

Abstract
Activating transcription factor 4 (ATF4) is a crucial mediator of the integrated stress response and a negative regulator of RET tyrosine kinase receptor in medullary thyroid carcinoma (MTC). However, the impact of genomic abnormalities in the ATF4 locus on MTC pathogenesis and response to tyrosine kinase inhibitor therapy remains unknown. Here, we evaluated ATF4 copy number variation and protein levels, with overall survival and response to TKIs in a clinical cohort of fifty-nine sporadic primary MTC. We assessed the somatic RETM918T mutation by sequencing, ATF4 copy number by a real-time polymerase chain reaction, and ATF4 protein levels using immunohistochemistry. This MTC cohort comprised 45 (76%) stage IV patients with a median follow-up of 100 months (interquartile range: 58-134 months). Somatic RETM918T was present in 23/57 (40%) tumors. Mono-allelic (36%; 21/59) and bi-allelic (5%; 3/59) loss of ATF4 was identified and was associated with low ATF4 protein expression (0-20%). Kaplan-Meier curves highlight low ATF4 protein or ATF4 loss alone had a significant negative impact on median survival compared to high protein expression (P<0.001) or diploid ATF4 (P=0.011), respectively. The combination of somatic RETM918T and low ATF4 protein levels further decreased overall survival. Both allelic loss and protein reduction were associated with worse overall survival (HR=3.79, 4.06 +RETM918T, and HR=10.64, 11.66 +RETM918T, respectively). Additionally, all 4 of the 11 patients treated with TKIs with a progressive disease by RECIST had low tumor ATF4 protein, with the two partial responder’s tumors having high ATF4 protein. These findings suggest that ATF4 may predict response to tyrosine kinase inhibitors, serve as a prognostic marker for personalized care, and a therapeutic target in MTC.
Keywords: RET, ATF4, loss of heterozygosity, tyrosine kinase inhibitors, medullary thyroid carcinoma
Introduction
MTC originates from the calcitonin secreting parafollicular cells (C-cells) in the thyroid gland accounting for 3-7% of all thyroid cancer [1]. Despite the low frequency, MTC causes a disproportionate number of thyroid cancer deaths. Somatic RET activating mutations are observed in 30-45% of sporadic MTC [2]. Overall 10-year survival for MTC patients with stage IV is 21% [3,4]. Fifty-75% of MTC patients have metastatic disease involving local lymph nodes and 10-15% with the lung, liver, bone, and brain at the time of diagnosis [5,6]. Oncogenic RET proteins activate a complex network of signal transduction pathways, including the Raf/MEK/ERK cascade and phosphatidylinositol-3 kinase (PI3K/AKT) pathway [7,8]. The M918T mutation modifies the kinase domain structure, thereby switching on the enzymatic function and altering RET’s substrate specificity [9].
Our previous report demonstrated that activated RET interacts with and phosphorylates ATF4, leading to ATF4 degradation and inhibition of ATF4 transcriptional activity [10]. Ectopic expression of ATF4 in MTC cells decreased cell survival, inhibited ERK activation, AKT, and mTOR signaling pathways, and causes ubiquitin-mediated degradation of RET [11]. Moreover, ATF4 knockdown decreased sensitivity to tyrosine kinase inhibitor (TKI)-induced apoptosis [11]. Atf4 heterozygote and knockout mice develop C-cell hyperplasia, a precursor lesion linked to hereditary MTC development [11]. The RET expression was increased in parafollicular C cells of the Atf4 heterozygote thyroid gland compared to wild-type mice that may mediate the proliferation of parafollicular C cells and develop C-cell hyperplasia [11].
ATF4 is a key mediator of the integrated stress response and apoptosis inducer under excessive oxidative stress [12]. We have shown that TKIs activates ATF4 and promote apoptotic cell death by induction of proapoptotic genes. The combination of eeyarestation, an inducer of ATF4 with TKIs, led to a synergistic increase of oxidative stress and cell death [13]. Small molecule ONC201 increases the mRNA expression of ATF4 and inhibits tumor growth in mice [14].
