

Abstract
People with obesity are often dyslipidemic and prescribed statins to prevent cardiovascular events. A common side effect of statin use is myopathy. This could potentially be caused by the reduction of selenoproteins that curb oxidative stress, in turn, affecting creatine metabolism. We determined if statins regulate hepatic and muscular selenoprotein expression, oxidative stress and creatine metabolism. Mice lacking selenocysteine lyase (Scly KO), a selenium-provider enzyme for selenoprotein synthesis, were fed a high-fat, Se-supplemented diet and treated with simvastatin. Statin improved creatine metabolism in females and oxidative responses in both sexes. Male Scly KO mice were heavier than females after statin treatment. Hepatic selenoproteins were unaffected by statin and genotype in females. Statin upregulated muscular Gpx1 in females but not males, while Scly loss downregulated muscular Gpx1 in males and Selenon in females. Osgin1 was reduced in statin-treated Scly KO males after AmpliSeq analysis. These results refine our understanding of the sex-dependent role of selenium in statin responses.
Keywords: selenium, selenocysteine lyase, obesity, statin
1. Introduction
Over the last decades, the prevalence of obesity has increased worldwide, reaching epidemic proportions [1]. Obesity currently affects 78.6 million people (33%) in the United States and is expected to increase to over 50% of the population by 2030 [2]. Dyslipidemia is frequently found in patients with obesity and often one of the first signals that metabolic dysfunction is taking place, doubling their heart disease risk [3].
Statins are the most commonly prescribed and effective pharmacological therapy for treating dyslipidemia and preventing cardiovascular events [4,5]. Despite their favorable overall safety profile, statins evoke distinct side-effects whose molecular origins have remained unsettled [5]. Muscle disorders are the most commonly reported adverse effects of statins, ranging from muscle weakness, fatigue, and pain to more severe conditions like rhabdomyolysis [6]. Individuals with obesity were shown to be predisposed to new or worsening muscle symptoms both while using statins, and after stopping treatment [7].
Statin-induced adverse events in the muscle may be triggered by a reduction in the expression and activity of selenium (Se)-containing selenoproteins. This reduction occurs due to the inhibition of the 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase step in the mevalonate pathway targeted by statins. HMG-CoA reductase inhibition reduces cholesterol synthesis and also curbs the production of downstream products such as the isoprenoid isopentenyl pyrophosphate [8], required for the post-transcriptional maturation of tRNAs such as the selenocysteine (Sec) tRNA, and consequently for selenoprotein synthesis [4,5,9,10].
Complete loss of selenoproteins is embryonically lethal [11]. However, mice lacking the gene for selenocysteine lyase (Scly), which encodes a Sec-decomposing enzyme that participates in selenoprotein synthesis, develop obesity with strong hypercholesterolemia and display differential impacts on selenoprotein levels, particularly in the liver [12–14]. This phenotype is aggravated by a Se-deficient diet [15,16] or by exposure to a Se-adequate, high-fat diet [16] and is not curbed after Se supplementation [17]. Interestingly, dietary Se supplementation mitigates statins’ side effects in individuals with obesity displaying an adequate Se intake [18], but in a Se-supplemented context, as is common in a significant portion of the American population, the regulatory factors responsible for triggering statin side effects are not fully clarified.
We utilized the Scly knockout (Scly KO) mouse as a model of obesity caused by a defect in Se metabolism and treated them with statins to further elucidate the role of Se metabolism and selenoproteins in statin-induced side effects in the liver and skeletal muscle.
2. Materials and Methods
2.1. Chemicals
All reagents are from Sigma-Aldrich/MilliporeSigma (Burlington, MA, USA) unless otherwise noted.
2.2. Animals
Age-matched, littermate homozygous C57BL/6N wild-type (WT; The Jackson Laboratory) and Scly KO mice of both sexes were born and raised in our vivarium and used in experiments after weaning following the Institutional Animal Care and Use Committee of the University of Hawaii (protocol n. 17–2616). Generation of the Scly KO mouse line has been previously described [19]. Body weights were recorded every two weeks after weaning. Animals were euthanized by CO2 asphyxiation at the same time of the day before collecting serum, liver, gonadal white adipose tissue (WAT), interscapular brown adipose tissue (BAT), soleus, and gastrocnemius skeletal muscles for further analyses.
2.3. Diets and drug treatment
Animals were fed a customized high-fat diet containing 45 kcal% fat as lard and 35 kcal% carbohydrates as a mixture of sucrose and corn starch (Research Diets, Inc., New Brunswick, NJ) supplemented with a blend of sodium selenite plus selenomethionine (previously described in [17]) for eight weeks. Diet was customized to mimic caloric intake of the American population with obesity, with additional Se supplementation. Mice received vehicle (Vehicle-Treated Mice – VTM) or simvastatin (Statin-Treated Mice - STM) (Enzo Life Sciences International, Farmingdale, NY) at a dose of 5 mg/kg body weight/day by oral gavage once daily for 21 days. Figure 1 describes the experimental design of the present study. Simvastatin was selected for this study because of its well-established link to adverse effects on skeletal muscle, along with its highest prescription rate among all statin drugs in the U.S. The selected simvastatin dose was both clinically relevant and physiologically appropriate to mice [5]. Simvastatin is insoluble in water; therefore, it was first dissolved in ethanol 100%, then diluted 60 times in water. VTM received a solution with the same concentration of ethanol.
Figure 1:
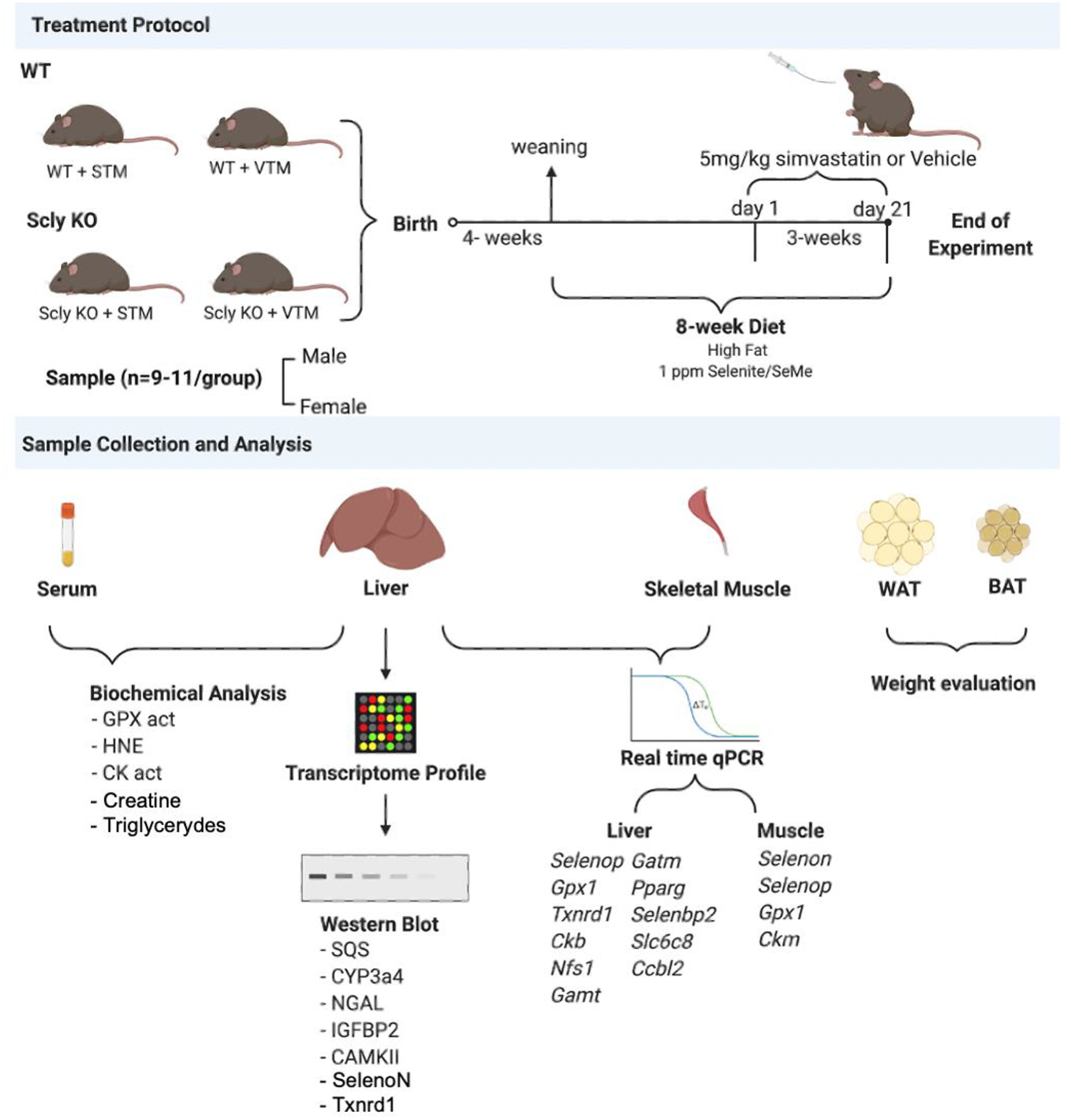
Treatment protocol.
2.4. Serum and liver assays
Serum cholesterol and triglycerides were assayed by commercial kits (Cayman Chemical Company, Ann Harbor, MI; and Abcam, Cambridge, MA) using colorimetry. Oxidative stress was gauged by measuring lipid peroxidation end products with an OxiSelect 4-hydroxynonenal (HNE)-His Adduct ELISA kit (Cell Biolabs, Inc., San Diego, CA). Creatine (Cr) and creatine kinase (CK) activity were determined using colorimetric assay kits (Sigma-Aldrich/MilliporeSigma). Total serum or liver glutathione peroxidase (GPX) activity was measured using a colorimetric assay kit (Abcam), where GPX oxidizes glutathione (GSH) to produce GSH dissulfide (GSSG) as part of the reaction in which it reduces cumene hydroperoxide. GSH reductase (GR) then reduces the GSSG to produce GSH, and in the same reaction, consumed NADPH. The decrease of NADPH measured at OD=340 nm is proportional to GPX activity. For all assays, we followed the manufacturer’s protocol.
