

ABSTRACT
Membrane proteins that are integrated into the outer membrane of Gram-negative bacteria typically contain a unique “β barrel” structure that serves as a membrane spanning segment. A conserved “β signal” motif is located at the C terminus of the β barrel of many outer membrane proteins (OMPs), but the function of this sequence is unclear. We found that mutations in the β signal slightly delayed the assembly of three model Escherichia coli OMPs by reducing their affinity for the barrel assembly machinery (Bam) complex, a heterooligomer that catalyzes β barrel insertion, and led to the degradation of a fraction of the protein in the periplasm. Interestingly, the absence of the periplasmic chaperone SurA amplified the effect of the mutations and caused the complete degradation of the mutant proteins. In contrast, the absence of another periplasmic chaperone (Skp) suppressed the effect of the mutations and considerably enhanced the efficiency of assembly. Our results reveal the existence of two parallel OMP targeting mechanisms that rely on a cis-acting peptide (the β signal) and a trans-acting factor (SurA), respectively. Our results also challenge the long-standing view that periplasmic chaperones are redundant and provide evidence that they have specialized functions.
KEYWORDS: Bam complex, β barrel, β signal, molecular chaperones, outer membrane proteins, protein targeting
INTRODUCTION
Gram-negative bacteria are a major class of organisms that have two cell membranes, a cytoplasmic or inner membrane (IM) and an outer membrane (OM). Proteins that are integrated into the OM mediate a variety of important physiological functions, including nutrient uptake, membrane homeostasis, and virulence (1). Unlike the integral membrane proteins found in most biological membranes, OMPs generally do not contain α-helical membrane spanning segments that readily partition into a hydrophobic environment. Instead, they contain a closed cylindrical structure composed of amphipathic β strands called a “β barrel” that must fold and expose a hydrophobic surface before they can insert stably into the OM (1). Despite sharing a common architecture, OMPs are variable in sequence and range in size from 8 to 36 β strands (2, 3). Although some β barrels are empty monomers, others contain an embedded polypeptide, and some form homodimers or homotrimers. A subset of OMPs also contain a periplasmic or extracellular domain linked to the β barrel domain.
After OMPs are translocated across the IM through the Sec machinery, they first interact with a variety of molecular chaperones, including Skp, SurA, and DegP, a protein that also functions as a protease (4–8). It has long been thought that the chaperones prevent aggregation and maintain OMPs in an insertion-competent state in the periplasm (9), but their exact function is unclear. Skp is a homotrimer that resembles a jellyfish in which α-helical “tentacles” protrude from a central cavity (10, 11). Nuclear magnetic resonance, fluorescence, and ion mobility spectroscopy have indicated that 8- to 16-stranded β barrels can be accommodated inside the cavity of Skp in a disordered but highly dynamic conformational ensemble (12–15). Interestingly, cross-linking studies performed in spheroplasts or intact cells suggest that Skp binds OMPs either before they are released from the Sec complex or at an early stage of assembly (7, 16, 17). SurA is a much larger protein that is composed of four domains, including two peptidyl-prolyl isomerase domains (18). Available evidence indicates that the chaperone recognizes peptides that contain the sequence ϕ-X-ϕ (where ϕ is an aromatic residue), which is prevalent in the “aromatic bands” of β barrels (19), and interacts with the N- and C-terminal regions of OMP β barrels (20). The finding that null mutations in surA and skp, as well as in surA and degP, are synthetically lethal has led to the long-standing view that SurA is a component of one pathway while Skp and DegP are components of a functionally redundant pathway (21, 22). Because the depletion of SurA (but not Skp or DegP) leads to a significant decrease in the level of a variety of OMPs and severe defects in the assembly of specific OMPs, most notably LamB, it is thought that SurA is the primary chaperone that guides periplasmic transit while Skp and DegP function in a backup pathway (21, 23, 24). The decreased abundance of many OMPs in surA deletion strains, however, has been attributed at least in part to a concomitant decrease in their mRNA levels that might result from the induction of the σE stress pathway (24). Furthermore, the observation that the efficient assembly of LptD and FhuA requires both Skp (or other OMP assembly factors such as FkpA) and SurA suggests that Skp is not simply a redundant chaperone for OMP biogenesis (25).
After OMPs transit the periplasm, their insertion into the OM is catalyzed by the barrel assembly machinery (Bam) complex (26, 27). In Escherichia coli, the Bam complex is composed of BamA, an OMP that contains a β barrel domain and five periplasmic polypeptide transport-associated (POTRA) domains, and four lipoproteins (BamB to BamE) that are bound to the POTRA domains (26, 28, 29). Only BamA and BamD are essential for viability and conserved in essentially all Gram-negative organisms (27, 30). Despite the fact that the structure of the complete Bam complex has been solved (31–34), the mechanism by which it promotes the assembly of client proteins is unclear. All of the current models are based on striking evidence that the first and last β strands of the BamA β barrel form an unstable seam that has the potential to open laterally (35, 36). The “threading” (or “budding”) model holds that OMPs pass through the pore of the BamA β barrel in an unfolded conformation and then insert into the lipid bilayer in a stepwise fashion through a lateral gate. In an alternative model (the “assisted” model), it has been proposed that the transient opening of the BamA β barrel promotes the insertion of folded or partially folded client proteins by disrupting the lipid bilayer. Evidence that at least some OMPs begin to fold prior to membrane integration (37–39), that BamA lowers the kinetic barrier for OMP insertion imposed by lipid head groups (40) and that BamA exerts a greater catalytic effect on OMP folding in thicker bilayers (41) is consistent with this model. The recent analysis of OMP assembly intermediates bound to the Bam complex, however, showed that the first and last β strands of the client protein form distinct interfaces with the open form of the BamA β barrel (42, 43). These studies strongly suggest that BamA facilitates OMP integration not just by perturbing the lipid bilayer but also by making direct contact with substrates. Interestingly, the observations that SurA can be cross-linked to BamA (21, 44) and that a ternary complex containing OMPs, SurA, and the Bam complex can be detected in living cells (20) suggest that the chaperone helps to deliver OMPs to the Bam complex regardless of its mechanism of action.
Here, we gained insight into the function of periplasmic chaperones and the Bam complex not by examining the properties of these factors per se but rather by analyzing a poorly understood conserved sequence motif (GXXϕXϕ) located at the extreme C terminus of β barrels known as the “β signal.” It was shown nearly 30 years ago that an aromatic residue is located at the C-terminal position of most OMPs and that mutating this residue impairs OMP biogenesis to some degree (45). More recent in vitro assays have provided evidence that this residue as well as other conserved residues in the motif promote OMP assembly by binding to BamA (40, 46). Consistent with these observations, the β signal of an OMP assembly intermediate was shown to form a stable, rigid interface with the first β strand of the BamA β barrel in vivo (42). To complicate matters, however, other in vivo experiments have suggested that the β signal binds to BamD (47), and the aforementioned evidence that SurA binds to peptides containing ϕXϕ suggests that the C-terminal motif might be a recognition signal for the chaperone. Furthermore, the β signal of at least one OMP (BamA itself) is located two β strands away from the C terminus (47). To clarify the function of the β signal, we mutated the aromatic residues in the C-terminal β strand of three E. coli OMPs, OmpA, EspP, and OmpC, and examined the effect of the mutations on assembly in vivo and in an in vitro assay in which the function of the Bam complex has been reconstituted. We obtained evidence that the mutations slow assembly by reducing the affinity of the OMPs for the Bam complex. Interestingly, we found that while the mutant proteins were rapidly degraded in a surA deletion strain, they were stabilized in a skp deletion strain and were assembled nearly as well as the wild-type protein. The results strongly suggest that the β signal and SurA promote targeting by parallel mechanisms and that the functions of Skp and SurA are much more distinct than previously recognized. Our results also raise the intriguing possibility that OMPs are recruited to the Bam complex through the recognition of multiple sequence or structural features rather than a single “signal sequence.”
RESULTS
Many but not all E. coli OMPs contain a C-terminal β signal.
Although the β signal has been discussed extensively in the literature (46–48), neither the prevalence nor significance of this motif has been rigorously determined. To address this issue, we first compiled a list of all of the proteins in E. coli strain MG1655 that clearly contain a complete β barrel domain using the β-barrel outer membrane protein predictor (BOMP) (49). We then identified a peptide that resembles previously described β signals in 37 of the 48 putative OMPs (77%). The β signal is located at the extreme C terminus (or at the C terminus of the known β barrel domain) of every protein except BamA and TamA (see Table S1A in the supplemental material). An alignment of these peptides revealed a conserved sequence pattern that extends seven residues from the C terminus (Fig. 1A). Two other OMPs also contain a C-terminal peptide that is conserved in a wide variety of porins found in Gammaproteobacteria that have been shown or predicted to transport sugars (see Table S1B). Although these peptides contain a C-terminal aromatic amino acid, however, they do not resemble canonical β signals (Fig. 1B). Interestingly, one of these proteins is LamB, an OMP that has been studied as a model protein for many years. The other nine OMPs are homologs of FimD (see Table S1C), an usher protein that requires a BamA paralog (TamA) for efficient assembly (50). Based on the crystal structure of FimD (51), the sequence of the C-terminal β strand of these and other closely related E. coli proteins is conserved but even more distantly related to the canonical β signal (Fig. 1C).
FIG 1.
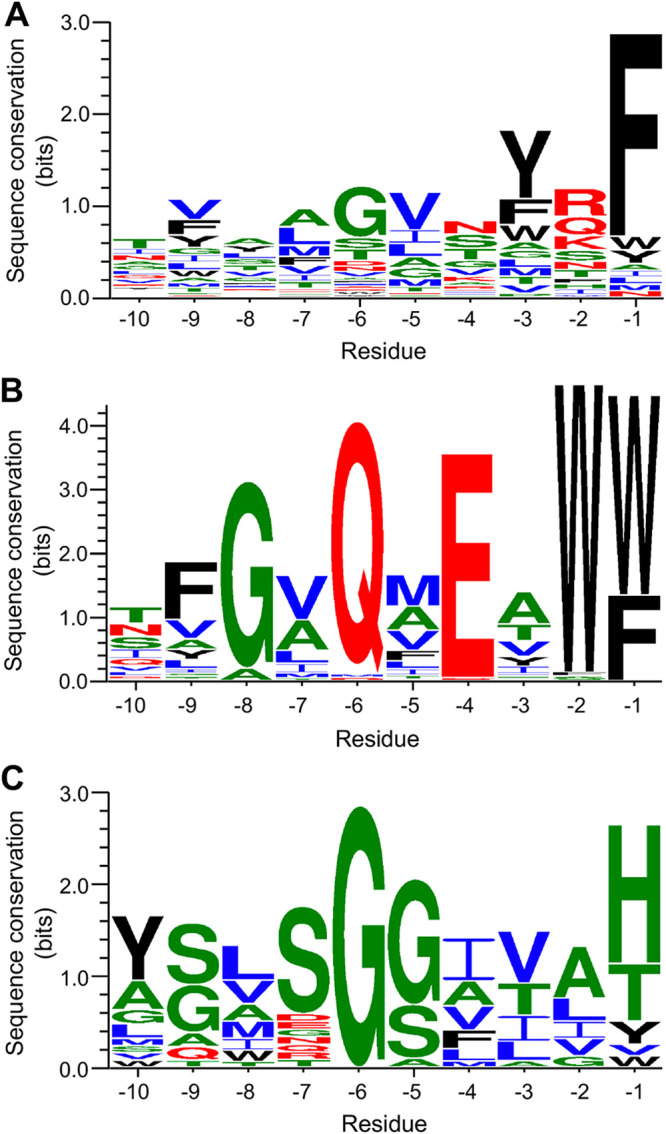
Sequence logo plots for the C-terminal 10 amino acids of 37 E. coli MG1655 OMP β barrels (A), 80 predicted trimeric porins that mediate carbohydrate uptake encoded in gammaproteobacterial genomes (B), and 20 predicted E. coli fimbrial usher proteins (C). The trimeric porins and usher proteins were identified based on homology to E. coli MG1655 LamB and FimD, respectively. Inputs are listed in Table S1. Black, aromatic amino acids; red, hydrophilic amino acids; green, neutral amino acids; blue, hydrophobic amino acids.
