containing aggressive and drug-resistant clones (1). However, the dynamics of how seeding occurs in specific organs is still unclear, as are the molecular features of aggressive seeders.
A recent study using a large dataset of matched primary tumors and metastases suggested that cells disseminating early can lack metastasis-specific driver mutations and that the clonality of metastases depends on the organ of dissemination and the presence of treatment (5). Several cellular and molecular processes may be accountable for the large degree of intra- and intermetastatic heterogeneity observed in breast cancer metastases. First, only a subset of cells present in primary tumors are able to spread to specific organs (6–8), and the selective growth of metastases is likely to be determined by an interplay between the intrinsic characteristics of metastatic clones and the properties of the metastatic niche. Second, disseminated cells can genetically evolve independently of primary tumors, a process known as parallel progression (9), and the genomic, transcriptomic, and epigenetic characteristics of metastases might be influenced by the tumor microenvironment and by standard therapies (5, 10, 11). Last, metastatic clones can interact within their niche, where both monoclonal and polyclonal seeding have been described (5, 10, 12, 13). These cellular interactions may confer a survival advantage or disadvantage to cancer cells, depending on whether clonal cooperation or competition is involved (14).
Understanding the clonal network that underpins tumor spread in different organs is of great clinical interest but remains a challenge when using patient samples. Tumor biopsies are often collected from patients who have previously received multiple rounds of treatment, which is likely to affect the clonality of metastases (5). Furthermore, needle biopsies may not always provide a complete representation of the overall tumor and metastatic spread. Mouse models in which metastases can be isolated from whole organs are a suitable alternative. In this context, single-cell tracking methods are emerging as a powerful tool to disentangle the complexity of intratumoral heterogeneity, as they provide a unique opportunity to track the fate of individual cancer cells and study the spatiotemporal evolution of their progenies. One such approach, based on genetic barcoding, allowed the detection of cancer cells with differential abilities to extravasate, survive, and grow in different organs. As an example, barcoding of cancer cells from the breast using libraries of thousands to millions of genetic tags has demonstrated that cell lines and patient-derived xenografts (PDXs) contain multiple clones of cancer cells with a wide range of characteristics that confer different cellular fates following orthotopic engraftment (15–19). Unexpectedly, primary tumors and metastases were highly heterogeneous despite having different evolutionary life spans and were found to contain both minor and dominant clones at varying frequencies (15–18).
Recently, an optical barcoding approach based on a multicolor panel of Lentiviral Gene Ontology (LeGO) vectors was developed to enable better spatiotemporal tracking of individual clones within heterogeneous populations of tumor cells (20–23). Despite the limited number of optical barcodes available, a key advantage of this approach over genetic barcoding is the ability to image, quantify, and sort labeled populations by microscopy and flow cytometry at the single-clone level for further molecular characterization. Cell transduction with specific viral titers results in cells expressing specific combinations of fluorescent tags, generating up to 63 colors and allowing the long-term in vitro tracking of labeled cells and their progeny (22, 24). However, the number of colors that can be used to track cancer cells in vivo is limited. In human cells, the combination of the three LeGO colors red-green-blue (RGB) resulting in seven colors was used to study the relapse of primary tumors in head and neck squamous cell carcinoma (25) and the spatiotemporal distribution of clones in colorectal primary tumors (26). However, increasing the number of color combinations that can be detected and quantified in vivo is a challenge because of the spectral overlap of fluorescent tags.
In this study, we used optical barcoding to explore metastatic heterogeneity in the context of triple-negative breast cancer (TNBC), as this molecular subtype is associated with poor prognosis and a higher risk of recurrence in the 5 years following diagnosis, compared to other subtypes (27). Mounting evidence suggests that clonal heterogeneity plays an important role in metastatic growth in this subtype (18, 28), with high rates of metastatic spread to visceral organs, such as lungs and liver (29, 30). To investigate the way TNBC cells interact and adapt to these organs, we optimized the LeGO technology to visualize and quantify in vivo 31 “barcoded clones” (defined here as subpopulations of cells arising from cancer cells and their progenies barcoded with a specific set of fluorescent tags, regardless of their genomic profile). Our results indicate that this cell tracking strategy allows us not only to compare the dynamics of metastatic seeding between organs using high-resolution imaging but also to identify new site-driven gene expression signatures associated with lung and liver metastases.
RESULTS
Labeling with BSVTK enables the tracking of 31 barcoded subclones at the single-cell resolution
Genetic barcoding has provided insight into clonal compositions at experimental end points. However, the lack of an endogenous fluorescent reporter means that individual clones cannot be directly visualized, nor isolated by flow cytometry to study their molecular characteristics. Meanwhile, optical barcoding based on RGB barcoding can only visualize a very small number of distinctly marked clones in vitro (20, 21) and primary tumors in vivo (25, 26). To address these limitations, we developed a combinatorial strategy based on the five fluorophores enhanced blue fluorescent protein 2 (eBFP2), tSapphire, Venus, tdTomato, and Katushka (BSVTK) and a corresponding visualization and quantification pipeline to analyze up to 31 barcoded subclones by confocal imaging as well as flow cytometry.
We used the MDA-MB-231 cell line that was established from a malignant pleural effusion of a patient with TNBC and therefore was likely to contain a large number of subclones able to metastasize in vivo. RNA sequencing (RNA-seq) analysis of 10,000 single cells inferred genomic heterogeneity based on extensive copy number variations (CNVs) (Fig. 1A), reminiscent of similar results obtained with other human cancer cell lines (31). We infected MDA-MB-231 cells with LeGO viruses containing the individual BSVTK tags and exploited a multiplicity of infection of 50 to 75% to allow individual cells to integrate different combinations of each fluorophore (Fig. 1B) (22, 24, 32). This resulted in the reproducible identification of 31 unique combinations, hereafter referred to as colors, and associated barcodes (fig. S1A). Spectral deconvolution was used to specifically separate the emission spectra of the five fluorescent BSVTK tags and generate five-channel confocal images (Fig. 1C and fig. S1, B and C). We processed the raw images by first thresholding the five channels and then attributing a value from 1 to 31 corresponding to the fluorescent protein compositions (fig. S1, A and C). The virtual 31-color image was quantified by measuring the volume of each color (fig. S1C) and calculating the relative contribution of each color to the entire imaged population (Fig. 1E). Using a second strategy, the same barcoded populations were analyzed by flow cytometry. Following optimization of gating thresholds, all cells were individually classified as either “positive” or “negative” for the expression of each fluorescent protein (fig. S1D). This resulted in 31 different gates corresponding to all possible combinations (Fig. 1D and fig. S1A). We confirmed that the quantification of color proportions within a given heterogeneous cell population using imaging and flow cytometry yielded similar results (Fig. 1E; Pearson correlation r = 0.8454). We also confirmed that expression of the fluorescent tags did not affect the proliferation of labeled subclones (fig. S1, E and F), nor their colony-forming ability in vitro (fig. S1, G and H), nor their sensitivity to chemotherapeutic drugs (fig. S1I).
Fig. 1. Heterogeneity of MDA-MB-231 cells highlighted by optical barcoding.
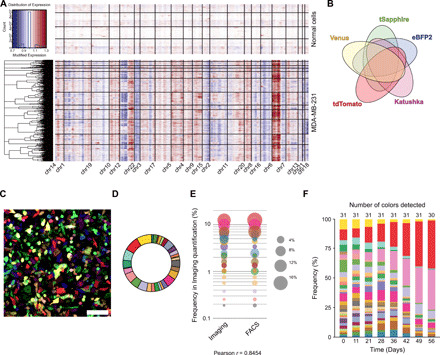
(A) Analysis of CNVs inferred from single-cell RNA-seq analysis from normal human mammary cells [top (66)] and MDA-MB-231 cells (bottom). Individual cells are represented on the y axis and the different genomic regions along the x axis. (B) Venn diagram representing the 31 possible combinations generated by expression of five fluorescent tags: eBFP2, tSapphire, Venus, tdTomato, and Katushka. (C) Representative confocal image of BSVTK-labeled cells. Scale bar, 100 μm. (D) Example of a pie chart representing the percentage [detected by fluorescence-activated cell sorting (FACS)] of each color-coded population in MDA-MB-231 cells transduced with optical barcodes for 48 hours. (E) Comparison between the quantification of each color-coded population obtained by either imaging or FACS. Each dot represents a subpopulation of cells with a given color. The size of the dot corresponds to the percentage of cells carrying this color within a population, analyzed by confocal imaging or FACS. (F) FACS analysis of the same population of cells maintained in 2D culture for 56 days. The frequency of each barcoded subclone is indicated on the y axis and the number of days on the x axis. The total number of barcoded subclones detected is indicated at the top.
To homogenize the population while increasing the genomic purity of each color-coded population, we collected 100 cells from each of the 31 differentially barcoded cells by flow cytometry (3100 cells in total), 48 hours after transduction with the lentiviruses. The resulting mixture was then propagated in two-dimensional (2D) tissue culture. Over the next 56 days, we observed a progressive clonal drift, with the number of optical tags decreasing and some barcoded subclones becoming dominant over multiple passages (Fig. 1F). This observation suggested that the BSVTK-labeled subclones displayed differential abilities to proliferate and expand in vitro. Overall, these results indicated that optically labeled MDA-MB-231 cells harbored some heterogeneity at both the genomic and phenotypic levels.
Dominant barcoded subclones in primary tumors remain dominant in metastases
To gain insight into the overall dynamics of clonal distribution during the metastatic process, we injected homogeneous batches of expanded BSVTK-labeled cells into the mammary fat pads of NOD-SCID-IL2Rγc−/− (NSG) mice and allowed metastatic outgrowth by resecting primary tumors when they reached 100 mm3 (fig. S2, A to C). We readily detected metastases in the lungs and liver (fig. S2, A and D) but occasionally also observed spread to the kidney and lymph nodes (not shown). To assess the inter- and intraclonal heterogeneity of the BSVTK-labeled metastatic subclones, we fluorescence-activated cell sorting (FACS)–purified cells from five different colors in the lungs and analyzed their genomic diversity based on CNVs inferred from single-cell RNA-seq (scRNA-seq) (fig. S2E). Our results indicated that these subclones had distinct CNV profiles and that cells of a given color were largely similar in terms of CNV profile, with few exceptions. These exceptions could be due to a lack of purity in the FACS or due to the fact that several cells that were genomically different received, by chance, the same color when transduced with the BSVTK lentiviruses. It could also be attributed to the genomic evolution of the barcoded subclones after in vitro and in vivo amplification, as previously described (31).