2.5. RNA Extraction
Liver and soleus were disrupted with disposable probes using the Qiagen TissueRuptor (Qiagen, Germantown, MD, USA). Total RNA for AmpliSeq and real-time semi-quantitative PCR (qPCR) was extracted using Qiagen AllPrep RNA Kit and EZNA Total RNA kit I (Omega Biotek, Norcross, GA), respectively. RNA sample quality was evaluated on an Agilent BioAnalyzer using the Nano RNA Kit (Agilent, Santa Clara, CA, USA).
2.6. Targeted Transcriptome Profiling
We used the Ion AmpliSeq Transcriptome Mouse Gene Expression panel (Thermo Fisher Scientific, Waltham, MA) for gene expression profiling of liver samples. The panel is designed to target 20,802 RefSeq genes. 100 ng of total RNA was used to construct sequencing libraries according to the manufacturer’s instructions. Indexed sequencing libraries were constructed using the Mouse Transcriptome AmpliSeq kit and quantified using the Ion Library Quantification kit on a StepOnePlus Real-Time PCR system (Thermo Fisher Scientific). Twenty-four multiplexed samples were templated using the Ion 540 Kit-Chef with a Chef Instrument (Thermo Fisher Scientific), enriched, loaded on two Ion 540 semiconductor sequencing chips (12 samples per chip), and analyzed on the Ion GeneStudio S5 System Sequencer using the Ion 540 Chip-Kit (Thermo Fisher Scientific). An Ion TorrentSuite was used for signal processing and base calling, and the Ion ampliSeqRNA plugin was used to obtain read counts for each targeted gene for each sample.
2.7. qPCR
One microgram of total RNA was reverse-transcribed using High-Capacity cDNA Reverse Transcription Kits (Thermo Fisher Scientific), with ten ng of resulting cDNA used for qPCR with PerfeCTa SYBR Green FastMix (Quantabio, Beverly, MA, USA) and 45 amplification cycles in a 384-well plate platform of a LightCycler 480 II (Roche, Basel, Switzerland). Samples ran in duplicates. Relative quantification used the Δ−CT method, normalized to hypoxanthine-guanine phosphoribosyltransferase (Hprt1) expression levels. All primers were used at 10 nM and evaluated for their efficiency before use in experiments according to MIQE guidelines [20]. Primer sequences used for qPCR can be found in Supplementary Table 1.
2.8. Western blotting
Liver and gastrocnemius tissues were pulverized and resuspended in CelLytic MT (Sigma-Aldrich/ MilliporeSigma) with protease inhibitors (ThermoFisher Scientific), sonicated, and centrifuged for 15 minutes, 12,000 x g at 4°C, and protein supernatant collected. Samples consisting of 10 μg of total protein were separated in 4–20% SDS-PAGE (Bio-Rad, Hercules, CA), transferred to Immobilon-FL polyvinylidene difluoride (IPFL) membranes (Sigma-Aldrich/MilliporeSigma), and probed for 1.5 h or overnight with 1:1,000 dilution of rabbit polyclonal anti-SQS (Acris Antibodies, San Diego, CA), rabbit polyclonal anti-CYP3a4 (Proteintech, Rosemont, IL), mouse monoclonal anti-beta-actin (Sigma-Aldrich), rabbit polyclonal anti-SelenoN (LifeSpan Biosciences, Seattle, WA), rabbit polyclonal anti-Txnrd1 (Novus Biologicals, Littleton, CO), rabbit polyclonal anti-Osgin1, rabbit polyclonal anti-IGFBP2, rabbit polyclonal anti-NGAL, and mouse monoclonal anti-CaMKII beta (Thermo Fisher Scientific). The protein expression levels were visualized using secondary infrared IRDyes antibodies (Li-Cor Biosciences, Lincoln, NE) incubated for 45 minutes at 1:10,000 dilution. Detection and analysis of Western blots were performed using an Odyssey CTx Infrared Imager (Li-Cor Biosciences), and bands were quantified using the ImageStudioLite software (Li-Cor Biosciences).
2.9. Bioinformatics analysis
Differential gene expression analysis of AmpliSeq was performed. The read counts were assigned to each amplicon for all samples using the R statistical programming language package DESeq2, which estimates variance-mean dependence in count data tests for differential expression using a model based on the negative binomial distribution. Significant expression differences were tested between the groups of samples. The ‘apeglm’ method was employed for log-fold change shrinkage and the Wald test to detect the differential expression of genes and 0.05 as a significance threshold for the adjusted P-values. Furthermore, functional and pathway analyses and gene ontology (GO) were done using the online Enrichr tool [21,22].
2.10. Statistical analyses
Data were plotted in GraphPad Prism software version 8.0 (GraphPad Software Inc., San Diego, CA). We analyzed males and females separately. Two-way analysis of variance (ANOVA) was carried out and followed by Bonferroni’s post hoc test. Comparisons were between genotypes, and vehicle or statin treatment. Differences were considered statistically significant if reaching P≤0.05.
3. Results
3.1. Effectiveness of statin treatment
Reduction in circulating cholesterol is the direct outcome of statin therapy in people. However, cholesterol levels in mice are notoriously resistant to robust decreases after statin use, and hepatic expression of squalene synthase (SQS) has been suggested as a more suitable biomarker of statin treatment effectiveness [23], as SQS is downstream the HMG-CoA reductase step in the cholesterol biosynthesis pathway that is the direct target of statins [8,24]. Treatment with statin reduced hepatic SQS levels in both WT and Scly KO mice (Figure 2), however a more substantial decrease occurred in females than in males. We also assessed for serum cholesterol before the start of the statin treatment and after three weeks on simvastatin and observed a decrease of 21% (males) and 27% (females) in the WT mice and 28% (males) and 20% (females) in the Scly KO mice (Supplementary Table 2A). Triglyceride levels were also lower in Scly KO mice treated with statins, while not affecting levels in WT mice (Supplementary Table 2B).
Figure 2: Hepatic squalene synthase (SQS) expression measured by Western blot and normalized by expression levels of β-actin.
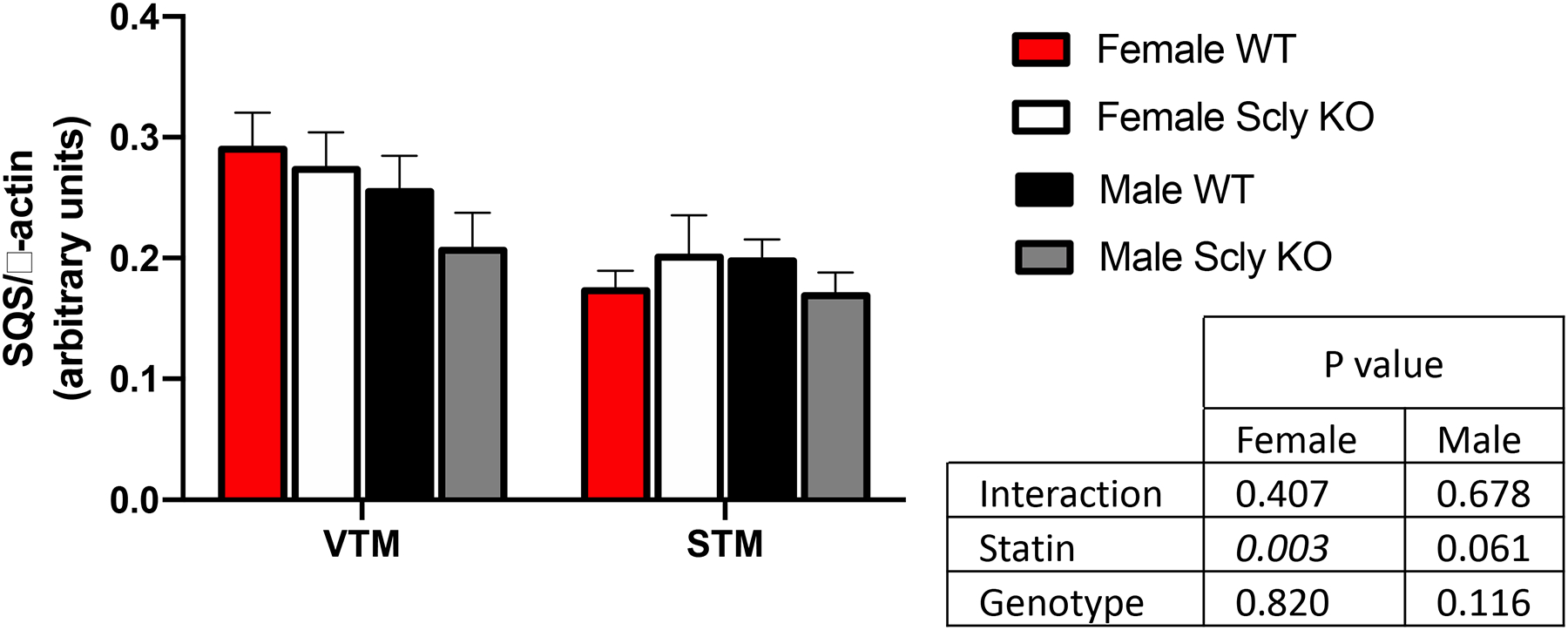
n = 3–4 per group. Two-way ANOVA was applied, considering the differences between treatments (VTM × STM) and/or between genotypes (WT × KO), for males and females separately, and P values are displayed in the inset table. VTM: vehicle-treated mice; STM: statin-treated mice; WT: wild type; KO: knockout.
3.2. Effect of statin treatment in body and adipose tissue weights
Mice were on a high-fat diet, a classic the paradigm to develop obesity in mice. We monitored their body weight throughout the experiment and did not observe differences in for WT mice with or without statin use, males or females. As previously reported [17], Scly KO mice fed a high-fat, Se-supplemented diet gained more weight than WT (Figure 3). This phenotype was aggravated by the use of simvastatin in male Scly KO mice (Figure 3A). Gonadal WAT weights were considerably higher in male Scly KO versus WT mice (Figure 3B). BAT mass was enlarged in male Scly KO mice, with further enlargement after statin treatment (Figure 3C). Interestingly, female Scly KO body weights were decreased by statin treatment (Figure 3D), but this reduction was not reflected in their WAT weights, which remained heavier (Figure 3E), or in their BAT mass in comparison to WT mice (Figure 3F).
Figure 3: Body and adipose depot weights in male (A–C) and (D–F) female WT and Scly KO mice.