C-terminal 10 amino acids of 37 E. coli MG1655 OMP β barrels (A), 80 predicted trimeric porins that mediate carbohydrate uptake in Gammaproteobacteria (B), and 20 predicted E. coli FimD family proteins (C). Download Table S1, PDF file, 0.6 MB (611.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The β signal maximizes the rate of OMP assembly.
We used three well characterized E. coli OMPs as model proteins to gain insight into the function of the β signal. One protein, OmpA, has an empty 8-stranded β barrel domain and a small C-terminal periplasmic domain (52). The second protein, EspP, is a member of the autotransporter family that has a large N-terminal extracellular (“passenger”) domain attached to a 12-stranded β barrel domain through an α-helical linker that traverses the barrel (53). The third protein, OmpC, has a 16-stranded barrel that forms a trimeric porin (54). The β barrel domains of all proteins contain a clear C-terminal β signal but are otherwise unrelated. To elucidate the function of the β signal, we mutated one or both of the highly conserved aromatic residues located at positions −3 and −1 to alanine. To focus on the effects of the mutations on β barrel assembly (and to avoid any complications that are due to the presence of periplasmic or extracellular domains), we analyzed a truncated form of OmpA that lacks the periplasmic domain (TM-OmpA, residues 21 to 195) or a truncated form of EspP that retains the linker but lacks the passenger domain (EspPΔ5, residues 998 to 1300) in many of our experiments. Both TM-OmpA and EspPΔ5 have been shown to assemble as efficiently as the full-length proteins in previous studies (55–57). OmpC was examined as an unmodified full-length protein.
Initially we examined the effect of the β signal mutations on the assembly of TM-OmpA in vivo in a wild-type E. coli strain. We first transformed XW100 (MC4100 ΔompA) with a plasmid that encodes TM-OmpA, TM-OmpAY189A, TM-OmpAF191A, or TM-OmpAY189A, F191A under the control of the IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible trc promoter. To obtain a semiquantitative assessment of protein folding, we exploited the observation that OmpA is the receptor for several T-even type bacteriophages including K3 (58). By monitoring the formation of plaques by serial dilutions of a phage K3 stock, we found that cells that expressed the TM-OmpA mutants were grossly as sensitive to infection as cells that expressed the wild-type protein (Fig. 2A). To further examine the effect of the mutations on TM-OmpA assembly, we grew cells to mid-log phase in Luria-Bertani (LB) medium and determined the level of the protein by Western blotting. Because fully folded OmpA is resistant to sodium dodecyl sulfate (SDS) denaturation and migrates more rapidly than its predicted molecular weight on SDS-PAGE in the absence of heat (59), one half of each sample was heated to 95°C, while the other half was left unheated. The results showed that although the single point mutations did not significantly affect the level or the folding of the protein, the double mutation strongly reduced the level of the protein. Presumably, the mutant protein was retained in the periplasm and rapidly degraded by periplasmic proteases (Fig. 2B, top blot; loading controls are shown in Fig. S1A). Interestingly, the residual TM-OmpAY189A, F191A retained the ability to migrate relatively rapidly in the absence of heat (Fig. 2B, top blot, lane 7). Taken together, with the results of the phage sensitivity assay, these data suggest that the double mutation strongly diminishes—but does not completely abolish—OmpA assembly under rapid growth conditions.
FIG 2.

TM-OmpA β signal mutations and a surA deletion synergistically impair folding in vivo. (A and B) XW100 (MC4100 ΔompA; WT) and isogenic surA and skp deletion strains were transformed with pTRC, pXW01 (Ptrc-TM-OmpA) or a derivative encoding a TM-OmpA β signal mutant. In panel A, the relative sensitivity of strains that contained each plasmid to infection by phage K3 was determined by plaque assay. R, resistant to K3 phage. In panel B, cells were grown in rich medium to mid-log phase, and lysates were analyzed by Western blotting with an antiserum against an OmpA loop 1 peptide. (C) Cells transformed with pXW04 (Prha-TM-OmpA) or pXW16 (Prha-TM-OmpAY189A, F191A) were grown in minimal medium and subjected to pulse-chase labeling. After the OM was permeabilized, half of each sample was treated with PK, and immunoprecipitations were performed using an antiserum against an OmpA loop 4 peptide. (D to F) Quantitation of pulse-chase data. The TM-OmpA signal in each lane was first normalized to an ∼46 kDa background band (see Fig. S1D). The percentage of TM-OmpA (blue squares) and TM-OmpAY189A, F191A (red circles) that was resistant to PK digestion and the percentage of the original pulse-labeled protein (i.e., total protein at t = 0) that remained are shown. PK resistance was defined as the +PK signal/–PK signal at the same time point except for TM-OmpAY189A, F191A in the surA deletion strain, where PK resistance was defined as the +PK signal/–PK signal at the 0-min time point. (The quantitation using data without normalization to the background band is shown in Fig. S2A.) Each plot is based on the average of at least three independent experiments. Error bars represent the standard deviations.
Loading controls used in Western blot and pulse-chase analysis. (A and B) Larger portions of the Western blots presented in Fig. 2B and 7B, respectively, are shown. An unidentified ∼45-kDa background band (arrow) was used as a reference to confirm that similar amounts of cell extracts were loaded in each lane. (C) XW100 (MC4100 ΔompA; WT) and isogenic surA and skp deletion strains transformed with pXW01 were grown to mid-log phase, and lysates were analyzed by Western blotting with antisera against an OmpA loop 1 peptide and a loading control, the cytoplasmic protein Ffh (H. D. Bernstein, D. Zopf, D. M. Freymann, and P. Walter, Proc Natl Acad Sci USA 90:5229–5233, 1993, 10.1073/pnas.90.11.5229). (D) In all pulse-chase experiments in which the stability and assembly kinetics of TM-OmpA or TM-OmpAY189A, F191A were measured, the TM-OmpA signal in each lane was first normalized to the signal generated by an unidentified ∼46-kDa background band to account for differences in loading. As an example, a larger portion of one of the gels presented in Fig. 2C (upper right gel) is shown. Download FIG S1, PDF file, 0.7 MB (673KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Alternative quantitation of pulse-chase results. (A) The percentage of TM-OmpA (blue squares) and TM-OmpAY189A, F191A (red circles) that was resistant to PK digestion and the percentage of the original pulse-labeled protein (i.e., total protein at t = 0) that remained in the experiments shown in Fig. 2 are plotted. The TM-OmpA signal in each lane was not normalized to an ∼46-kDa background band (see Fig. S1D). (B) The percentage of the original pulse-labeled EspPΔ5 (blue squares) and EspPΔ5Y1298A, F1300A (red circles) (i.e., total protein at t = 0) that was cleaved at each timepoint in the experiments shown in Fig. 3 is plotted. Error bars represent the standard deviations. Download FIG S2, PDF file, 0.3 MB (349.5KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To obtain additional insights into the function of the β signal, we next examined the fate of newly synthesized TM-OmpA and TM-OmpAY189A, F191A over time under slower growth conditions. XW100 transformed with a plasmid encoding either the wild-type or mutant protein were grown in M9 minimal medium. Presumably by slowing cell physiology, minimal media often create a relatively permissive condition that reduces the effect of mutations on biological processes (60). When cultures reached early-log-phase cells were subjected to pulse-chase labeling and collected by centrifugation. The OM was then permeabilized and proteinase K (PK) was added to one half of each sample to digest unintegrated TM-OmpA. Finally, we performed immunoprecipitations using an antiserum generated against an OmpA extracellular loop 4 peptide and resolved proteins by SDS-PAGE. The observation that almost all of the pulse-labeled TM-OmpA (0 min) was resistant to PK digestion indicated that the protein assembled very rapidly (Fig. 2C, top left gel; Fig. 2D, blue squares; a background band used to normalize the signal in each lane is illustrated in Fig. S1D; the quantitation without normalization is shown in Fig. S2A). In contrast, most of the TM-OmpAY189A, F191A was sensitive to PK digestion at early time points, but became resistant to protease treatment after a 5-min chase (Fig. 2C, top right gel; Fig. 2D, red circles). Taken together with the Western blot analysis described above, these results strongly suggest that the double mutation slows the membrane integration of TM-OmpA and thereby exposes the protein to degradation, but that under permissive conditions most of the protein is eventually assembled correctly.
An analysis of the effect of β signal mutations on EspPΔ5 and OmpC biogenesis generated somewhat different results, but led to similar conclusions. In our experiments, we used a plasmid encoding wild-type EspPΔ5 under the control of a rhamnose-inducible promoter and a derivative encoding the β signal double mutant EspPΔ5Y1298A, F1300A. AD202 (MC4100 ompT::kan) were transformed with one of the plasmids, and rhamnose was added to cultures grown to early log phase in M9 medium to induce EspPΔ5 expression. Cells were then subjected to pulse-chase labeling, and immunoprecipitations were conducted using an antiserum against a C-terminal EspP peptide. Membrane insertion was assessed by monitoring a gel shift that results from the autoproteolytic processing of proEspPΔ5, a precursor form of the protein in which the linker and β barrel domain are covalently bound. The intrabarrel cleavage reaction, which leads to the release of the linker, has been shown to require the complete folding of the β barrel domain (61, 62). Consistent with previous results (57, 61), more than 90% of the wild-type protein was processed within 2 min (Fig. 3A, top gel, lanes 1 to 5; Fig. 3B, blue squares). Curiously, more than half of the EspPΔ5Y1298A, F1300A was degraded within 1 min (Fig. 3A, top gel, compare lanes 6 and 7; Fig. 3B, red circles). Furthermore, although essentially all of the remaining protein was assembled, there was an ∼1-min delay in proteolytic processing (Fig. 3A, top gel, lanes 6 to 10; Fig. 3B; Fig. S2B). The results suggest that the EspPΔ5 β signal mutation, like the cognate mutation in TM-OmpA, delayed membrane insertion. Presumably because EspPΔ5Y1298A, F1300A is recognized by periplasmic proteases more readily than TM-OmpAY189A, F191A, the delay led to the rapid degradation of a significant fraction of the protein. A similar β signal mutation (Y365A, F367A) exerted an even more dramatic effect on OmpC assembly. Presumably due to an even longer delay in membrane integration, >80% of the protein was degraded during the 10 min chase (see Fig. S3).
FIG 3.