Comparison of the barcode repertoire in primary tumors and metastases by imaging (Fig. 2A) and flow cytometry (Fig. 2, B and C) revealed a large degree of variability between minor and dominant barcoded subclones. Consistent with previous studies using genetic barcoding in PDXs (16) and cell lines (17), we found that the number of barcoded subclones (fig. S2F) and the Shannon diversity indexes (accounting for the abundance and evenness of the subclones) (fig. S2G) were lower in metastatic sites compared to primary tumors. Unexpectedly, however, we found that some of the less abundant barcoded subclones in vitro became dominant in vivo, with the most dominant subclone in the primary tumor and metastatic sites (Fig. 2B, light blue) accounting for less than 2% of all the cells propagated in vitro (Fig. 2, B and C, and fig. S2H).
Fig. 2. Visualization and quantification of metastatic burden at single-cell resolution in whole organs.
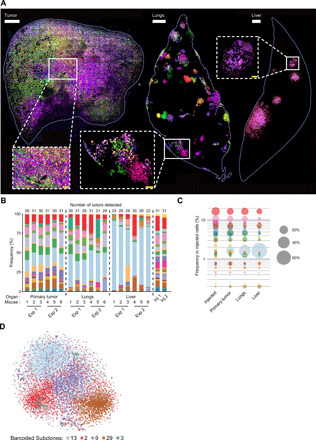
(A) Representative images obtained by confocal imaging of the primary tumor (left), the lungs (middle), and the liver (right). Insets, magnified images. The white bars represent 500 μm and the yellow bars 100 μm. (B) Stacked histogram indicating the frequency (%) of each color-coded subclone detected in the primary tumors and lung and liver metastases of six different recipient mice. These results summarize two independent experiments (Exp 1 and Exp 2), with the cell population analyzed at the time of injection on both occasions (Inj 1 and Inj 2). Organs were harvested at metastatic end point and digested before quantification using flow cytometry. (C) Average subclonal frequency in the injected population, the primary tumor, and the lung and liver metastases from the six mice shown in (B). Each dot represents a color-coded subclone, and the size of the dot represents the frequency of the subclone. The y axis represents the frequency of each subclone, ranked according to their frequency in the injected population (D) t-distributed stochastic neighbor embedding (t-SNE) (perplexity = 50) of 10,418 single cells from five barcoded subclones (subclone 13, 3224 cells; 2, 2712 cells; 9, 2566 cells; 29, 1778 cells; 3, 140 cells) representing a feature set (table S1) that was derived through a differential expression analysis between the dominant subclone 13 and the minor subclone 29.
Comparison of the barcode repertoire in lung and liver metastases revealed a notable difference in clonal heterogeneity between these organs. While we observed an overall strong correlation in subclonal frequency between lung and liver metastases (fig. S2I; Pearson correlation of 0.908), the total number of barcodes (fig. S2F) and associated Shannon diversity index (fig. S2G) were smaller in the liver compared to the lungs. We observed a large variability between the frequencies of dominant and minor barcoded subclones in the liver, while the frequencies of the subclones were more equally distributed in the lungs (Fig. 2, B and C, and fig. S2G). These results suggested that the subclonal composition of metastases was related to the nature of the metastatic site, the liver being more selective than the lungs. In addition, the analysis across our entire cohort of recipients revealed a notably similar distribution in each organ (primary tumors, liver, or lungs) in terms of numbers and size of barcoded subclones, when comparing different mice (Fig. 2B and fig. S2F). These results indicated that the ability of individual subclones to survive, proliferate, and spread in a given organ were likely to be intrinsically programmed rather than stochastic, as described previously in murine breast cancer cells (17).
To confirm that individual barcoded subclones had different transcriptomic programs, we first compared the scRNA-seq profile of the most aggressive barcoded subclone (subclone 13) to that of a minor subclone (subclone 29). The resulting feature set, including 609 differentially expressed genes (table S1), was then applied to the five barcoded subclones selected from lung metastases (13, 2, 29, 3, and 9) and was expected to capture transcriptional differences across barcoded subclones, spanning from minor to dominant subclones. This feature set was sufficient to cluster cells according to their clonal origin (Fig. 2D). While the number of cells from barcoded subclone 3 was low and might be insufficient to provide robust information, we found that subclones 13, 2, 9, and 29 showed distinct transcriptomic programs, and some of these differences might account for their phenotypic differences.
Intrametastastic heterogeneity depends on the metastatic site
Because a large number of barcoded subclones were able to metastasize to the lungs and liver, we investigated their ability to cluster spatially within individual metastases of three mice. We quantified individual barcoded subclones in a total of 1992 lung and 510 liver metastases (Fig. 3, A and B) by confocal imaging using the analytic pipeline described in fig. S3A and validated this approach against flow cytometry enumeration of metastatic cells dissociated from the same organs (fig. S3, B and C).
Fig. 3. Analysis of the intrametastatic heterogeneity of lung and liver metastases.
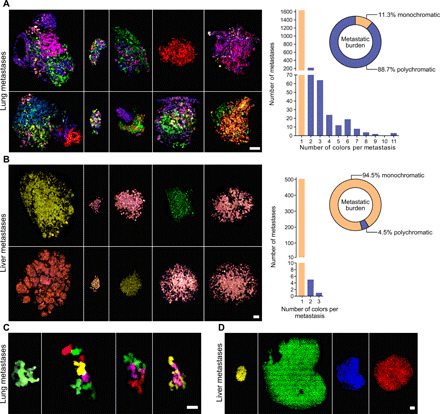
(A and B) Representative confocal images (left) and color quantification (right) for metastases detected in the lungs (A) or the liver (B) of mice engrafted with BSVTK MDA-MB-231 cells. Left: Scale bars, 100 μm. Right: Bar graph represents the number of metastases containing one (yellow bars) or multiple (blue bars) colors; pie charts represent the proportion of total metastatic burden associated with monochromatic (yellow) or polychromatic (blue) metastases. (C and D) Representative confocal images of some of the largest metastases detected in the lungs (C) and the liver (D) of mice engrafted with cancer cells from the BSVTK-barcoded PDX-0066. Scale bars, 20 μm (C) and 200 μm (D).
The analysis of the resulting dataset revealed that most of the lung and liver metastases were monochromatic, although we noted that the percentage of polychromatic metastases was significantly higher in the lungs (17%) compared to the liver (1.2%) (Fig. 3, A and B). In addition, the polychromatic lung metastases contained up to 11 colors, whereas the few polychromatic liver metastases were mostly bicolor, when analyzing comparable overall metastasis volumes between the two organs (fig. S3D). Notably, the 17% polychromatic lung metastases contributed to 88.7% of the overall metastatic burden in lungs (Fig. 3A). This result indicated that despite their smaller number, polychromatic metastases were the main contributors to metastatic burden in this organ. This distribution was not observed in the liver, with polychromatic metastases contributing to only 4.5% of the metastatic burden. This observation was reproduced across all mice, highlighting the reproducibility and the nonstochasticity of these results (fig. S3, E and F).
To extend the validity of the results obtained on the established MDA-MB-231 cell line, we used two TNBC PDXs. PDX-0066 and PDX KCC-P-4295 were generated by infection of tumor cells with BSVTK lentiviruses, before transplantation in the mammary fat pad of NSG mice. PDX-0066 metastasized to the liver and, to a lesser extent, the lungs (fig. S4A). Although the number and size of metastases in lungs remained small (fig. S4A), we confirmed using imaging that 35.4% of the lung metastases were polychromatic (fig. S4B), while 99.996% of liver metastases were monochromatic (fig. S4C). Likewise, analysis of the PDX KCC-P-4295–derived lung metastases confirmed their extensive polyclonality (85.5%; fig. S4, D and E), although this PDX did not metastasize to the liver. It is likely that the low percentage of polyclonality observed in the lung metastases of PDX-0066 is due to their small size.
Individual barcoded subclones have different intrinsic abilities to interact within lung metastases
Previous studies indicated that cancer clones were able to interact in polyclonal metastases (14, 33). First, we investigated whether the extent of heterogeneity observed in individual lung and liver metastases correlated with their overall tumor mass (Fig. 4A). Using the large imaging dataset generated with MDA-MB-231–labeled cells (described in fig. S3A), we observed a positive correlation between the size of the lung lesions and the number of colors per lesion (Fig. 4A). By contrast, this correlation was not observed in the liver, where only a small number of polychromatic metastases were detected, regardless of the size of the lesions (Figs. 3B and 4A).
Fig. 4. Mapping clonal interactions within metastases.
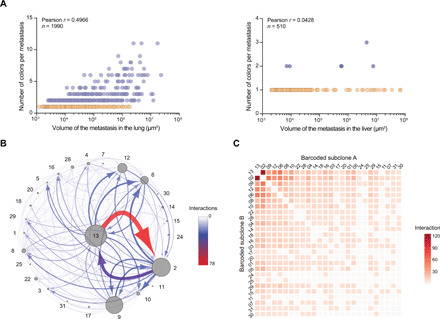
(A) Correlation between the number of colors detected per metastasis and the volume of the metastases in the lungs (left) and liver (right). Each dot represents a metastasis. Monochromatic metastases are represented in orange and polychromatic metastases in blue. The number of metastases included in this dataset (from six mice) and the Pearson correlation coefficient are indicated in the figure. (B) Interactome map summarizing the frequency of colocalization events between barcoded subclones within lung metastases. Each subclone is represented by a dot and a number. The size of each dot correlates with the total volume occupied by the corresponding subclone in the dataset analyzed by confocal imaging. Arrows linking two dots indicate that the corresponding two barcoded subclones are colocalized within individual metastases, with the thickness and the color of the line (as indicated in the scale) reflecting the number of times that this colocalization event was detected across all metastases, and the arrows pointing from the dominant to the minor subclone. (C) Heatmap representing the number of colocalization events between all the barcoded subclones detected in lungs. The number of times two subclones were colocalized was quantified from the imaging dataset, and the values are represented in this heatmap.
Second, we interrogated our data for the ability of individual barcoded subclones to cluster within given lung metastases (Fig. 4, B and C). Among the three most overrepresented barcoded subclones, we observed a preferred colocalization of subclone 13 with subclone 2 rather than subclone 9, although both occurred at similar abundance (Fig. 4B). This observation suggested that while abundance was a major determinant for clustering of specific barcoded subclones, the nature of the subclones was also likely to have an impact (Fig. 4, B and C). Furthermore, this pattern of clustering was nonstochastic as we consistently observed it across different recipient mice (fig. S3G). Although the barcoded subclones 2 and 13 were also highly abundant in the liver, we never observed their colocalization in this organ. Collectively, these results indicated that the probability of colocalization of subclones within metastases is likely dependent on their abundance, their identity, and their metastatic site.
