

Abstract
Acute myeloid leukemia (AML) is a highly heterogenous and aggressive disease with a poor prognosis, necessitating further improvements in treatment therapies. Recently, several targeted therapies have become available for specific AML populations. To identify potential new therapeutic targets for AML, we analyzed published genome wide CRISPR-based screens to generate a gene essentiality dataset across a panel of 14 human AML cell lines while eliminating common essential genes through integration analysis with core fitness genes among 324 human cancer cell lines and DepMap databases. The key glutathione metabolic enzyme, glutamate-cysteine ligase catalytic subunit (GCLC), met the selection threshold. Using CRISPR knockout, GCLC was confirmed to be essential for the cell growth, survival, clonogenicity, and leukemogenesis in AML cells but was comparatively dispensable for normal hematopoietic stem and progenitor cells (HSPCs), indicating that GCLC is a potential therapeutic target for AML. In addition, we evaluated the essentiality of GCLC in solid tumors and demonstrated that GCLC represents a synthetic lethal target for ARID1A-deficient ovarian and gastric cancers.
Keywords: Acute myeloid leukemia (AML), synthetic lethality, glutathione metabolic enzyme, CRISPR, glutamate-cysteine ligase catalytic subunit (GCLC)
Introduction
Acute myeloid leukemia (AML) is a genetically heterogenous myeloid malignancy that is characterized by aberrant clonal proliferation of myeloid progenitor cells found within the bone marrow [1]. In adults, AML is the most common acute leukemia type with an incidence rate of 80% of all acute leukemia cases and a 5 year survival rate of approximately 29% [1]. While recent advances have been made in understanding the biology of the disease, poor overall survival rates indicate the need for additional treatment options [2]. Therefore, the development of new therapeutic strategies, focusing on fundamental physiological properties that differ between cancer and normal cells will help to selectively eradicate cancer cells and significantly improve AML patient outcomes.
Altered oxidative stress and redox balance have been characterized as key properties underlying cancer development, progression, and metastasis [3,4]. Reactive oxygen species (ROS) at reduced levels are able to act as signaling molecules for proliferation and survival [5], while at moderately increased levels can act to promote mutagenesis [6]. Conversely, high ROS levels have been found to exert oxidative stress leading to cell death or senescence [7]. However, AML cells are capable of tolerating relatively high ROS levels through increased activity of antioxidant pathways [8]. Thus, identification of targets regulating oxidative stress would provide potential therapeutic strategies in AML.
Glutathione (GSH) is a tripeptide comprised of cysteine, glycine and glutamate, and has been found to be the most abundant antioxidant within all cells [5]. The first step of GSH synthesis is rate limiting and consists of the ligation of glutamate with cysteine by the glutamate-cysteine ligase (GCL) holoenzyme [5]. GCL is composed of catalytic and modifier subunits (GCLC: glutamate-cysteine ligase catalytic subunit and GCLM: glutamate-cysteine ligase modifier) [5]. In patients with therapeutic resistance and tumor progression, levels of GSH and the rate limiting metabolite cysteine have been found to increased [6]. It has been established that AML cancer cells have aberrant glutathione regulation and metabolism and are sensitive to agents that disrupt the glutathione pathway [9]. Therefore, targeting glutathione metabolism in AML represents a potential promising strategy.
Large scale perturbational CRISPR-screening provides the ability to systemically identify therapeutic vulnerabilities and to pinpoint genetic liabilities specific to AML. In this study, by mining published genome-wide CRISPR-based screens [10-12] we identified GCLC as a key enzyme in targeting glutathione metabolism for anti-leukemic activity. In order to validate the selective dependency on GCLC in AML, we utilized a CRISPR knockout system to confirm the differential sensitivity in AML versus normal hematopoietic stem and progenitor cells. To identify the underlying mechanism that mediates cell growth inhibition caused by GCLC depletion, we confirmed that GCLC knockout induces apoptosis and differentiation in addition to inhibiting colony formation. We further confirmed in vivo that GCLC knockout inhibited AML tumor growth and progression. Ogiwara et al. [13] revealed metabolic dependency of GSH synthesis in cancers with ARID1A mutations. We further confirmed that, in gastric and ovarian cancers, ARID1A mutations sensitize these cancer cells to GCLC depletion, demonstrating that GCLC represents a synthetic lethal target in ARID1A deficient cancers.
Materials and methods
Cell culture and reagents
We cultured 293T cells in Dulbecco’s Modified Eagle Medium (DMEM) medium and supplemented with 10% fetal bovine serum (FBS, Corning) and 100 U/mL penicillin/streptomycin. Human leukemia cell lines, EOL-1, MOLM-13, THP-1, HEL, U937, and P31/FUJ, were cultured in RPMI-1640 medium supplemented with 10% FBS. Additional human leukemia cell lines were cultured in either RPMI 1640 medium supplemented with 20% FBS (PL-21), Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FBS (MV4;11) or Minimum Essential Medium supplemented with 20% FBS (OCI-AML2, OCI-AML3 and OCI-AML5) plus 100 U/mL penicillin/streptomycin. Human cord blood CD34+ cells were purchased from STEMCELL Technologies (Catalog # 70008.5) and maintained in StemSpan SFEM II media (STEMCELL Technologies) supplemented with TPO, Flt3L, SCF, IL-3, IL-6, (100 ng/mL; PeproTech), SR1 (0.75 μM; Cellagen Technology), and UM171 (35 nM; STEMCELL Technologies) [14]. Solid tumor cell lines were cultured in RPMI 1640 medium (MKN-1, NUGC-4, SNU-16, SNU-668, MKN-74, SNU-620, 23132/87, SNU-1, KURAMOCHI, OVKATE, IGROV1, OVISE, EF027, and SKOV3), DMEM medium (OCUM-1 and COV318), IMDM (SNU-5 and KATOIII), F-12K Medium (AGS), or 1:1 MCDB105/Medium 199 (TOV112D and TOV21G) supplemented with 10-20% FBS and 100 U/mL penicillin/streptomycin. Piperlongumine (PLM), parthenolide (PTM), and doxycycline were obtained from Sigma-Aldrich. Ferrostatin-1 (Fer-1), Erastin, and RSL-3 were purchased from ApexBio.
Cas9-sgRNA (RNP) complex transfection
Target-specific sgRNAs were designed and synthesized by Synthego (The Gene Knockout Kit v2, Synthego). Sequences for sgRNAs are listed in Table 1. To generate Cas9-sgRNA RNPs, synthetic sgRNAs were incubated with Cas9 protein for at least 15 minutes at room temperature. 200,000 cells were resuspended in Buffer R (ThermoFisher) and electroporated with the Neon Transfection System. The electroporation programs used for solid tumor and AML cell lines were 1200 V, 30 ms, 2 pulses and 1600 V, 10 ms, 3 pulses, respectively.
Table 1.