EspPΔ5 β signal mutations and a surA deletion synergistically impair folding in vivo. (A) AD202 (WT) and isogenic surA and skp deletion strains transformed with pJH207 (Prha-espPΔ5) or pJH208 (Prha-espPΔ5Y1298A, F1300A) were grown in minimal medium and subjected to pulse-chase labeling. Immunoprecipitations were performed using an antiserum raised against an EspP C-terminal peptide. (B to D) Quantitation of pulse-chase results. The percentage of EspPΔ5 (blue squares) and EspPΔ5Y1298A, F1300A (red circles) observed at each time point that was cleaved and the percentage of the original pulse-labeled protein (i.e., total protein at t = 0) that remained are shown. (The percentage of the original pulse-labeled protein that was cleaved is shown in Fig. S2B). Each plot is based on the average of at least three independent experiments. Error bars represent the standard deviations.
OmpC β signal mutations and a surA deletion synergistically impair folding in vivo. Cells transformed with pJH220 (Prha-OmpC) or a derivative containing the OmpC Y365A F367A mutation were subjected to pulse-chase labeling. After the OM was permeabilized, half of each sample was treated with PK, and immunoprecipitations were performed using an antiserum against an OmpC peptide. Download FIG S3, PDF file, 0.6 MB (653.9KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
A plausible explanation for the delay in the membrane insertion of EspPΔ5Y1298A, F1300A emerged from cross-linking experiments. Based on evidence that the β signal promotes the binding of client proteins to the BamA β barrel or BamD (40, 42, 46, 47), we hypothesized that the double mutation delays membrane insertion by impairing a key interaction between EspPΔ5 and the Bam complex. To test this possibility, we exploited the observation that upon the incorporation of the photoactivatable amino acid analog p-benzoyl-l-phenylalanine (Bpa) into specific positions in the β barrel of full-length EspP using a well-established amber suppression method (63), interactions with components of the Bam complex can be detected by photo-cross-linking (17). AD202 were transformed with plasmids encoding the amber suppression system (pDULE) and either wild-type EspP or EspPY1298A, F1300A containing an amber mutation at residue 1214. Bpa introduced at this position has been shown to form a cross-link with BamD at early stages of EspP biogenesis (17, 57, 61, 64). Cells were subjected to pulse-chase labeling, and half of each sample was UV irradiated. In the presence of UV light, two bands that were slightly larger than proEspP (the unprocessed form of the protein in which the passenger and β barrel domains are covalently linked) were immunoprecipitated from cells that expressed wild-type EspP with the anti-EspP C-terminal antiserum (Fig. 4A, lanes 5 to 8). These bands were most prominent at early time points and disappeared as the protein folded, disassociated from the Bam complex, and underwent autoproteolytic processing. Consistent with previous results, the larger band was immunoprecipitated with an anti-BamD antiserum and corresponded to a proEspP-BamD cross-linking product. In contrast, no cross-linking between EspPY1298A, Y1300A and BamD could be detected; only the smaller band, which corresponds to a cross-linking product between proEspP and at least one uncharacterized protein was observed (Fig. 4B, top gel) (17, 57). Like EspPΔ5Y1298A, F1300A, EspPY1298A, F1300A was assembled more slowly than the wild-type protein and was consequently more sensitive to periplasmic proteases. These results suggest that the double mutation reduces the affinity of the EspP β barrel domain for the Bam complex (or at least affects the interaction of the EspP β barrel with BamD).
FIG 4.

Mutations in the EspP β signal disrupt the binding of proEspP to the Bam complex. AD202 transformed with pRI22 (Plac-10His-espP) harboring an amber codon at residue 1214 (A) or a derivative containing the Y1298A F1300A mutation (B) were subjected to pulse-chase labeling. Half of the cells were UV irradiated, and immunoprecipitations were performed with antisera against an EspP C-terminal peptide or BamD. For clarity, an expanded region of lanes 5 and 6 in the top gels is shown.
We next examined the effect of β signal mutations on the interaction of OMPs with the Bam complex directly using an in vitro assembly assay. This assay is based on the observation that the purified Bam complex catalyzes the folding of urea-denatured OMPs into proteoliposomes (55, 65, 66). Initially we incubated OmpA, OmpAY189A, F191A and OmpAV98R, L100R, a mutant that contains two surface exposed arginine residues that are predicted to impair assembly (64), with proteoliposomes consisting solely of the Bam complex and a synthetic phospholipid at 30°C. Samples were removed at various time points, and proteins were resolved by SDS-PAGE in the absence of heat. OmpA was then detected by Western blotting, and assembly was assessed by monitoring the accumulation of a rapidly migrating form of the protein. When the Bam complex was added at a 2.5-fold excess over OmpA, the fraction of OmpA and OmpAY189A, F191A that folded and the kinetics of assembly were similar (Fig. 5, top left blot). As expected, the OmpAV98R, L100R mutant did not fold (Fig. 5, top right blot). The addition of stoichiometric or substochiometric amounts of the Bam complex, however, led to a reduction in both the fraction of OmpA that assembled as well as the rate of assembly, but nearly completely abolished OmpAY189A, F191A assembly (Fig. 5, middle and bottom blots). The simplest explanation of these results is that the mutation impairs the binding of OmpA to the Bam complex and increases the tendency of the protein to lose insertion competence when the availability of the Bam complex is reduced.
FIG 5.

A β signal defect impairs the assembly of OmpA in vitro. Urea-denatured OmpA, OmpAY189A, F191A or OmpAV98R, L100R (0.2 μM) was incubated with proteoliposomes containing the Bam complex and POPC at 30°C. The amount of proteoliposomes added to the reaction was varied to achieve the indicated OmpA/Bam complex ratios. Unheated samples were subjected to SDS-PAGE, and OmpA assembly was analyzed by Western blotting with an antiserum raised against an OmpA C-terminal peptide. The percentage of folded OmpA was defined as folded OmpA/total OmpA (OmpA at t = 0).
In a second set of experiments, we tested whether mutations in the β signal have a similar effect on the assembly of EspPΔ5. Urea-denatured EspPΔ5, EspPΔ5Y1298A, F1300A, or EspPΔ5I1119R, a mutant that does not assemble in vivo (64), were mixed with proteoliposomes containing the Bam complex at a 1:1 ratio and SurA, a chaperone that is required for EspPΔ5 assembly in vitro (55). Samples were removed at various time points and assembly was measured by monitoring the proteolytic processing of proEspPΔ5. A smaller fraction of the EspPΔ5Y1298A, F1300A mutant was assembled than wild-type EspPΔ5, and the time required to reach 50% maximal assembly (t1/2) was considerably longer (Fig. 6). Because the efficiency of assembly was not affected by the concentration of SurA (see Fig. S4), the data suggest that the mutation reduces the affinity of EspPΔ5 for the Bam complex and thereby increases the opportunity for the protein to misfold in solution. Taken together, the in vitro results are consistent with the hypothesis that the β signal promotes efficient binding of OMPs to the Bam complex.
FIG 6.

A β signal defect impairs the assembly of EspPΔ5 in vitro. (A) Urea-denatured EspPΔ5, EspPΔ5Y1298A, F1300A, or EspPΔ5I1119R (0.2 μM) was incubated with proteoliposomes containing the Bam complex (0.2 μM) in POPC and 2 μM SurA at 30°C. Protein cleavage during a 30-min time course was analyzed by Western blotting with an antiserum against an EspP C-terminal peptide. (B) The average fraction of protein cleaved at each time point in at least three independent experiments is shown. Curves were fitted to the data and a rate constant (K) and the time required to reach 50% maximal folding (t1/2) were calculated using Prism 8 software. Error bars represent the standard deviations.
A β signal mutation does not affect the interaction of EspPΔ5 with SurA. Urea-denatured EspPΔ5 or EspPΔ5Y1298A, F1300A (0.2 μM) was incubated with proteoliposomes containing the Bam complex (0.2 μM) in POPC and the indicated amount of SurA at 30°C for 30 min. Protein cleavage was visualized by Western blotting with an antiserum raised against an EspP C-terminal peptide. Download FIG S4, PDF file, 0.5 MB (480.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
surA and skp deletions differentially affect the biogenesis of β signal mutants.
We next wished to determine whether defects in the β signal might affect the requirement for periplasmic chaperones in OMP biogenesis. To this end, we repeated the experiments described above using XW101 (MC4100 surA::cm ompA::kan) transformed with a plasmid that encodes TM-OmpA or a β signal mutant. We found that the absence of SurA did not affect the sensitivity of cells that expressed wild-type TM-OmpA to infection by K3 phage. In contrast, surA-negative cells that expressed TM-OmpAY189A or TM-OmpAF191A were at least 1 to 2 orders of magnitude less sensitive to infection than surA+ cells, while surA-negative cells that expressed TM-OmpAY189A, F191A were 3 to 4 orders of magnitude less sensitive (Fig. 2A). surA-negative cells that expressed TM-OmpAF191A were presumably more resistant to phage infection than those that expressed TM-OmpAY189A because the more highly conserved C-terminal residue plays a more critical role in β signal function (Fig. 1A). Consistent with these results, Western blot analysis showed that the disruption of surA did not affect the level of TM-OmpA in cells grown in LB medium but progressively destabilized the single and double β signal mutants (Fig. 2B, middle blot; see Fig. S1A and C in the supplemental material). Likewise, the examination of TM-OmpA assembly in radiolabeled cells grown in M9 medium, permeabilized, and treated with PK revealed that the absence of SurA only slightly delayed the membrane integration of the wild-type protein but led to the complete degradation of TM-OmpAY189A, F191A (Fig. 2C, middle gels, and Fig. 2E; see also Fig. S2A).
We obtained similar results in complementary experiments in which we analyzed the effect of disrupting surA on the assembly of EspPΔ5 and OmpC and their respective β signal mutants. HDB130 (AD202 surA::cm) transformed with a plasmid encoding either the wild-type or mutant protein were subject to pulse-chase radiolabeling as described above. The absence of SurA did not discernably affect the assembly of EspPΔ5, but led to the rapid degradation of EspPΔ5Y1298A, F1300A and impaired the assembly of the remaining protein (Fig. 3A, middle gel and Fig. 3C; see also Fig. S2B). Furthermore, although EspP containing Bpa at residue 1214 was cross-linked to BamD in cells that lacked SurA following UV irradiation, no cross-linking between EspPY1298A, F1300A and BamD was observed (see Fig. S5A). The mutant protein, however, was preferentially cross-linked to Skp (see Fig. S5A and C). Furthermore, although the assembly of OmpC was slightly delayed in a surA deletion strain, the OmpCY365A, F367A mutant was completely degraded within ∼1 min (see Fig. S3, middle gels). Taken together, the results strongly suggest that the lack of SurA exerts a slight or no effect on the biogenesis of the three OMPs we tested, but strongly exacerbates assembly defects associated with mutations in their respective β signals. These effects were specific to the β signal mutations; four control proteins in which aromatic residues located outside the β signal in TM-OmpA or EspPΔ5 were mutated to alanine (TM-OmpAL104A, Y106A, TM-OmpAY162A, W164A, EspPΔ5Y1157A, Y1159A, and EspPΔ5Y1229A, F1231A) folded as efficiently as the wild-type proteins in the absence of SurA (see Fig. S6). The results suggest that the β signal mutations impair OMP assembly by perturbing a cis-acting targeting signal rather than by altering the thermodynamic stability of the protein previously associated with aromatic residues (67).