Karyotyping and chromosomal-based comparative genomic hybridization (CGH) studies demonstrated various chromosomal alterations in MTC [15-22]. These studies revealed allelic loss to be more prevalent than allelic gains, and allelic loss was most frequent in four loci-7q36.1, 12p13.31, 13q12.11, 19p13.3, and large chromosomal losses involved 22q and 1p [23]. Chromosome 22 is a common region of loss of heterozygosity in many cancers and is the locus of ATF4 [24-30]. However, the impact of genomic abnormalities in the ATF4 locus on pathogenesis and response to therapy in MTC remains unknown. This study aims to investigate the copy number variation of ATF4 and whether the loss of function of ATF4 is associated with poor overall survival and predicts response to TKI-induced cell death.
Materials and methods
MTC patients and clinical data
We performed a retrospective study of 59 patients with MTC who underwent surgery between 1986 to 2015 at The University of Texas MD Anderson Cancer Center with approval from the Institutional Review Board. A pathologist confirmed all diagnoses at the institution. The primary tumor’s pathology variables included tumor size (greatest dimension) and extrathyroidal extension, including macroscopic involvement based on the surgeon’s operative report. TNM staging was based on the American Joint Committee on Cancer 7th edition staging criteria [31]. Nodal status was assigned as NX if no lymph nodes were removed at surgery. RET germline mutation (exon 16) status was determined by sequencing. Disease status was censored at the last evaluation or time of death. Overall survival was measured from the date of diagnosis, defined as the date of initial surgery, until death from any cause. Of these 59 patients, the 11 patients who were treated with a TKI or a combination of different TKIs (e.g., sunitinib, vandetanib, cabozantinib) were analyzed for the response to treatment. Response Evaluation Criteria In Solid Tumors (RECIST) criteria were used to determine radiographic tumor response rate. Progressive disease (PD) was defined as the following: a >20% increase in the sum of diameters of target lesions and an absolute increase of at least 5 mm relative to the smallest such sum measured; the appearance of 1 or more new lesions; and/or unequivocal progression of non-target lesions. Stable disease (SD) was defined as neither sufficient shrinkage to qualify as a partial response nor sufficient increase to qualify as progression disease, relative to the smallest sum of diameters measured. Partial response (PR) was defined as at least a 30% decrease in the sum of the longest diameters of target lesions, compared with the baseline sum of diameters.
Patients who were treated with TKIs also underwent biochemical analysis for tumor biomarkers calcitonin at the time of diagnosis, during treatment, and through the follow-up period. A >50% decrease from baseline (pre-treatment) calcitonin levels was defined as a partial response (PR), and a 20%-50% change from baseline was defined as stable disease (SD).
Copy number analysis
Genomic DNA was extracted from paraffin-embedded tissue following macrodissection to contain at least 80% tumor using the QIAamp DNA FFPE tissue kit (QIAGEN) according to the manufacturer’s instructions. DNA quality (260:280 ratio) and concentration were assessed by NanoDrop (ND-100 spectrophotometer, Thermo Fisher Scientific) and then diluted to generate a uniform DNA concentration (5 ng/μl per replicate/reaction). We used a TaqMan-based copy number assay specific for ATF4 (NCBI location: Chr 22: 39519709-3952268; assay gene location: exon 2; cytoband: 22q13.1; assay reference genome location: Chr 22: 39521668 on NCBI build GRCh38.2) (assay ID: Hs01046325_cn, Life Technologies). We used the copy number assay human RNAase-P (Life Technologies, #4401631) as a reference. Briefly, each 20-μl reaction mixture was run in a 96-well plate with four replicates per sample, each containing five μl of DNA sample at five ng/ml, ten μl of Taqman master, one microliter of copy number assay for ATF4, and one μl of reference assay RNAase-P. The real-time polymerase chain reaction system’s program was 10 minutes of polymerase activation at 95°C, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. We verified that amplification curves for the reference assay (VIC signal) and the copy number assay (FAM signal) have a distinct, linear amplification phase. The data were imported to CopyCaller Software V2.0 (Life Technologies) for comparative Ct (delta-delta Ct) relative quantification analysis of the real-time data. The comparative CT (delta-delta CT) method first calculates the difference (delta CT) between the threshold cycles of the target and reference assay. Then, the method compares the delta CT values of the test samples to a calibrator sample that contains a known number of copies of the target sequence (human genomic DNA, male and female (Catalog #G1521, G1571, Promega). The analysis parameters for VIC-CT detection threshold were set to 32 cycles, and the calibrator sample copy number was set at two. The calculated and predicted copy number is shown.