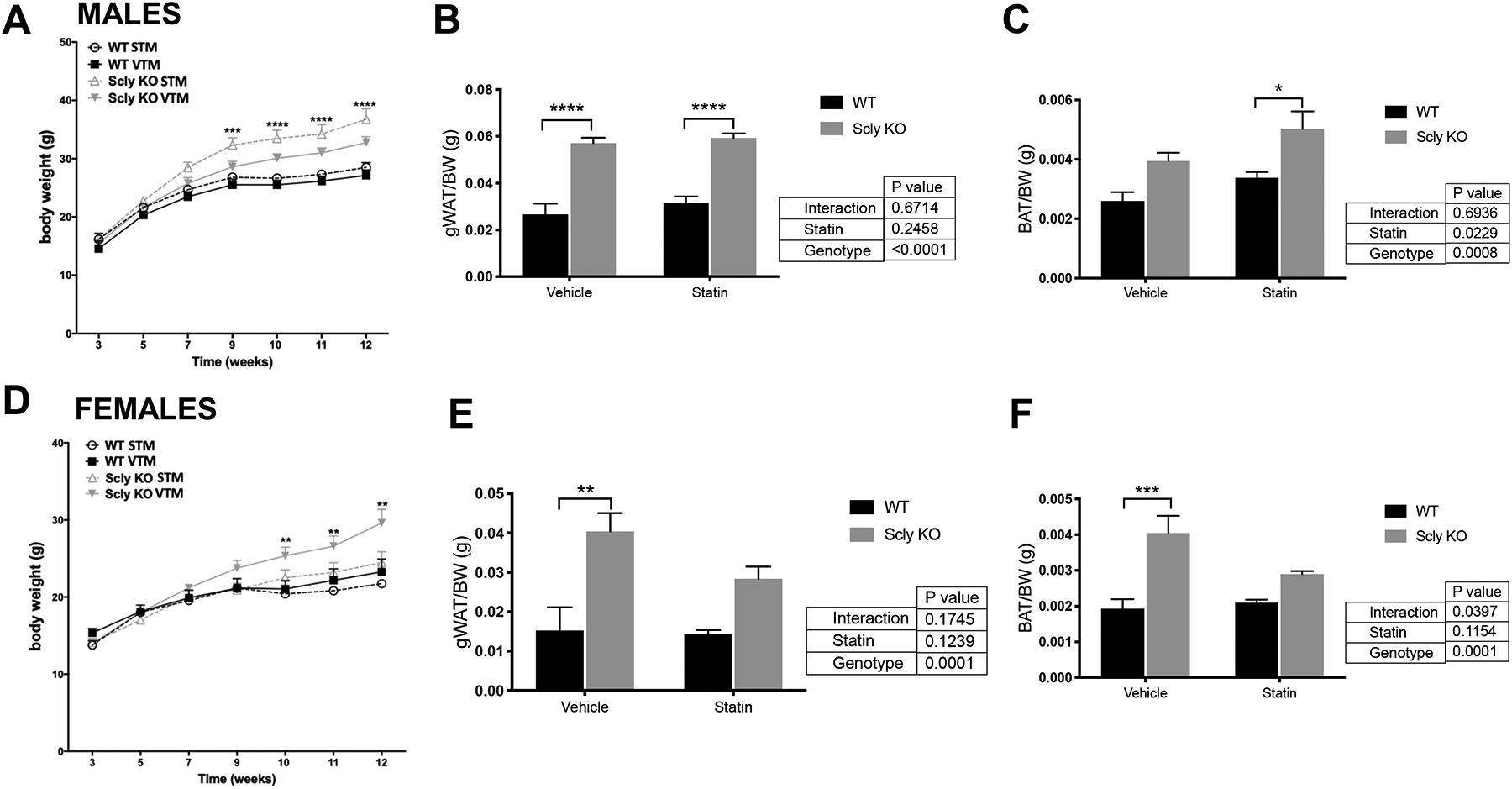
Body weights from male (A) and female (D) mice from 3 to 12 weeks of age were monitored biweekly. Male white gonadal (B) and brown adipose tissue (C), and female white gonadal (E) and brown adipose tissue (F) weights were measured after the end of experiment. Values are mean + SEM and in (B), (C), (E), and (F) were normalized by total body weight at 12 weeks for each mouse. Two-way ANOVA was applied, followed by Bonferroni’s post-hoc test, considering the differences between treatments (VTM × STM) and/or between genotypes (WT × Scly KO), for males and females separately. *, P < 0.05; **, P < 0.01; ***, P < 0.001, n=9–11 per genotype. gWAT, gonadal white adipose tissue; BAT, brown adipose tissue, VTM: vehicle-treated mice; STM: statin-treated mice; WT: wild type; KO: knockout. Black bars, WT; gray bars, Scly KO.
3.3. Effect of statin treatment in oxidative stress parameters
Statins are known to improve responses to oxidative stress [25], an effect that is partially dependent on the GPX class of selenoproteins. We observed that Scly KO mice had about 9% higher circulating GPX activity than WT mice. Statin treatment reduced circulating GPX activity for female, but not male, Scly KO mice (Table 1), but no reduction was detected in males. Hepatic GPX activity was unchanged in all tested conditions. Interestingly, when assessing lipid peroxidation levels using the HNE-adduct protocol, we observed that statin treatment improved hepatic oxidative status in WT and Scly KO mice for both sexes, while circulating oxidative stress was increased in the Scly KO mice. However, this increase was independent of statin treatment (Table 1).
Table 1.
Serum and hepatic oxidative stress markers in WT and Scly KO mice fed a high-fat, Se-supplemented diet, and treated with vehicle or statin.
| MALES | Vehicle | Statin | 2-way ANOVA | ||||
|---|---|---|---|---|---|---|---|
| WT | Scly KO | WT | Scly KO | Pint | PStatin | PGen | |
| GPX act serum (nmol/mL−1) | 40.7±1.5 | 43.0±2.4 | 41.0±1.0 | 41.9±1.0 | 0.342 | 0.6052 | 0.0399 |
| GPX act liver (nmol/mg1) | 1.24±0.04 | 1.20±0.04 | 1.17±0.03 | 1.23±0.05 | 0.0132 | 0.2036 | 0.6066 |
| HNE serum (μg/mL) | 3.4±1.6 | 8.5±8.2* | 2.6±1.6 | 5.0±3.4*# | 0.4234 | 0.2151 | 0.0322 |
| HNE liver (μg/mL) | 12.8±5.4# | 18. 9±10.9 | 6.2±3.5 | 7.3±4.1# | 0.3531 | 0.0023 | 0.1874 |
| FEMALES | Vehicle | Statin | 2-way ANOVA | ||||
| WT | Scly KO | WT | Scly KO | Pint | PStatin | PGen | |
| GPX act serum (nmol/mL−1) | 49.4±1.2 | 52.0±1.0 | 46.7±0.9 | 50.9±1.9* | 0.1945 | 0.0053 | <0.0001 |
| GPX act liver (nmol/mg1) | 1.33±0.11 | 1.25±0.04 | 1.26±0.07 | 1.23±0.04 | 0.3226 | 0.1726 | 0.093 |
| HNE serum (μg/mL) | 3.9±1.4 | 7.7±4.4* | 3.4±0.8 | 5.6±2.2*# | 0.4948 | 0.2437 | 0.0157 |
| HNE liver (μg/mL) | 14.5±14.2 | 31.7±16.7* | 11.1±3.7 | 9.4±3.6# | 0.0504 | 0.014 | 0.1148 |
Values are mean ± SEM. Two-way analysis of variance (ANOVA) was applied, followed by a Bonferroni post hoc test. Italicized numbers represent P<0.05 after 2-way ANOVA.
represents P <0.05 between Scly KO and WT, and
represents within the same genotype groups after post hoc test. P-values under 0.05 deemed significant; n = 5–8 per group. WT: wild type, KO: knockout; GPX: glutathione peroxidase, HNE: 4-Hydroxy-Trans-2-Nonenal; Act: activity; Int: interaction; Gen: genotype.
3.4. Effect of statin treatment in the hepatic transcriptomic profile
We first opted to examine a broad overview of the transcriptomic changes occurring in the liver following treatment with statin. Ampli-Seq analysis revealed differentially expressed genes in WT or Scly KO mice’s liver after statin treatment (Supplementary Tables 3A–3F). Three genes were differentially expressed in male mice (Supplementary Table 3A) and 12 genes in female mice (Supplementary Table 3B). Genes differentially expressed in male mice were cytochrome P450, family 3, subfamily a, polypeptide 11 (Cyp3a11), cytochrome P450, family 3, subfamily a, polypeptide 44 (Cyp3a44), insulin-like growth factor-binding protein 2 (Igfbp2), transcript variant 1. In female mice, genes differentially expressed included calcium/calmodulin-dependent protein kinase II beta (Camk2b), AT-rich interactive domain 5B (MRF1-like) (Arid5b), transcript variant X3, and lipocalin 2 (Lcn2).
Interestingly, we found a downregulation of oxidative stress-induced growth inhibitor 1 (Osgin1) in WT males (Supplementary Table 3C) and females (Supplementary Table 3E) after statin treatment. Male WT mice also had upregulation of cytochrome P450, family 3, subfamily a, polypeptides 11 (Cyp3a11) and 44 (Cyp3a44), while WT females did not show variations in the expression of these genes, having instead an upregulation of alcohol dehydrogenase 4 (class II), pi polypeptide (Adh4), and downregulation of WD repeat and SOCS box-containing 1 (Wsb1) and connective tissue growth factor (Ctgf). Genes downregulated by statin treatment in Scly KO mice included insulin-like growth factor-binding protein 2 (Igfbp2) in males (Supplementary Table 3D) and lipocalin 2 (Lcn2) in females (Supplementary Table 3F).
Correspondent protein levels of top differentially regulated genes were then validated by Western Blot analysis. We selected these genes to validate as they presented the highest effect between experimental groups. We found that levels of NGAL, the product of Lcn2 gene, were higher in male Scly KO than WT mice, but not female (Figure 4A and F). For Osgin1, changes were influenced by genotype (P=0.04) and statin treatment (P=0.04) in male (Figure 4B), while in female only a genotype effect was observed (Figure 4G). CaMK2b (Figure 4C and 4H) was significantly higher in Scly KO female mice than WT when treated with vehicle and, interestingly, these differences disappeared after statin treatment. Igfbp2 was unchanged in male mice (Figure 4E), while only an interaction effect was found in female, with higher levels in the Scly KO mice restored after statin treatment (Figure 4J).