Effect of β signal mutations on the binding of proEspP to the Bam complex and Skp. HDB130 (surA deletion strain) (A) or HDB131 (skp deletion strain) (B) transformed with pRI22 (Plac-10His-espP) harboring an amber codon at residue 1214 or a derivative containing the Y1298A F1300A mutation were subjected to pulse-chase labeling. Half of the cells were UV irradiated, and immunoprecipitations were performed with antisera against an EspP C-terminal peptide or BamD. For clarity, an expanded region of lanes 5 and 6 in the top gels is shown. (C) An aliquot of the UV-irradiated samples generated in the experiments shown in panels A and B, and Fig. 4 was subjected to immunoprecipitation with an antiserum against Skp. A background band that migrates slightly faster than the proEspP-Skp crosslinking product is denoted with an asterisk. Download FIG S5, PDF file, 1.1 MB (1.1MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The mutation of aromatic residues outside the β signal does not affect OMP assembly in a surA deletion strain. XW101 (MC4100 ompA::kan surA::cm) transformed with pXW19 (Prha-TM-OmpAL104A, Y106A) or pXW20 (Prha-TM-OmpAY162A, W164A) (A) or HDB130 (AD202 surA::cm) transformed with pJH209 (Prha-espPΔ5L1157A, Y1159A) or pJH210 (Prha-espPΔ5Y1298A, F1300A) (B) were subjected to pulse-chase labeling. After the OM was permeabilized, half of each sample was treated with PK, and immunoprecipitations were performed with an antiserum against an OmpA loop 4 peptide or an EspP C-terminal peptide. Download FIG S6, PDF file, 0.6 MB (604KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Remarkably, despite the fact that SurA and Skp have been proposed to be central components of redundant chaperone pathways (22), we found that the disruption of the genes that encode the two proteins exerted opposite effects on the assembly of β signal mutants. Experiments conducted using strain XW102 (XW100 Δskp) showed that the absence of Skp, unlike the absence of SurA, did not affect the sensitivity of cells that expressed TM-OmpA β signal mutants to infection by K3 phage (Fig. 2A). Interestingly, while similar amounts of TM-OmpAY189A and TM-OmpAF191A were detected by Western blotting in wild-type and skp deletion strains grown in LB medium, an even higher level of TM-OmpAY189A, F191A was detected in the skp deletion strain (Fig. 2B; see also Fig. S1A and C). Consistent with this finding, experiments performed in M9 medium showed that the loss of Skp not only failed to produce the assembly defects observed in the surA deletion strain but also almost completely suppressed the delay in the assembly of TM-OmpAY189A, F191A and TM-OmpA observed in wild-type and surA deletion strains, respectively (Fig. 2C, bottom gels, and Fig. 2F). Likewise, the examination of EspPΔ5 and OmpC assembly in skp deletion strains showed that the absence of Skp largely suppressed the degradation of EspPΔ5Y1298A, F1300A and OmpCY365A, F367A seen in wild-type cells (Fig. 3A, bottom gel, and Fig. 3D; see also Fig. S2B and Fig. S3). In addition, a moderate amount of EspPY1298A, F1300A containing Bpa at residue 1214 was cross-linked to BamD in a skp deletion strain following UV irradiation (see Fig. S5B). Because the absence of SurA—but not Skp—triggers the σE stress response (24), it is conceivable that the differential effects we observed were an indirect consequence of an increase in the level of periplasmic proteases in the surA deletion strain. To test this possibility, we examined the assembly of EspPΔ5Y1298A, F1300A in AD202 transformed with a plasmid that encodes rpoE under the control of an IPTG-inducible promoter (68) or an empty vector. Quantitative RT-PCR and Western blot analysis confirmed that the addition of IPTG elevated the expression of rpoE and increased the level of both DepP and SurA (see Fig. S7A and B in the supplemental material). Interestingly, consistent with the notion that the absence of SurA directly exacerbates assembly defects associated with β signal mutations, the activation of σE slightly enhanced the assembly of EspPΔ5Y1298A, F1300A (see Fig. S7C).
Activation of the σE stress response does not affect EspPΔ5Y1298A, F1300A assembly. AD202 transformed with pJH208 (Prha-espPΔ5Y1298A, F1300A) and either pTrc or pLC45 (Ptrc-rpoE) were incubated with IPTG for 25 min and then rhamnose for an additional 5 min. Cells were collected for qRT-PCR (A) and Western blot analysis (B) and EspPΔ5Y1298A, F1300 assembly was analyzed by pulse-chase labeling and immunoprecipitation using an antiserum raised against an EspP C-terminal peptide (C). In panel A, rpoE expression was normalized to the expression of the housekeeping gene rssA (black bar, average). In panel B, Western blots were performed with antisera raised against the indicated proteins. Download FIG S7, PDF file, 0.6 MB (596.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The simplest interpretation of these results is that SurA and Skp play distinct roles in the biogenesis of both proteins we analyzed. The data indicate that the efficient assembly of TM-OmpA and EspPΔ5 requires either the presence of a fully functional β signal or SurA as a trans-acting chaperone. That is, both the β signal and SurA likely play at least partially redundant roles in targeting OMPs to the Bam complex. In contrast, Skp promotes the destabilization of β signal mutants that have not been correctly integrated into the OM. In the absence of Skp, mislocalized β signal mutants have a greater window of opportunity to assemble (possibly by reaching the OM more rapidly) and tend to escape the degradation that is observed in wild-type and surA deletion strains.
Mutations in the β signal affect the assembly of a circularly permuted β barrel.
To obtain further insight into the function of the β signal, we exploited the observation that a “circularly permuted” derivative of OmpA in which the position of the four N-terminal and four C-terminal β strands are switched can fold and insert efficiently into the E. coli OM (69). In this variant [which we designate TM-OmpA(34)(12) to indicate the order of the loops] the native β signal is located in the middle of the β barrel and the C-terminal sequence does not clearly match the β signal consensus motif (Fig. 7A). Based on the results described above, we surmised that because TM-OmpA(34)(12) lacks a typical C-terminal β signal, its assembly might require SurA. Western blot analysis showed that the levels of the native and circularly permuted forms of TM-OmpA produced in wild-type cells grown in LB were similar (see Fig. S1A and B in the supplemental material). Surprisingly, however, the level of TM-OmpA(34)(12) was not significantly affected by the absence of either SurA or Skp (Fig. 7B). Furthermore, only a slight delay in assembly was observed in the surA deletion strain grown in M9 (see Fig. S8A). To determine whether the results could be explained by the ability of the β signal to function as an internal targeting signal, we changed the conserved aromatic residues in the motif to alanine and tested the effect of the mutations on the assembly of TM-OmpA(34)(12). Consistent with our hypothesis, we found that the Y189A and F191A mutations and the Y189A F191A double mutation progressively reduced the level of the protein in wild-type cells grown in LB medium (Fig. 7B, top blot). Our idea was further supported by the observation that the absence of SurA further destabilized the mutants, while the absence of Skp slightly increased their stability (Fig. 7B, middle and bottom blots). Interestingly, we also found that the introduction of the last three amino acids of the OmpA β signal into the C terminus of β strand 2, 4, or 6 of TM-OmpAY189A, F191A slightly improved the assembly of the protein in cells that lacked SurA (see Fig. S8B and C). Taken together, our data raise the intriguing possibility that internally located β signals can promote OMP assembly at least to some degree.
FIG 7.

Mutations in the β signal of a circularly permuted version of TM-OmpA affect protein assembly in vivo. (A) The structure of TM-OmpA (PDB 2GE4) (52) and the structure of TM-OmpA(34)(12) predicted using the EzMol server (84) are shown. The β signal is colored red. The diagrams show the arrangement of the β strands and loops of the native and circularly permuted proteins, as well as the amino acid sequence of the last β strand. (B) XW100 (MC4100 ΔompA; WT) and isogenic surA and skp deletion strains transformed with pXW02 or a related plasmid encoding the indicated TM-OmpA(34)(12) mutant were grown to mid-log phase, and lysates were analyzed by Western blotting with an antiserum raised against an OmpA loop 1 peptide.
Internally located β signals partially enhance the assembly of a circularly permuted version of TM-OmpA. (A) XW100 (MC4100 ΔompA; WT) and isogenic surA and skp deletion strains transformed with pXW17 [Prha-TM-OmpA(34)(12)] were subjected to pulse-chase labeling. After the OM was permeabilized, half of each sample was treated with PK, and immunoprecipitations were performed using an antiserum against an OmpA loop 4 peptide. These results show that the disruption of surA or skp does not significantly affect the assembly of circularly permuted TM-OmpA. (B) The last three residues of the OmpA β-signal motif (YRF) were introduced into TM-OmpAY189A, F191A β-strand 2, 4, or 6 by mutagenesis as shown. (C) XW101 (MC4100 ompA::kan surA::cm) were transformed with a derivative of pXW01 (Ptrc-TM-OmpA) that encodes one of the mutants and grown in LB medium. Samples were collected at mid-log phase, and cell lysates were heated at 95°C or incubated on ice. The folding of the TM-OmpA mutants was visualized by Western blotting with an antiserum against an OmpA loop 1 peptide. The results show that internal mutations that recreate the OmpA β-signal motif partially restore the folding of TM-OmpAY189A, F191A in a surA deletion strain. Download FIG S8, PDF file, 0.7 MB (753.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
In this report we describe evidence that the conserved “β signal” located at the C terminus of bacterial OMPs promotes interactions between the Bam complex and its client proteins. All of our experiments were conducted using three model proteins that consist of an empty β barrel or a β barrel with an embedded α-helical segment. Under rapid growth conditions, the mutation of the two most highly conserved residues in the β signal of TM-OmpA (Y189 and F191) led to its degradation in wild-type cells. Presumably because biological pathways are less sensitive to perturbation under slow growth conditions, the same mutation only delayed the assembly of TM-OmpA when cells were grown in minimal medium. Taken together with the finding that the efficient assembly of the double mutant in a reconstituted in vitro assay required a high Bam complex/OmpA ratio, these results strongly suggest that the mutation reduces the affinity of the OmpA β barrel for the Bam complex. The assembly of variants of two other model proteins (EspPΔ5 and OmpC) that contain mutations at the equivalent positions in the β signal was also delayed under slow growth conditions, but a large fraction of the protein was rapidly degraded. The EspPΔ5 mutant was clearly assembled more slowly than the wild-type protein in vitro, and photo-cross-linking experiments provided direct evidence that the mutation impairs the binding of EspPΔ5 to the Bam complex. While the results obtained with all of the model proteins indicate that the β signal facilitates recognition by the Bam complex, the finding that the EspPΔ5 and OmpC mutants were relatively unstable in wild-type cells suggests that OMPs vary in the efficiency of their interaction with the Bam complex and/or in their susceptibility to periplasmic proteases.