Single guide RNA sequences used for knockout experiments
| Gene Name | Guide RNA Sequence |
|---|---|
| GART | CUACCUCAAAAUGAAGCUGC, UAGGGAUUGUUGGGAACCUG, AUGUCUGUCCAUAAACUCUU |
| LIPT2 | GCGCAGCCCGGCCGUAUACA, UACUGGGGCUGCAGGACCGC, GGCCCCGUCGGGGACUGAGG |
| PPCDC | CAAAGCUUUUGGACAUUCCU, CCGUGACACCCACAAGAACA, CAGACCCCACAUGGAACCAA |
| CAD | CCAGAACCAUGGGUUUGCUG, AACCAGGCUCACCUGAAGAA, CUCUUCACCAACGCCAAUGA |
| GCLC | GCCAUGGGGCUGCUGUCCCA, GGAUCCCGUGCCGCCGCACG, ACUUGAGAACGUCCUUGUGC |
| MTHFD1 | GCCCAUCUGGAUGAGGAGGU, CGAUGCAUGACUUGCUUCUG, CAGGGGUGCCGAUUGCCGGA |
| PPCS | GCUCGCUUCGCGGCCAGGCU, GGACCCCGUAGCCGGCGGCU, AAGUUGUCCAGGAAGCGCAC |
| CDK13 | UGACUGGGGAAAACGCUGCG, CUAUAUUUGAUAGAAUAUGU, CGGACAAGUUUACAAAGCCA |
| GFPT1 | AACGGGUAUGAGCUAUUCCA, CAGCGCUCUGAUAAAAAUAA, UUCUUCAGAGCAACAAGAUA |
| OGDH | UGCUAAGUUGAGGCCAUUGA, GACUAGUUCGAACUAUGUGG, CUAGGACAUUUCAACAGAUU |
| RFK | UCCCCGCUCCCCACGUGACC, GGCACCUGCCUUACUUCUGC, AAGCAGCUGGGCAUCCCCAC |
| CDS2 | AGGCUCUUCUCUCUGGACCA, UCCUUUACUCUCUAUCUAAU, AGAAGAAAUAGUUUACACAC |
| GSS | GUGUUGGGAUGGCCACCAAC, GGCACGGCAGGCCGUGGACC, ACAGGAGCCCACUUCCUCGG |
| PAICS | CUACCUGUUUGUCUUUCUGU, CUACCUGUUUGUCUUUCUGU, UGCUGUUUCCAGAUUGAAUU |
| SYVN1 | GUCAUCCCGAAAAACGGUGA, GCAACUGAGGGCAGCAGAGA, AGCACCUUCUGGAACGUUCC |
| PFAS | ACACUCGGAGGAAACUGCAA, CGAACAUAGAAGUGAAGGAC, AACUGUGCUACAACGUGAAC |
| CTAZ | ACGGUGAUGAGGGGCGUGGC, GCCCGGGUACCCCAGAGAUG, CCUCCCUGUUGUGCACGGUC |
| DHODH | GAGUCUUGAAAUCUGGCCCG, GCCCCUGCAGAGUCGGCAUC, CGCCUCCUACCUGAUGGCCA |
| PGM3 | UGAUCAUGUCAUGUUUCGCA, AUGGACUGAUCCUUCAAUAC, AAUUUUGUAGACAUGGAUUU |
| TRIM28 | GGCGGCAGCCUCGGCAGCAG, CCGCCAGCGGAGCCCUCGCC, CGCGGCGUCGUCGCCCGCGG |
| DLD | GAUGUAACAGUUAUAGGUUC, GUACUUUGCAGGCAAAAACA, ACCUUACCUUGAAGCCUAAC |
| LIAS | CACCAGAUACAAAAUCUUGA, UAAAACGCCAGAAAGGAGAA, AAGGAGCUUAACGGUCUGAC |
| PMVK | CUGACCCAGGCUUCUUUUGC, AUGGAGCCCUCUUACCCAGA, CUUGAACUUCCAGAGACUCC |
| UMPS | UCGACCGCGUCUUCUGAGUC, GGGGACUUCGUGCUGAAGAG, GGCGGUCGCUCGUGCAGCUU |
| LIG3 | UCUCAAACAUGCAUUUAAUG, GUGAAGGGCGUAUGCCGAAU, UGUGUGGACUAUGCCAAGCG |
| PPAT | UGAUAGCAGUAGGACAAUAA, CAUCUCUGUGCAUUAUAAGC, GUGUCUGAUAUAAAUGACAA |
| NPM1 | GTAAGGGCACTTACATACTT |
| KMT2A | UGACGAAGACGAAGACGAGG, GGUGGGCCCGGGCUUCGACG, CGCUCUCCCCAAACACGGCC |
| Non-targeting control | GCACUACCAGAGCUAACUCA |
Colony-forming assay
Human AML cell lines were electroporated with sgRNAs against GCLC (sgGCLC) or non-targeting control (sgNC). Four days after electroporation, 2,000 cells were cultured in 1 mL methylcellulose medium (H4230, STEMCELL Technologies) and colonies were counted after 8 days of incubation. Images were taken and analyzed by the IncuCyte S3 Live-Cell Analysis System (Essen BioScience).
Cell growth assay
The CellTiter-Glo (G7570, Promega) Luminescent Cell Viability Assay (CTG assay) was used to determine the cell proliferation in culture. Normally, CTG reagent was added into the cell culture and incubated for 10 minutes at room temperature on the shaker. The CTG signals were measured by luminescence measurement with Synergy Neo2 (BioTek).
Caspase 3/7 activity assay
We used the Caspase-Glo 3/7 assay kit (G8090, Promega) to detect caspase 3/7 activity. Briefly, cells were electroporated with sgGCLC or sgNC and cultured in RPMI 1640 medium with supplements. 24, 48 or 72 hours after electroporation, cells were harvested, and Caspase 3/7 activity was determined based on the manufacturer’s instructions. Luminescence was measured with Synergy Neo2 (BioTek).
Cell-cycle analysis
The cell-cycle status was analyzed by flow cytometry using bromodeoxyuridine (BrdU)/7-AAD incorporation assay kit (#559619, BD Pharmingen) following the manual instructions. Proliferating cells were stained for incorporated BrdU against total DNA content using 7-AAD. BrdU was detected by FITC-labeled anti-BrdU antibodies.
Real-time qPCR (RT-qPCR)
Total RNA was isolated using a RNAeasy Mini Kit (Qiagen) and the first-strand cDNA was synthesized with iScript Reverse Transcription Supermix (Bio-Rad) according to the manufacturer’s protocol. TaqMan Master mix (Applied Biosystems) and probes (Taqman Gene Expression Assays, ThermoFisher Scientific) were added to prepare the reaction mix and qPCR analysis was performed using a QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific). The RT-qPCR signals were quantified using the ΔCt method. The relative gene expression level was calculated and normalized to β-actin levels. TaqMan probes used in the reaction are Hs00921938_m1 (SLC7A11) and Hs01060665_g1 (β-actin).
shRNA constructions, lentiviral production, and cell infection
shRNAs targeting GCLC were designed and cloned into a lentiviral vector, pRSIEGP-U6-sh-EF1-TagGFP2-2A-Puro vector (Cellecta). shRNA sequences for shGCLC1 and shGCLC2 are CGGCACAAGGACGTTCTCAAG and CCTCCAGTTCCTGCACATCTA, respectively. Lentivirus were generated using the Lenti-vpak packaging kit (TR30037, ORIGENE). Human CD34+ and leukemia cells were transduced with lentivirus by spinoculation at 2,500 rpm for 3 hours [15]. Cells were selected with 1 µg/mL puromycin for 3 days before performing functional assays.