Figure 4: Hepatic protein levels in male (A–E) and female (F–J) mice.
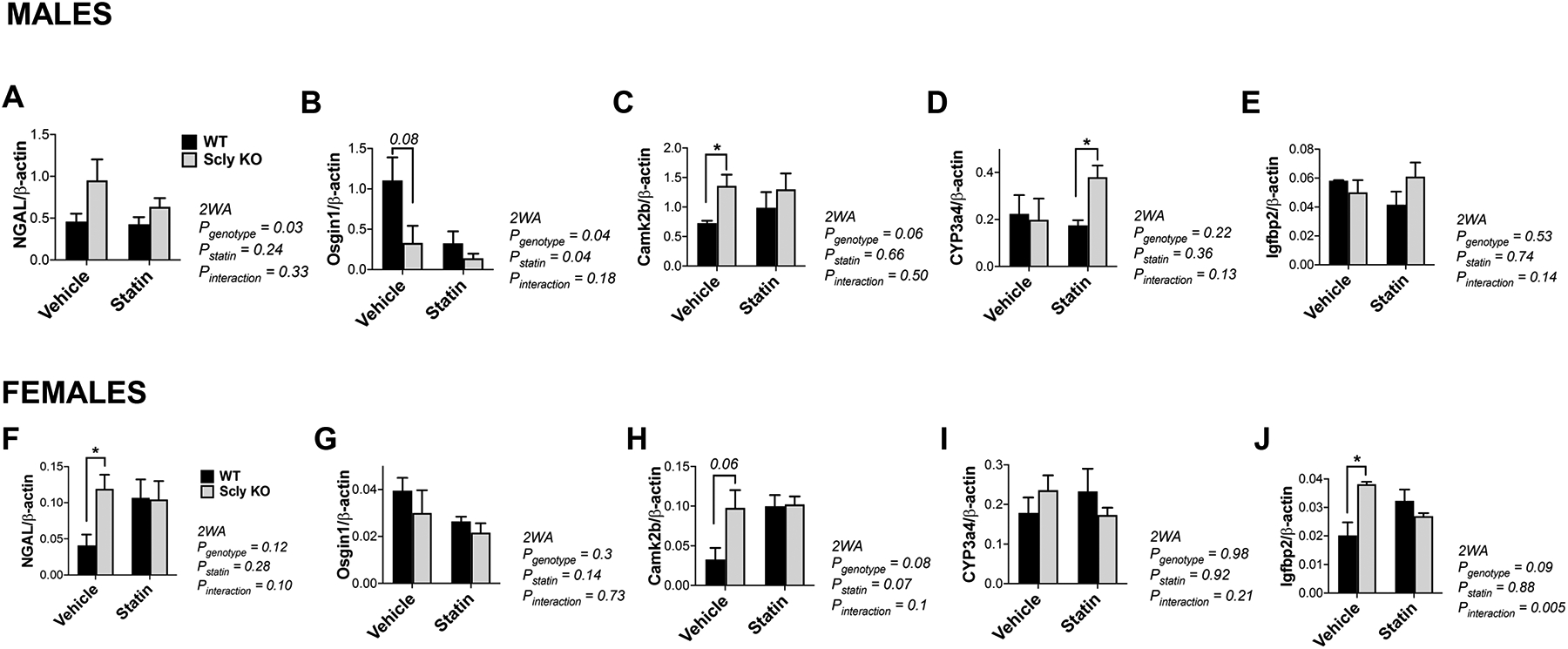
Levels of (A, F) NGAL, (B, G) Osgin1, (C, H) Camk2b, (D, I) CYP3a4, (E, J) Igfbp2 were assessed by Western Blot, with results normalized by the expression of β-actin. Data are mean ± SEM. Two-way ANOVA (2WA) was performed and *, ** and *** represent P<0.05, P<0.01, and P<0.001 respectively, after Bonferroni’s post-test; n=4 per group. Black bars, WT; gray bars, Scly KO.
Based on the fold-change differences observed for up- or downregulated genes found using the Ampli-Seq approach, enrichment analysis of gene ontologies and pathways was performed. For males, WT, and Scly KO mice, gene ontology (GO) of the biological process revealed regulation of insulin-like growth factor receptor signaling pathway to have the highest score between vehicle and statin-treated mice. GO of molecular function indicated insulin-like growth factor II binding with the highest score between these same groups (Table 2). For females, both WT and Scly KO mice, GO of biological process revealed negative regulation of glucocorticoid receptor signaling pathway and GO of molecular function indicated phosphorylase activity to have the highest score between vehicle and statin-treated mice (Table 2).
Table 2.
Pathways exhibiting the highest transcript enrichment between vehicle- and statin-treated WT and Scly KO mice unveiled by online Enrichr tool.
| Males - WT and Scly KO (Vehicle × Statin) | |||||
|---|---|---|---|---|---|
| GO Biological Process | |||||
| Term | Overlap | Gene | P-adj | Odds ratio | Combined score |
| regulation of insulin-like growth factor receptor signaling pathway (GO:0043567) | 1/19 | Igfbp2 | 1 | 392.1568 | 2342.1 |
| GO Molecular Function | |||||
| Term | Overlap | Gene | P-adj | Odds ratio | Combined score |
| insulin-like growth factor II binding (GO:0031995) | 1/7 | Igfbp2 | 1 | 952.3810 | 6532.7 |
| Females - WT and Scly KO (Vehicle × Statin) | |||||
| GO Biological Process | |||||
| Term | Overlap | Gene | P-adj | Odds ratio | Combined score |
| negative regulation of glucocorticoid receptor signaling pathway (GO:2000323) | 1/6 | Per1 | 1 | 277.77778 | 1563.3921 |
| GO Molecular Function | |||||
| Term | Overlap | Gene | P-adj | Odds ratio | Combined score |
| phosphorylase activity (GO:0004645) | 1/6 | Tymp | 1 | 277.77778 | 1563.3921 |
WT: wild type, KO: knockout.
3.5. Statin effect on the hepatic expression of genes for selenoproteins and enzymes of the creatine metabolism
As statins are postulated to impact selenoprotein expression, particularly in the liver, we surprisingly uncovered that statin alone had no effect on gene expression of Selenop, Gpx1, and Txnrd1 (Table 3), both in male and female mice. An effect of the interaction between genotype and statin treatment in the expression of the same genes occurred in males only. Further measurement of Txnrd1 protein levels in the liver showed no changes in this selenoprotein (Supplementary Figure 1).
Table 3.
Hepatic expression of genes for selenoproteins and enzymes involved in Se, creatine, or energy metabolism in WT and Scly KO mice after vehicle or statin treatment.
| MALES | Vehicle | Statin | 2-way ANOVA (P-value) | ||||
|---|---|---|---|---|---|---|---|
| WT | Scly KO | WT | Scly KO | Interaction | Statin | Genotype | |
| Gpx1 | 40.0±11.1 | 54.8±7.5* | 48.5±7.7 | 44.1±11.4# | 0.0001 | 0.062 | 0.2543 |
| Txnrd1 | 1.8±0.13 | 2.2±0.7 | 2.1±0.4 | 1.7±0.2 | 0.0116 | 0.5111 | 0.9703 |
| Selenop | 332.4±65.7 | 438.7±125.8 | 366.7±74 | 315.0±74.4 | 0.0321 | 0.2075 | 0.4348 |
| Nfs1 | 0.5±0.06 | 0.6±0.1 | 0.4±0.1 | 0.6±0.1 | 0.1354 | 0.3523 | 0.0627 |
| Ccbl2 | 2.74±0.16 | 3.51±0.95 | 2.67±1.3 | 2.26±0.2 | 0.1674 | 0.1264 | 0.6602 |
| Selenbp2 | 9.7±3.1 | 3.8±0.7* | 7.9±4.8 | 3.9±1.4* | 0.5188 | 0.5401 | 0.0016 |
| Ck-b | 0.10±0.04 | 0.13±0.03 | 0.14±0.03 | 0.07±0.02 | 0.0015 | 0.3496 | 0.1708 |
| Gamt | 4.4±1.0 | 6.2±0.7* | 6.0±1.3 | 4.3±1.2* | 0.0019 | 0.8037 | 0.9861 |
| Gatm | 0.22±0.04 | 0.25±0.03 | 0.25±0.02 | 0.17±0.02* | 0.0002 | 0.0611 | 0.0895 |
| Slc6a8 | 0.04±0.01 | 0.06±0.02 | 0.04±0.01 | 0.045±0.02 | 0.2253 | 0.2253 | 0.1858 |
| Pparg | 0.15±0.02 | 0.39±0.1* | 0.2±0.09 | 0.5±0.1* | 0.902 | 0.1206 | <0.0001 |
| FEMALES | Vehicle | Statin | 2-way ANOVA (P-value) | ||||
| WT | Scly KO | WT | Scly KO | Interaction | Statin | Genotype | |
| Gpx1 | 66.8±11.5 | 63.7±15.2 | 72.4±20.2 | 76.9±22.8 | 0.6367 | 0.2431 | 0.9308 |
| Txnrd1 | 2.2±0.4 | 2.8±0.8 | 2.3±0.7 | 2.1±0.4 | 0.2105 | 0.2478 | 0.4508 |
| Selenop | 642.8±68.8 | 586.1±141 | 510.9±74.8 | 573.5±134 | 0.2384 | 0.1572 | 0.9496 |
| Nfs1 | 0.8±0.3 | 0.9±0.2 | 0.8±0.2 | 0.8±0.2 | 0.6895 | 0.9824 | 0.8083 |
| Ccbl2 | 4.7±1.5 | 4.3±0.8 | 3.1±0.6 | 4.4±0.6 | 0.0664 | 0.0827 | 0.3379 |
| Selenbp2 | 5.1±0.3 | 3.3±0.6* | 3.6±0.5# | 4.11±1.1* | 0.0022 | 0.2602 | 0.05 |
| Ck-b | 0.2±0.03 | 0.3±0.1 | 0.2±0.04 | 0.3±0.1 | 0.8664 | 0.7909 | 0.0783 |
| Gamt | 9.7±2.8 | 7.1±1.7* | 6.5±2.1 | 12.8±1.8* | 0.0002 | 0.2045 | 0.0646 |
| Gatm | 0.5±0.2 | 0.4±0.2 | 0.3±0.04 | 0.4±0.1 | 0.1441 | 0.1151 | 0.8755 |
| Slc6a8 | 0.07±0.02 | 0.05±0.01 | 0.06±0.01 | 0.05±0.02 | 0.3279 | 0.4441 | 0.1639 |
| Pparg | 0.3±0.07 | 0.4±0.08 | 0.2±0.05 | 0.4±0.1* | 0.1350 | 0.8019 | 0.0006 |
Values are mean ± SEM and were normalized to Hprt1 mRNA levels. Two-way ANOVA was applied followed by Bonferroni’s post hoc test. P-values under 0.05 were deemed significant. Italicized numbers represent P<0.05 after 2-way ANOVA.
represents P<0.05 between Scly KO and WT, and
represent P<0.05 between vehicle and statin treatment after post hoc test; n = 4–6 per group. WT: wild type; KO: knockout.