Metastatic barcoded subclones acquire a reversible, site-specific gene expression profile
As the same barcoded subclones (subclones 2 and 13) contributed to many metastases in lungs and liver, we then investigated whether these subclones retained the same expression profile across the two sites. Principal components analysis (PCA) of these samples showed that the main dimension of variation separated the two subclones (seen on principal component 1), while the second dimension of variation separated each of the subclones by the site of metastasis (principal component 2) (Fig. 5A and fig. S5A). We therefore compared the gene expression pattern of barcoded subclones 2 and 13 in lungs versus liver across three mice to identify a site-driven signature of 1366 differentially expressed genes, referred to as BSVTK feature set (Fig. 5B, fig. S5B, and table S2). We noted that, when taking into consideration all the genes included in this feature set, the profile of the primary tumors was closer to the profile of liver metastases compared to lung metastases (Fig. 5B).
Fig. 5. Impact of the tumor microenvironment on the transcriptomic landscape of metastatic subclones.
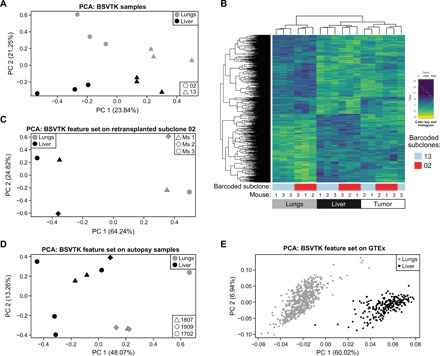
(A) PCA plot demonstrating the transcriptomic differences between subclones 2 (round) and 13 (triangle) depending on the organs (lungs in gray and liver in black) from which they were isolated. (B) Heatmap of the differentially expressed genes in barcoded subclones 2 (red) and 13 (blue) between lung and liver metastases, in three mice. The expression level of these genes (referred to as BSVTK feature set) was also depicted in primary tumors from matching animals. (C) PCA plot showing the separation of lung and liver metastases based on genes of the BSVTK feature set, from three second-generation mice (Ms 1, Ms 2, and Ms 3), generated by mammary fat pad retransplantation of cells from subclone 2 that were isolated from lung metastasis of first-generation mice. (D) PCA plot representing the analysis of differentially expressed genes from the BSVTK feature set in lung (gray) and liver (black) metastases collected at autopsy from three patients with metastatic breast cancer (patients 1807, 1909, and 1702). These data were subset to the differentially expressed genes in the BSVTK feature set, demonstrating the ability of this feature set to distinguish between lung and liver samples. (E) PCA plot representing the analysis of the differentially expressed genes from the BSVTK feature set in GTEx normal lung (gray) and liver (black) tissue. This subset of the BSVTK feature set enabled the separation of these normal tissues by organ.
A testable hypothesis is that this site-specific BSVTK feature set should be reversible. Thus, we FACS-purified subclone 2 cells recovered from lung metastases and injected them into the mammary fat pad of NSG hosts. These mice became sick from lung and liver metastases, suggesting that the polyclonality observed previously was not a requirement for the metastatic spread of this barcoded subclone (fig. S5, C and D). PCA of transcriptomic data from these newly developed metastases showed that the BSVTK feature set (Fig. 5B and table S2) clearly separated these samples according to the two metastatic locations (Fig. 5C). We inferred from these results that the transcriptomic differences between lung and liver metastases were less likely to result from clonal selection but rather dictated by the tumor microenvironment, which acts on the inherently plastic subclones.
To confirm the relevance of this site-dependent signature in human hosts, we collected four lung and seven liver metastases from three patients with breast cancer enrolled in the breast origin cancer tissue donated (BROCADE) rapid autopsy program. We found that the BSVTK feature set (table S2) separated patient metastases according to the organ from which they were recovered (Fig. 5D) and that this signature might not only be restricted to TNBC (i.e., patient 1702) but also apply to estrogen receptor–positive breast cancer (i.e., patients 1807 and 1909) (table S3). Therefore, these results confirmed that these genes were also differentially expressed between lung and liver in a human context. Furthermore, when applied to the transcriptome of healthy human lung and liver tissues from the publicly available genotype-tissue expression (GTEx) dataset (34), this set of genes was also sufficient to separate the samples on the basis of their tissue of origin in a PCA (Fig. 5E). This demonstrated that genes that distinguished lung and liver metastases (Fig. 5, A to D) also enabled the discrimination between phenotypically normal lung and liver tissues (Fig. 5E). These results suggest that breast cancer cells shared some of the transcriptomic patterns of the microenvironment in which they are placed.
The tumor necrosis factor–α pathway is strongly up-regulated in lung metastases
To better understand how the transcriptomic response of the metastases might relate to biological processes specific to given organs, we used gene ontology (GO) enrichment of differentially expressed genes between lung and liver metastases (fig. S6A) or genes selectively up-regulated in one type of metastases when compared to primary tumor samples (fig. S6, B and C). We found that genes involved in extracellular matrix organization or cell junction were up-regulated in the liver compared to the lungs (fig. S6A). When comparing metastases to primary tumors, the GO analysis revealed an enrichment of genes associated with oxidative phosphorylation in lungs (fig. S6B) and with carbohydrate metabolic process in the liver (fig. S6C). These results, corroborating and extending results from previous studies (35, 36), suggested that these metabolic changes might be driven by the tumor microenvironment. The hallmark analysis revealed a significant up-regulation of the tumor necrosis factor–α (TNFα) signaling pathway associated with canonical nuclear factor κB (NFκB) target genes in lungs when compared to liver metastases using the BSVTK dataset (Fig. 6A), the BROCADE dataset (Fig. 6B), and the aforementioned retransplantation experiment involving subclone 2 (Fig. 6C). Likewise, all three datasets also displayed elevated expression of the inflammatory response gene set that is part of TNFα- and NFκB-mediated inflammatory response (37). Comparing the genes from the enriched gene sets shared between these datasets (Fig. 6D and fig. S7, A and B), we found that a large number of genes from the TNFα pathway overlapped between the BSVTK and BROCADE datasets (Fig. 6D) and, to a lesser extent, between the BSVTK-labeled cells and the retransplantation dataset (fig. S7B). Furthermore, an enrichment analysis against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database confirmed that the corresponding TNFα pathway was also significantly up-regulated in lung metastases compared to liver metastases (Fig. 6E), in the BSVTK and BROCADE datasets (fig. S7C).
Fig. 6. Enrichment analyses of the molecular signatures associated with transcriptomic changes in different metastatic sites.
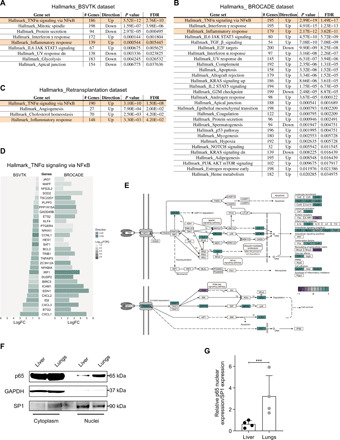
(A to C) Competitive gene set enrichment tests were performed using the hallmark gene set collection for lung versus liver comparison in the BSVTK feature set (A), autopsy dataset (B), and retransplantation dataset (C). The results were filtered by P value and false discovery rate (FDR) (<0.05), where the number of genes and the directionality in the gene set are also presented (“up” means up-regulated in the lungs and “down” means down-regulated in the lungs). Highlighted gene sets were common to all three datasets. (D) The absolute log fold changes (logFC) were plotted for genes present in the TNFα-related hallmark gene set that were differentially expressed between lungs and liver and common to both the BSVTK and BROCADE autopsy datasets. The opacity indicates the log10(FDR) for each gene, and the color represents the directionality of the log(FC) in the differential expression analysis between lungs and liver. (E) Representation of the KEGG’s TNFα signaling pathway illustrating the up- and down-regulated genes between lungs (green) and liver (purple). This was produced using the Pathview Bioconductor package (https://bioconductor.org/packages/release/bioc/html/pathview.html). p, phosphate; u, ubiquitin. (F) Western blots showing the level of p65 in the cytoplasm and nuclei of lung and liver metastases from a mouse with a MDA-MB-231 tumor (representative of four experiments). Glyceraldehyde phosphate dehydrogenase (GAPDH) was used as a loading control for the cytoplasm fraction and specificity protein 1 (SP1) for the nucleus fraction. (G) Relative quantification of the p65 protein level in the nuclei of lung and liver metastases from four individual mice normalized to SP1 expression. Means ± SD are shown, paired t test, ***P = 0.0008.
To gain some insights into the level of interclonal heterogeneity in terms of TNFα pathway activation, we interrogated the expression of the genes involved in the TNFα signaling pathway [KEGG and molecular signatures database (MSigDB) hallmark] in the scRNA-seq data of barcoded subclones 13, 2, 29, 3, and 9, isolated from lung metastases (as described in Fig. 2D and fig. S2E). Visualizing these gene expression profiles in a t-distributed stochastic neighbor embedding (t-SNE) (fig. S7E), we observed a lack of discrimination between the subclones, indicating that overall, TNFα-related genes did not contribute to any significant variation between the subclones.
To investigate whether NFκB activation was stronger in lung metastases compared to liver metastases, we analyzed by Western blot the level of nuclear p65 proteins in lung and liver metastases from four mice with MDA-MB-231 tumors. We observed that the amount of p65 was significantly higher in the nuclei of lung metastases compared to liver metastases (Fig. 6, F and G), and this result indicated a substantial difference in NFκB activation between these two metastatic sites.
Given the prominent induction of a TNFα response in the lungs, we next assessed TNFα expression in normal tissues. While the TNFα pathway was not significantly dysregulated in the GTEx dataset (fig. S7D), we observed a higher expression of TNFα in naïve lungs compared to the naïve liver (fig. S7F). Collectively, these results suggested that the up-regulation of the TNFα pathway in lung metastases may be due to host-derived TNFα and may lead to a tumor-specific vulnerability amenable for therapeutic interference.
Inhibition of TNFα pathway has a significant impact on the diversity of lung metastases
To further investigate the role of exogenous TNFα/NFκB pathway on the survival and clonality of metastases in lungs and liver, we used two pharmacological inhibitors of this pathway, etanercept and birinapant. Etanercept is a fusion protein consisting of the extracellular domain of the human TNF receptor linked to the Fc section of immunoglobulin G1 (38). After injection of the BSVTK MDA-MB-231 population in NSG hosts and resection of the primary tumors, mice were treated with vehicle (saline) or etanercept (10 mg/kg). At this dose, the drug is known to sequester human and mouse TNFα in vivo (39). When mice developed the first symptoms of metastatic disease (occurring at similar times in control and etanercept groups), all animals were euthanized, and we analyzed the impact of the drug on metastatic burden and clonality. Flow cytometry analysis of the metastases in lungs and liver indicated that the etanercept treatment did not reduce the number of cancer cells in these organs (Fig. 7, A and B) nor the number (Fig. 7, C and D) or frequency (Fig. 7E) of barcoded subclones. However, 3D imaging of metastases from three mice (randomly chosen from two independent experiments) revealed that TNFα blockade had a significant impact on the Shannon diversity index in lung metastases (Fig. 7F). We then determined the number of colors per metastasis and found that mice from the etanercept group had a significantly lower number of metastases containing more than three colors compared to the control group (Fig. 7G), despite the volume of metastases being comparable (fig. S8A). In particular, we observed an increase in the number of monochromatic metastases (fig. S8B). This was associated with a decrease in the number of metastases containing a high number of colors (4 to 11; fig. S8B), resulting in an overall reduction in the number of colors per cubic micrometer (fig. S8C).