While our results implicate the β signal in targeting, they also provide clear evidence that this motif is not absolutely required for recognition by the Bam complex. The data are therefore incompatible with “threading” models in which an interaction between the β signal and BamA is required to initiate a stepwise membrane integration process. Interestingly, although a stable interface between the β signal of the EspP β barrel and the first β strand of BamA has been detected in vivo when the assembly of an EspP derivative is arrested (42), available crystal structures of the BamA and EspP β barrels (31–34, 53) do not reveal potential chemical bonds that would explain this strong interaction. In light of the previous results and the results presented here, we propose that BamA recognizes multiple sequence or structural elements that result from the partial folding of OMPs in the periplasm. In this scenario, the β signal would be an especially important component of the β barrel “signature” that facilitates high-affinity interactions. The surprising finding that the β signal might function as an internal targeting signal under some conditions, however, raises the possibility that this segment is important because it mediates an uncharacterized interaction between OMPs and BamD and/or the BamA POTRA domains that has been suggested by previous biochemical and genetic results (47, 70). In any case, it is interesting to note that although signal peptides are generally thought to initiate interactions between presecretory proteins and the Sec complex, intriguing evidence that they are not completely essential for secretion in an E. coli strain that contains a specific secY mutation has been reported (71). In addition, it was recently shown that the mature domains of many E. coli presecretory proteins share the common ability to form long-lived, loosely packed folding intermediates in vitro (72). These results suggest a possible amendment to the classical “signal hypothesis” (73) in which targeting signals function primarily to enhance the speed and efficiency of an intracellular localization process that is guided in part by general chemical or structural properties of specific classes of proteins.
The observation that mutations in the β signal and the deletion of surA create synergistic effects on OMP assembly strongly suggests that the conserved sequence and the chaperone direct proteins to the Bam complex by two parallel mechanisms. Likewise, the finding that the deletion of skp suppresses assembly defects associated with β signal mutations challenges the notion that SurA and Skp mediate redundant functions. Taken together, our results suggest a new two-step model to explain the early stages of OMP biogenesis. Based on previous cross-linking studies (7, 16, 17, 20), we propose that Skp binds to OMPs as (or shortly after) they traverse the IM (Fig. 8A, left). This interaction presumably maintains assembly competence and may even promote early steps in protein folding but is not essential for OMP biogenesis. Subsequently, Skp releases its client proteins and transfers them to SurA. As suggested by the observation that SurA can bind and stabilize OMPs and that SurA can form a ternary complex with OMPs and BamA (21, 44, 74, 75), the chaperone then escorts OMPs to the Bam complex. The presence of the β signal as a cis-acting sequence maximizes the affinity of the interaction of the OMP with the Bam complex and promotes rapid assembly. SurA still delivers β signal mutants and native OMPs that lack a β signal to the Bam complex, but the variants can dissociate and, as suggested by our photo-cross-linking data rebind Skp (Fig. 8A, right; see also Fig. S5C). Although these proteins can be rescued, OMPs that effectively remain bound to Skp are eventually delivered to DegP and degraded. In the absence of a SurA escort OMPs reach the Bam complex slightly more slowly, but in many cases the relatively strong targeting function of the β signal suffices to promote efficient assembly (Fig. 8B, left). In contrast, OMPs that lack a β signal can neither be escorted to the Bam complex nor form a stable interaction. As a consequence, these proteins remain in the periplasm and are completely degraded in the Skp-dependent process described above (Fig. 8B, right). It should be noted that the synergistic effect of the β signal mutations and the surA deletion implies that SurA does not simply recognize the β signal but targets OMPs to the Bam complex in an independent reaction that involves the recognition of internal sequences. Finally, in a skp deletion strain SurA binds directly to OMPs and escorts them rapidly to the Bam complex (Fig. 8C). In the absence of Skp β signal mutants are free of a quality control factor that delays their assembly through repeated rebinding and targets them for degradation. The opportunities to interact productively with the Bam complex are thereby enhanced, and the mutants are assembled almost as rapidly and efficiently as their wild-type counterparts.
FIG 8.

Model of early events in OMP assembly. (A) In wild-type cells, OMPs bind to Skp immediately after they are transported into the periplasm. Skp maintains the assembly competence of its client proteins and may promote initial steps of folding. OMPs are subsequently transferred to SurA, which escorts them to the Bam complex. The β signal promotes a strong interaction with the Bam complex and thereby stimulates efficient assembly. Mutants that lack the β signal and native OMPs that lack a β signal are escorted to the OM by SurA but do not bind efficiently to the Bam complex and can rebind Skp. A prolonged association with Skp increases the probability that mislocalized OMPs will be transferred to DegP and degraded. (B) In the absence of SurA, OMPs may reach the OM more slowly, but a strong interaction between the β signal and the Bam complex is often sufficient to promote efficient assembly. Mutants that lack the β signal, however, continue to rebind Skp and are targeted to DegP for degradation. (C) In the absence of Skp, OMPs bind directly to SurA. Mutants that lack the β signal, however, cannot be targeted for degradation and have a greater time window to interact productively with the Bam complex. In this model the β signal and SurA target OMPs to the Bam complex by two parallel mechanisms while Skp and DegP function as quality control factors.
In light of our results, we propose that deletions of surA and either skp or degP are synthetically lethal because SurA and Skp are components of complementary pathways and not redundant pathways as originally proposed (21, 22). In this scheme SurA is an OMP escort, while Skp is a factor that both promotes the earliest steps of OMP assembly and acts as a timer. In the absence of SurA, many OMPs are still assembled relatively effectively because the β signal functions as a cis-acting targeting signal. Molecules that remain in the periplasm (perhaps because they lack β signals or contain weak β signals) are bound by Skp and eventually delivered to DegP for degradation. In the absence of both SurA and Skp or DegP, however, toxic aggregates build up in the periplasm because the quality control system is compromised. As suggested by previous results (21, 22), SurA is the more important of the two factors because it is involved in the OMP assembly process per se, whereas Skp is needed primarily (or only) under specific conditions in which one or more OMPs are not localized effectively.
Two parallel mechanisms that both target OMPs to the Bam complex may have evolved at least in part because of functional or physiological constraints on the sequence of a subset of OMPs. Porins that transport carbohydrates, for example, contain a conserved C-terminal motif that does not resemble a β signal and do not appear to contain an internal β signal. The observation that the most highly conserved residue in the motif (the tryptophan at position −2) is required for the maltoporin transport activity of LamB (76), however, might explain the presence of the unique C-terminal sequence. Consistent with our model, the assembly of LamB is highly dependent on SurA (6). Interestingly, members of the FimD family not only contain a unique C-terminal motif (Fig. 1C) but also appear to be assembled by a distinct mechanism that involves both the Bam complex and a BamA homolog called TamA (50). Indeed, it is conceivable that these proteins are highly dependent on SurA to escort them to the OM and to enable them to access the alternate assembly pathway.
MATERIALS AND METHODS
Sequence logo plots.
A Python script was run to identify proteins in a comprehensive list of E. coli MG1655 exported and secreted proteins (77) that are localized in the OM and that contain a signal peptidase I cleavage site. Proteins that passed this initial screen were further analyzed using the BOMP server to predict OMPs (49). Specialized OMPs (CsgG, CusC, GspD, and TolC) that have been shown to form a β barrel from multiple subunits were excluded. The C-terminal 10 amino acids of the β barrel of other known OMPs that have been characterized structurally and/or functionally and putative OMPs whose β barrel positions could be determined are listed in Table S1A in the supplemental material. Porins encoded in the genomes of Gammaproteobacteria that are predicted to transport carbohydrates were identified by using E. coli LamB as a query sequence in a BLASTp search (see Table S1B). Fimbrial usher proteins produced by E. coli strains were identified from the UniProt database (see Table S1C). Sequence logos were plotted using Weblogo 3 (78).
Strains, antibiotics, and antisera.
The E. coli K-12 strains used in this study were MC4100 [araD139 Δ(argF-lac)169 λ-e14-flhD5301 Δ(fruK-yeiR)725(fruA25) relA1 rpsL150 rbsR22 Δ(fimB-fimE)632(::IS1) deoC1], AD202 (MC4100 ompT::kan), XW100 (MC4100 ΔompA), XW101 (MC4100 ompA::kan surA::cm), XW102 (MC4100 ΔompA Δskp), XW103 (MC4100 ΔompC), XW104 (MC4100 ΔompC surA::cm), XW105 (MC4100 ΔompC Δskp), HDB130 (AD202 surA::cm) (62), and HDB131 (AD202 Δskp) (62). To create the ΔompA and ΔompC strains, the ompA::kan and ompC::kan alleles from strain JW0940-6 and strain JW2203-1 were introduced into MC4100, respectively, by P1 transduction and the kanamycin resistance gene was removed using pCP20 (79). Ampicillin (100 μg/ml), kanamycin (30 μg/ml), chloramphenicol (25 μg/ml), and trimethoprim (50 μg/ml) were added to media as needed. Rabbit polyclonal antisera were generated against a peptide derived from OmpA extracellular loop 1 (NH2-CQYHDTGFINNNGPTHENQ-COOH) and the OmpA C terminus (NH2-CAPDRRVEIEVKGIKDVVTQPQA-COOH). Rabbit polyclonal antisera generated against an OmpA extracellular loop 4 peptide, an EspP C-terminal peptide, OmpC, BamD, Skp, SurA, DegP, and Ffh have been described (57, 65, 80, 81).
Plasmid construction.
The plasmids used in this study are listed in Table S2. Plasmid pJH207, which encodes espPΔ5 under the control of the rhaB2 promoter, pRI22 (Plac-10His-espP) harboring an amber codon at residue 1214, and pET28b::espPΔ5 have been described previously (55, 62, 64). To construct pXW01 (Ptrc-TM-OmpA), a DNA fragment encoding the OmpA β barrel was amplified by PCR using E. coli MC4100 genomic DNA and primers XW01 and XW02 (oligonucleotide primers used in this study are listed in Table S2). The PCR product was then ligated to pTrc99a by Gibson assembly (82). To construct pXW02 [Ptrc-TM-OmpA(34)(12)], a plasmid that we used to produce a “circularly permuted” form of OmpA (69), the primer pairs XW01/XW17, XW18/XW19, and XW20/XW21 were used to amplify DNA fragments that encode the OmpA signal peptide, and the C- and N-terminal fragments of the OmpA β barrel, respectively. The three PCR products were ligated to pTrc99a by Gibson assembly (82). To construct pXW03 (PT7-OmpA22–346), the mature region of ompA was amplified by PCR using E. coli MC4100 genomic DNA and the primer pair XW15/XW16, and the resulting PCR product was ligated to pET28b using Gibson assembly. To construct pXW04 (Prha-TM-OmpA), an NdeI site was introduced into pXW01 in front of the TM-OmpA ribosome binding site using the primers XW11 and XW12. The DNA fragment encoding TM-OmpA was then subcloned into pSCrhaB2 (83) using the NdeI and BamHI restriction sites. To construct pJH220 (Prha-OmpC), the primer pair JH300/JH301 was used to amplify ompC by PCR. The resulting fragment was digested with NdeI and HindIII and cloned into the cognate sites of pSCrhaB2. Mutations were introduced into each of the above plasmids using the QuikChange mutagenesis kit (Agilent) with appropriate primers.
Plasmids (A) and oligonucleotides (B) used in this study. Download Table S2, PDF file, 0.6 MB (639.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Phage assays.