We additionally observed no changes in the expression of cysteine desulfurase Nfs1 and cysteine conjugate beta lyase 2 Ccbl2, involved in the metabolism of sulfur aminoacids. The gene for selenium-binding protein 2 (Selenbp2), found previously to be downregulated in the liver of Scly KO mice [15], was again downregulated in Scly KO mice, with statin only affecting its expression in female WT mice (Table 3). Expression assessment of creatine kinase (CK) isoform b (Ck-b), sodium- and chloride-dependent creatine transporter 1 (Slc6a8), guanidinoacetate N-methyltransferase (Gamt), and glycine amidinotransferase (Gatm) genes, known to participate in creatine metabolism in the liver [26], revealed that neither genotype nor statin alone affected the regulation of these genes (Table 3). An interaction effect of genotype and statin was found for the expression of Ckb, Gamt and Gatm in male Scly KO mice, while in female only Gamt was affected. We also found that genotype affected the expression of nuclear transcription factor and lipid metabolism regulator peroxisome proliferator-activated receptor-gamma (Pparg), previously shown to be upregulated in Scly KO mice [15], was also upregulated in this genotype after high-fat diet, but not altered by statin treatment.
3.6. Gene expression changes in the skeletal muscle after statin treatment
Appealing selenoprotein candidates for participation in statin adverse effects are SelenoN, a selenoprotein, whose loss-of-function is linked to skeletal muscle disorders, such as myalgia and weakness [4,27]; SelenoP, produced in the liver and responsible to distribute Se to extra-hepatic tissues [28,29]; and GPX1, that detoxifies hydrogen peroxide (H2O2) and organic peroxides, protecting cells against oxidative injury [28]. Soleus mRNA expression of selenoprotein genes Selenon, Selenop, and Gpx1 revealed that statin treatment upregulated Gpx1 expression in females of both genotypes. Gpx1 was also upregulated in male Scly KO mice treated with vehicle, and this effect was abrogated after treatment with statin (Table 4). Selenop expression was unchanged in male mice, while female Scly KO mice upregulated Selenop in response to statin. Both male and female mice had an interaction effect of statin and genotype on Selenon expression, with female Scly KO mice upregulating its expression after statin treatment, while male Scly KO mice maintained its expression (Table 4). As SelenoN has been hypothesized as a primary candidate for the muscular side effects of statin, we further measured its levels in the gastrocnemius of mice and found that SelenoN levels were similar between WT and Scly KO mice treated with statin (Supplementary Figure 1).
Table 4.
Expression of genes for selenoprotein and creatine metabolic enzymes in soleus muscle of wild-type (WT) and Scly KO mice as assessed by qPCR.
| MALES | Vehicle | Statin | 2-way ANOVA (P-value) | ||||
|---|---|---|---|---|---|---|---|
| WT | Scly KO | WT | Scly KO | Interaction | Statin | Genotype | |
| Selenon | 0.097±0.03 | 0.138±0.06 | 0.162±0.03 | 0.125±0.04 | 0.0497 | 0.1697 | 0.9106 |
| Selenop | 6.6±1.3 | 8.2±3.0 | 8.6±1.7 | 8.7±02.2 | 0.4367 | 0.1910 | 0.3785 |
| Gpx1 | 0.55±0.08 | 0.96±0.1* | 0.77±0.1 | 0.59±0.1*# | <0.0001 | 0.1738 | 0.0464 |
| Ck-m | 142.1±23.4 | 203.5±42.3 | 177.5±50.1 | 180±45.7 | 0.1351 | 0.7538 | 0.1074 |
| FEMALES | Vehicle | Statin | 2-way ANOVA (P-value) | ||||
| WT | Scly KO | WT | Scly KO | Interaction | Statin | Genotype | |
| Selenon | 0.12±0.02 | 0.10±0.02* | 0.09±0.01 | 0.15±0.01* | 0.0003 | 0.2436 | 0.0115 |
| Selenop | 10.3±2.1 | 6.2±1.0* | 9.0±2.8 | 10.4±2.0# | 0.0112 | 0.1379 | 0.1841 |
| Gpx1 | 0.54±0.14 | 0.48±0.07 | 0.74±0.30# | 0.73±0.24# | 0.7524 | 0.0269 | 0.6926 |
| Ck-m | 126.7±15.6 | 128.5±28.1 | 144.9±39.7 | 187.4±27*# | 0.1363 | 0.0087 | 0.1067 |
Values are mean ± SEM and were normalized to Hprt1 mRNA levels. Two-way ANOVA was applied and P-values under 0.05 deemed significant. Bonferroni’s post hoc test was performed. Italicized numbers represent P<0.05 after 2-way ANOVA.
represents P<0.05 between Scly KO and WT mice, and
represents P<0.05 between vehicle and statin treatent within the same genotype group after post hoc test; n = 4–6 per group. WT: wild type, KO: knockout.
Moreover, we assessed the soleus for the gene expression of the homodimeric muscle CK isozyme (Ckm) that resides within the myocyte cytosol [30]. We found that Ckm was upregulated in female, but not male mice treated with statin (Table 4).
3.7. Effect of statin treatment in the creatine metabolism
Creatine metabolism is altered in people taking statins and displaying muscular side effects [31]. To determine whether statin treatment was affecting whole-body creatine metabolism in our experimental model, we measured CK activity in the serum, liver and skeletal muscle, and creatine levels in the liver and skeletal muscle of mice. Statin treatment differentially affected creatine metabolism in male and female skeletal muscle (Table 5). After statin treatment, a reduction of CK activity was observed in the soleus of male mice (~40% in WT and ~25% in Scly KO) without changes in CK activity in the liver or in the serum. Female mice displayed an elevation of CK activity in the serum after statin treatment without significant changes in the soleus. Interestingly, female Scly KO mice presented lower hepatic CK activity than WT animals. Hepatic creatine levels were unchanged in male mice, while female Scly KO mice displayed higher creatine levels at baseline than WT mice, with statin treatment restoring it back to similar levels. In the gastrocnemius, creatine levels were also unchanged in male mice, while statin reduced creatine levels in female mice of both genotypes (Table 5).
Table 5.
Creatine Kinase (CK) activity and creatine (Cr) levels in WT and Scly KO mice treated with vehicle or statin.
| MALES | Vehicle | Statin | 2-way ANOVA | ||||
|---|---|---|---|---|---|---|---|
| WT | Scly KO | WT | Scly KO | Pint | PStatin | PGen | |
| CK act serum (U/L) | 102.3±69.8 | 118.0±43.7* | 101.0±44.8 | 165.1±31.2*# | 0.2471 | 0.2738 | 0.0637 |
| CK act liver (U/L) | 236.3±15.2 | 225.6±37.1 | 228.4±36.0 | 248.5±20.8 | 0.1878 | 0.5154 | 0.6809 |
| CK act soleus (U/L) | 224.0±31.1 | 213.2±34.1* | 138.3±62.0# | 159.6±36.3*# | 0.3999 | 0.0015 | 0.7799 |
| Cr liver | 129.2±13.1 | 131.4±15.1 | 139.5±27.1 | 142.5±23.1 | 0.9685 | 0.2577 | 0.7766 |
| Cr gastrocnemius | 205.2±75.8 | 174.9±51.0 | 174.9±20.7 | 252.5±66.5 | 0.0521 | 0.3707 | 0.3719 |
| FEMALES | Vehicle | Statin | 2-way ANOVA | ||||
| WT | Scly KO | WT | Scly KO | Pint | PStatin | PGen | |
| CK act serum (U/L) | 92.5±56.2 | 101.6±66.2* | 185.7±60.5# | 203.2±48.2*# | 0.8739 | 0.0016 | 0.6174 |
| CK act liver (U/L) | 154.1±34.4 | 82.7±57.7 | 167.2±56.5 | 80.2±43.0 | 0.7214 | 0.808 | 0.0019 |
| CK act soleus (U/L) | 240.9±31.9 | 192.2±34.8 | 206.1±44.0 | 205.5±68.3 | 0.2395 | 0.5918 | 0.2288 |
| Cr liver | 119.3±17.4 | 138.5±13.9* | 137.1±9.9 | 131.9±6.6 | 0.0461 | 0.335 | 0.2331 |
| Cr gastrocnemius | 203.7±25.9 | 166.6±49.3 | 147.4±36.4 | 152.1±39.8 | 0.2449 | 0.05 | 0.3638 |
Values are mean ± SEM. Two-way analysis of variance (ANOVA) was applied with Bonferroni’s post hoc test, and P-values under 0.05 were deemed significant. Italicized numbers represent P<0.05 after 2-way ANOVA.
represents P <0.05 between Scly KO and WT, and
between vehicle and statin treatment within same genotype after post hoc test. n = 5–8 per group. WT: wild type; KO: knockout; Act: activity; int: interaction; gen: genotype.
4. Discussion
In the present study, we determined the consequences of statin treatment using an animal model of obesity related to disruptions of Se metabolism and in the context of a high-fat diet with Se supplementation, which mimics a significant part of the American population’s dietary pattern. As Se and selenoproteins are known to participate in antioxidant mechanisms, it is notable that statin effects on oxidative stress are conflicting [32]. Hence, Se supplementation used in our study helps to rule out increased oxidative stress due to this micronutrient deficits. Interestingly, we previously demonstrated that Scly KO mice were more susceptible to oxidative stress after a high-fat diet with Se supplementation [17], and now we uncovered that the use of statin mitigates hepatic oxidative stress not only in the WT mice but also in this mouse model, regardless of sex. Mitigation of oxidative stress after statin treatment is likely triggered by downregulation of Osgin1, particularly in males, and this transcript has been positively correlated with improved oxidative stress responses in hepatic ischemic postconditioning recovery [33].