Inducible sgRNA cell line constructions and edit efficiency validation
The RSGTEBleo-U6Tet-sg-EF1-TetRep-2A-Bleo lentiviral vector (Cellecta) was used for the construction of inducible expression of sgRNAs. sgRNA sequences targeting GCLC are AGGCCAACATGCGAAAACGC (sgGCLC1), CAATGTCTGACACATAGCCT (sgGCLC2), ATTGCCCATTCCAAATCCCA (sgGCLC3) and AGAAATATCCGACATAGGAG (sgGCLC4). Indel frequency and knockout efficiency were analyzed by Sanger sequencing [16] and ICE analysis (Synthego). Primers used for PCR and Sanger sequencing are: 5’ primer: ACTGAGTGCACTCACCACGG/3’ primer: CCCCATACTTCATGTCCTCAC (sgGCLC-1), 5’ primer: AAACAGCCATCAGCACTTCC/3’ primer: GTGACCAATTTATGACGTTTGG (sgGCLC-2) 5’ primer: ACTGAGTGCACTCACCACGG/3’ primer: GGACCCAATACTTGTTGACCC (sgGCLC-3) and 5’ primer: GGAGACAGCAATTGCCCATTC/3’ primer: GTAAGTTACAGGGAGCCTTGG (sgGCLC-4). AML cells transduced with lentivirus expressing inducible GCLC sgRNAs were selected with 200 µg/mL Zeocin (InvivoGen). 1 µg/mL Doxycycline was used for the induction (Sigma).
Capillary western blot (Wes) analysis
Harvested cells were lysed in cell lysis buffer (#9803, Cell Signaling) that was supplemented with protease inhibitors (Roche). Protein targets were detected using primary antibodies and immunoprobed with HRP-conjugated secondary antibodies using 12-230 kDa separation module in the capillary-based western blot analysis system (WES, ProteinSimple). The chemiluminescent signal was detected and analyzed by Compass software (ProteinSimple) and displayed as traditional blot-like image and electropherogram. Antibodies used for Wes analysis were GAPDH (G9545, Sigma), GCLC (ab181839, Abcam), ARID1A (ab242377, Abcam), Caspase 3 (#9668, Cell Signaling), cleaved caspase 3 (#9664, Cell Signaling), and LC3B (#2775, Cell Signaling).
Luciferase activity assay
MV4;11 cells were genetically modified to express enhanced firefly luciferase by lentiviral transduction with RediFect Red-FLuc-Puromycin Lentiviral Particles (PerkinElmer). After transduction, 1 ug/mL puromycin was used to select stable luciferase expression cells. The ONE-Glo luciferase assay system (Promega) was used to measure luciferase activity.
Flow cytometry
After treatment, harvested cells were washed with PBS, and then resuspended in PBS containing 5% BSA (staining buffer). Antibodies against CD11b (Clone M1/70, BD Bioscience) and CD14 (Clone M5E2, BD Bioscience) were added to the cell suspension. After 30 minutes of incubation, cells were washed once, resuspended in staining buffer, and analyzed using Cytek Aurora flow cytometer (Cytek Biosciences). To detect AML cell engraftment, mouse bone marrow cells were harvested and incubated with lysis buffer for red blood cells (eBioscience) for 5 minutes before being detected with anti-human CD45 antibody (BioLegend). FlowJo Software version 10 (BD Life Sciences) was used to analyze the flow cytometry data.
Mouse studies and in vivo imaging
In vivo studies were conducted according to the IACUC guidelines and AbbVie standard operating procedures. Mice were maintained under specific pathogen-free conditions, housed with a 12-hour light/dark cycle under consistent room temperature and given free access to food and water. MV4;11 cells with enhanced firefly luciferase (MV4;11.FLuc) were injected intravenously into NOD-scid IL2Rgnull (NSG) mice (The Jackson Laboratory, Bar Harbor, ME). In vivo bioluminescence imaging (BLI) to monitor disease progression was conducted using Lago X in vivo instrument (Spectral Instruments Imaging, AZ). Eight or 18 days after implantation, the mice were randomized into two separate groups according to luminescence intensity and started to be fed with control diet (Control Diet) or doxycycline-contained diet (Dox Diet). Tumor progression was monitored once or twice per week by BLI. Tumor growth curve was established by quantification of luminescence intensity.
Statistical analyses
GraphPad Prism 8.3 was used to perform the statistical analyses and Student t-test was used to calculate the statistical significance.
Results
Identification and validation of GCLC dependency in AML cells
To identify novel therapeutic targets for AML, we analyzed previously published and publicly available data from genome-wide CRISPR-Cas9 screens that were conducted across a panel of 14 human AML cell lines [10]. Genes with a CRISPR score less than -1 in more than 90% of tested AML cell lines (Frequency > 0.9) were selected. Common essential genes were eliminated by performing integration analysis with core fitness genes identified from the 324 human cancer cell lines [11] and DepMap databases (https://depmap.org/portal/). In order to select for potential druggable targets, we narrowed our focus to targets with enzymatic activity and identified 26 genes including the glutathione metabolic enzyme GCLC for further CRISPR-based validation in AML cells (Figure 1A). Utilizing 3 AML cell lines (OCI-AML3, EOL-1, and MOLM-13) and CD34+ hematopoietic stem and progenitor cells (HSPCs) from human cord blood, we transfected the Cas9-sgRNA complex targeting these individual genes and evaluated their effects on cell growth. The CD34+ HSPCs served as our normal cell controls and our reference point to determine therapeutic index. We included two AML driver genes as positive controls, NPM1 and KMT2A, which have been previously reported to be affiliated with poor patient prognosis and play crucial roles in AML [17,18]. GCLC depletion resulted in significantly reduced cell growth in the AML cells (95%) as compared to our control HSPCs cells (40%), demonstrating an enhanced dependency of AML cells on GCLC (Figure 1B). We confirmed the knockdown of GCLC protein in AML and control HSPCs by capillary Wes analysis (Figure 1C). In addition, we used shRNA knockdown as an orthogonal approach to GCLC depletion. Similar GCLC protein knockdown efficiency was achieved by two different shRNAs targeting GCLC in both EOL-1 and CD34+ HSPCs (Figure 1D) resulting in significant reduction in cell growth in EOL-1 cells as compared to CD34+ HSPCs (Figure 1E). To study the potential effects pharmacological GCLC inhibition in AML cells, we utilized two naturally occurring small molecules Pathenolide (PTL) and Piperlongumine (PLM) that have been reported to deplete cellular glutathione [9]. We treated a panel of AML cell lines (MV4;11, EOL-1, OCI-AML2, OCI-AML3, OCI-AML5, THP-1, MOLM-13, HEL, PL-21, U937, and P31/FUJ) and CD34+ HSPCs from 4 donors with PTL and PLM for 3 days and found that AML cells were significantly more sensitive to glutathione depletion with lower EC50s (Figure 1F; Table 2). Together, these results indicate that AML cells, but not normal HSPCs strongly depend on GCLC for survival.
Figure 1.

Identification and validation of GCLC dependency in AML cells. A. Summary of AML new target identification and validation strategies. B. CRISPR validation of potential target genes in OCI-AML3, MOLM-13, EOL-1 and normal CD34+ HSPCs. Four-day CTG assay was performed to evaluate the effect of target gene depletion on cell growth. Arrows point to the GCLC sgRNAs transduced samples. The data were normalized to the readout from Cas9 only samples and shown as average from triplicates (error bars indicate SEM). C. Depletion of GCLC protein by sgRNAs was confirmed using capillary Wes analysis. sgNC: non-targeting control; sgGCLC: GCLC sgRNA. D. Reduction in GCLC protein level by shRNAs was confirmed by capillary Wes analysis. shLuc: luciferase shRNA control; shGCLC: GCLC shRNAs. E. GCLC shRNAs induced cell growth suppression was determined in EOL-1 and CD34+ HSPCs. F. AML cells and CD34+ HSPCs were treated with GCLC inhibitors, Piperlongumine (PLM) and Parthenolide (PTL), for 3 days and the CTG EC50s were shown in the boxplots (*p-value < 0.005; **p-value < 0.0001).
Table 2.