Immunohistochemistry
Sequential sections from formalin-fixed, paraffin-embedded tissue blocks were fixed to charged slides. After deparaffinization, slides were treated with citrate buffer solution, and endogenous peroxidases were inactivated in a solution of 3% hydrogen peroxidase (Sigma-Aldrich) for 15 minutes and incubated for 1 hour with blocking buffer (Avidin/Biotin Blocking Kit, Vector Laboratories). Tumor samples were stained with ATF4 antibody (ab23760, Abcam, dilution 1:100) for 1 hour at room temperature, followed by rabbit horseradish peroxidase-conjugated secondary antibody and peroxidase substrate (Vector Laboratories) for detection. ATF expression was evaluated as a percent of tumor cells with positive nuclear expression. An ATF4 score of less than 20% of nuclei positive for ATF4 was considered low ATF4 expression, regardless of intensity. After a review of the slides and observation of a breakpoint in the expression pattern, the threshold was determined. The images were taken with a Leica microscope at 20× and 40× magnification.
Statistical analysis
Categorical variables were summarized by frequencies and percentages and compared between groups using Fisher exact tests; continuous variables were summarized using means, standard deviations, medians, and ranges and compared between groups by 2-sample t-tests or analysis of variance if more than two subgroups were compared. Unadjusted survival distributions were estimated by the Kaplan-Meier method and compared using the log-rank test. Cox proportional hazards regression models were used to evaluate the associations between overall survival and covariates of interest. Multivariable Cox regression models were fitted for covariate of interests (ATF4 protein, copy number, and combination with RET mutation), adjusting age and N stage (with a p-value less than 0.1 in univariable analysis). The proportional hazards (PH) assumptions were checked with Schoenfeld Residuals, and no PH violation was observed. Firth’s penalized Cox regression models were fitted when there was no observed death event for a covariate-defined subgroup. All statistical analyses were performed using R version 3.6.3, including R packages of survival (URL: https://CRAN.R-project.org/package=survival) and coxphf (https://CRAN.R-project.org/package=coxphf). All statistical tests used a significance level of 5%.
Results
Patient characteristics
The median age at diagnosis was 52 years (range 23-73 years). Twenty-six (44%) were male, and 33 (56%) were female. Forty-five (76%) patients were stage IV. Twenty-four (41%) patients presented with distant metastasis at diagnosis. The median follow-up time was 100 months (interquartile range 58-134 months). All patients were clinically verified as non-hereditary through genetic testing. Twenty-three (40%) of the sporadic MTCs in this cohort showed a somatic RETM918T mutation (Table 1).
Table 1.
Summary of patient and tumor characteristics overall and by ATF4 copy number
| Variable | N | Overall | ATF4 gene copy number | p-value | ||
|---|---|---|---|---|---|---|
|
| ||||||
| 0 copy (0N) (n=3) | 1 copy (1N) (n=21) | 2 copies (2N) (n=35) | ||||
| Age, median (range), years | 59 | 52 (23-73) | 38 (34-40) | 54 (28-73) | 52 (23-72) | 0.199 |
| Sex, N (%) | 59 | 0.414 | ||||
| Female | 33 (56) | 1 (33) | 10 (48) | 22 (63) | ||
| Male | 26 (44) | 2 (67) | 11 (52) | 13 (37) | ||
| RET M918T mutation, N (%) | 57 | 0.811 | ||||
| Positive | 23 (40) | 1 (33) | 10 (48) | 12 (36) | ||
| Negative | 34 (60) | 2 (67) | 11 (52) | 21 (64) | ||
| pT category, N (%) | 57 | 0.599 | ||||
| T1 | 13 (23) | 0 (0) | 3 (15) | 10 (29) | ||
| T2 | 12 (21) | 0 (0) | 4 (20) | 8 (24) | ||
| T3 | 16 (28) | 1 (33) | 6 (30) | 9 (26) | ||
| T4a | 16 (28) | 2 (67) | 7 (35) | 7 (21) | ||
| pN category, N (%) | 57 | 0.853 | ||||
| N0 | 13 (23) | 0 (0) | 4 (19) | 9 (27) | ||
| N1/N1a | 9 (16) | 0 (0) | 4 (19) | 5 (15) | ||
| N1b | 35 (61) | 3 (100) | 13 (62) | 19 (58) | ||
| M category, N (%) | 59 | 0.742 | ||||
| M0 | 35 (59) | 1 (33) | 13 (62) | 21 (60) | ||
| M1 | 24 (41) | 2 (67) | 8 (38) | 14 (40) | ||
| Extrathyroidal invasion, N (%) | 45 | 0.258 | ||||
| No | 27 (60) | 2 (67) | 6 (43) | 19 (68) | ||
| Yes | 18 (40) | 1 (33) | 8 (57) | 9 (32) | ||
| Distant metastases, N (%) | 52 | 0.466 | ||||
| No | 28 (54) | 1 (33) | 8 (44) | 19 (61) | ||
| Yes | 24 (46) | 2 (67) | 10 (56) | 12 (39) | ||
| Stage, N (%)^ | 59 | 0.571 | ||||
| I | 5 (8) | 1 (33) | 0 (0) | 4 (11) | ||
| II | 4 (7) | 0 (0) | 1 (5) | 3 (9) | ||
| III | 5 (8) | 0 (0) | 2 (10) | 3 (9) | ||
| IVa | 16 (27) | 0 (0) | 6 (29) | 10 (29) | ||
| IVc | 29 (49) | 2 (67) | 12 (57) | 15 (43) | ||
N = total number per variable;
AJCC 7th edition.