Fig. 7. Effect of etanercept and birinapant treatments on the survival and heterogeneity of metastases.
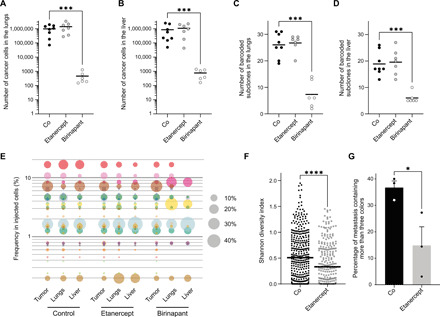
Mice with BSVTK-labeled MDA-MB-231 tumors were treated with saline (Co), etanercept (10 mg/kg) three times weekly, or birinapant (30 mg/kg) three times weekly, after resection of the primary tumors. Number of cells detected by flow cytometry in (A) lungs and (B) liver of mice treated with saline, etanercept, or birinapant. Number of colors detected by flow cytometry in (C) lungs and (D) liver. For (A) to (D), each dot represents a mouse. Mann-Whitney test, ***P = 0.0007. (E) Bubble plot indicating the frequency of the barcoded subclones (ranked according to their frequency in the injected cells). Each dot is a barcoded subclone, and its size correlates to its frequency, determined by flow cytometry. (A to E) The number of mice is n = 8 for the control, 7 for the etanercept, and 6 for the birinapant. (F) Shannon diversity indexes of individual lung metastases, quantified by imaging. Each dot is a metastasis in the control (n = 3 mice, three sections per lung, 388 metastases) or etanercept (n = 3 mice, three sections per lung, 312 metastases) group. (G) Percentage of metastases containing more than three colors in the control or etanercept group. Each dot represents an average of the percentages obtained for an individual mouse (n = 3 mice, three sections per lung). Error bars represent SEM. (F and G) Unpaired t test. ****P < 0.0001 and *P = 0.0418.
We then tested the impact of birinapant, an inhibitor of inhibitor of apoptosis (IAP) proteins, known to induce apoptotic cell death downstream of TNFα by the inactivation of the NFκB survival pathway. This class of drug can induce apoptosis in MDA-MB-231 via the autocrine secretion of TNFα (40, 41). Given our finding that cancer cells expressed a similar level of the TNF gene in lungs and liver (despite differences in the regulation of other genes from this TNFα pathway) (table S2), we hypothesized that this drug should kill metastases from both sites. Treatment with birinapant significantly decreased the number of cells and barcoded subclones in both organs (Fig. 7, A to D). The treatment also changed the frequency of barcoded subclones detected by flow cytometry in lung and liver metastases (Fig. 7E). However, these residual cells were hardly detectable by imaging because of their low number, which precluded further analyses of intrametastatic heterogeneity. Overall, these results indicated that blocking TNFα with etanercept affected the cellular clonality of lung metastases without affecting subclone survival. In contrast, targeting this pathway downstream of TNFα using birinapant proved to be highly efficient at reducing metastatic burden. These findings suggest that birinapant may have a high clinical potential in the treatment of patients with metastatic breast cancer.
DISCUSSION
We used optical barcoding to accurately detect, visualize, and quantify minor and dominant barcoded subclones across different metastatic organs in vivo. We found that specific subclones can display different behaviors in two clinically relevant metastatic sites, lungs and liver, in terms of clonal dynamics and transcriptomic profile. Overall, our results suggest that the fitness of the barcoded subclones and their ability to colocalize with other subclones within individual metastases were influenced by the nature of both the subclones and the metastatic niche. We found that the barcode repertoire detected in the lungs is highly heterogeneous compared to the liver, not only at the level of inter-organ heterogeneity, as previously shown (12, 16–18), but also at the level of intrametastatic heterogeneity. By mapping the interaction of the barcoded subclones within individual lung and liver metastases, we found that lung metastases were highly polychromatic compared to liver metastases. While abundant subclones were frequently colocalized with those that were less abundant in lung metastases, this was not the case in the liver. This suggests that the tissue-specific tumor microenvironment may dictate the degree of clonal heterogeneity of the corresponding metastases. The relative abundance and colocalization patterns of barcoded subclones within lung and liver metastases were highly reproducible between animals, indicating that these processes were not random. We also confirmed that the difference in polyclonality between lung and liver metastases observed using the barcoded MDA-MB-231 xenografts was corroborated in PDX models. The presence of both monochromatic and polychromatic lung metastases has been previously described in the context of breast cancer (12, 33). Furthermore, a recent analysis of metastases collected from patients confirmed that untreated metastases in the liver were mostly monoclonal (5). In this study, however, the number of treatment-naïve lung metastases was insufficient to study differences in polyclonality between lung and liver metastases. While further investigations are required to precisely determine the timing of polyclonal seeding and its importance in metastatic outgrowth, polyclonal metastases are more likely than monoclonal metastases to contain clones capable of surviving standard therapies (1, 5).
Several mechanisms have been reported to underpin polyclonal seeding in lung metastases, including circulating tumor cell (CTC) clustering and active corecruitment of subclones to distant sites (42). Although still controversial, the presence of CTC clusters is thought to be associated with poor prognosis (43, 44), metastasis polyclonality (12, 33), and increased lung tropism of metastases (12, 44–46). In addition, metastatic cells have been shown to compete or cooperate in lungs (14, 47, 48). Recruitment mechanisms include interleukins (ILs) and growth factors such as IL-11 and vascular endothelial growth factor D, recently shown to favor the polyclonal growth of metastases (28, 48, 49). In the future, detailed longitudinal studies involving CTC detection, time course analysis of metastasis “patchiness,” and molecular characterization of microenvironmental factors capable of fostering polyclonal metastasis recruitment or growth may fully elucidate the metastasis seeding process in breast cancer models. Regardless, our results indicate that these processes are unlikely to occur in the liver where the vast majority of macrometastases is monochromatic, although the barcoded subclones are the same as those found in the lungs. This disparity may be due to the anatomy of each organ, including differences in the architecture of the vasculature, resulting in different dynamics of extravasation between both organs (50). Alternatively, this difference could be due to the nature of the tumor microenvironment itself, inducing changes in the transcriptome, proteome, epigenome, and secretome of metastatic cells, resulting in differences in the clonal seeding of metastases.
Here, the optical barcoding system allowed us to identify the transcriptomic changes induced by the microenvironment on metastatic subclones. We derived a niche-associated signature, distinguishing genes differentially expressed in lung versus liver metastases. We confirmed that this signature was sufficient to distinguish samples freshly collected from the lungs and liver of three patients with metastatic breast cancer, showing that its relevance extends to human disease. Some of the genes making up this signature [such as inhibitor of DNA binding 1 (ID1), c-x-c motif chemokine ligand 1 (CXCL1), and angiopoietin like 4 (ANGPTL4) in the lungs] were identified previously using serial passages of lung metastases to select clones from the same cell line that have a preferential tropism for this organ (6–8). The site-specific gene signature that we derived from barcoded subclones that metastasized to both liver and lungs was conserved after the lung metastasis retransplantation experiment, indicating that these genes represent a feature of niche adaptation by fundamentally plastic cells. Furthermore, genes from this signature were also differentially expressed in normal lung and liver tissues from healthy donors, suggesting that the transcriptional programs selectively displayed by MDA-MB-231 and patient’s breast cancer cells in given metastatic locations shared similarities with those of the microenvironment in which they are located. This indicates that tumor cells have the ability to adapt their gene expression landscape in response to their microenvironment, a feature reminiscent of organ mimicry.
Among these mechanisms of cellular adaptation, we found that the TNFα pathway was most significantly up-regulated in lung compared to liver metastases. The dysregulation of this pathway was confirmed in both retransplantation and autopsy datasets. Some of the genes involved in these pathways may be responsible for the cellular interactions, survival, polyclonality, and proliferation specifically observed in lung metastases. c-x-c motif chemokine ligand 8 (CXCL8), IL-11, and IL-6, for instance, which are known targets of the TNFα and inflammatory response pathways and were significantly up-regulated in lungs according to our BSVTK feature set, have previously been shown to foster polyclonal seeding and metastatic growth in lungs (14, 28, 48, 51, 52). While our results suggest that the involvement of these specific genes may be model dependent, the up-regulation of the TNFα and inflammatory response pathways in lungs was confirmed in autopsy samples and could be due to the presence of TNFα in the lung microenvironment. Although etanercept-mediated TNFα inhibition did not have any significant impact on the survival of the subclones, it significantly decreased the diversity of metastases in lungs. This effect might be mediated by changes in the tumor microenvironment and/or may be directly attributed to changes in the transcriptome of the barcoded subclones. On the other hand, the inhibition of IAPs using birinapant, known to induce TNFα-dependent apoptosis, killed both lung and liver metastases. From a pharmacological point of view, both TNFα inhibitors and SMAC mimetics are likely to be used in the clinic. Anti-TNFα drugs have been proposed to be used in combination with immunotherapy (53). Because the role of TNFα as an anti- or procancer cytokine is still a matter of debate [as reviewed in (54)], it will be important to precisely characterize its function in the context of tumor immunology and heterogeneity. Last, birinapant has shown some efficacy in targeting TNBC primary tumors (55), and our results now suggest that this compound could also be used to target metastases.
In conclusion, we have used an innovative optical barcoding approach to longitudinally monitor the clonal makeup of developing metastases and to identify a set of cellular and molecular features that are directly regulated by the metastatic niche. It will be interesting to investigate whether some of the pathways unraveled by this gene set may contribute to the differential sensitivity of metastatic cells to given drugs. This analysis pipeline holds promise for investigations of the impact of other targeted therapies on clonal cooperation and tumor survival across different microenvironments.
MATERIALS AND METHODS
Cell culture
The human cell line MDA-MB-231 was purchased from the American Type Culture Collection (ATCC; #HTB-26). Cells were maintained in RPMI 1640 medium with Hepes (#22400089, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Moregate Biotech) and penicillin-streptomycin (10,000 U/ml) (#15140122, Thermo Fisher Scientific) at 37°C in a humidified atmosphere with 5% CO2.