EC50s of Piperlongumine and Parthenolide in AML cell lines and CD34+ HSPCs
| AML cell lines | EC50 (µM) of Piperlongumine | EC50 (µM) of Parthenolide |
|---|---|---|
| EOL-1 | 0.42 | 0.87 |
| HEL | 1.61 | 2.60 |
| P31/PUJ | 0.60 | 1.45 |
| MV4;11 | 0.64 | 2.39 |
| MOLM-13 | 0.70 | 0.99 |
| OCI-AML2 | 1.06 | 2.57 |
| OCI-AML3 | 0.72 | 1.97 |
| OCI-AML5 | 1.21 | 3.82 |
| PL21 | 1.38 | 1.74 |
| U937 | 1.21 | 1.82 |
| THP-1 | 1.59 | 4.16 |
| CD34+ HSPCs | ||
| CD34+ HSPCs-1 | 3.02 | 4.47 |
| CD34+ HSPCs-2 | 3.95 | 4.28 |
| CD34+ HSPCs-3 | 4.66 | 5.22 |
| CD34+ HSPCs-4 | 3.50 | 4.72 |
Data mining of GCLC dependency across different indications
To further evaluate the contribution of glutathione metabolism to AML cell survival, we reanalyzed the gene essentiality datasets across a panel of 14 human AML cell lines generated by Wang et al. [10] and compared the CRISPR scores of genes involved in this regulation pathway. AML cells showed a significant dependency on glutathione pathway genes: GPX4, GCLC, GSS, and SOD1 (Figure 2A). GPX4 and SOD1 showed dependency in a broad range of cancer cells lines [11] suggesting core fitness and common essentiality of these genes (Figure 2B). However, GCLC and GSS were selectively dependent in a subset of cell lines (Figure 2B). CRISPR and RNAi screening data from the DepMap database across over 800 cell lines provide the gene effect (shown as CERES and DEMETER2) of specific gene loss or transcript depletion on cell growth (Figure 2C). A lower score indicates higher dependency or requirement for survival of the gene in a designated cell line. A score of zero indicates no guide depletion while a score of -1 corresponds to the median depletion of all common essential genes for a given cell line [12]. GCLC is strongly selective in CRISPR screens, indicating that its dependency is at least 100 folds more likely to have been sampled from a skewed distribution compared to a normal distribution (Figure 2C). We also analyzed the gene effect generated by CRISPR and RNAi genome-wide screens performed in different indications and identified that depletion of GCLC resulted in significant impact on growth of cell lines across heme malignancies and a subset of solid tumors (Figure 2D, 2E).
Figure 2.
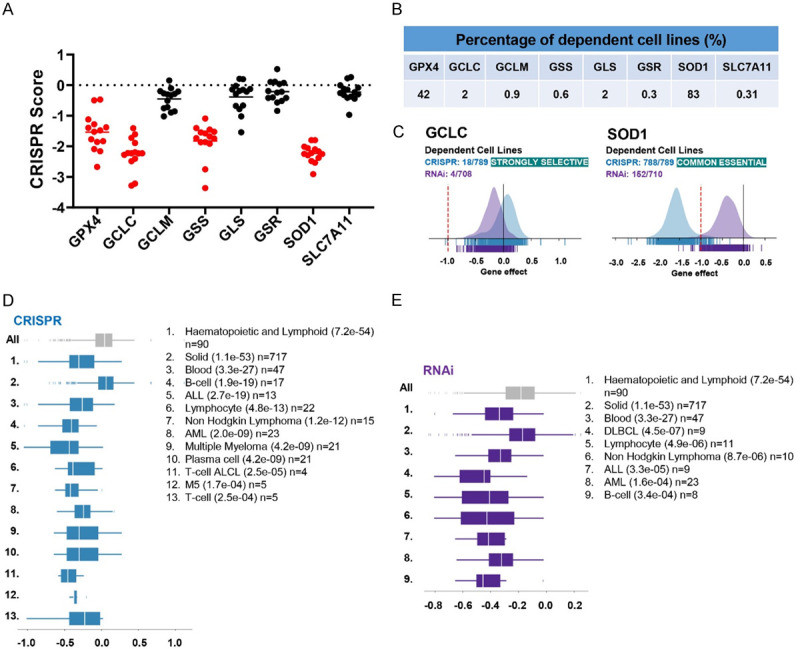
Data mining publicly available screening data of GCLC dependency across different indications. (A) CRISPR scores from gene essentiality dataset across 14 human AML cell lines were compared among molecules involving in glutathione metabolism [10]. (B) Dataset from genome-wide CRISPR-Cas9 fitness screens in 324 cancer cell lines was analyzed and the percentage of cell lines with dependency on indicated genes was shown in the table [11]. (C) DepMap analysis of GCLC or SOD1 dependency of tumor cell line panels in CRISPR (blue) and RNAi (violet) databases. (D, E) DepMap representation of lineage enrichment in the analysis of dependency of CRISPR (D) and RNAi (E) panels on GCLC. Enriched lineages have p-values < 0.0005 (shown in parentheses). n = indicates the number of cell lines plotted in that lineage.
Characterization of GCLC function in cell cycle regulation, cell differentiation, cell death and clonogenicity
To elucidate how GCLC contributes to AML cell survival, we investigated the cell cycle state after GCLC depletion in AML cells using BrdU and 7-AAD. In both MOLM-13 and EOL-1 cells, GCLC sgRNA transduced cells had lower BrdU incorporation (MOLM-13, 6.89% and EOL-1, 20.2%) than control (sgNC) cells (MOLM-13, 31.7% and EOL-1, 30.2%), indicating impaired proliferation of GCLC depleted cells (Figure 3A). A concomitant increase in the percentage of GCLC depleted cells was found in the G2/M gated populations (MOLM-13, 4.7% to 10.4% and EOL-1, 10.1% to 15.3%), indicative of growth arrest. In addition to inhibiting cell cycle progression, depletion of GCLC by sgRNAs also induced myeloid differentiation in OCI-AML3 cells compared to sgNC cells as demonstrated by the significant induction of myelomonocytic markers, CD11b and CD14 (Figure 3B). Together, these results indicate that GCLC influences the cell cycle progression and the differentiation states of AML cells.
Figure 3.

Characterization of GCLC function in the regulation of cell cycle, cell differentiation, cell death and clonogenicity. (A) Effect of GCLC sgRNAs on cell cycle was determined by BrdU/7-AAD incorporation assays. (B) Expression levels of surface CD11b and CD14 in OCI-AML3 cells were assessed by flow cytometry following sgRNA-mediated GCLC knockout. (C, D) Effect of GCLC depletion by sgRNAs on apoptotic or autophagic cell death was detected by caspase 3/7 assay (C) and capillary Wes analysis with antibodies specific to cleaved caspase 3 or LC3B (D). EOL-1 cells were collected at time-points 24 h, 48 h, and 72 h. (E) MV4;11 cells were treated with ferroptosis inducer Erastin and GCLC inhibitors Parthenolide and Piperlongumine for 24 h. Gene expression of SLC7A11 was determined by quantitative real-time PCR (qPCR) and normalized to the level of β-ACTIN (ACTB). (F) MV4;11 cells were transduced with GCLC sgRNAs and the effect of GCLC depletion on SLC7A11 gene expression was determined by qPCR on day 3 with the normalization to ACTB level. (G) MV4;11 and EOL-1 cells were treated with ferroptosis inhibitor (1 µM Ferrostatin-1, Fer-1), GPX4 inhibitor (1 µM RSL3), or the combination. The effect on cell viability was determined after 24 h of incubation by CTG assay. (H) MV4;11 and EOL-1 cells were transduced with GCLC or control sgRNAs (sgGCLC or sgNC) for 48 h and Fer-1 was added to block ferroptosis for additional 24 h. The effect on cell growth was determined by CTG assay. (I) AML cell lines were transduced with sgGCLC or sgNC and plated in the methylcellulose media. The images of colonies and the quantification were determined by IncuCyte live-cell analyses.