K3 phage sensitivity was determined by modifying a previously described protocol (69). Cells from an overnight culture (100 μl) were mixed with 4 ml of 0.7% (wt/vol) LB agar containing 100 μg/ml ampicillin and poured onto an LB agar plate containing the same concentration of ampicillin. A K3 phage stock was serially diluted 10-fold to a maximum dilution of 107-fold. An aliquot (5 μl) of each phage dilution was spotted onto the plates, and the plates were incubated at 30°C overnight. The dilution at which a plaque was observed on each strain was then normalized to the dilution at which a plaque was observed on MC4100 (106-fold dilution) to determine the relative phage sensitivity. The values we obtained are based on the relative phage sensitivity observed in three independent experiments.
Analysis of OmpA levels at steady state.
Strains XW100-XW102 transformed with pXW01 or a pXW01 derivative were grown in LB medium to an optical density at 600 nm (OD600) of 0.45 to 0.55. Cells (1 OD600 equivalent) were collected by centrifugation (3,000 × g, 6 min, 4°C), washed with 1 ml of cold PBS, resuspended in 100 μl of BugBuster Master Mix (Novagen) containing EDTA-free protease inhibitors (Roche), and incubated on ice for 20 min. The lysate was then mixed with 4× LDS sample buffer (Thermo Fisher). Half of the lysate was heated at 95°C for 15 min, while the other half was kept on ice. Proteins were resolved on 12% Bis-Tris minigels (Thermo Fisher) using MES buffer, and folded and unfolded forms of the OmpA β barrel were detected by Western blotting with an antiserum raised against an OmpA loop 1 peptide.
Pulse-chase labeling and photo-cross-linking.
MC4100-based strains transformed with a derivative of pXW04 or pJH220, or AD202-based strains transformed with a pJH207 derivative, were grown at 37°C overnight in M9 containing 0.2% glycerol and all of the l-amino acids except methionine and cysteine (40 μg/ml). The overnight cultures were diluted into fresh medium at OD550 = 0.02 and grown to OD550 ∼ 0.2 to 0.25. Rhamnose was then added to a final concentration of 0.2% to induce expression of TM-OmpA, OmpC, or EspPΔ5. After 5 min, pulse-chase labeling was performed as previously described (62). To monitor the assembly of TM-OmpA or OmpC, 1 ml aliquots were pipetted over ice at each time point. Cells were collected by centrifugation (3,000 × g, 6 min, 4°C) and resuspended in spheroplast buffer (33 mM Tris [pH 8.0], 40% sucrose). The OM was then permeabilized by incubating the cells with 100 μg/ml lysozyme and 2 mM EDTA on ice for 20 min. Half of each sample was mixed with 10% trichloroacetic acid (TCA) to precipitate proteins. The other half was incubated for 20 min on ice with 200 μg/ml PK. After 2 mM phenylmethylsulfonyl fluoride was added to stop the PK digestions, proteins were precipitated with 10% TCA. To monitor the assembly of EspPΔ5, aliquots obtained at each time point were added directly to 10% TCA. Immunoprecipitations were then performed as described using antisera raised against a peptide derived from OmpA loop 4, OmpC loop 7, or the EspP C terminus. Proteins were resolved on 12% Bis-Tris or 8 to 16% Tris-glycine minigels (Thermo Fisher) to monitor the assembly of TM-OmpA and EspPΔ5, respectively. Radioactive proteins were detected using a Fuji BAS-2500 or FLA-9000 phosphorimager. To correct for loading disparities the TM-OmpA was normalized to an ∼46-kDa background band (see Fig. S1D). Photo-cross-linking experiments were conducted essentially as described previously (17), except that when cultures reached an OD550 of 0.2 only 1 mM Bpa was added. After 5 min, 200 μM IPTG was added to induce EspP synthesis, and pulse-chase labeling was conducted 10 min later.
Analysis of the effect of σE pathway activation on OMP assembly.
AD202 transformed with pJH208 and either a reconstructed version of pLC45 (68) or pTrc99A were grown in M9 medium as described above. When cultures reached an OD550 of ∼0.2, they were treated with 10 μM IPTG for 25 min to induce rpoE expression and then 0.2% rhamnose for 5 min to induce EspPΔ5Y1298A, F1300A synthesis. EspPΔ5Y1298A, F1300A assembly was analyzed by pulse-chase labeling as described above, and samples were collected for qRT-PCR and Western blotting. For qRT-PCR, cells (1 OD550 equivalent) were collected, and RNA was prepared by using an RNeasy minikit (Qiagen). RNA preps were treated with DNase I (NEB) and used as the templates to synthesize cDNA using the SuperScript III first-strand synthesis system (Thermal Fisher). qPCR was performed using Power-Up SYBR Green Master Mix (Thermal Fisher) and Bio-Rad CFX-96 real-time PCR detection system to determine the expression of rpoE and the housekeeping gene rssA as a normalization reference. For Western blots, proteins were TCA precipitated.
Purification of urea denatured OmpA and EspPΔ5.
E. coli BL21 transformed with pXW03 or pET28b::espPΔ5 or one of their derivatives were grown in 10 ml of LB medium at 37°C. When cultures reached an OD600 of ∼0.7, the expression of wild-type and mutant forms of OmpA and EspPΔ5 was induced by adding 0.5 mM IPTG. After 3 h, the cells were harvested, washed with 10 ml of PBS, and resuspended in 0.5 ml of BugBuster Master Mix with protease inhibitors. After a 20 min of incubation at room temperature, inclusion bodies were isolated by centrifugation (16,000 × g, 20 min, 4°C). The pellets were washed four times with 1 ml of 0.1× BugBuster Master Mix with protease inhibitors and once with 1 ml of H2O. The washed pellets were then resuspended in 0.5 ml 8 M urea, followed by incubation at room temperature for 1 h to solubilize proteins. Purified proteins were analyzed by SDS-PAGE, flash-frozen, and stored at −80°C.
Bam complex reconstitution and OMP assembly assays.
The Bam complex was expressed in BL21 transformed with pYG120, purified, and reconstituted into liposomes containing 1-palmitoy-2-oleoyl-glycero-3-phosphocholinel (POPC; Avanti Polar Lipids) as described previously (55, 65). SurA was also expressed and purified using a published method (55). OmpA and EspPΔ5 assembly assays were performed at 30°C essentially as described using 0.2 μM urea-denatured protein (55, 65). OmpA assembly assays contained 0.1 to 0.5 μM Bam complex, while EspPΔ5 assembly assays contained 0.2 μM Bam complex and 2 μM SurA. Samples were collected at different time points and mixed with 2 × SDS-PAGE sample buffer. Proteins were then resolved on 8 to 16% Tris-glycine minigels, and OMP assembly was visualized by Western blotting as described previously (65).
ACKNOWLEDGMENTS
We thank Sunyia Hussain for generating a plasmid that encodes the EspPΔ5 I1119R mutant and purifying the protein SurA, and we thank Matt Doyle for providing insightful comments on the manuscript.
This study was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
X.W. and H.D.B. designed the research and analyzed data, X.W. and J.H.P. performed the research, and X.W. and H.D.B. wrote the paper.
We declare there are no competing interests.
Footnotes
Citation Wang X, Peterson JH, Bernstein HD. 2021. Bacterial outer membrane proteins are targeted to the Bam complex by two parallel mechanisms. mBio 12:e00597-21. https://doi.org/10.1128/mBio.00597-21.
Contributor Information
Harris D. Bernstein, Email: harris_bernstein@nih.gov.
M. Stephen Trent, University of Georgia.
REFERENCES
- 1.Koebnik R, Locher KP, Gelder PV. 2000. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol 37:239–253. doi: 10.1046/j.1365-2958.2000.01983.x. [DOI] [PubMed] [Google Scholar]
- 2.Fairman JW, Noinaj N, Buchanan SK. 2011. The structural biology of β-barrel membrane proteins: a summary of recent reports. Curr Opin Struct Biol 21:523–531. doi: 10.1016/j.sbi.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lauber F, Deme JC, Lea SM, Berks BC. 2018. Type 9 secretion system structures reveal a new protein transport mechanism. Nature 564:77–82. doi: 10.1038/s41586-018-0693-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen R, Henning U. 1996. A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol Microbiol 19:1287–1294. doi: 10.1111/j.1365-2958.1996.tb02473.x. [DOI] [PubMed] [Google Scholar]
- 5.Lazar SW, Kolter R. 1996. SurA assists the folding of Escherichia coli outer membrane proteins. J Bacteriol 178:1770–1773. doi: 10.1128/jb.178.6.1770-1773.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rouvière PE, Gross CA. 1996. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev 10:3170–3182. doi: 10.1101/gad.10.24.3170. [DOI] [PubMed] [Google Scholar]
- 7.Schäfer U, Beck K, Müller M. 1999. Skp, a molecular chaperone of gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J Biol Chem 274:24567–24574. doi: 10.1074/jbc.274.35.24567. [DOI] [PubMed] [Google Scholar]
- 8.Krojer T, Sawa J, Schäfer E, Saibil HR, Ehrmann M, Clausen T. 2008. Structural basis for the regulated protease and chaperone function of DegP. Nature 453:885–890. doi: 10.1038/nature07004. [DOI] [PubMed] [Google Scholar]
- 9.Mas G, Thoma J, Hiller S. 2019. The periplasmic chaperones Skp and SurA, p 169–186. In Kuhn A (ed), Bacterial cell walls and membranes. Springer International Publishing, Cham, Switzerland. [DOI] [PubMed] [Google Scholar]
- 10.Walton TA, Sousa MC. 2004. Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol Cell 15:367–374. doi: 10.1016/j.molcel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Korndörfer IP, Dommel MK, Skerra A. 2004. Structure of the periplasmic chaperone Skp suggests functional similarity with cytosolic chaperones despite differing architecture. Nat Struct Mol Biol 11:1015–1020. doi: 10.1038/nsmb828. [DOI] [PubMed] [Google Scholar]
- 12.Walton TA, Sandoval CM, Fowler CA, Pardi A, Sousa MC. 2009. The cavity-chaperone Skp protects its substrate from aggregation but allows independent folding of substrate domains. Proc Natl Acad Sci U S A 106:1772–1777. doi: 10.1073/pnas.0809275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qu J, Behrens-Kneip S, Holst O, Kleinschmidt JH. 2009. Binding regions of outer membrane protein A in complexes with the periplasmic chaperone Skp: a site-directed fluorescence study. Biochemistry 48:4926–4936. doi: 10.1021/bi9004039. [DOI] [PubMed] [Google Scholar]
- 14.Burmann BM, Wang C, Hiller S. 2013. Conformation and dynamics of the periplasmic membrane-protein-chaperone complexes OmpX-Skp and tOmpA-Skp. Nat Struct Mol Biol 20:1265–1272. doi: 10.1038/nsmb.2677. [DOI] [PubMed] [Google Scholar]
- 15.Schiffrin B, Calabrese AN, Devine PWA, Harris SA, Ashcroft AE, Brockwell DJ, Radford SE. 2016. Skp is a multivalent chaperone of outer-membrane proteins. Nat Struct Mol Biol 23:786–793. doi: 10.1038/nsmb.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harms N, Koningstein G, Dontje W, Muller M, Oudega B, Luirink J, de Cock H. 2001. The early interaction of the outer membrane protein PhoE with the periplasmic chaperone Skp occurs at the cytoplasmic membrane. J Biol Chem 276:18804–18811. doi: 10.1074/jbc.M011194200. [DOI] [PubMed] [Google Scholar]
- 17.Ieva R, Tian P, Peterson JH, Bernstein HD. 2011. Sequential and spatially restricted interactions of assembly factors with an autotransporter beta domain. Proc Natl Acad Sci U S A 108:E383–E391. doi: 10.1073/pnas.1103827108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bitto E, McKay DB. 2002. Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10:1489–1498. doi: 10.1016/s0969-2126(02)00877-8. [DOI] [PubMed] [Google Scholar]