Lentivirus production, purification, and titration
Lentiviruses were produced by transient cotransfection of human embryonic kidney–293T cells with the LeGO vector of interest and the packaging plasmids (LeGO-EBFP2, Addgene, #85213; LeGO-S2, Addgene, #85211; LeGO-V2, Addgene, #27340; LeGO-T2, Addgene, #27342; LeGO-dKatushka2, Addgene, #85214; pCMVR8.74, Addgene, #22036; pMD2.G, Addgene, #12259) (56), using a FuGENE HD transfection reagent (#E2311, Promega) following the manufacturer’s recommendations. The transfection and virus production were performed in serum-free Opti-MEM (#31985070, Thermo Fisher Scientific). After 48 hours, the supernatant of the cells was collected and concentrated using an Amicon Ultra-15 100-kDa centrifugal filter unit (#UFC910024, Merck). The virus titer was then measured using the fluorescence titrating assay and the formula TU/ml = (Number of cells transduced × % fluorescent)/(Virus volume in ml).
Establishment of the BSVTK-labeled MDA-MB-231 population
Human MDA-MB-231 breast cancer cells (passage 39 from ATCC) were infected with a mixture of five lentiviruses coding for eBFP2, tSapphire, Venus, tdTomato, and dKatushka2. The combination of the five different fluorophores resulted in 31 different colors. Viral titration was predetermined and normalized for each virus to achieve 60 to 75% of positive cells. The cells were harvested and sorted by flow cytometry, 48 hours after the infection. For each color, 100 cells were sorted, and the mixture of the 31 color-coded populations (3100 cells in total) was plated in a 96-well plate [day 0 (D0)]. As the frequency of MDA-MB-231 clones able to engraft in NSG mice was shown to vary between 1 of 7 and 1 of 3333 (15), we sorted 100 cells per color to increase the purity of individual color-coded populations while maintaining a low likelihood of having multiple “genetically diverse subclones” of the same color successfully engrafting in vivo. This population was then expanded for 18 days (D18) and sorted by flow cytometry but only on the basis of positivity this time to ensure the absence of negative cells. After this second sort, 180,000 cells were plated in a six-well plate and expanded for 10 days (D28) before being frozen into several aliquots to allow multiple experiments to be completed using the same stock. A portion of the cells was kept in culture to observe the population drift.
Cell proliferation and viability assays
MTT assay or CellTiter-Glo was used to determine the proliferation of the cells. For both assays, 1500 cells were seeded per well in a 96-well plate in 100 μl of medium. To avoid the “edge effect” and evaporation, peripheral wells were filled with 200 μl of Dulbecco’s phosphate-buffered saline (DPBS). For each condition, three technical replicates were prepared. For the MTT assay, 10 μl of MTT (5 mg/ml) (#M5655, Sigma-Aldrich) in DPBS was added to each well and incubated for 4 hours at 37°C. To solubilize formazan crystals, 100 μl of solubilization buffer (0.1 N of hydrochloric acid in isopropanol) was added to each well, followed by 1 hour of incubation at room temperature. After homogenization, plates were analyzed with the EnSight Multimode Plate Reader by measuring the absorbance at 590 nm. For the CellTiter-Glo assay (#G7571, Promega), 50 μl of CellTiter-Glo lysis buffer were added to the cells, and the plates were incubated at room temperature with gentle mixing for 15 min. The luminescence was measured using the EnSight plate reader.
Colony formation assays
MDA-MB-231 parental cells and BSVTK-labeled MDA-MB-231 cells were plated at low density (1000 cells per well in six-well plates) with three technical replicates per experiment. After 1 week of incubation, the colonies were stained with crystal violet overnight at 4°C. After wash steps, the plates were scanned and colonies manually counted using FiJi software. For soft agar colony formation, 3 ml of bottom agarose layer, containing 0.75% agarose (#50101, Lonza) in RPMI medium (#23400-021, Thermo Fisher Scientific) was supplemented with FBS and penicillin-streptomycin. Once the bottom layer was solidified, 25,000 MDA-MB-231 parental or LeGO cells were mixed in top agarose layer (0.45% agarose) and seeded on top of the bottom layer. The wells were covered with 3 ml of culture media and incubated 2 weeks at 37°C in a humidified atmosphere with 5% CO2. After 2 weeks, 140 μl of MTT (5 mg/ml) in PBS was added into each well, and the plates were incubated for 4 hours. Last, the plates were scanned and manually counted using FiJi software.
Establishment of the BSVTK-labeled PDX cells
PDX-0066 was established at the Olivia Newton-John Cancer Research Institute (ONJCRI) from a malignant pleural effusion from a patient with breast cancer 1 (BRCA1)–mutated breast cancer, previously treated with Nab-paclitaxel. KCC-P-4295 model, generated from a drug-naïve TNBC primary tumor at the Kinghorn Cancer Centre and Garvan Institute, was obtained from BROCADE. The study was approved by the Austin Health Human Ethics Research Committee. Single-cell suspensions from early-passage PDXs (passage 3 for PDX-0066 and passage 4 for PDX KCC-P-4295) were prepared as described below. The cells were plated at the density of 300,000 cells per well in a 24-well flat-bottom ultralow attachment plate (#734-1584, Corning) and were kept in culture for 48 hours in 300 μl of mammosphere media containing Dulbecco’s modified Eagle’s medium F12 (#10565042, Thermo Fisher Scientific), B-27 supplement 1X (#17504001, Thermo Fisher Scientific), penicillin-streptomycin (100 U/ml) (#15140122, Thermo Fisher Scientific), insulin (5 μg/ml) (#11376497001, Sigma-Aldrich), hydrocortisone (1 μg/ml) (#H0396-100MG, Sigma-Aldrich), heparin (0.8 U/ml) (#H0878-100KcU, Sigma-Aldrich), basic fibroblast growth factor (20 ng/ml) (#01-106, Merck-Millipore), and epidermal growth factor (20 ng/ml) (#E9644, Sigma-Aldrich) in the presence of the BSVTK viruses. Cancer cells were then sorted before transplantation in NSG mice, as described below.
Mammary fat pad transplantation of BSVTK-labeled cancer cells
The BSVTK MDA-MB-231 population was thawed and expanded for 3 days before injection of 100,000 cells into the exposed fat pad from the fourth mammary gland of 7-week-old NSG female mice. The cells were resuspended in 10 μl of injection buffer: 25% growth factor–reduced Matrigel (#734-1101, Corning) 42.5% DPBS (#14190144, Thermo Fisher Scientific), 30% FBS (Moregate Biotech), and 2.5% Trypan blue (Bio-Rad Laboratories) for injection. For the retransplantation experiment, barcoded subclone 2 was isolated from the lungs of a mouse reaching ethical end point after being initially injected with the BSVTK population. Cells were sorted three times to ensure the purity of the population, and 10,000 cells were injected into the mammary fat pads of three NSG mice. For PDX-0066, 150,000 fluorescent cells were transplanted in the mammary fat pads of three mice. For PDX KCC-P-4295, 20,000 labeled cells were injected per mammary fat pad in two recipient mice. All animal procedures included in this manuscript were approved by the Austin Animal Ethics Committee.
Mouse monitoring, tumor resection, and organ collection
Tumor volume was measured twice weekly using electronic calipers. The tumor volume was estimated using the following formula: (minimum diameter)2 × (maximum diameter)/2. Tumors were resected when they reached a volume of about 100 mm3. For all experiments, the resection was completed approximately 28 days after injection. When the tumor was resected, it was cut into two pieces; one half was processed for imaging, and the other half was used for single-cell suspension preparation. The weight of the mice was also measured twice weekly or daily once they lost 10% of their body weight. When the weight loss reached 15%, the animals were euthanized by CO2 excess. To limit the background autofluorescence of tissues for imaging, the animals were transcardially perfused with a freshly made, ice-cold solution containing 0.45% NaCl (Sigma-Aldrich) and 10 U of heparin (Sigma-Aldrich) per milliliter in Milli-Q water. For imaging, three slices of 2-ml thickness were manually cut in three lobes of lungs and three lobes of liver and immediately put in freshly made, ice-cold 4% paraformaldehyde (#28908, Thermo Fisher Scientific) in DPBS. The rest of the organs were kept in ice-cold DPBS before single-cell suspension preparation and flow cytometry analysis.
In vivo drug testing with etanercept and birinapant
Mice were injected with the BSVTK MDA-MB-231 population (as described above), and the primary tumors were resected when they reached 100 mm3 before the treatment. For the etanercept experiment (two independent experiments), the control group received saline as vehicle; the treated group received etanercept (Enbrel) (10 mg/kg), three times weekly until the end of the experiment. For the birinapant experiment (one experiment), the treated group received birinapant (#S7015, Selleck Chemicals) at the dose of 30 mg/kg, three times weekly until the end of the experiment. The mice were euthanized when the control groups died from metastatic disease. Metastases were analyzed by imaging and flow cytometry, as described below.
Single-cell suspension preparation
The tissues were manually chopped into small pieces (about 1 mm by 1 mm) and resuspended in the digestion medium: collagenase IA (300 U/ml) (#C9891, Sigma-Aldrich), hyaluronidase (100 U/ml) (#H3506, Sigma-Aldrich), and deoxyribonuclease I (DNase I) (100 U/ml) (#LS002139, Worthington) in RPMI 1640 medium with Hepes (#22400089, Thermo Fisher Scientific). The tumor was resuspended in 5 ml of digestion medium, the lungs in 5 ml of digestion medium, and the liver in 10 ml of digestion medium. Samples were first incubated for 25 min at 37°C with agitation, then passed through an 18-gauge (G) needle three times, and incubated for another 25 min at 37°C with agitation before being passed through a 20G needle three times. The cell suspension was then filtered through a 70-μm cell strainer and spun down for 5 min at 500g.
Sample preparation for confocal imaging
Fixed samples were washed in DPBS and embedded in 3% low–melting point agarose (#1613111, Bio-Rad Laboratories). Sections (200 μm) were cut using a vibratome (#VT1000S, Leica Biosystems) and mounted on slides (#J1800AMNT, Thermo Fisher Scientific) with high-performance coverslips (#474030-9000-000, Carl Zeiss Microscopy) and antifade mounting medium (#P36961, Thermo Fisher Scientific). Images were acquired using LSM 880 (Carl Zeiss) and LSM 980 (Carl Zeiss) confocal microscopes equipped with 20×/0.8 objectives. Online fingerprinting was used to spectrally unmix the BSVTK fluorophores with reference spectra acquired in cancer cells expressing a single color only. The entire section was acquired as a tile scan, with a resolution of 0.47 μm in xy and 3 μm in z.