We also uncovered a genetic basis for the side effects of statin in obese, Se-supplemented mice, findings that can inform future research in human populations. For example, male mice, but not females, had a differential expression of Cyp3a11 and Cyp3a44 after treatment with a statin. CYPs are a superfamily of phase-I enzymes that play a leading role in drug metabolism and detoxification. Of all CYP enzymes, CYP3A4 (Cyp3a11 in mice) catalyzes the initial step of many foreign compounds’ detoxification pathway, including statin drug simvastatin [34–36]. In the present study, upregulation of Cyp3a11 and Cyp3a44 expression could represent activation of statin detoxification pathways by the liver. Nevertheless, the lack of differences in the levels of Cyp3a4 between WT and Scly KO mice indicate, in fact, that statin is possibly being detoxified at similar rates, and its effects on skeletal muscle were not due to slower clearance by the liver.
Mice overexpressing human IGFBP2 had reduced susceptibility to obesity and improved insulin sensitivity [37]. Concentration of IGFBP-2, a protein that binds with high affinity to the IGFs, correlate inversely with body mass index and lower adiposity in humans [38]. We did observe male WT mice upregulating Igfbp2 expression in response to statin. However, protein levels did not follow this elevation, suggesting that either Igfbp2 transcript is decayed, or the excess protein is preferentially targeted to degradation. This study reiterated our previous findings that mice lacking Scly become obese after a high-fat diet [23], with Se supplementation exacerbating obesity [17], particularly in males. Yet, we paradoxically observed an upregulation of Igfbp2 in male Scly KO mice at baseline, despite the phenotype of this mouse model. Furthermore, statin treatment led to a downregulation of Igfbp2 that could be connected with the worsened obesity state observed. The GO analyses also confirmed the biological activity of IGF, possibly leading to the observed metabolic differences after statin administration, and future studies may be required to untangle the relationship between Igfbp2 and the consequences of Scly loss.
We previously demonstrated that loss of Scly led to downregulation of Lcn2 only in female mice fed normocaloric diet [15]. Interestingly, female Scly KO mice fed a high-fat, Se-supplemented diet upregulated Lcn2 in comparison to WT mice, an effect reversed by statin treatment. LCN2, also known as neutrophil gelatinase-associated lipocalin (NGAL), is an osteoblast-derived, iron-carrier hormone recently revealed as a biomarker of liver cirrhosis [39], with anorexigenic properties in primates, and abolished regulation in obesity [40]. Female Lcn2 KO mice are resistant to diet-induced obesity with larger BAT and improved thermogenic capacity [41]. In our studies, BAT of female Scly KO mice were larger at baseline without affecting its susceptibility to high-fat diet, and statin treatment reduced BAT and body weight while also downregulating Lcn2. Notably, while male Lcn2 KO mice were not affected by high-fat diet, male Scly KO mice were more susceptible to high-fat diet, with higher levels of NGAL at baseline, with statin treatment in fact worsening obesity and maintaining NGAL levels. As statin treatment enhances selenoprotein type 2 deiodinase (Dio2) activity in BAT of male mice [42], an indicator of improved thermogenic responses, it is puzzling that male Scly KO mice are even more susceptible to diet-induced obesity, which reflects impaired BAT responses. The relationship between phenotypical responses of these two mouse models to hypercaloric diets can be correlational only, however it is also possible that Se metabolism may directly interfere in the effects of this hormone, particularly to BAT thermogenesis, in a sex-dependent manner.
Regarding creatine metabolism, CK catalyzes the reversible reaction of creatine and ATP forming phosphocreatine and ADP [43], playing a significant role in energy homeostasis of cells with intermittently high energy demand, such as myocytes. It is known that decreased CK activity in the muscle and higher serum CK activity after statin treatment could reduce energy generation and muscle contractility [46]. While we only observed reduced CK activity in the muscle of male mice treated with statins, this effect was absent in female skeletal muscle, which contained similar creatine levels. Notably, female serum CK activity was elevated after statin treatment, but not in males. As statin possibly first affects creatine metabolism in the liver, which cascades then into the serum and skeletal muscle CK and creatine changes, the observed compartmentalized responses to statin in each sex reflect either a temporal difference, with a delayed muscular response in females, or a difference in statin clearance rate in the liver, which is a possibility not corroborated by the hepatic Cyp3a4 results herein obtained. Such differential creatine metabolism may also point to molecular mechanisms that lead being female as a risk factor for statin-triggered myopathies [44]. It is also possible that impairment in another factor regulating the creatine-phosphocreatine (Cr-PCr) system, such as coenzyme q10 [45], may play a role in the differential statin responses.
Selenoprotein expression depends on tRNA[Ser]Sec [46–49]. Mammalian tRNA[Ser]Sec population consists of two major isoforms [49]. One isoform contains methylcarboxymethyl-5′-uridine (mcm5U) at position 34 and is the precursor of mcm5Um [47]. The addition of this methyl group marks the final step in tRNA[Ser]Sec maturation [47]. Efficient methylation of mcm5U requires prior synthesis of each modified base within the anticodon loop, including isopentenyladenosine at position 37 [47,48]. Changes in the anticodon loop of tRNA[Ser]Sec, as promoted by statins for the supply of the isopentenyl adduct to the tRNA[Ser]Sec, could affect efficiency of selenoprotein expression [47,48]. Interestingly, Se status influences both the steady-state levels and distributions of the two Sec tRNA[Ser]Sec isoforms [46–49]. The presence of Se elevates the levels of the Sec tRNA[Ser]Sec population with the mcm5Um form, while the reverse is true under conditions of Se deprivation [47,48]. Our results highlight that statin, when taken in combination with a Se supplemented diet, does not affect selenoprotein expression equally. In this study, supplementation with higher doses of Se, simulating a Se-rich diet of the United States population, possibly influenced the enrichment of the Sec tRNA[Ser]Sec population towards predominance of a methylated isoform. Thus, selenoprotein expression was likely not disrupted. Furthermore, we uncovered sex differences in the selenoprotein expression response. Statin or loss of Scly did not affect hepatic selenoprotein expression in female mice; however, in male Scly KO mice, statin treatment significantly downregulated hepatic selenoproteins Gpx1, Txnrd1, and Selenop, while slightly increasing GPX activity and not affecting Txnrd1 levels. This suggests that the Gpx1 transcript may be more prone to be targeted to nonsense-mediated decay (NMD) mechanisms, while its expression is favored by the presence of methylated tRNA[Ser]Sec isoform due to the supplementation with Se. Se deficiency preferentially targets Gpx1 mRNA degradation, reducing its levels, however Se supplementation may be contributing to stabilization of the protein, slowed degradation or increased activity rates, explaining the paradoxal pattern [50]. It also suggests that members of the GPX system are actively involved in the statin response, possibly to alleviate oxidative stress. Nevertheless, there are considerable limitations of using the Scly KO mice, with its lower hepatic Se levels [13], to tackle a mechanism involving selenoprotein synthesis that relies on the downstream tRNA[Ser]Sec integrity. Yet, we have previously showed that, under similar Se supplementation, GPX activity in the Scly KO mice is restored, while downregulating the Gpx1 transcript in females, but not males [17]. In this study, the increase in GPX activity observed in male Scly KO mice suggests the GPX system is responding preferentially to statin in these mice, but not to the Se supplementation.
Notably, maintenance of Selenop expression in the liver may indicate that Se distribution to other tissues is unaffected. Assuming that adequate Se is reaching the skeletal muscle, it is intriguing that male Scly KO mice lowered Gpx1 expression, while in females the opposite occurred. Moreover, muscular Selenon was downregulated by statin in female WT mice, while downregulated at baseline, but upregulated by statin in female Scly KO mice, which suggests statin regulation of its expression is impaired when Scly-dependent Se delivery is compromised, and independent of dietary Se levels, since mice are Se-supplemented. SelenoN regulates the replenishment of endoplasmic reticulum (ER) calcium stores mediated by sarcoplasmic/ER calcium ATPase, a crucial mechanism for excitation-contraction coupling in skeletal muscle [51]. Selenon KO mouse model presented redox and calcium storage impairment, sensitizing skeletal muscle to oxidative insult and leading to chronic ER stress [51]. Selenon deficiency in mice could also lead to oxidation of various proteins, promote aggregation or degradation, and corrupt several cellular functions, contributing to the replacement of muscle cells by connective tissue [27]. As SelenoN has been speculated to be involved in statin responses [52], the downregulation of Selenon observed in male Scly KO mice could suggest muscular injury mechanisms are at play, while female upregulation of the same transcript in female Scly KO mice may indicate protective responses against muscular injury. Nevertheless, SelenoN levels were unaffected by statin treatment regardless of genotype, potentially ruling out its participation in muscle disorders driven by statin treatment, at least due to its protein levels.
Interestingly, the upregulation of Selenop expression in the muscle of female Scly KO mice after statin treatment may indicate a detrimental role of statin even in a repleted Se condition. It is possible that Selenop upregulation is a compensatory response to due to impairment in the processing of the several Se-containing residues present in SelenoP reaching the muscle, compromising intracellular Se homeostasis. SelenoP has an essential role in maintaining Se concentrations in muscular tissues of mice, while inhibiting resistance to exercise despite its antioxidative capacity [53,54]. Se supplementation could ensure the synthesis of SelenoN or other unrecognized selenoproteins to improve muscular responses to statin regardless of the disruption in Scly, but compensatory increases in SelenoP synthesis could compromise muscular physiology by acting upon other pathways, such as controlling AMPK phosphorylation [54].
Our findings using an animal model of obesity treated with statins resonate with the clinical setting, given the widespread use of statins and the prevalence of obesity in humans. Supplementation with one unit of Brazil nut was sufficient to increase the Se status and contribute to the improvement of oxidative stress parameters in populations with an already adequate Se intake [18], which has been directly associated with decreased serum CK activity that could benefit the muscle homeostasis of patients using statins. However, Se supplementation still did not improve statin side effects in Se-deficient people [55]. In the present study we demonstrated that, in fact, statin responses are strongly sex-dependent, with a refined regulation between expression of selenoproteins in the liver and skeletal muscle, particularly Selenon, Gpx1 and Selenop. It should be noted, though, that Scly mutations have not yet been connected with obesity in humans, and this poses a limitation to the translational applicability of our study model. Currently, only one single nucleotide polymorphism (SNP) of the human SCLY gene has been associated with low-density lipoprotein, apolipoprotein B, total cholesterol and atherosclerosis prevalence in Mexican-Americans [12], however whether this SNP confers improved resistance to statin side-effects remains untested.