Allelic loss of ATF4 in medullary thyroid carcinoma
Previously, using array-based CGH in MTC patients, we found losses at 22q13.1, to which ATF4 maps (Start: 38,246,515; Stop: 38,248,637) in 40% of sporadic MTCs and accompanied by RETM918T mutations [23]. We examined 59 MTC cases for ATF4 copy number variation to validate the CGH data, using real-time quantitative polymerase chain reaction (Figure 1A). Of the 59 patients, 3 (5%) showed bi-allelic loss of ATF4 (0N), and 21 (35%) showed mono-allelic loss of ATF4 (1N) with the remaining 35 tumors consistent with diploid genotype (2N) (Figure 1; Table 1). There were no cases with ambiguous results or with amplification. Clinicopathologic factors, including age at diagnosis, sex, TNM stage, and somatic RETM918T mutation status, did not significantly differ between cases with or without ATF4 allelic loss (Table 1).
Figure 1.

Somatic ATF4 copy number loss associated with decreased protein levels in primary medullary thyroid carcinomas (MTC). A. Copy number variation analysis of MTC tumors using real-time polymerase chain reaction and CopyCaller Software V2.0 (n=59). The top plot displays the calculated copy number of each sample, and bars indicate the minimum and maximum copy number (CN) calculated for the sample replicate group. The bottom plot displays the predicted copy number. B. Immunohistochemical analysis of ATF4, Hematoxylin, and eosin staining in primary MTCs. The representative of ATF4 staining of primary tumors with 0 copy, one copy loss, two copies, a case with 2.5 ATF4 copies, and corresponding H&E staining. A negative control (spleen tissue) and positive control (normal thyroid) for ATF4 are shown.
ATF4 allelic loss is associated with low ATF4 protein levels and poor overall survival of MTC patients
To determine whether somatic ATF4 gene copy loss at the chromosomal level is associated with decreased ATF4 protein levels, we analyzed ATF4 protein expression in the tumors using immunohistochemical analysis. We observed that 33 (56%) of the analyzed MTCs (n=59) had low ATF4 protein expression, defined as less than 20% nuclear staining (Figure 1B; Table 2). Among cases with low ATF4 protein levels, 9 of the 33 patients (27%) had retained diploid ATF4 (2 copies). To investigate the functional role of ATF4 through allelic loss and/or decreased ATF4 protein expression in MTC, we correlated these factors with patients’ overall survival. We found that MTC patients whose tumor samples had somatic ATF4 loss (1 or 2 copy loss) had markedly worse overall survival than patients whose tumors had normal copy numbers of ATF4 (HR=3.26 [95% CI: 1.25-8.54]; P=0.016) in univariable Cox analysis (Figure 2A; Table 3). In the multivariable Cox analysis adjusting age and N stage, patients with somatic ATF4 loss also showed worse OS (HR=2.24, 95% CI: 0.91-6.15), although results were not statistically significant (P=0.08). The median overall survival time was 111.3 months (95% CI: 84.9 months-not reached) in patients whose tumors with chromosomal loss of ATF4 (1N or 0N) but was not reached in patients whose tumors had two copies of ATF4 (P=0.011, Figure 2A). Similarly, patients whose tumors had low or no ATF4 protein expression had markedly worse overall survival than patients whose tumors had high ATF4 expression (HR=6.21 [95% CI: 1.81-21.28]; P=0.004) in univariable Cox analysis (Figure 2B; Table 3). These results were confirmed by the multivariable Cox analysis adjusting age and N stage (HR=4.03 [95% CI: 1.42-15.45]; P=0.007). The median overall survival time for low ATF4 protein was 111.3 months (95% CI: 84.9 months-not reached), and the median overall survival was not reached for high ATF4 (P<0.001, Figure 2B).