- 19.Bitto E, McKay DB. 2003. The periplasmic molecular chaperone protein SurA binds a peptide motif that is characteristic of integral outer membrane proteins. J Biol Chem 278:49316–49322. doi: 10.1074/jbc.M308853200. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Wang R, Jin F, Liu Y, Yu J, Fu X, Chang Z. 2016. A supercomplex spanning the inner and outer membranes mediates the biogenesis of β-barrel outer membrane proteins in bacteria. J Biol Chem 291:16720–16729. doi: 10.1074/jbc.M115.710715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev 21:2473–2484. doi: 10.1101/gad.1581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rizzitello AE, Harper JR, Silhavy TJ. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J Bacteriol 183:6794–6800. doi: 10.1128/JB.183.23.6794-6800.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denoncin K, Schwalm J, Vertommen D, Silhavy TJ, Collet J-F. 2012. Dissecting the Escherichia coli periplasmic chaperone network using differential proteomics. Proteomics 12:1391–1401. doi: 10.1002/pmic.201100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vertommen D, Ruiz N, Leverrier P, Silhavy TJ, Collet J-F. 2009. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9:2432–2443. doi: 10.1002/pmic.200800794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwalm J, Mahoney TF, Soltes GR, Silhavy TJ. 2013. Role for Skp in LptD assembly in Escherichia coli. J Bacteriol 195:3734–3742. doi: 10.1128/JB.00431-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J. 2003. Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299:262–265. doi: 10.1126/science.1078973. [DOI] [PubMed] [Google Scholar]
- 28.Sklar JG, Wu T, Gronenberg LS, Malinverni JC, Kahne D, Silhavy TJ. 2007. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc Natl Acad Sci U S A 104:6400–6405. doi: 10.1073/pnas.0701579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Malinverni JC, Sliz P, Silhavy TJ, Harrison SC, Kahne D. 2007. Structure and function of an essential component of the outer membrane protein assembly machine. Science 317:961–964. doi: 10.1126/science.1143993. [DOI] [PubMed] [Google Scholar]
- 30.Malinverni JC, Werner J, Kim S, Sklar JG, Kahne D, Misra R, Silhavy TJ. 2006. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol 61:151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- 31.Iadanza MG, Higgins AJ, Schiffrin B, Calabrese AN, Brockwell DJ, Ashcroft AE, Radford SE, Ranson NA. 2016. Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM. Nat Commun 7:12865. doi: 10.1038/ncomms12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, Stansfeld PJ, Wang Z, Zhang Y, Wang W, Dong C. 2016. Structural basis of outer membrane protein insertion by the BAM complex. Nature 531:64–69. doi: 10.1038/nature17199. [DOI] [PubMed] [Google Scholar]
- 33.Han L, Zheng J, Wang Y, Yang X, Liu Y, Sun C, Cao B, Zhou H, Ni D, Lou J, Zhao Y, Huang Y. 2016. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol 23:192–196. doi: 10.1038/nsmb.3181. [DOI] [PubMed] [Google Scholar]
- 34.Bakelar J, Buchanan SK, Noinaj N. 2016. The structure of the β-barrel assembly machinery complex. Science 351:180–186. doi: 10.1126/science.aad3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, Lithgow T, Buchanan SK. 2013. Structural insight into the biogenesis of β-barrel membrane proteins. Nature 501:385–390. doi: 10.1038/nature12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Noinaj N, Kuszak AJ, Balusek C, Gumbart JC, Buchanan SK. 2014. Lateral opening and exit pore formation are required for BamA function. Structure 22:1055–1062. doi: 10.1016/j.str.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ieva R, Skillman KM, Bernstein HD. 2008. Incorporation of a polypeptide segment into the β-domain pore during the assembly of a bacterial autotransporter. Mol Microbiol 67:188–201. doi: 10.1111/j.1365-2958.2007.06048.x. [DOI] [PubMed] [Google Scholar]
- 38.Lee J, Xue M, Wzorek JS, Wu T, Grabowicz M, Gronenberg LS, Sutterlin HA, Davis RM, Ruiz N, Silhavy TJ, Kahne DE. 2016. Characterization of a stalled complex on the β-barrel assembly machine. Proc Natl Acad Sci U S A 113:8717–8722. doi: 10.1073/pnas.1604100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sikdar R, Peterson JH, Anderson DE, Bernstein HD. 2017. Folding of a bacterial integral outer membrane protein is initiated in the periplasm. Nat Commun 8 doi: 10.1038/s41467-017-01246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gessmann D, Chung YH, Danoff EJ, Plummer AM, Sandlin CW, Zaccai NR, Fleming KG. 2014. Outer membrane β-barrel protein folding is physically controlled by periplasmic lipid head groups and BamA. Proc Natl Acad Sci U S A 111:5878–5883. doi: 10.1073/pnas.1322473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schiffrin B, Calabrese AN, Higgins AJ, Humes JR, Ashcroft AE, Kalli AC, Brockwell DJ, Radford SE. 2017. Effects of periplasmic chaperones and membrane thickness on BamA-catalyzed outer-membrane protein folding. J Mol Biol 429:3776–3792. doi: 10.1016/j.jmb.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doyle MT, Bernstein HD. 2019. Bacterial outer membrane proteins assemble via asymmetric interactions with the BamA β-barrel. Nat Commun 10:3358. doi: 10.1038/s41467-019-11230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomasek D, Rawson S, Lee J, Wzorek JS, Harrison SC, Li Z, Kahne D. 2020. Structure of a nascent membrane protein as it folds on the BAM complex. Nature 583:473–478. doi: 10.1038/s41586-020-2370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vuong P, Bennion D, Mantei J, Frost D, Misra R. 2008. Analysis of YfgL and YaeT interactions through bioinformatics, mutagenesis, and biochemistry. J Bacteriol 190:1507–1517. doi: 10.1128/JB.01477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Struyvé M, Moons M, Tommassen J. 1991. Carboxy-terminal phenylalanine is essential for the correct assembly of a bacterial outer membrane protein. J Mol Biol 218:141–148. doi: 10.1016/0022-2836(91)90880-F. [DOI] [PubMed] [Google Scholar]
- 46.Robert V, Volokhina EB, Senf F, Bos MP, Gelder PV, Tommassen J. 2006. Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific C-terminal motif. PLoS Biol 4:e377. doi: 10.1371/journal.pbio.0040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hagan CL, Wzorek JS, Kahne D. 2015. Inhibition of the β-barrel assembly machine by a peptide that binds BamD. Proc Natl Acad Sci U S A 112:2011–2016. doi: 10.1073/pnas.1415955112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paramasivam N, Habeck M, Linke D. 2012. Is the C-terminal insertional signal in Gram-negative bacterial outer membrane proteins species-specific or not? BMC Genomics 13:510. doi: 10.1186/1471-2164-13-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berven FS, Flikka K, Jensen HB, Eidhammer I. 2004. BOMP: a program to predict integral β-barrel outer membrane proteins encoded within genomes of Gram-negative bacteria. Nucleic Acids Res 32:W394–W399. doi: 10.1093/nar/gkh351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stubenrauch C, Belousoff MJ, Hay ID, Shen H-H, Lillington J, Tuck KL, Peters KM, Phan M-D, Lo AW, Schembri MA, Strugnell RA, Waksman G, Lithgow T. 2016. Effective assembly of fimbriae in Escherichia coli depends on the translocation assembly module nanomachine. Nat Microbiol 1:1–8. doi: 10.1038/nmicrobiol.2016.64. [DOI] [PubMed] [Google Scholar]
- 51.Phan G, Remaut H, Wang T, Allen WJ, Pirker KF, Lebedev A, Henderson NS, Geibel S, Volkan E, Yan J, Kunze MBA, Pinkner JS, Ford B, Kay CWM, Li H, Hultgren S, Thanassi DG, Waksman G. 2011. Crystal structure of the FimD usher bound to its cognate FimC:FimH substrate. Nature 474:49–53. doi: 10.1038/nature10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pautsch A, Schulz GE. 1998. Structure of the outer membrane protein A transmembrane domain. Nat Struct Biol 5:1013–1017. doi: 10.1038/2983. [DOI] [PubMed] [Google Scholar]
- 53.Barnard TJ, Dautin N, Lukacik P, Bernstein HD, Buchanan SK. 2007. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat Struct Mol Biol 14:1214–1220. doi: 10.1038/nsmb1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baslé A, Rummel G, Storici P, Rosenbusch JP, Schirmer T. 2006. Crystal structure of osmoporin OmpC from Escherichia coli at 2.0 Å. J Mol Biol 362:933–942. doi: 10.1016/j.jmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 55.Roman-Hernandez G, Peterson JH, Bernstein HD. 2014. Reconstitution of bacterial autotransporter assembly using purified components. Elife 3:e04234. doi: 10.7554/eLife.04234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koebnik R. 1999. Structural and functional roles of the surface-exposed loops of the β-Barrel membrane protein OmpA from Escherichia coli. J Bacteriol 181:3688–3694. doi: 10.1128/JB.181.12.3688-3694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pavlova O, Peterson JH, Ieva R, Bernstein HD. 2013. Mechanistic link between β barrel assembly and the initiation of autotransporter secretion. Proc Natl Acad Sci U S A 110:E938–E947. doi: 10.1073/pnas.1219076110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morona R, Krämer C, Henning U. 1985. Bacteriophage receptor area of outer membrane protein OmpA of Escherichia coli K-12. J Bacteriol 164:539–543. doi: 10.1128/JB.164.2.539-543.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Freudl R, Schwarz H, Stierhof YD, Gamon K, Hindennach I, Henning U. 1986. An outer membrane protein (OmpA) of Escherichia coli K-12 undergoes a conformational change during export. J Biol Chem 261:11355–11361. doi: 10.1016/S0021-9258(18)67391-0. [DOI] [PubMed] [Google Scholar]
- 60.Kumamoto CA, Beckwith J. 1985. Evidence for specificity at an early step in protein export in Escherichia coli. J Bacteriol 163:267–274. doi: 10.1128/JB.163.1.267-274.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peterson JH, Hussain S, Bernstein HD. 2018. Identification of a novel post-insertion step in the assembly of a bacterial outer membrane protein. Mol Microbiol 110:143–159. doi: 10.1111/mmi.14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ieva R, Bernstein HD. 2009. Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc Natl Acad Sci U S A 106:19120–19125. doi: 10.1073/pnas.0907912106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Farrell IS, Toroney R, Hazen JL, Mehl RA, Chin JW. 2005. Photo-cross-linking interacting proteins with a genetically encoded benzophenone. Nat Methods 2:377–384. doi: 10.1038/nmeth0505-377. [DOI] [PubMed] [Google Scholar]
- 64.Peterson JH, Plummer AM, Fleming KG, Bernstein HD. 2017. Selective pressure for rapid membrane integration constrains the sequence of bacterial outer membrane proteins. Mol Microbiol 106:777–792. doi: 10.1111/mmi.13845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hussain S, Bernstein HD. 2018. The Bam complex catalyzes efficient insertion of bacterial outer membrane proteins into membrane vesicles of variable lipid composition. J Biol Chem 293:2959–2973. doi: 10.1074/jbc.RA117.000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagan CL, Kim S, Kahne D. 2010. Reconstitution of outer membrane protein assembly from purified components. Science 328:890–892. doi: 10.1126/science.1188919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hong H, Park S, Jiménez RHF, Rinehart D, Tamm LK. 2007. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J Am Chem Soc 129:8320–8327. doi: 10.1021/ja068849o. [DOI] [PubMed] [Google Scholar]