In conclusion, mice fed a high-fat, Se-supplemented diet and lacking Scly treated with statin revealed that male mice were more susceptible to obesity, with enhanced responses to oxidative stress, while females improved creatine metabolism. We also revealed genes involved in hepatic statin clearance and metabolic homeostasis related to statin treatment in these animals. Our results emphasize and improve our understanding of sex differences in the response to a widely used therapy in humans and may serve as basis to refine future guidelines of protocols of patient’s prescription.
Supplementary Material
Supplementary Figure 1. Selenoprotein levels in Scly KO mice treated with statin.
Supplementary Table 1. PCR primer sequences.
Supplementary Table 2A. Serum cholesterol in WT and Scly KO mice before and after vehicle or statin treatment. Supplementary Table 2B. Serum triglyceride content in WT and Scly KO mice after statin or vehicle treatment.
Supplementary Table 3A. Differentially expressed genes in male WT and Scly KO mice after statin treatment. Supplementary Table 3B. Differentially expressed genes in female WT and Scly KO mice after statin treatment. Supplementary Table 3C. Differentially expressed genes in male WT mice after statin treatment. Supplementary Table 3D. Differentially expressed genes in male Scly KO mice after statin treatment. Supplementary Table 3E. Differentially expressed genes in female WT mice after statin treatment. Supplementary Table 3F. Differentially expressed genes in female Scly KO mice after statin treatment.
Supplementary Figure 4A. KEGG pathway analysis implicated in statin responses in the liver of WT and Scly KO mice. Supplementary Figure 4B. Wiki pathways implicated in statin responses in the liver of WT and Scly KO mice. Supplementary Figure 4C. Gene ontologies uncovered by AmpliSeq analysis in the liver of WT and Scly KO mice treated with statin. AmpliSeq results are found in: http://epigenomics.jabsom.from-hi.com/core/proj-kFczBs2yE6cxwKXz/differential.html. Additional enrichment analysis with gene ontologies can be accessed in: http://epigenomics.jabsom.from-hi.com/core/proj-kFczBs2yE6cxwKXz/enrichment.html#enrichment_analysis.
Highlights.
Male mice lacking Scly fed a high-fat, Se-supplemented diet and statin-treated were more susceptible to obesity and oxidative stress.
Female mice lacking Scly submitted to the same diet and treatment improved creatine metabolism.
Statin treatment affected the selenoproteins expression in different tissues in a sex-dependent manner.
A genetic basis for the side effects of statin in obese, Se-supplemented mice was uncovered and can assist future research in human populations.
Acknowledgments
We thank the Epigenomics Core Facility of Hawaii from the University of Hawaii John A. Burns School of Medicine (UH-JABSOM) for Ampli-Seq analysis assistance. This core was partially supported by the National Institutes of Health (NIH) grant P20GM113134 - Centers of Biomedical Research Excellence (COBRE) in Diabetes, which also provided partial support AmpliSeq analysis via the Diabetes Dollars Program to LAS. Further support was provided by the Hawaii Community Foundation grant 20ADVC-102166 to LAS; NIH grants R01DK047320 to MJB; U54MD007601 – Subproject 5544 and R01DK128390 to LAS; F32DK124963 and a Research Supplement to Promote Diversity in Health-Related Research, R01DK047320-22S2 to DJT; and an Administrative Supplement for Research on Dietary Supplements from the Office of the Director (OD) and co-funded by the Office of Dietary Supplements (ODS), R01DK047320-22S1 to MJB; an Associate Dean for Research Core Credits Program from UH-JABSOM to LAS; and fellowship 2018/09478-4 from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) to LMW. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We are grateful to Natália Yumi Noronha (University of São Paulo) in the preparation of Figure 1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflicts of interest.
References
- [1].Vecchié A, Dallegri F, Carbone F, Bonaventura A, Liberale L, Portincasa P, et al. Obesity phenotypes and their paradoxical association with cardiovascular diseases. European Journal of Internal Medicine 2018;48:6–17. 10.1016/j.ejim.2017.10.020. [DOI] [PubMed] [Google Scholar]
- [2].Andolfi C, Fisichella PM. Epidemiology of Obesity and Associated Comorbidities. Journal of Laparoendoscopic and Advanced Surgical Techniques 2018;28:919–24. 10.1089/lap.2018.0380. [DOI] [PubMed] [Google Scholar]
- [3].Taylor BA, Thompson PD. Statin-Associated Muscle Disease: Advances in Diagnosis and Management. Neurotherapeutics 2018. 10.1007/s13311-018-0670-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Moosmann B, Behl C. Selenoprotein synthesis and side-effects of statins. Lancet 2004;363:892–4. 10.1016/S0140-6736(04)15739-5. [DOI] [PubMed] [Google Scholar]
- [5].Southern WM, Nichenko AS, Shill DD, Spencer CC, Jenkins NT, McCully KK, et al. Skeletal muscle metabolic adaptations to endurance exercise training are attainable in mice with simvastatin treatment. PLoS ONE 2017. 10.1371/journal.pone.0172551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kromer A, Moosmann B. Statin-induced liver injury involves cross-talk between cholesterol and selenoprotein biosynthetic pathways. Molecular Pharmacology 2009;75:1421–9. 10.1124/mol.108.053678. [DOI] [PubMed] [Google Scholar]
- [7].Brinton EA, Maki KC, Jacobson TA, Sponseller CA, Cohen JD. Metabolic syndrome is associated with muscle symptoms among statin users. Journal of Clinical Lipidology 2016. 10.1016/j.jacl.2016.05.003. [DOI] [PubMed] [Google Scholar]
- [8].Davidson MH. Squalene synthase inhibition: A novel target for the management of dyslipidemia. Current Atherosclerosis Reports 2007;9:78–80. 10.1007/BF02693932. [DOI] [PubMed] [Google Scholar]
- [9].Kromer A, Moosmann B. Statin-induced liver injury involves cross-talk between cholesterol and selenoprotein biosynthetic pathways. Molecular Pharmacology 2009;75:1421–9. 10.1124/mol.108.053678. [DOI] [PubMed] [Google Scholar]
- [10].Grover HS, Luthra S, Maroo S. Are statins really wonder drugs? Journal of the Formosan Medical Association 2014. 10.1016/j.jfma.2013.05.016. [DOI] [PubMed] [Google Scholar]
- [11].Carlson BA, Yoo MH, Tsuji PA, Gladyshev VN, Hatfield DL. Mouse models targeting selenocysteine tRNA expression for elucidating the role of selenoproteins in health and development. Molecules 2009;14. 10.3390/molecules14093509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Seale LA. Selenocysteine β-lyase: Biochemistry, regulation and physiological role of the selenocysteine decomposition enzyme. Antioxidants 2019;8. 10.3390/antiox8090357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Seale LA, Hashimoto AC, Kurokawa S, Gilman CL, Seyedali A, Bellinger FP, et al. Disruption of the Selenocysteine Lyase-Mediated Selenium Recycling Pathway Leads to Metabolic Syndrome in Mice. Molecular and Cellular Biology 2012. 10.1128/mcb.00293-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kurokawa S, Takehashi M, Tanaka H, Mihara H, Kurihara T, Tanaka S, et al. Mammalian selenocysteine lyase is involved in selenoprotein biosynthesis. Journal of Nutritional Science and Vitaminology 2011. 10.3177/jnsv.57.298. [DOI] [PubMed] [Google Scholar]
- [15].Seale LA, Khadka VS, Menor M, Xie G, Watanabe LM, Sasuclark A, et al. Combined omics reveals that disruption of the selenocysteine lyase gene affects amino acid pathways in mice. Nutrients 2019. 10.3390/nu11112584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Seale LA, Gilman CL, Hashimoto AC, Ogawa-Wong AN, Berry MJ. Diet-induced obesity in the selenocysteine lyase knockout mouse. Antioxidants and Redox Signaling 2015;23:761–74. 10.1089/ars.2015.6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Watanabe LM, Hashimoto AC, Torres DJ, Berry MJ, Seale LA. Effects of selenium supplementation on diet-induced obesity in mice with a disruption of the selenocysteine lyase gene. Journal of Trace Elements in Medicine and Biology 2020;62:126596. 10.1016/j.jtemb.2020.126596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Watanabe LM, Fernandes de Lima L, Ferraz-Bannitz R, Takaara D, Coimbra Romano B, Braga Costa TM, et al. Association between creatine kinase activity, oxidative stress and selenoproteins mRNA expression changes after Brazil nut consumption of patients using statins. Clinical Nutrition 2020. 10.1016/j.clnu.2020.02.012. [DOI] [PubMed] [Google Scholar]
- [19].Raman AV, Pitts MW, Seyedali A, Hashimoto AC, Seale LA, Bellinger FP, et al. Absence of selenoprotein P but not selenocysteine lyase results in severe neurological dysfunction. Genes, Brain and Behavior 2012. 10.1111/j.1601-183X.2012.00794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Whale AS, De Spiegelaere W, Trypsteen W, Nour AA, Bae Y-K, Benes V, et al. The Digital MIQE Guidelines Update: Minimum Information for Publication of Quantitative Digital PCR Experiments for 2020. Clinical Chemistry 2020. 10.1093/clinchem/hvaa125. [DOI] [PubMed] [Google Scholar]
- [21].Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research 2016. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yu P, Xiong T, Tenedero CB, Lebeau P, Ni R, Macdonald ME, et al. Rosuvastatin Reduces Aortic Sinus and Coronary Artery Atherosclerosis in SR-B1 (Scavenger Receptor Class B Type 1)/ApoE (Apolipoprotein E) Double Knockout Mice Independently of Plasma Cholesterol Lowering. Arteriosclerosis, Thrombosis, and Vascular Biology 2018;38:26–39. 10.1161/ATVBAHA.117.305140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tansey TR, Shechter IH. Squalene Synthase : Structure and Regulation diphosphate (FPP), in a two-step reaction (Fig. 2A). Initially, two molecules rearranged and reduced by NADPH to form squalene (9, IO). as cholesterol precursors, both PSPP and squalene have additiona. Progress in Nucleic Acid Research and Molecular Biology 2001;65:157–95. [DOI] [PubMed] [Google Scholar]