Table 2.
Summary of characteristics overall and by ATF4 protein levels
| Variable | N | Overall | High (n=26) | Low (n=33) | p-value |
|---|---|---|---|---|---|
| Age (range), years | 59 | 52 (23-73) | 52 (23-70) | 53 (28-73) | 0.862 |
| Sex, N (%) | 59 | 1.000 | |||
| Female | 33 (56) | 15 (58) | 18 (55) | ||
| Male | 26 (44) | 11 (42) | 15 (45) | ||
| RET M918T mutation, N (%) | 57 | 0.786 | |||
| Positive | 23 (40) | 11 (44) | 12 (38) | ||
| Negative | 34 (60) | 14 (56) | 20 (62) | ||
| pT category, N (%) | 57 | 0.036 | |||
| T1 | 13 (23) | 8 (32) | 5 (16) | ||
| T2 | 12 (21) | 8 (32) | 4 (12) | ||
| T3 | 16 (28) | 6 (24) | 10 (31) | ||
| T4a | 16 (28) | 3 (12) | 13 (41) | ||
| pN category, N (%) | 57 | 0.644 | |||
| N0 | 13 (23) | 7 (29) | 6 (18) | ||
| N1/N1a | 9 (16) | 3 (12) | 6 (18) | ||
| N1b | 35 (61) | 14 (58) | 21 (64) | ||
| M category, N (%) | 59 | 0.795 | |||
| M0 | 35 (59) | 16 (62) | 19 (58) | ||
| M1 | 24 (41) | 10 (38) | 14 (42) | ||
| Extrathyroidal invasion, N (%) | 45 | 0.371 | |||
| No | 27 (60) | 13 (68) | 14 (54) | ||
| Yes | 18 (40) | 6 (32) | 12 (46) | ||
| Distant metastases, N (%) | 52 | 0.269 | |||
| No | 28 (54) | 14 (64) | 14 (47) | ||
| Yes | 24 (46) | 8 (36) | 16 (53) | ||
| Stage, N (%)^ | 59 | 0.504 | |||
| I | 5 (8) | 3 (12) | 2 (6) | ||
| II | 4 (7) | 3 (12) | 1 (3) | ||
| III | 5 (8) | 2 (8) | 3 (9) | ||
| IVa | 16 (27) | 8 (31) | 8 (24) | ||
| IVc | 29 (49) | 10 (38) | 19 (58) |
N = total number per variable;
AJCC 7th edition.
Figure 2.
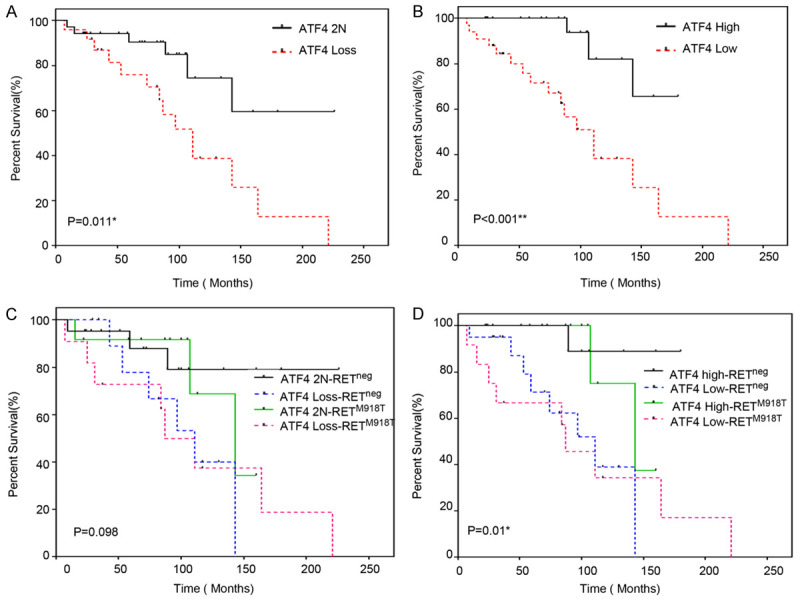
Overall survival (OS) of MTC patients with ATF4 loss. A. Overall survival based on somatic ATF4 copy number normal (2N) versus loss (0 or 1 copy). Log-rank test, *P=0.011. The median OS for patients with ATF4 copy number loss was 111.3 months (95% CI: 84.9 months to not reached), 2N: 2 copies of ATF4). B. Overall survival based on ATF4 protein levels (High ≥20% nuclear expression; Low, <20% nuclear expression) in primary tumors by immunohistochemical analysis. Log-rank test, **P<0.001. The median OS time for low ATF4 protein was 111.3 months (95% CI: 84.9 months to not reached). C. Overall survival of MTC patients according to somatic ATF4 copy number in combination with somatic RETM918T mutation status (RET neg negative for RETM918T mutation vs. RETM918T present; ATF4 loss (only one or no copies of ATF4). The median OS time for ATF42N/RETneg was not reached; the median OS for ATF4 loss/RETM918T was 87.7 (95% CI: 84.9-Not reached); the median OS for ATF4 loss /RETneg, was 111.3 (95% CI: 74.6-Not reached); the median OS time for ATF4 2N/RETM918T was 143.7 (95% CI: 107.5-Not reached)). Log-rank test, P=0.098. D. Overall survival based on ATF4 protein levels and somatic RETM918T mutation status. ATF4-High/RETneg, median OS time was not reached; the median OS time for ATF4-High/RETM918T was 143.7 (95% CI: 107.5-Not reached); the median OS time for low/RETM918T was 87.7 (95% CI: 31.4-Not reached) and the median OS time for low/RETneg was 113.3 (95% CI: 74.6-Not reached); Log-rank test, *P=0.01.