Flow cytometry
All samples were sorted/analyzed using a BD FACSAria III cell sorter with BD FACSDiva 8.0.2 software (Becton Dickinson and Company, BD Biosciences, San Jose, CA, USA), equipped with four lasers and capable of detecting up to 17 parameters. Samples were analyzed using the following excitation lasers and emission filters: eBFP2 (405 nm; 450/40), tSapphire (405 nm; 510/50), Venus (488 nm; 530/30), propidium iodide (488 nm; 695/40), tdTomato (561 nm; 582/15), and Katushka (561 nm; 670/14). For sorting, a 100-μm nozzle tip was used with a sheath pressure of 138 kPa and a drop drive frequency of 30 kHz. BD FACSFlow (BD Biosciences, San Jose, CA, USA) sheath fluid was used. The sample and collection tubes were maintained at 4°C.
Subclone identification and measurement in the imaging dataset
First, a 3D median filter (with radius in xy = 8 and radius in z = 2) was applied to all channels to reduce the noise and homogenize the intensity. A manually determined hysteresis thresholding was then applied to the five channels, and clone identity was reconstructed according to the signal combination of the five channels. Two thresholds were set, one to determine signal voxels with high confidence and one to determine signal voxels with medium confidence—i.e., above background noise—that may belong to residual noise or signal. The hysteresis procedure will consider medium-confidence voxels as positive if they are connected to high-confidence voxels. Medium-confidence voxels not connected to high-confidence voxels were labeled as background. 3D quantification of subclones was performed using algorithms and tools from the 3D ImageJ suite (57). For untreated mice, structures with a volume of less than 1000 μm3 were discarded, this volume being too small to correspond to more than one cell. For the etanercept-treated and control experiments, structures with a volume of less than 100,000 μm3 were discarded to focus on macrometastases. Individual metastases were defined as populations of directly connected voxels within the dataset. Volumes of each subclone within each metastasis were then computed. Images were stored within an OMERO database (58); processing and analysis were then automated using a custom-built analysis suite called towards an automated processing and analysis system (TAPAS) (59). Results were validated by two observers.
Interaction analysis in the imaging dataset
An “interactome” was obtained by computing how often specific pairs of subclones were detected within all metastases. We also computed a “dominant” subclone for each interaction by comparing volumes of the two subclones within the metastasis. Analyses were completed using R software (60), and interactome visualizations were completed within Cytoscape (61).
Histology
Samples were fixed in 4% paraformaldehyde for 12 hours before embedding in paraffin. Sections were then stained with hematoxylin and eosin and scanned using an Aperio AT2 (Leica Biosystems).
Single-cell capture for scRNA-seq
To analyze the heterogeneity of the parental MDA-MB-231 population, a cell suspension of MDA-MB-231 was prepared using the TrypLE Express Enzyme (#12604021, Thermo Fisher Scientific). Using a Chromium controller machine (#120223, 10x Genomics) and a Chromium single-cell gel bead kit (#1000075, 10x Genomics), 10,000 cells were captured in gel bead in emulsion. The complementary DNA was then isolated, and the library was prepared as recommended by the manufacturer for the 3′ V3 kit (#1000078, 10x Genomics). Sequencing runs were completed on a NextSeq machine (Illumina) with the following specifications: Read1, 28 cycles; Read2, 91 cycles; Index Read, 8 cycles.
To verify the purity of the metastatic barcoded subclones and to investigate whether they were genomically distinct, subclones 13, 2, 9, 29, and 3 were FACS-purified from lung metastases. The number of cells sorted for single-cell analysis varied between the subclones on the basis of their frequency. For the dominant subclones 13, 2, and 9, 20,000 cells were sorted. For the minor subclones 29 and 3, 7000 and 1000 cells were sorted, respectively.
Identifying genomic heterogeneity using scRNA-seq and inferred CNVs
To investigate the genomic heterogeneity of the original MDA-MB-231 cell line, we (i) probed cells with single-cell mRNA sequencing, (ii) identified cell clusters on the basis of their transcriptional profiles, and (iii) inferred copy number profiles in clusters. For cell clustering, the software suite Seurat3 was used (62). Briefly, the raw reads were aligned to the hg38 reference genome (63), and the transcript counts were inferred using CellRanger (10x Genomics: Resolving biology to advance human health, 10x Genomics; http://10xgenomics.com) with default parameters. The postprocessing analyses were conducted using Seurat3. Droplets were excluded from further analyses if including less than 200 detected transcripts or if they included a fraction of mitochondrial derived reads higher than 0.1. Cell cycle state was inferred for each cell using the CellCycleScoring function. The transcript abundance was adjusted for cell cycle stage and for mitochondrial sequences. PCA was used to reduce the dimensionality of the data. Cells were assigned to clusters using the shared nearest neighbor (SNN) method (64) using a resolution of 0.7. For copy number inference, the algorithm inferCNV (infercnv, Bioconductor; https://bioconductor.org/packages/release/bioc/html/infercnv.html) (65) was used with the parameters scale_data = TRUE and cutoff = 0.1, providing as input the raw transcript abundance matrix as well as cell cluster labels. The dataset gencode_v21 was used as reference transcript location within the genome. The scRNA-seq dataset (GSE113197) including material from four noncancerous epithelial samples was used as a benign reference for inferCNV inference (66, 67).
To investigate the genomic purity of the five barcoded subclones, we (i) probed cells with single-cell mRNA sequencing, (ii) aligned the sequencing reads to the hg38 reference genome and the transcript counts were inferred using CellRanger with default parameters as described above, (iii) inferred the copy number profile for each cell using inferCNV, and (iv) defined five cell clusters (as five clones were sequenced) on the basis of hierarchical similarity.
mRNA extraction and bulk RNA-seq
mRNA was extracted from sorted cells using an miRNEasy kit (#217084, Qiagen) according to the manufacturer’s recommendations. Briefly, mRNA was isolated using a guanidinium thiocyanate-phenol-chloroform extraction approach. The mRNA is then bound to an exchange column where the remaining genomic DNA was digested by the RNase-Free DNase (#79254, Qiagen) on the column. The RNA was finally washed and eluted in water. The quantity and the quality of the isolated mRNA were assessed using the TapeStation (4200 TapeStation System, Agilent), and 100 ng of the mRNA was used as an input for the library preparation using the TruSeq RNA Library Prep Kit (Illumina) according to the manufacturer’s recommendations. The indexed libraries were pooled and diluted to 1.5 pM for paired-end sequencing (2 × 81 cycles) on a NextSeq 500 instrument using the v2 150 cycle high output kit (Illumina) as per the manufacturer’s instructions. The base calling and quality scoring were determined using Real-Time Analysis on board software v2.4.6, while the FASTQ file generation and demultiplexing used bcl2fastq conversion software v2.15.0.4.
Collection of autopsy samples
Patients were recruited by their oncologists to the BROCADE rapid autopsy program (www.petermac.org/research/research-cohort-studies/brocade), approved by the Human Research Ethics Committee of the Peter MacCallum Cancer Centre, Melbourne. Patients with advanced breast cancer provided written informed consent. Lungs and liver metastases were collected soon after death for three patients (table S3), and the mRNA was extracted and sequenced as described above.
Analysis of NFκB activation using subcellular fractionation and Western blotting
Metastatic cells were sorted from the lungs and liver as described above. The subcellular fractionation experiments were performed using the subcellular protein fractionation kit for Cultured Cells (#78840, Thermo Fisher Scientific) according to the manufacturer’s instructions. The cytoplasmic and nuclear fractions were then analyzed by Western blotting using the following primary antibodies: anti-p65 (#8242, Cell Signaling Technology), anti-specificity protein 1 (SP1) (#9389, Cell Signaling Technology), and anti–glyceraldehyde phosphate dehydrogenase (GAPDH) (#5174, Cell Signaling Technology). The bands were quantified using Image Lab (V6.0.1, Bio-Rad Laboratories), and the relative nuclear expression of p65 was determined by comparing the liver nuclear expression to the lung nuclear expression, normalized for SP1 expression.
Comparing the transcriptome of barcoded subclones at single-cell resolution
To demonstrate intrasubclone homogeneity and intersubclone heterogeneity, we clustered scRNA-seq from five barcoded subclones (2, 3, 9, 13, and 29) and performed differential expression analysis on pseudo-bulked samples to derive a gene signature. The dimensionality of the log-normalized gene counts was reduced, and to pseudo-bulk each subclone, reads were aggregated across cells into pseudo-bulk samples for each K-nearest neighbor (KNN) cluster. KNN clusters were identified separately for each barcoded subclone (k = 20) by applying the walktrap method (68) to SNN graphs. Subclone pseudo-bulk samples were filtered and normalized using edgeR (69), where samples with low library sizes were discarded or merged and genes required a logCPM greater than or equal to 0.5 in at least 10% of pseudo-bulk samples. As above, differentially expressed genes were identified with P value and false discovery rate (FDR) thresholds of 0.5, Benjamini and Hochberg (BH) correction, and using a generalized log-linear model. To apply the feature set, log-normalized counts were subset to these differentially expressed genes and visualized using a t-SNE. In Fig. 2D, genes from the TNF signaling pathway (KEGG) and TNFα signaling by NFκB (hallmark) were used as a feature set. In fig. S7E, genes that are differentially expressed between barcoded subclones 13 and 29 (table S1) were used as a feature set.
Data processing for the LeGO, autopsy, and GTEx datasets
Read pairs were filtered using cutadapt (70), mapped to the GRCh38 genome using STAR (71), and quantified using featureCounts (72). Genes without a current ensemble gene ID were removed. Preprocessing was completed using edgeR (69), including gene filtering and summing over technical replicates. However, for the autopsy data, genes were only retained if they achieved a count per million (CPM) of 0.5 in at least four samples. Batch effect correction to remove the mouse effect (for the xenograft experiments) was performed using limma (73), and counts were converted to logCPM with a prior count of 2 for visualization. The GTEx count data downloaded from https://gtexportal.org/home/datasets were log-transformed and subset to liver and lung samples according to the associated annotation files.
Statistical analyses of the LeGO and autopsy datasets
Three separate subsets of the LeGO dataset were processed and analyzed in parallel, according to the same methodology. The first dataset included all the LeGO primary and metastatic samples, another dataset containing only the lung and liver metastases, and another for the metastases from the retransplanted subclone 2 tumors. Differential expression analyses were completed in edgeR (60), all of them fitting a generalized log-linear model to uncorrected data using the glmQLfit function. P values were adjusted using BH correction and were filtered using 0.05 thresholds for both P value and FDR. Data were scaled for the heatmap visualizations, and mitochondrial genes were excluded.