- 68.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the sigmaE stress response in related genomes. PLoS Biol 4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koebnik R, Krämer L. 1995. Membrane assembly of circularly permuted variants of the E. coli outer membrane protein OmpA. J Mol Biol 250:617–626. doi: 10.1006/jmbi.1995.0403. [DOI] [PubMed] [Google Scholar]
- 70.Ricci DP, Hagan CL, Kahne D, Silhavy TJ. 2012. Activation of the Escherichia coli β-barrel assembly machine (Bam) is required for essential components to interact properly with substrate. Proc Natl Acad Sci U S A 109:3487–3491. doi: 10.1073/pnas.1201362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Derman AI, Puziss JW, Bassford PJ, Beckwith J. 1993. A signal sequence is not required for protein export in prlA mutants of Escherichia coli. EMBO J 12:879–888. doi: 10.1002/j.1460-2075.1993.tb05728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsirigotaki A, Chatzi KE, Koukaki M, De Geyter J, Portaliou AG, Orfanoudaki G, Sardis MF, Trelle MB, Jørgensen TJD, Karamanou S, Economou A. 2018. Long-lived folding intermediates predominate the targeting-competent secretome. Structure 26:695–707.e5. doi: 10.1016/j.str.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 73.Blobel G, Dobberstein B. 1975. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J Cell Biol 67:835–851. doi: 10.1083/jcb.67.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Calabrese AN, Schiffrin B, Watson M, Karamanos TK, Walko M, Humes JR, Horne JE, White P, Wilson AJ, Kalli AC, Tuma R, Ashcroft AE, Brockwell DJ, Radford SE. 2020. Inter-domain dynamics in the chaperone SurA and multi-site binding to its outer membrane protein clients. Nat Commun 11:2155. doi: 10.1038/s41467-020-15702-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marx DC, Leblanc MJ, Plummer AM, Krueger S, Fleming KG. 2020. Domain interactions determine the conformational ensemble of the periplasmic chaperone SurA. Protein Sci 29:2043–2053. doi: 10.1002/pro.3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Denker K, Orlik F, Schiffler B, Benz R. 2005. Site-directed mutagenesis of the greasy slide aromatic residues within the LamB (maltoporin) channel of Escherichia coli: effect on ion and maltopentaose transport. J Mol Biol 352:534–550. doi: 10.1016/j.jmb.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 77.Marani P, Wagner S, Baars L, Genevaux P, de Gier J-W, Nilsson I, Casadio R, von Heijne G. 2006. New Escherichia coli outer membrane proteins identified through prediction and experimental verification. Protein Sci 15:884–889. doi: 10.1110/ps.051889506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Crooks GE, Hon G, Chandonia J-M, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Szabady RL, Peterson JH, Skillman KM, Bernstein HD. 2005. An unusual signal peptide facilitates late steps in the biogenesis of a bacterial autotransporter. Proc Natl Acad Sci U S A 102:221–226. doi: 10.1073/pnas.0406055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yap M-NF, Bernstein HD. 2013. Mutations in the Escherichia coli ribosomal protein L22 selectively suppress the expression of a secreted bacterial virulence factor. J Bacteriol 195:2991–2999. doi: 10.1128/JB.00211-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 83.Cardona ST, Valvano MA. 2005. An expression vector containing a rhamnose-inducible promoter provides tightly regulated gene expression in Burkholderia cenocepacia. Plasmid 54:219–228. doi: 10.1016/j.plasmid.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 84.Reynolds CR, Islam SA, Sternberg MJ. 2018. EzMol: a web server wizard for the rapid visualization and image production of protein and nucleic acid structures. J Mol Biol 430:2244–2248. doi: 10.1016/j.jmb.2018.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C-terminal 10 amino acids of 37 E. coli MG1655 OMP β barrels (A), 80 predicted trimeric porins that mediate carbohydrate uptake in Gammaproteobacteria (B), and 20 predicted E. coli FimD family proteins (C). Download Table S1, PDF file, 0.6 MB (611.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Loading controls used in Western blot and pulse-chase analysis. (A and B) Larger portions of the Western blots presented in Fig. 2B and 7B, respectively, are shown. An unidentified ∼45-kDa background band (arrow) was used as a reference to confirm that similar amounts of cell extracts were loaded in each lane. (C) XW100 (MC4100 ΔompA; WT) and isogenic surA and skp deletion strains transformed with pXW01 were grown to mid-log phase, and lysates were analyzed by Western blotting with antisera against an OmpA loop 1 peptide and a loading control, the cytoplasmic protein Ffh (H. D. Bernstein, D. Zopf, D. M. Freymann, and P. Walter, Proc Natl Acad Sci USA 90:5229–5233, 1993, 10.1073/pnas.90.11.5229). (D) In all pulse-chase experiments in which the stability and assembly kinetics of TM-OmpA or TM-OmpAY189A, F191A were measured, the TM-OmpA signal in each lane was first normalized to the signal generated by an unidentified ∼46-kDa background band to account for differences in loading. As an example, a larger portion of one of the gels presented in Fig. 2C (upper right gel) is shown. Download FIG S1, PDF file, 0.7 MB (673KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Alternative quantitation of pulse-chase results. (A) The percentage of TM-OmpA (blue squares) and TM-OmpAY189A, F191A (red circles) that was resistant to PK digestion and the percentage of the original pulse-labeled protein (i.e., total protein at t = 0) that remained in the experiments shown in Fig. 2 are plotted. The TM-OmpA signal in each lane was not normalized to an ∼46-kDa background band (see Fig. S1D). (B) The percentage of the original pulse-labeled EspPΔ5 (blue squares) and EspPΔ5Y1298A, F1300A (red circles) (i.e., total protein at t = 0) that was cleaved at each timepoint in the experiments shown in Fig. 3 is plotted. Error bars represent the standard deviations. Download FIG S2, PDF file, 0.3 MB (349.5KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
OmpC β signal mutations and a surA deletion synergistically impair folding in vivo. Cells transformed with pJH220 (Prha-OmpC) or a derivative containing the OmpC Y365A F367A mutation were subjected to pulse-chase labeling. After the OM was permeabilized, half of each sample was treated with PK, and immunoprecipitations were performed using an antiserum against an OmpC peptide. Download FIG S3, PDF file, 0.6 MB (653.9KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
A β signal mutation does not affect the interaction of EspPΔ5 with SurA. Urea-denatured EspPΔ5 or EspPΔ5Y1298A, F1300A (0.2 μM) was incubated with proteoliposomes containing the Bam complex (0.2 μM) in POPC and the indicated amount of SurA at 30°C for 30 min. Protein cleavage was visualized by Western blotting with an antiserum raised against an EspP C-terminal peptide. Download FIG S4, PDF file, 0.5 MB (480.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Effect of β signal mutations on the binding of proEspP to the Bam complex and Skp. HDB130 (surA deletion strain) (A) or HDB131 (skp deletion strain) (B) transformed with pRI22 (Plac-10His-espP) harboring an amber codon at residue 1214 or a derivative containing the Y1298A F1300A mutation were subjected to pulse-chase labeling. Half of the cells were UV irradiated, and immunoprecipitations were performed with antisera against an EspP C-terminal peptide or BamD. For clarity, an expanded region of lanes 5 and 6 in the top gels is shown. (C) An aliquot of the UV-irradiated samples generated in the experiments shown in panels A and B, and Fig. 4 was subjected to immunoprecipitation with an antiserum against Skp. A background band that migrates slightly faster than the proEspP-Skp crosslinking product is denoted with an asterisk. Download FIG S5, PDF file, 1.1 MB (1.1MB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The mutation of aromatic residues outside the β signal does not affect OMP assembly in a surA deletion strain. XW101 (MC4100 ompA::kan surA::cm) transformed with pXW19 (Prha-TM-OmpAL104A, Y106A) or pXW20 (Prha-TM-OmpAY162A, W164A) (A) or HDB130 (AD202 surA::cm) transformed with pJH209 (Prha-espPΔ5L1157A, Y1159A) or pJH210 (Prha-espPΔ5Y1298A, F1300A) (B) were subjected to pulse-chase labeling. After the OM was permeabilized, half of each sample was treated with PK, and immunoprecipitations were performed with an antiserum against an OmpA loop 4 peptide or an EspP C-terminal peptide. Download FIG S6, PDF file, 0.6 MB (604KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Activation of the σE stress response does not affect EspPΔ5Y1298A, F1300A assembly. AD202 transformed with pJH208 (Prha-espPΔ5Y1298A, F1300A) and either pTrc or pLC45 (Ptrc-rpoE) were incubated with IPTG for 25 min and then rhamnose for an additional 5 min. Cells were collected for qRT-PCR (A) and Western blot analysis (B) and EspPΔ5Y1298A, F1300 assembly was analyzed by pulse-chase labeling and immunoprecipitation using an antiserum raised against an EspP C-terminal peptide (C). In panel A, rpoE expression was normalized to the expression of the housekeeping gene rssA (black bar, average). In panel B, Western blots were performed with antisera raised against the indicated proteins. Download FIG S7, PDF file, 0.6 MB (596.7KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Internally located β signals partially enhance the assembly of a circularly permuted version of TM-OmpA. (A) XW100 (MC4100 ΔompA; WT) and isogenic surA and skp deletion strains transformed with pXW17 [Prha-TM-OmpA(34)(12)] were subjected to pulse-chase labeling. After the OM was permeabilized, half of each sample was treated with PK, and immunoprecipitations were performed using an antiserum against an OmpA loop 4 peptide. These results show that the disruption of surA or skp does not significantly affect the assembly of circularly permuted TM-OmpA. (B) The last three residues of the OmpA β-signal motif (YRF) were introduced into TM-OmpAY189A, F191A β-strand 2, 4, or 6 by mutagenesis as shown. (C) XW101 (MC4100 ompA::kan surA::cm) were transformed with a derivative of pXW01 (Ptrc-TM-OmpA) that encodes one of the mutants and grown in LB medium. Samples were collected at mid-log phase, and cell lysates were heated at 95°C or incubated on ice. The folding of the TM-OmpA mutants was visualized by Western blotting with an antiserum against an OmpA loop 1 peptide. The results show that internal mutations that recreate the OmpA β-signal motif partially restore the folding of TM-OmpAY189A, F191A in a surA deletion strain. Download FIG S8, PDF file, 0.7 MB (753.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Plasmids (A) and oligonucleotides (B) used in this study. Download Table S2, PDF file, 0.6 MB (639.4KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.