Disruption of GSH homeostasis results in a ROS imbalance, cellular stress, and cell death [6]. Since GCLC is the rate limiting step in GSH synthesis, we sought to assess any additional impact of GCLC depletion on programmed cell death, apoptosis, autophagy, and ferroptosis. Time-dependent increase of caspase 3/7 activity (Figure 3C) and cleaved caspase 3 (Figure 3D) were detected in GCLC depleted EOL-1 cells, indicating that GCLC depletion induced cell apoptosis. To examine if GCLC plays a role in autophagy, we assessed the formation of lipidated LC3-II from cytosolic LC3B-I as a marker of autophagosome formation. However, there was no induction of LC3B-II in our GCLC knockout sgGCLC cells, suggesting that autophagy doesn’t play a role in the cell growth inhibition observed in these cells (Figure 3D). Ferroptosis is a newly defined form of regulated cell death initiated by lipid peroxidation that has been shown in GSH deficient cells [19]. To evaluate the role of GCLC on ferroptosis, we tested whether GCLC depletion induces the expression of SLC7A11, which is a critical subunit of cystine-glutamate antiporter system Xc- (xCT) [19]. As expected, the inhibition of xCT by the control compound Erastin resulted in a compensatory transcriptional up-regulation of SLC7A11 (Figure 3E). In contrast, treatment with glutathione modulating agents (PTL and PLM) or GCLC depletion in MV4;11 cells had no impact on SLC7A11 expression (Figure 3E, 3F). We further applied the GPX4 inhibitor RSL3 to induce ferroptosis. Growth suppression by RSL3 in both MV4;11 and EOL-1 cells were blocked by the ferroptosis inhibitor, Fer-1. In contrast, Fer-1 did not protect cells from sgGCLC-mediated growth inhibition (Figure 3G, 3H). Thus, GCLC depletion induced apoptosis but not autophagy or ferroptosis in AML cells. Finally, we tested if GCLC is required for clonogenicity. It has been suggested that only a small subset of leukemia stem cells is capable of self-renewal with sufficient numbers of cell divisions in order to form colonies in semisolid medium. These leukemic colony-forming cells may act as progenitor cells and help to maintain the rest of the leukemic cell population in vivo [20]. Therefore, transduction of GCLC sgRNA caused a decrease in colony forming ability of a broad range of AML cell lines (Figure 3I), suggesting a critical role for GCLC in AML stem cell function.
Validation of inducible GCLC sgRNA systems and in vivo knockout efficacy
To elucidate the in vivo requirement for GCLC in AML, an inducible Cas9-CRISPR approach was used to inactivate GCLC in MV4;11 cells. The MV4;11 cells were then transduced with Cas9 and firefly luciferase expressing lentivirus to generate stable cells (MV4;11.Fluc). MV4;11.FLuc cells were then transduced with 4 different doxycycline-inducible sgRNAs targeting GCLC and treated with doxycycline (dox) to induce sgRNA expression. MV4;11.FLuc.sgGCLC1 and MV4;11.FLuc.sgGCLC3 were selected for in vivo evaluation as they exhibited the most robust edit efficiency (Figure 4A) and GCLC depletion (Figure 4B). We confirmed that dox-induced GCLC depletion significantly suppressed in vitro cell growth and colony formation (Figure 4C, 4D). For PD studies, MV4;11.FLuc.sgNC, MV4;11.FLuc.sgGCLC1 and MV4;11.FLuc.sgGCLC3 cells were intravenously injected into mice. Mice were placed on dox containing diet to induce GCLC knockout after randomization on Day 18. Bone marrow cells were collected after 5 or 7 days on dox diet to measure AML cell populations and to validate the GCLC knockout efficiency (Figure 4E). In the MV4;11.Fluc.sgGCLC1 injected mice, analysis of bone marrow from control diet animals revealed 31.7% AML cells in the bone marrow, while the CD45+ AML cell population was nearly undetectable in the bone marrow of mice on doxycycline diet (Figure 4F). Consistently, there was no detectable human GCLC PCR product in samples from doxycycline diet groups (Figure 4H). In MV4;11.FLuc.sgGCLC3 model, significant reduction of CD45+ AML cell population was detected in the bone marrow with doxycycline diet comparing to those with control diet (6-7% vs 16%) (Figure 4G). Notably, gene editing efficiency was further confirmed in the human GCLC PCR products (Figure 4I, 4J).
Figure 4.
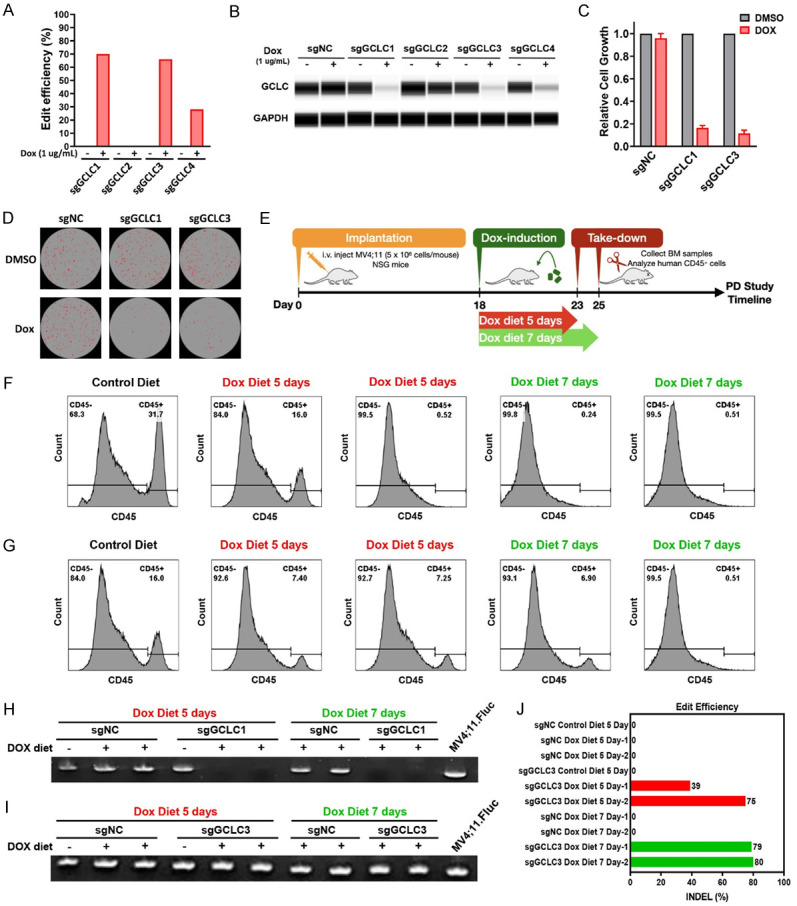
Validation of GCLC inducible sgRNA systems and in vivo knockout efficacy. (A, B) MV4;11.FLuc cells were stably transduced with Cas9 and indicated inducible GCLC sgRNAs. After 4 days of induction with 1 µg/ml Doxycycline (Dox), GCLC edit efficiency was analyzed by Sanger sequencing and ICE analysis (A) and GCLC protein level was detected using capillary Wes analysis (B). (C, D) MV4;11.FLuc cells transduced with sgNC, sgGCLC1, or sgGCLC3 were induced with 1 µg/ml Doxycycline. Cell growth was measured using CTG assay on day 6 (C) and colony formation was detected on day 8 (D). (E) NSG mice were intravenously injected with either MV4;11.FLuc.sgNC, MV4;11.FLuc.sgGCLC1, or MV4;11.FLuc.sgGCLC3 cells (5 × 106 cells/mouse), followed by feeding control or Dox diet starting from 18 days after implantation. Mouse BM cells were collected 5 or 7 days after dosing for PD studies. (F, G) Human CD45+ cell populations in BM samples from mice implanted with MV4;11.FLuc.sgGCLC1 (F) or MV4;11.FLuc.sgGCLC3 (G) were detected by flow cytometry. (H, I) Human GCLC gene was detected by PCR. (J) Genomic sequencing was performed to evaluate GCLC knockout efficiency in BM samples from mice transplanted with MV4;11.FLuc.sgNC or MV4;11.FLuc.sgGCLC3 cells and fed with control or Dox diet.