Competitive gene set enrichment tests were performed in limma (73) using the hallmark gene set collection from MSigDB (74). Here, FDR and P value thresholds of 0.05 were also used for each of the two-group comparisons, after excluding genes that did not map to Entrez Gene identifiers.
Overrepresentation analysis was used to identify GO terms from the “biological process (BP)” subontology that were enriched in three differential expression analyses. Using the same model, primary tumors were compared to lung and liver metastasis separately, and then the metastases were compared to each other. Enriched terms were visualized for the genes up-regulated in the two metastases compared to the primary tumors (636 genes for lungs and 314 for liver), as well as the differentially expressed genes between the metastases (464 for lung and 656 for liver). The GO enrichment plots were generated using the clusterProfiler (75) by applying BH correction, P value, and q value cutoffs of 0.05. The minimum and maximum number of genes in a gene set to be tested were 10 and 500, respectively.
Applying the BSVTK feature set to other datasets
The BSVTK feature set was derived using the second data subset, comparing only lung (650 up-regulated genes) and liver (716 up-regulated genes) metastases, according to the differential expression analysis methods above. To demonstrate that the differentially expressed genes between liver and lung metastases can result in a separation of lung and liver samples in other datasets, a PCA was performed on these data subsets to the BSVTK feature set (1366 genes). This was applied to the retransplantation and autopsy datasets after batch effect correction (logCPM with a prior count of 1) and was also applied to the lung and liver samples from the GTEx (34) dataset.
Acknowledgments
We are extremely grateful to the donors and their families for choosing to donate samples to the BROCADE program and the ONJCRI-Breast Cancer Biobank. We thank C. Bell, N. Lalaoui, D. Liew, and C. Shembrey for insightful discussions and technical support. We are grateful for the support of the BROCADE Rapid Autopsy Program, funded by the Australian National Breast Cancer Foundation (NBCF) under infrastructure grant IF-14-001. We also acknowledge the Victorian Institute of Forensic Medicine that conducted the rapid autopsies; the Peter MacCallum, Kinghorn Centre, Garvan Institute, and Tobin Brothers Funeral Directors for providing transport of donors; and L. Devereux, who manages BROCADE and who helped with the sample collection and processing. We acknowledge the Kinghorn Cancer Centre and Garvan for the KCC-P-4295 model. The authors and the Olivia Newton-John Cancer Research Institute gratefully acknowledges the generous support of the Love Your Sister Foundation. We acknowledge the ACRF Centre for Imaging the Tumour Environment at the Olivia Newton-John Cancer Research Institute for providing microscopy support. The Olivia Newton-John Cancer Research Institute acknowledges the support of the Operational Infrastructure Program of the Victorian Government. Funding: D.M., F.H., and B.P. are supported by the NBCF (Investigator Initiated Research Grant IIRS-19-082). D.M. is supported by Susan G. Komen and Cancer Australia (CCR19606878). F.H. is supported by the National Health and Medical Research Council of Australia (grant no. 1164081) and by a Senior Research Grant from the Tour de Cure Foundation. D.M., M.J.D., and B.Y. are supported by the Grant-in-Aid Scheme administered by Cancer Council Victoria. D.M., B.Y., and R.L.A. are supported by the Love Your Sister Foundation. R.L.A. and M.J.D. also acknowledge fellowship support from NBCF. The contents of the published material are solely the responsibility of the individual authors and do not reflect the views of Cancer Australia or other funding agencies. Author contributions: All the authors were involved in the data analysis and interpretation. J.B., V.C.W., K.L.R., F.H., and D.M. designed the experiments and wrote the paper. J.B. performed the experiments and analyzed the results. V.C.W., A.C.P., and K.L.R. optimized the imaging strategy. T.B. developed the image analysis software. T.B., J.B., and V.C.W. analyzed the imaging dataset. H.J.W., S.M., A.T.P., and M.J.D. analyzed the scRNA-seq and bulk RNA-seq results. A.S., M.M., and F.E.S. helped with the in vitro and in vivo experiments. D.B. provided some expert advice on flow cytometry analysis. B.Y., M.E., and R.L.A. contributed expertise and patient samples. J.W., S.W., and B.P. helped with the sequencing of the samples. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. BROCADE PDX KCC-P-4295 and autopsy samples can be provided by BROCADE pending scientific review and a completed material transfer agreement. Requests should be submitted to the BROCADE Manager: lisa.devereux@petermac.org. PDX-0066 can be provided by the ONJCRI, depending on availability and completing a material transfer agreement.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/28/eabf4408/DC1
REFERENCES AND NOTES
- 1.Dagogo-Jack I., Shaw A. T., Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81–94 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Wang Y., Waters J., Leung M. L., Unruh A., Roh W., Shi X., Chen K., Scheet P., Vattathil S., Liang H., Multani A., Zhang H., Zhao R., Michor F., Meric-Bernstam F., Navin N. E., Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512, 155–160 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim C., Gao R., Sei E., Brandt R., Hartman J., Hatschek T., Crosetto N., Foukakis T., Navin N. E., Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 173, 879–893.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casasent A. K., Schalck A., Gao R., Sei E., Long A., Pangburn W., Casasent T., Meric-Bernstam F., Edgerton M. E., Navin N. E., Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell 172, 205–217.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu Z., Li Z., Ma Z., Curtis C., Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 52, 701–708 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta G. P., Minn A. J., Kang Y., Siegel P. M., Serganova I., Cordon-Cardo C., Olshen A. B., Gerald W. L., Massague J., Identifying site-specific metastasis genes and functions. Cold Spring Harb. Symp. Quant. Biol. 70, 149–158 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Chen W., Hoffmann A. D., Liu H., Liu X., Organotropism: New insights into molecular mechanisms of breast cancer metastasis. NPJ Precis. Oncol. 2, 4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minn A. J., Kang Y., Serganova I., Gupta G. P., Giri D. D., Doubrovin M., Ponomarev V., Gerald W. L., Blasberg R., Massague J., Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Invest. 115, 44–55 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein C. A., Selection and adaptation during metastatic cancer progression. Nature 501, 365–372 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Siegel M. B., He X., Hoadley K. A., Hoyle A., Pearce J. B., Garrett A. L., Kumar S., Moylan V. J., Brady C. M., Van Swearingen A. E., Marron D., Gupta G. P., Thorne L. B., Kieran N., Livasy C., Mardis E. R., Parker J. S., Chen M., Anders C. K., Carey L. A., Perou C. M., Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J. Clin. Invest. 128, 1371–1383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marusyk A., Janiszewska M., Polyak K., Intratumor heterogeneity: The Rosetta Stone of therapy resistance. Cancer Cell 37, 471–484 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung K. J., Padmanaban V., Silvestri V., Schipper K., Cohen J. D., Fairchild A. N., Gorin M. A., Verdone J. E., Pienta K. J., Bader J. S., Ewald A. J., Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. U.S.A. 113, E854–E863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamamoto N., Yang M., Jiang P., Xu M., Tsuchiya H., Tomita K., Moossa A. R., Hoffman R. M., Determination of clonality of metastasis by cell-specific color-coded fluorescent-protein imaging. Cancer Res. 63, 7785–7790 (2003). [PubMed] [Google Scholar]
- 14.Tabassum D. P., Polyak K., Tumorigenesis: It takes a village. Nat. Rev. Cancer 15, 473–483 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Nguyen L. V., Cox C. L., Eirew P., Knapp D. J. H. F., Pellacani D., Kannan N., Carles A., Moksa M., Balani S., Shah S., Hirst M., Aparicio S., Eaves C. J., DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nat. Commun. 5, 5871 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merino D., Weber T. S., Serrano A., Vaillant F., Liu K., Pal B., Di Stefano L., Schreuder J., Lin D., Chen Y., Asselin-Labat M. L., Schumacher T. N., Cameron D., Smyth G. K., Papenfuss A. T., Lindeman G. J., Visvader J. E., Naik S. H., Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 10, 766 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagenblast E., Soto M., Gutiérrez-Ángel S., Hartl C. A., Gable A. L., Maceli A. R., Erard N., Williams A. M., Kim S. Y., Dickopf S., Harrell C. J., Smith A. D., Perou C. M., Wilkinson J. E., Hannon G. J., Knott S. R. V., A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 520, 358–362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Echeverria G. V., Powell E., Seth S., Ge Z., Carugo A., Bristow C., Peoples M., Robinson F., Qiu H., Shao J., Jeter-Jones S. L., Zhang X., Ramamoorthy V., Cai S., Wu W., Draetta G., Moulder S. L., Symmans W. F., Chang J. T., Heffernan T. P., Piwnica-Worms H., High-resolution clonal mapping of multi-organ metastasis in triple negative breast cancer. Nat. Commun. 9, 5079 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nolan-Stevaux O., Tedesco D., Ragan S., Makhanov M., Chenchik A., Ruefli-Brasse A., Quon K., Kassner P. D., Measurement of cancer cell growth heterogeneity through lentiviral barcoding identifies clonal dominance as a characteristic of in vivo tumor engraftment. PLOS ONE 8, e67316 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian G., Wu Y., Ferrari D., Qiao P., Hollande F., Semisupervised clustering by iterative partition and regression with neuroscience applications. Comput. Intell. Neurosci. 2016, 4037380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiao P., Molck C., Ferrari D., Hollande F., A spatio-temporal model and inference tools for longitudinal count data on multicolor cell growth. Int. J. Biostat. 14, 20180008 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Mohme M., Maire C. L., Riecken K., Zapf S., Aranyossy T., Westphal M., Lamszus K., Fehse B., Optical barcoding for single-clone tracking to study tumor heterogeneity. Mol. Ther. 25, 621–633 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malide D., Metais J.-Y., Dunbar C. E., In vivo clonal tracking of hematopoietic stem and progenitor cells marked by five fluorescent proteins using confocal and multiphoton microscopy. J. Vis. Exp., e51669 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber K., Thomaschewski M., Warlich M., Volz T., Cornils K., Niebuhr B., Tager M., Lutgehetmann M., Pollok J. M., Stocking C., Dandri M., Benten D., Fehse B., RGB marking facilitates multicolor clonal cell tracking. Nat. Med. 17, 504–509 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Roh V., Abramowski P., Hiou-Feige A., Cornils K., Rivals J.-P., Zougman A., Aranyossy T., Thielecke L., Truan Z., Mermod M., Monnier Y., Prassolov V., Glauche I., Nowrouzi A., Abdollahi A., Fehse B., Simon C., Tolstonog G. V., Cellular barcoding identifies clonal substitution as a hallmark of local recurrence in a surgical model of head and neck squamous cell carcinoma. Cell Rep. 25, 2208–2222.