- [25].Yilmaz MI, Baykal Y, Kilic M, Sonmez A, Bulucu F, Aydin A, et al. Effects of statins on oxidative stress. Biological Trace Element Research 2004;98. 10.1385/BTER:98:2:119. [DOI] [PubMed] [Google Scholar]
- [26].Nabuurs CI, Choe CU, Veltien A, Kan HE, van Loon LJC, Rodenburg RJT, et al. Disturbed energy metabolism and muscular dystrophy caused by pure creatine deficiency are reversible by creatine intake. Journal of Physiology 2013. 10.1113/jphysiol.2012.241760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Marino M, Stoilova T, Giorgi C, Bachi A, Cattaneo A, Auricchio A, et al. SEPN1, an endoplasmic reticulum-localized selenoprotein linked to skeletal muscle pathology, counteracts hyperoxidation by means of redox-regulating SERCA2 pump activity. Human Molecular Genetics 2014;24:1843–55. 10.1093/hmg/ddu602. [DOI] [PubMed] [Google Scholar]
- [28].Burk RF, Hill KE. Regulation of Selenium Metabolism and Transport. Annual Review of Nutrition 2015. 10.1146/annurev-nutr-071714-034250. [DOI] [PubMed] [Google Scholar]
- [29].Duntas LH, Benvenga S. Selenium: an element for life. Endocrine 2015;48:756–75. 10.1007/s12020-014-0477-6. [DOI] [PubMed] [Google Scholar]
- [30].Nuss JE, Amaning JK, Bailey CE, DeFord JH, Dimayuga VL, Rabek JP, et al. Oxidative modification and aggregation of creatine kinase from aged mouse skeletal muscle. Aging 2009;1:557–72. 10.18632/aging.100055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Balestrino, Adriano. Creatine as a Candidate to Prevent Statin Myopathy. Biomolecules 2019;9:496. 10.3390/biom9090496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fuhrmeister J, Tews M, Kromer A, Moosmann B. Prooxidative toxicity and selenoprotein suppression by cerivastatin in muscle cells. Toxicology Letters 2012;215:219–27. 10.1016/j.toxlet.2012.10.010. [DOI] [PubMed] [Google Scholar]
- [33].Zhang P, Ming Y, Cheng K, Niu Y, Ye Q. Gene expression profiling in ischemic post conditioning to alleviate mouse liver ischemia/reperfusion injury. International Journal of Medical Sciences 2019;16. 10.7150/ijms.29393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thompson PD, Panza G, Zaleski A, Taylor B. Statin-associated side effects. Journal of the American College of Cardiology 2016;67:2395–410. 10.1016/j.jacc.2016.02.071. [DOI] [PubMed] [Google Scholar]
- [35].Adhyaru BB, Jacobson TA. Safety and efficacy of statin therapy. Nature Reviews Cardiology 2018;15:757–69. 10.1038/s41569-018-0098-5. [DOI] [PubMed] [Google Scholar]
- [36].Beltowski J, Wojcicka G, Jamroz-Wisniewska A. Adverse Effects of Statins - Mechanisms and Consequences. Current Drug Safety 2009;4:209–28. 10.2174/157488609789006949. [DOI] [PubMed] [Google Scholar]
- [37].Haywood NJ, Slater TA, Matthews CJ, Wheatcroft SB. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Molecular Metabolism 2019;19:86–96. 10.1016/j.molmet.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].DeMambro VE, Clemmons DR, Horton LG, Bouxsein ML, Wood TL, Beamer WG, et al. Gender-specific changes in bone turnover and skeletal architecture in Igfbp-2-null mice. Endocrinology 2008;149:2051–61. 10.1210/en.2007-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Law IKM, Xu A, Lam KSL, Berger T, Mak TW, Vanhoutte PM, et al. Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes 2010;59:872–82. 10.2337/db09-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Petropoulou PI, Mosialou I, Shikhel S, Hao L, Panitsas K, Bisikirska B, et al. Lipocalin-2 is an anorexigenic signal in primates. ELife 2020. 10.7554/eLife.58949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ishii A, Katsuura G, Imamaki H, Kimura H, Mori KP, Kuwabara T, et al. Obesity-promoting and anti-thermogenic effects of neutrophil gelatinase-associated lipocalin in mice. Scientific Reports 2017;7:1–11. 10.1038/s41598-017-15825-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Miller BT, Ueta CB, Lau V, Jacomino KG, Wasserman LM, Kim BW. Statins and downstream inhibitors of the isoprenylation pathway increase type 2 iodothyronine deiodinase activity. Endocrinology 2012;153. 10.1210/en.2012-1117. [DOI] [PubMed] [Google Scholar]
- [43].McLeish MJ, Kenyon GL. Relating structure to mechanism in creatine kinase. Critical Reviews in Biochemistry and Molecular Biology 2005. 10.1080/10409230590918577. [DOI] [PubMed] [Google Scholar]
- [44].Moßhammer D, Schaeffeler E, Schwab M, Mörike K. Mechanisms and assessment of statin-related muscular adverse effects. British Journal of Clinical Pharmacology 2014;78:454–66. 10.1111/bcp.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Caso G, Kelly P, McNurlan MA, Lawson WE. Effect of Coenzyme Q10 on Myopathic Symptoms in Patients Treated With Statins. American Journal of Cardiology 2007;99. 10.1016/j.amjcard.2006.12.063. [DOI] [PubMed] [Google Scholar]
- [46].Diamond AM, Choi In Shon, Crain PF, Hashizume T, Pomerantz SC, Cruz R, et al. Dietary selenium affects methylation of the wobble nucleoside in the anticodon of selenocysteine tRNA([Ser]Sec). Journal of Biological Chemistry 1993;268. 10.1016/s0021-9258(19)85229-8. [DOI] [PubMed] [Google Scholar]
- [47].Moustafa ME, Carlson BA, El-Saadani MA, Kryukov G v., Sun Q-A, Harney JW, et al. Selective Inhibition of Selenocysteine tRNA Maturation and Selenoprotein Synthesis in Transgenic Mice Expressing Isopentenyladenosine-Deficient Selenocysteine tRNA. Molecular and Cellular Biology 2001;21. 10.1128/mcb.21.11.3840-3852.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schweizer U, Bohleber S, Fradejas-Villar N. The modified base isopentenyladenosine and its derivatives in tRNA. RNA Biology 2017;14. 10.1080/15476286.2017.1294309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chittum HS, Hill KE, Carlson BA, Lee BJ, Burk RF, Hatfield DL. Replenishment of selenium deficient rats with selenium results in redistribution of the selenocysteine tRNA population in a tissue specific manner. Biochimica et Biophysica Acta - Molecular Cell Research 1997;1359. 10.1016/S0167-4889(97)00092-X. [DOI] [PubMed] [Google Scholar]
- [50].Moriarty PM, Reddy CC, Maquat LE. Selenium Deficiency Reduces the Abundance of mRNA for Se-Dependent Glutathione Peroxidase 1 by a UGA-Dependent Mechanism Likely To Be Nonsense Codon-Mediated Decay of Cytoplasmic mRNA. Molecular and Cellular Biology 1998;18. 10.1128/mcb.18.5.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chernorudskiy A, Varone E, Colombo SF, Fumagalli S, Cagnotto A, Cattaneo A, et al. Selenoprotein N is an endoplasmic reticulum calcium sensor that links luminal calcium levels to a redox activity. Proceedings of the National Academy of Sciences of the United States of America 2020;117:21288–98. 10.1073/pnas.2003847117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Castets P, Lescure A, Guicheney P, Allamand V. Selenoprotein N in skeletal muscle: From diseases to function. Journal of Molecular Medicine 2012;90:1095–107. 10.1007/s00109-012-0896-x. [DOI] [PubMed] [Google Scholar]
- [53].Hill KE, Zhou J, McMahan WJ, Motley AK, Burk RF. Neurological Dysfunction Occurs in Mice with Targeted Deletion of the Selenoprotein P Gene. Journal of Nutrition, 2004. 10.1093/jn/134.1.157. [DOI] [PubMed] [Google Scholar]
- [54].Misu H, Takayama H, Saito Y, Mita Y, Kikuchi A, Ishii KA, et al. Deficiency of the hepatokine selenoprotein P increases responsiveness to exercise in mice through upregulation of reactive oxygen species and AMP-activated protein kinase in muscle. Nature Medicine 2017. 10.1038/nm.4295. [DOI] [PubMed] [Google Scholar]
- [55].Fedacko J, Pella D, Fedackova P, Hänninen O, Tuomainen P, Jarcuska P, et al. Coenzyme Q10 and selenium in statin-associated myopathy treatment. Canadian Journal of Physiology and Pharmacology 2013;91:165–70. 10.1139/cjpp-2012-0118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Selenoprotein levels in Scly KO mice treated with statin.
Supplementary Table 1. PCR primer sequences.
Supplementary Table 2A. Serum cholesterol in WT and Scly KO mice before and after vehicle or statin treatment. Supplementary Table 2B. Serum triglyceride content in WT and Scly KO mice after statin or vehicle treatment.
Supplementary Table 3A. Differentially expressed genes in male WT and Scly KO mice after statin treatment. Supplementary Table 3B. Differentially expressed genes in female WT and Scly KO mice after statin treatment. Supplementary Table 3C. Differentially expressed genes in male WT mice after statin treatment. Supplementary Table 3D. Differentially expressed genes in male Scly KO mice after statin treatment. Supplementary Table 3E. Differentially expressed genes in female WT mice after statin treatment. Supplementary Table 3F. Differentially expressed genes in female Scly KO mice after statin treatment.
Supplementary Figure 4A. KEGG pathway analysis implicated in statin responses in the liver of WT and Scly KO mice. Supplementary Figure 4B. Wiki pathways implicated in statin responses in the liver of WT and Scly KO mice. Supplementary Figure 4C. Gene ontologies uncovered by AmpliSeq analysis in the liver of WT and Scly KO mice treated with statin. AmpliSeq results are found in: http://epigenomics.jabsom.from-hi.com/core/proj-kFczBs2yE6cxwKXz/differential.html. Additional enrichment analysis with gene ontologies can be accessed in: http://epigenomics.jabsom.from-hi.com/core/proj-kFczBs2yE6cxwKXz/enrichment.html#enrichment_analysis.