Suppression of AML tumor growth by GCLC knockout
To define the role of GCLC in AML tumor progression in vivo, MV4;11.FLuc.sgNC, MV4;11.FLuc.sgGCLC1 and MV4;11.FLuc.sgGCLC3 cells were intravenously injected into mice. Dox-containing diet was administrated to mice to induce GCLC molecular knockout after randomization on Day 8. AML tumor progression was monitored by bioluminescence intensity for 21 days. Bone marrow cells were collected to detect AML cell populations and validate the GCLC knockout efficiency (Figure 5A). Doxycycline-induced GCLC depletion significantly suppressed MV4;11.FLuc.sgGCLC1 and MV4;11.FLuc.sgGCLC3 tumor progression (Figure 5B, bioluminescence intensity quantification; Figure 5C, in vivo images of individual mice). In contrast, there was no tumor growth difference between control or dox diet groups in mice with MV4;11.FLuc.sgNC. To further assess disease burden, we analyzed human CD45+ cells in mouse bone marrow samples using flow cytometry to quantitate AML cell populations. Consistent to the result of bioluminescence intensity, administration of doxycycline-contained diet significantly reduced AML cell populations in the mice with MV4;11.FLuc.sgGCLC1 (3.14% vs 12.41%) or MV4;11.FLuc.sgGCLC3 tumors (4.63% vs 26.51%), supporting the role of GCLC in AML progression (Figure 5D, 5E). Whole genome sequencing revealed that the surviving AML cells in the dox-treated mice showed little to no editing of GCLC (Figure 5F). Together, these data indicate that GCLC is essential for AML cell survival and disease progression in vivo.
Figure 5.

Suppression of AML tumor growth by GCLC knockout. (A) NSG mice were intravenously injected with either MV4;11.FLuc.sgNC, MV4;11.FLuc.sgGCLC1, or MV4;11.FLuc.sgGCLC3 cells (5 × 106 cells/mouse), followed by feeding Control or Dox diet starting from 8 days after implantation. (B) At indicated time points after implantation, tumor progression was monitored, and luminescence signals were quantified (*p-value < 0.0005). (C) Mice were repeatedly imaged to record luminescence signals. There were 9 mice in each group and 1 naïve mouse without tumor bearing was included as background control. (D) Bone marrow samples at endpoints were collected and human CD45+ cells were detected by flow cytometry as indication of AML populations. (E) Quantification data from (D) were shown as barograph (*p-value < 0.005; **p-value < 0.0001). (F) Genomic sequencing was performed to evaluate GCLC knockout efficiency in AML cells.
Differential sensitivity of ARID1A-wildtye (WT) and -mutant (Mut) ovarian and gastric cancer cells to GCLC depletion
Our analysis of the DepMap data also identified a subset of solid tumor cell lines with enhanced dependence on GCLC. A previous report from Ogiwara et al. [13] demonstrated that ARID1A-deficient cancer cells are dependent on GCLC due to impaired transcriptional activation of SLC7A11 and a limited supply of cysteine (Figure 6A). ARID1A, encodes a SWI/SNF chromatin-remodeling factor, which has been reported to be frequently mutated in many cancer types [21]. We evaluated 181 TCGA and non-TCGA studies with no overlapping samples in the cBioPortal database to determine the mutation frequency of ARID1A across different cancer types (Figure 6B). Given the high frequency of ARID1A mutation and its association with poor prognosis in gastric and ovarian cancers [21], we evaluated GCLC dependency in ARID1A-WT and -Mut cell lines from these indications. Ovarian and gastric cancer cell lines were collected and ARID1A mutation profiles were analyzed through the COSMIC (the Catalogue of Somatic Mutations in Cancer) database (data not shown). ARID1A protein loss in ARID1A-Mut cell lines was confirmed by capillary Wes analysis (Figure 6C). We observed significant growth suppression in cells with ARID1A mutation but not ARID1A wild type cells upon CRISPR-mediated GCLC depletion despite similar reduction in the level of GCLC protein (Figure 6D, 6E), demonstrating a selective dependency on GCLC in ARID1A-Mut cells.
Figure 6.
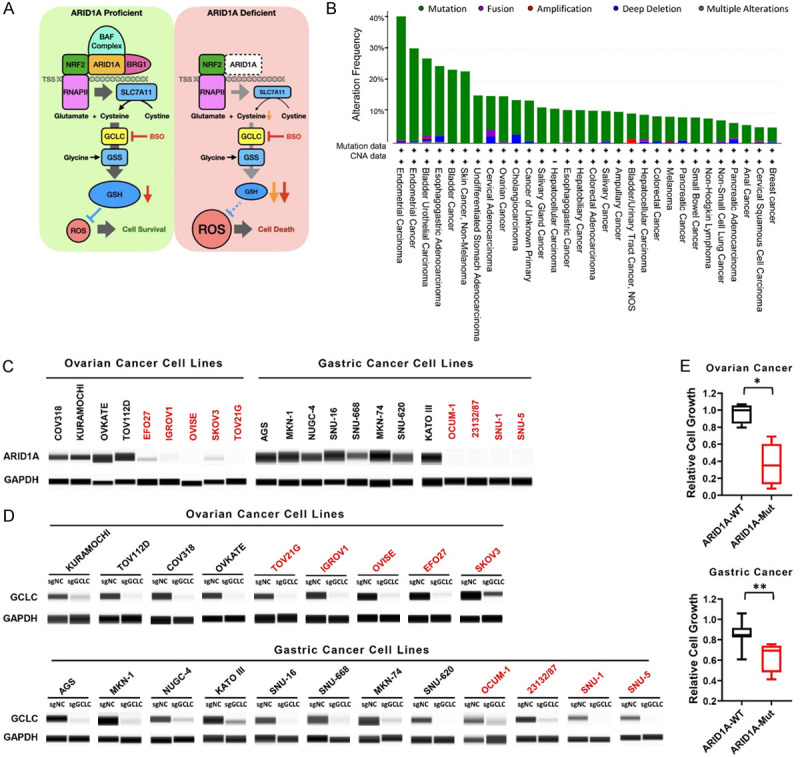
Differential sensitivity of ARID1A wild-type (WT) and mutant (Mut) ovarian and gastric cancer cells to GCLC depletion. A. The graph depicts mechanistic relationship of GCLC synthetic lethality in ARID1A-deficient cancers (modified from Cancer Cell 2019) [13]. B. The alteration frequency of ARID1A in various types of cancers was analyzed by cBioPortal database. Mutations are represented in green, fusion in purple, amplification in red, deep deletion in blue, and multiple alterations in grey. C. ARID1A protein expression in ovarian and gastric cancer cells was detected by capillary Wes analysis. Black indicates ARID1A-WT cells and red indicates ARID1A-Mut cells. D. Capillary Wes analysis indicates the depletion of GCLC protein by sgRNAs. GAPDH was used as loading control. E. The effect of GCLC depletion by sgRNAs in ovarian cancer or gastric cancer cells with WT- or Mut-ARID1A was determined by CTG assay (*p-value < 0.005, **p-value < 0.05).