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 26.van der Heijden M., Miedema D. M., Waclaw B., Veenstra V. L., Lecca M. C., Nijman L. E., van Dijk E., van Neerven S. M., Lodestijn S. C., Lenos K. J., de Groot N. E., Prasetyanti P. R., Arricibita Varea A., Winton D. J., Medema J. P., Morrissey E., Ylstra B., Nowak M. A., Bijlsma M. F., Vermeulen L., Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc. Natl. Acad. Sci. U.S.A. 116, 6140–6145 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pogoda K., Niwinska A., Murawska M., Pienkowski T., Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Med. Oncol. 30, 388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janiszewska M., Tabassum D. P., Castano Z., Cristea S., Yamamoto K. N., Kingston N. L., Murphy K. C., Shu S., Harper N. W., Del Alcazar C. G., Aleckovic M., Ekram M. B., Cohen O., Kwak M., Qin Y., Laszewski T., Luoma A., Marusyk A., Wucherpfennig K. W., Wagle N., Fan R., Michor F., McAllister S. S., Polyak K., Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat. Cell Biol. 21, 879–888 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liedtke C., Mazouni C., Hess K. R., Andre F., Tordai A., Mejia J. A., Symmans W. F., Gonzalez-Angulo A. M., Hennessy B., Green M., Cristofanilli M., Hortobagyi G. N., Pusztai L., Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 26, 1275–1281 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Perez J. E., Machiavelli M., Leone B. A., Romero A., Rabinovich M. G., Vallejo C. T., Bianco A., Rodriguez R., Cuevas M. A., Alvarez L. A., Bone-only versus visceral-only metastatic pattern in breast cancer: Analysis of 150 patients. A GOCS study. Grupo Oncológico Cooperativo del Sur. Am. J. Clin. Oncol. 13, 294–298 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Ben-David U., Siranosian B., Ha G., Tang H., Oren Y., Hinohara K., Strathdee C. A., Dempster J., Lyons N. J., Burns R., Nag A., Kugener G., Cimini B., Tsvetkov P., Maruvka Y. E., O’Rourke R., Garrity A., Tubelli A. A., Bandopadhayay P., Tsherniak A., Vazquez F., Wong B., Birger C., Ghandi M., Thorner A. R., Bittker J. A., Meyerson M., Getz G., Beroukhim R., Golub T. R., Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber K., Thomaschewski M., Benten D., Fehse B., RGB marking with lentiviral vectors for multicolor clonal cell tracking. Nat. Protoc. 7, 839–849 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Tiede S., Kalathur R. K. R., Luond F., von Allmen L., Szczerba B. M., Hess M., Vlajnic T., Muller B., Canales Murillo J., Aceto N., Christofori G., Multi-color clonal tracking reveals intra-stage proliferative heterogeneity during mammary tumor progression. Oncogene 40, 12–27 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Consortium G. T., The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis R. T., Blake K., Ma D., Gabra M. B. I., Hernandez G. A., Phung A. T., Yang Y., Maurer D., Lefebvre A. E. Y. T., Alshetaiwi H., Xiao Z., Liu J., Locasale J. W., Digman M. A., Mjolsness E., Kong M., Werb Z., Lawson D. A., Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat. Cell Biol. 22, 310–320 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Dupuy F., Tabaries S., Andrzejewski S., Dong Z., Blagih J., Annis M. G., Omeroglu A., Gao D., Leung S., Amir E., Clemons M., Aguilar-Mahecha A., Basik M., Vincent E. E., St-Pierre J., Jones R. G., Siegel P. M., PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 22, 577–589 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Liu T., Zhang L., Joo D., Sun S. C., NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2, 17023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peppel K., Crawford D., Beutler B., A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J. Exp. Med. 174, 1483–1489 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pido-Lopez J., Tanudjojo B., Farag S., Bondulich M.-K., Andre R., Tabrizi S. J., Bates G. P., Inhibition of tumour necrosis factor alpha in the R6/2 mouse model of Huntington’s disease by etanercept treatment. Sci. Rep. 9, 7202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petersen S. L., Wang L., Yalcin-Chin A., Li L., Peyton M., Minna J., Harran P., Wang X., Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 12, 445–456 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varfolomeev E., Blankenship J. W., Wayson S. M., Fedorova A. V., Kayagaki N., Garg P., Zobel K., Dynek J. N., Elliott L. O., Wallweber H. J., Flygare J. A., Fairbrother W. J., Deshayes K., Dixit V. M., Vucic D., IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131, 669–681 (2007). [DOI] [PubMed] [Google Scholar]
- 42.San Juan B. P., Garcia-Leon M. J., Rangel L., Goetz J. G., Chaffer C. L., The complexities of metastasis. Cancers (Basel) 11, 1575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hou J.-M., Krebs M. G., Lancashire L., Sloane R., Backen A., Swain R. K., Priest L. J. C., Greystoke A., Zhou C., Morris K., Ward T., Blackhall F. H., Dive C., Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 30, 525–532 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Aceto N., Bardia A., Miyamoto D. T., Donaldson M. C., Wittner B. S., Spencer J. A., Yu M., Pely A., Engstrom A., Zhu H., Brannigan B. W., Kapur R., Stott S. L., Shioda T., Ramaswamy S., Ting D. T., Lin C. P., Toner M., Haber D. A., Maheswaran S., Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore G. E., Sandberg A. A., Watne A. L., The comparative size and structure of tumor cells and clumps in the blood, bone marrow, and tumor imprints. Cancer 13, 111–117 (1960). [DOI] [PubMed] [Google Scholar]
- 46.Liotta L. A., Saidel M. G., Kleinerman J., The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res. 36, 889–894 (1976). [PubMed] [Google Scholar]
- 47.Marusyk A., Almendro V., Polyak K., Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 12, 323–334 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Marusyk A., Tabassum D. P., Altrock P. M., Almendro V., Michor F., Polyak K., Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514, 54–58 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnstone C. N., Chand A., Putoczki T. L., Ernst M., Emerging roles for IL-11 signaling in cancer development and progression: Focus on breast cancer. Cytokine Growth Factor Rev. 26, 489–498 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Martin M. D., Kremers G. J., Short K. W., Rocheleau J. V., Xu L., Piston D. W., Matrisian L. M., Gorden D. L., Rapid extravasation and establishment of breast cancer micrometastases in the liver microenvironment. Mol. Cancer Res. 8, 1319–1327 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gross A. C., Cam H., Phelps D. A., Saraf A. J., Bid H. K., Cam M., London C. A., Winget S. A., Arnold M. A., Brandolini L., Mo X., Hinckley J. M., Houghton P. J., Roberts R. D., IL-6 and CXCL8 mediate osteosarcoma-lung interactions critical to metastasis. JCI Insight 3, e99791 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim M.-Y., Oskarsson T., Acharyya S., Nguyen D. X., Zhang X. H.-F., Norton L., Massague J., Tumor self-seeding by circulating cancer cells. Cell 139, 1315–1326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montfort A., Colacios C., Levade T., Andrieu-Abadie N., Meyer N., Segui B., The TNF paradox in cancer progression and immunotherapy. Front. Immunol. 10, 1818 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lebrec H., Ponce R., Preston B. D., Iles J., Born T. L., Hooper M., Tumor necrosis factor, tumor necrosis factor inhibition, and cancer risk. Curr. Med. Res. Opin. 31, 557–574 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Lalaoui N., Merino D., Giner G., Vaillant F., Chau D., Liu L., Kratina T., Pal B., Whittle J. R., Etemadi N., Berthelet J., Grasel J., Hall C., Ritchie M. E., Ernst M., Smyth G. K., Vaux D. L., Visvader J. E., Lindeman G. J., Silke J., Targeting triple-negative breast cancers with the Smac-mimetic birinapant. Cell Death Differ. 27, 2768–2780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weber K., Bartsch U., Stocking C., Fehse B., A multicolor panel of novel lentiviral "gene ontology" (LeGO) vectors for functional gene analysis. Mol. Ther. 16, 698–706 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Ollion J., Cochennec J., Loll F., Escude C., Boudier T., TANGO: A generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 29, 1840–1841 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allan C., Burel J. M., Moore J., Blackburn C., Linkert M., Loynton S., Macdonald D., Moore W. J., Neves C., Patterson A., Porter M., Tarkowska A., Loranger B., Avondo J., Lagerstedt I., Lianas L., Leo S., Hands K., Hay R. T., Patwardhan A., Best C., Kleywegt G. J., Zanetti G., Swedlow J. R., OMERO: Flexible, model-driven data management for experimental biology. Nat. Methods 9, 245–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.L. Whitehead, V. Wimmer, F. Lafouresse, D. Ratnayake, P. Currie, J. Groom, K. Rogers, T. Boudier, paper presented at the International Microscopy Congress IMC 19, 2018.
- 60.R. C. Team, R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2018);www.R-project.org/.
- 61.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. III, Hao Y., Stoeckius M., Smibert P., Satija R., Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider V. A., Graves-Lindsay T., Howe K., Bouk N., Chen H.-C., Kitts P. A., Murphy T. D., Pruitt K. D., Thibaud-Nissen F., Albracht D., Fulton R. S., Kremitzki M., Magrini V., Markovic C., McGrath S., Steinberg K. M., Auger K., Chow W., Collins J., Harden G., Hubbard T., Pelan S., Simpson J. T., Threadgold G., Torrance J., Wood J. M., Clarke L., Koren S., Boitano M., Peluso P., Li H., Chin C.-S., Phillippy A. M., Durbin R., Wilson R. K., Flicek P., Eichler E. E., Church D. M., Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu R., Wang H., Yu X., Shared-nearest-neighbor-based clustering by fast search and find of density peaks. Inform. Sci. 450, 200–226 (2018). [Google Scholar]
- 65.Patel A. P., Tirosh I., Trombetta J. J., Shalek A. K., Gillespie S. M., Wakimoto H., Cahill D. P., Nahed B. V., Curry W. T., Martuza R. L., Louis D. N., Rozenblatt-Rosen O., Suva M. L., Regev A., Bernstein B. E., Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nguyen Q. H., Pervolarakis N., Blake K., Ma D., Davis R. T., James N., Phung A. T., Willey E., Kumar R., Jabart E., Driver I., Rock J., Goga A., Khan S. A., Lawson D. A., Werb Z., Kessenbrock K., Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 9, 2028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen W., Morabito S. J., Kessenbrock K., Enver T., Meyer K. B., Teschendorff A. E., Single-cell landscape in mammary epithelium reveals bipotent-like cells associated with breast cancer risk and outcome. Commun. Biol. 2, 306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Csardi G., Nepusz T., The igraph software package for complex network research. InterJournal Complex Systems 1695 5, 1–9 (2006). [Google Scholar]
- 69.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011). [Google Scholar]
- 71.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J. P., Tamayo P., The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu G., Wang L.-G., Han Y., He Q.-Y., clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/28/eabf4408/DC1