Table 3.
Univariable and multivariable Cox analysis of overall survival by clinical and tumor characteristics
| Prognostic Factor | N | Events | HR | 95% CI | p-value |
|---|---|---|---|---|---|
| Age | |||||
| ≤52 (ref) | 30 | 10 | - | - | - |
| >52 | 29 | 10 | 1.39 | (0.56, 3.47) | 0.478 |
| Sex | |||||
| Female (ref) | 33 | 9 | - | - | - |
| Male | 26 | 11 | 0.93 | (0.35, 2.44) | 0.883 |
| ATF4 protein | |||||
| High (ref) | 26 | 3 | - | - | - |
| Low | 33 | 17 | 6.21 | (1.81, 21.28) | 0.004 |
| ATF4 copy number | |||||
| 2N (ref) | 35 | 6 | - | - | - |
| Loss, 0N/1N | 24 | 14 | 3.26 | (1.25, 8.54) | 0.016 |
| RET M918T mutation | |||||
| Negative (ref) | 34 | 9 | - | - | - |
| Positive | 23 | 11 | 1.49 | (0.62, 3.61) | 0.376 |
| pT category | |||||
| T1 (ref) | 13 | 2 | - | - | - |
| T2 | 12 | 2 | 0.57 | (0.08, 4.14) | 0.581 |
| T3 | 16 | 5 | 1.51 | (0.29, 7.92) | 0.626 |
| T4a | 16 | 10 | 2.54 | (0.53, 12.08) | 0.242 |
| pN category | |||||
| N0 (ref) | 13 | 0 | - | - | - |
| N1 | 44 | 20 | 11.52 | (1.57, 1467.52) | 0.009 |
| M category | |||||
| M0 (ref) | 35 | 8 | - | - | - |
| M1 | 24 | 12 | 1.50 | (0.59, 3.84) | 0.395 |
| Extrathyroidal invasion | |||||
| No (ref) | 27 | 6 | - | - | - |
| Yes | 18 | 6 | 1.26 | (0.40, 3.96) | 0.692 |
| Stage^ | |||||
| I (ref) | 5 | 2 | - | - | - |
| II | 4 | 0 | 0.12 | (0.00, 1.49) | 0.103 |
| III | 5 | 1 | 0.57 | (0.05, 4.31) | 0.578 |
| IVa | 16 | 4 | 0.24 | (0.05, 1.51) | 0.120 |
| IVc | 29 | 13 | 0.56 | (0.16, 2.91) | 0.437 |
| ATF4 protein/RET mutation status* | |||||
| High/negative (ref) | 14 | 1 | - | - | - |
| High/M918T | 11 | 2 | 2.97 | (0.26, 33.17) | 0.378 |
| Low/negative | 20 | 8 | 10.64 | (1.30, 87.10) | 0.027 |
| Low/M918T | 12 | 9 | 11.66 | (1.47, 92.29) | 0.020 |
| ATF4 copy number/RET mutation status* | |||||
| 2N/negative (ref) | 21 | 3 | - | - | - |
| 2N/M918T | 12 | 3 | 1.68 | (0.33, 8.47) | 0.528 |
| 0N or 1N/negative | 13 | 6 | 3.79 | (0.93, 15.45) | 0.063 |
| 0N or 1N/M918T | 11 | 8 | 4.06 | (1.07, 15.41) | 0.040 |
0N: homozygous deletion, 1N: one copy, 2N: diploid, Low: <20% expression, High: >20% expression, HR: hazard ratio, M918T: RETM918T present, negative: absent RETM918T mutation, Ref: reference;
AJCC 7th edition;
RET mutation status specifically for M918T.