Discussion
In this study, we analyzed public, genome-wide CRISPR screening datasets to identify putative targets in AML [10-12] and validated a GCLC dependency in AML. These studies demonstrate that depletion of GCLC significantly reduces cell growth and inhibits colony formation by inducing apoptosis and differentiation in AML cells. We extended these findings and confirmed a synthetic lethality relationship between GCLC and ARID1A deficiency in gastric and ovarian cancer cells.
The GSH pathway is known to be involved in many processes regulating tumor progression and sensitivity to cancer therapy [7]. Several recent studies have shown aberrant glutathione metabolism in leukemic cells with elevated expression of multiple glutathione regulatory proteins [9,22]. Our method identified the glutathione pathway genes, GPX4, GSS, SOD1 and GCLC as dependencies in AML cell lines. To identify targets with the potential for a clinical therapeutic window, we included several approaches to eliminate common essential genes (Figure 2B, 2C), highlighting GCLC as a potential therapeutic target. In AML, the gene and protein expression of glutathione pathway components, including GSS, GCLC, GCLM, GPX1 and GSR, were found to be upregulated [9]. The reported upregulation coupled with our results demonstrating that GCLC is essential for cell growth in AML but not in CD34+ HSPCs, suggest that inhibition of GCLC could target AML cells while sparing normal hematopoietic cells. Additionally, glutathione depleting agents, PTM and PTL, were able to significantly induce cell death in AML cell lines but had limited effect and toxicity on normal CD34+ bone marrow cells [9]. Therefore, these data show that the AML cells have an enhanced dependency on the glutathione synthesis pathway, providing support for the pharmacological targeting of GCLC.
We examined the possible cellular mechanisms by which GCLC depletion modulates proliferation in AML cells. Our data demonstrate that GCLC is required for cell cycle progression. Many previous studies have shown that GSH levels are associated with an early proliferative response and that sufficient levels are essential for cells to enter S phase [23,24]. This increase in GSH corresponded with an increase in GCLC mRNA observed during the shift from G0 to G1 phase [25]. Indeed, our results confirm that GCLC knockdown in AML cells caused a decrease in S phase cells and an increased percentage of cells in the G2/M phase. Defects in G2/M cell cycle arrest may allow damaged cells to enter mitosis and eventually undergo apoptosis, consistent to our observation of increased apoptosis signals in GCLC depleted cells.
Increased ROS levels can trigger cell death through the apoptotic, autophagic and ferroptosis pathways. Our data shows that GCLC depletion induces apoptosis but not autophagy or ferroptosis in AML cells. Knockdown of GCLC in our studies induced apoptosis as evidenced by increased caspase 3/7 activity and cleavage of caspase 3. These results are consistent with several studies demonstrating that a decrease in cellular GSH concentration activates the apoptosis cell death cascade [26]. Depletion of GSH depletion can lead cells to apoptosis or directly trigger cell death by regulating the activation of execution caspases [27]. Since GCLC is the rate limiting step of GSH synthesis, it is the key factor that suppresses apoptosis via the antioxidant system. Our results are also consistent with several studies demonstrating that the inhibition of GCLC in lung cancer cells [28] and in liver [29] induced apoptosis. Depletion of GSH, mainly through the inhibition of GPX4 and system xc, has been shown to predispose to ferroptosis, a cellular death process induced by excessive lipid peroxidation [19]. However, in our GCLC knockdown AML cells, the observed cell death was not due to ferroptosis as shown by the inability of the lipid targeted antioxidant ferrostatin-1 to rescue the cell viability. Further investigation is required for better understanding of the connections between GCLC inhibition, GSH depletion and ferroptosis.
A subpopulation of leukemic cells have stem cell features and self-renewing potential, are less sensitive to chemotherapy and are responsible for the recurrence of AML [20], and therefore regarded as a crucial subpopulation to target. Previous studies have provided evidence that glutathione and redox regulatory processes are involved in myeloid differentiation [30]. An increase in ROS levels is requisite to prime multipotent haematopoietic progenitors for differentiation into more mature myeloid cells, while suppression of elevated ROS levels is able to retard differentiation [30]. Inhibition of GCLC leads to an imbalance in ROS resulting in cellular stress. In our studies, we report that GCLC knockdown in AML cells led to a significant increase in myeloid differentiation markers as determined by upregulated CD11b and CD14 expression in AML cells. Simultaneous eradication of both bulk AML cells and stem cells is critical to achieving better clinical outcomes. We show that GCLC knockdown in several AML cell lines reduced the ability for colony formation, which is indicative of the potential to inhibit leukemia stem cells. Similarly, Pei et al. [9] reported that glutathione inhibition by PTL in combination with cytarabine was able to reduce CD34+ AML cell viability. Since standard AML chemotherapy agents such as cytarabine have limited toxicity toward CD34+ AML cell populations [31], combination therapy with GCLC inhibition could represent a promising treatment strategy.
Over 20% of all human cancers are found to carry deleterious mutations in genes that encode the subunits of the SWI/SNF chromatin remodeling complexes. These mutations are thought to promote tumorigenesis by impairing chromatin remodeling in transcribed genes and DNA repair [32]. ARID1A, is one of the most frequently mutated subunits of the SWI/SNF chromatin remodeling complexes [33]. Defects in ARID1A are thought to confer advantages to tumor cell growth. Ogiwara et al. [13] has reported that ARID1A-deficient cancer cells are sensitive to the inhibition of the GSH synthesis pathway and specifically to GCLC across multiple cancer cell lines. Mechanistically, ARID1A occupies the transcription start site of SLC7A11 [13]. SLC7A11 encodes a subunit of the cystine/glutamate transporter Xc that imports the amino acid cystine, a critical source in the glutathione mechanism. The predisposition of ARID1A-deficient cancer cells may result from the reduction of GSH caused by the impairment of SLC7A11 expression making those cancer cells reliant on GCLC. In our study we showed that in ARID1A-mutated ovarian and gastric tumors, with no ARID1A protein expression, knockdown of GCLC reduced cell growth [13,34]. However, the mechanism by which GSH/GCLC inhibition induces cell death in the absence of ARID1A still remains to be elucidated.
In sum, we propose that targeting GSH metabolism may have therapeutic potential in AML as well and ARID1A-deficient solid tumors. The exact mechanism by which AML cells have a heightened dependency on GSH metabolism remains to be fully elucidated. Additionally, we extended previous findings to show the potential of targeting GCLC in ARID1A-deficient ovarian and gastric cancer cell lines. Given the systemic importance of GSH metabolism, our studies suggest that targeting GCLC may provide an approach to preferentially target cancer cells while sparing normal cells from the deleterious effects of excessive ROS accumulation and subsequent DNA damage.
Acknowledgements
We would like to thank Michael Flister and Joshua Plotnik of AbbVie for the valuable scientific discussions and to thank Neema Parikh, Leanne Goon, and Mandefro Tensae of AbbVie for research material acquisition. Special thanks to Jesse Anguiano of AbbVie for helping in the publication process.
Disclosure of conflict of interest
None.