RETM918T mutation and ATF4 loss
Thirty-three of the 59 (56%) MTCs had a loss of ATF4 protein, of which twelve had a concurrent RETM918T mutation (Table 2). The combination of RETM918T mutation and ATF4 copy loss (HR=4.06 [95% CI 1.07-15.41]; P=0.040) or ATF4 protein loss (HR=11.66 [CI 1.47-92.29]; P=0.02) were observed to be a significant predictor of the outcome in univariable Cox analysis (Table 3). The multivariable Cox analysis, adjusting age and N stage, also showed significance when low ATF4 protein levels were combined with RETM918T mutation (HR=5.80 [CI 1.29-54.93]; P=0.019). Notably, in univariable analysis, RETM918T mutation alone was not associated with worse survival in this cohort (HR=1.49 [CI 0.62-3.61]; P=0.376) (Table 3). Grouping patients by the ATF copy number and RETM918T status, the median overall survival was determined (Table 3). The median overall survival time of MTC patients with two ATF4 copies or high ATF4 expression but no RETM918T was not reached but was 143.7 months [95% CI: 107.5-Not reached] in those with two ATF4 copies and RETM918T mutation, and was 111.3 months [95% CI: 74.6-Not reached] in those with low ATF4 protein expression and no RETM918T (Figure 2C, 2D). The median overall survival of patients with both ATF4 allelic loss (1N or 0N) and a somatic RETM918T mutation was 87.7 months [95% CI: 84.9-NA] (P=0.098) (Figure 2C). The median overall survival of patients with both ATF4 low protein expression and a somatic RETM918T mutation was 87.7 months [95% CI: 31.4-Not reached] (P=0.01) (Figure 2D).
ATF4 loss and response to TKIs
To determine whether the degree of ATF4 protein loss contributes to adverse outcomes after TKI treatment in MTC patients, we analyzed the response to TKIs according to RECIST criteria and tumor biomarker levels. In this cohort, 11 patients were treated with TKIs, including cabozantinib (n=4), vandetanib (n=4), sunitinib (n=1), cabozantinib combined with sorafenib (n=1), and sunitinib combined with pazopanib (n=1). Four patients showed high ATF4 protein levels in their tumors, and 7 showed low tumor ATF4 levels. Based on serum calcitonin levels, 5 of the 11 patients had a PR, 2 had SD, and 4 had PD (Table 4). According to the RECIST criteria of the tumor by imaging, two patients had a PR, 5 had SD, and 4 had PD (Table 4). Three of the five patients who showed a PR by calcitonin levels were considered to have SD according to RECIST criteria (Table 4). Of the five patients with PR according to calcitonin levels, four tumors expressed high ATF4 by immunohistochemistry analysis, and one tumor showed low ATF4 expression. All four patients with PD by rising calcitonin levels had tumors with low ATF4 protein levels (P=0.018; Table 4). Of the two patients with PR according to RECIST criteria, both had high tumor ATF4 protein levels, and among the five patients with SD according to RECIST, two had high tumor ATF4 protein, and three had low tumor ATF4 protein levels by immunohistochemical analysis (Table 4). All four patients with PD, according to RECIST, had low ATF4 levels (P=0.05) (Table 4). Somatic RET mutation status was not significantly associated with response to TKIs (Table 4). These results suggest that low ATF4 protein levels may predict resistance to TKIs, and RETM918T mutation status is non-contributory.