References
- 1.Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 2.Winer ES, Stone RM. Novel therapy in acute myeloid leukemia (AML): moving toward targeted approaches. Ther Adv Hematol. 2019;10:2040620719860645. doi: 10.1177/2040620719860645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Er TK, Tsai SM, Wu SH, Chiang W, Lin HC, Lin SF, Wu SH, Tsai LY, Liu TZ. Antioxidant status and superoxide anion radical generation in acute myeloid leukemia. Clin Biochem. 2007;40:1015–1019. doi: 10.1016/j.clinbiochem.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Zhou FL, Zhang WG, Wei YC, Meng S, Bai GG, Wang BY, Yang HY, Tian W, Meng X, Zhang H, Chen SP. Involvement of oxidative stress in the relapse of acute myeloid leukemia. J Biol Chem. 2010;285:15010–15015. doi: 10.1074/jbc.M110.103713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meister A. Selective modification of glutathione metabolism. Science. 1983;220:472–447. doi: 10.1126/science.6836290. [DOI] [PubMed] [Google Scholar]
- 6.Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217:2291–2298. doi: 10.1083/jcb.201804161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, Elia A, Berger T, Cescon DW, Adeoye A, Brustle A, Molyneux SD, Mason JM, Li WY, Yamamoto K, Wakeham A, Berman HK, Khokha R, Done SJ, Kavanagh TJ, Lam CW, Mak TW. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Li M, Sun C, Francisco L, Chakraborty S, Sabado M, McDonald T, Gyorffy J, Chang K, Wang S, Fan W, Li J, Zhao LP, Radich J, Forman S, Bhatia S, Bhatia R. Altered hematopoietic cell gene expression precedes development of therapy-related myelodysplasia/acute myeloid leukemia and identifies patients at risk. Cancer Cell. 2011;20:591–605. doi: 10.1016/j.ccr.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, O’Dwyer KM, Li Z, Shi L, Greninger P, Settleman J, Benes C, Hagen FK, Munger J, Crooks PA, Becker MW, Jordan CT. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem. 2013;288:33542–33558. doi: 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic ras. Cell. 2017;168:890–903. e815. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, Ansari R, Harper S, Jackson DA, McRae R, Pooley R, Wilkinson P, van der Meer D, Dow D, Buser-Doepner C, Bertotti A, Trusolino L, Stronach EA, Saez-Rodriguez J, Yusa K, Garnett MJ. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568:511–516. doi: 10.1038/s41586-019-1103-9. [DOI] [PubMed] [Google Scholar]
- 12.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, Goodale A, Lee Y, Ali LD, Jiang G, Lubonja R, Harrington WF, Strickland M, Wu T, Hawes DC, Zhivich VA, Wyatt MR, Kalani Z, Chang JJ, Okamoto M, Stegmaier K, Golub TR, Boehm JS, Vazquez F, Root DE, Hahn WC, Tsherniak A. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779–1784. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogiwara H, Takahashi K, Sasaki M, Kuroda T, Yoshida H, Watanabe R, Maruyama A, Makinoshima H, Chiwaki F, Sasaki H, Kato T, Okamoto A, Kohno T. Targeting the vulnerability of glutathione metabolism in ARID1A-deficient cancers. Cancer Cell. 2019;35:177–190. e178. doi: 10.1016/j.ccell.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Lin CH, Wang Z, Duque-Afonso J, Wong SH, Demeter J, Loktev AV, Somervaille TCP, Jackson PK, Cleary ML. Oligomeric self-association contributes to E2A-PBX1-mediated oncogenesis. Sci Rep. 2019;9:4915. doi: 10.1038/s41598-019-41393-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong SH, Goode DL, Iwasaki M, Wei MC, Kuo HP, Zhu L, Schneidawind D, Duque-Afonso J, Weng Z, Cleary ML. The H3K4-methyl epigenome regulates leukemia stem cell oncogenic potential. Cancer Cell. 2015;28:198–209. doi: 10.1016/j.ccell.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15:334–346. doi: 10.1038/nrc3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heath EM, Chan SM, Minden MD, Murphy T, Shlush LI, Schimmer AD. Biological and clinical consequences of NPM1 mutations in AML. Leukemia. 2017;31:798–807. doi: 10.1038/leu.2017.30. [DOI] [PubMed] [Google Scholar]
- 19.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129:1627–1635. doi: 10.1182/blood-2016-10-696039. [DOI] [PubMed] [Google Scholar]
- 21.Luchini C, Veronese N, Solmi M, Cho H, Kim JH, Chou A, Gill AJ, Faraj SF, Chaux A, Netto GJ, Nakayama K, Kyo S, Lee SY, Kim DW, Yousef GM, Scorilas A, Nelson GS, Köbel M, Kalloger SE, Schaeffer DF, Yan HB, Liu F, Yokoyama Y, Zhang X, Pang D, Lichner Z, Sergi G, Manzato E, Capelli P, Wood LD, Scarpa A, Correll CU. Prognostic role and implications of mutation status of tumor suppressor gene ARID1A in cancer: a systematic review and meta-analysis. Oncotarget. 2015;6:39088–39097. doi: 10.18632/oncotarget.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregory MA, Nemkov T, Park HJ, Zaberezhnyy V, Gehrke S, Adane B, Jordan CT, Hansen KC, D’Alessandro A, DeGregori J. Targeting glutamine metabolism and redox state for leukemia therapy. Clin Cancer Res. 2019;25:4079–4090. doi: 10.1158/1078-0432.CCR-18-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamilos DL, Zelarney P, Mascali JJ. Lymphocyte proliferation in glutathione-depleted lymphocytes: direct relationship between glutathione availability and the proliferative response. Immunopharmacology. 1989;18:223–235. doi: 10.1016/0162-3109(89)90020-9. [DOI] [PubMed] [Google Scholar]
- 24.Messina JP, Lawrence DA. Cell cycle progression of glutathione-depleted human peripheral blood mononuclear cells is inhibited at S phase. J Immunol. 1989;143:1974–1981. [PubMed] [Google Scholar]
- 25.Huang ZZ, Chen C, Zeng Z, Yang H, Oh J, Chen L, Lu SC. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2001;15:19–21. doi: 10.1096/fj.00-0445fje. [DOI] [PubMed] [Google Scholar]
- 26.Circu ML, Aw TY. Glutathione and apoptosis. Free Radic Res. 2008;42:689–706. doi: 10.1080/10715760802317663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varghese J, Khandre NS, Sarin A. Caspase-3 activation is an early event and initiates apoptotic damage in a human leukemia cell line. Apoptosis. 2003;8:363–370. doi: 10.1023/a:1024121017841. [DOI] [PubMed] [Google Scholar]
- 28.Liu M, Zhao Y, Zhang X. Knockdown of glutamate cysteine ligase catalytic subunit by siRNA causes the gold nanoparticles-induced cytotoxicity in lung cancer cells. PLoS One. 2015;10:e0118870. doi: 10.1371/journal.pone.0118870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramani K, Tomasi ML, Yang H, Ko K, Lu SC. Mechanism and significance of changes in glutamate-cysteine ligase expression during hepatic fibrogenesis. J Biol Chem. 2012;287:36341–36355. doi: 10.1074/jbc.M112.370775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, Fukata M, Miyamoto T, Lyons B, Ohshima K, Uchida N, Taniguchi S, Ohara O, Akashi K, Harada M, Shultz LD. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 32.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sasaki M, Chiwaki F, Kuroda T, Komatsu M, Matsusaki K, Kohno T, Sasaki H, Ogiwara H. Efficacy of glutathione inhibitors for the treatment of ARID1A-deficient diffuse-type gastric cancers. Biochem Biophys Res Commun. 2020;522:342–347. doi: 10.1016/j.bbrc.2019.11.078. [DOI] [PubMed] [Google Scholar]