Table 4.
ATF4 expression may predict response to tyrosine kinase inhibitor (TKI) therapy. ATF4 protein level and response rate to TKI according to serum calcitonin levels and RECIST criteria are shown
| Variable | Total (n=11) | Response according to calcitonin levels | Response according to RECIST criteria | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| PR (n=5) | SD (n=2) | PD (n=4) | p-value | PR (n=2) | SD (n=5) | PD (n=4) | p-value | ||
| ATF4 protein, N (%) | 0.018 | 0.05 | |||||||
| High | 4 (37) | 4 (80) | 0 | 0 | 2 (100) | 2 (40) | 0 (0) | ||
| Low | 7 (63) | 1 (20) | 2 (100) | 4 (100) | 0 (0) | 3 (60) | 4 (100) | ||
| RET mutation, N (%) | |||||||||
| M918T negative | 9 (82) | 5 (100) | 2 (100) | 2 (50) | 0.127 | 2 (100) | 5 (100) | 2 (50) | 0.117 |
| M918T positive | 2 (18) | 0 | 0 | 2 (50) | 0 (0) | 0 (0) | 2 (50) | ||
N: total number per variable, PR: partial response, SD: stable disease, PD: progressive disease.
Discussion
In this study, we demonstrated genomic loss of ATF4 occurs in a large fraction of primary MTC tumors (24/59, 41%) and that ATF4 loss was associated with shorter overall survival of MTC patients. The combination of low ATF4 protein levels with a somatic RETM918T mutation further decreased overall survival. We observed a high degree of overlap between mono-allelic ATF4 loss and loss of ATF4 protein expression; However, a subset of MTC tumors showed loss of ATF4 protein despite the presence of normal copy numbers of ATF4 (9/33, 27%), suggesting an additional mechanism of ATF4 inactivation, such as RET-mediated degradation of ATF4, as previously described [10,11,32].
While RET has been known as a critical driver of tumorigenesis in MTC, altered in ~60% of sporadic tumors, identification of other tumorigenesis drivers is warranted. Frequent loss of heterozygosity on the long arm of chromosome 22 occurs in various cancers, including thyroid, breast, colon, ovary, pancreatic, oral cavity, stomach, liver, and lung cancer [24-30]. Several candidate tumor suppressor genes have been identified in this region, including NF2 (22q13), BIK (22q13.3), and EP300 (22q13) [30,33,34]. An analysis of NF2 allelic loss localized on chromosome 22q13.1 showed a high rate of loss of heterozygosity (44%) [35]. In our study, the incidence of ATF4 allelic loss (40%) is higher than the reported incidence of loss of chromosome 22q13.1 regions that contain ATF4 (range 20-35%) in MTC that could be related to the higher number of advanced cases analyzed in this study [15,16,20-23,36-38]. Our findings demonstrate that genomic and protein loss of ATF4 contributes to the pathogenesis of MTC. The role of ATF4 allelic loss in MTC tumorigenesis is further supported by mouse knockout of ATF4, showing that one copy loss of ATF4 in mice causes C-cell hyperplasia, a precancerous lesion that precedes the development of MTC, through increased expression of RET in parafollicular C cells [11]. Our previous and current clinical study suggests that decreased ATF4 expression contributes to tumorigenesis and is associated with a more aggressive form of MTC, supporting a tumor suppressor role of ATF4.
In recent years, the US Food and Drug Administration approved the multi-kinase inhibitors vandetanib, and cabozantinib, and specific RET inhibitor selpercatinib to treat advanced MTC [39-42]. However, biomarkers of response and resistance mechanisms are needed to optimize treatment decisions and improve clinical outcomes. During this time, studies have shown that ATF4 is a master regulator of the cellular stress response, regulating the expression of a large number of target genes involved in signaling pathways, including cell survival/apoptosis, oxidative stress, autophagy, translation, and inflammation [43-45]. Previous studies from our laboratory indicated that overexpression of ATF4 in MTC cells induces apoptosis and that the shRNA knockdown of ATF4 causes resistance to TKI-induced cell death [11]. Furthermore, the induction of ATF4 in combination with TKIs induced irresolvable oxidative stress, leading to cell death [13]. Our current cohort included a subset of MTC patients treated with TKIs. We retrospectively analyzed responses to treatment according to ATF4 genomic status and at the protein level. Despite the small number of patients, we found that MTCs with low ATF4 protein expression tended to respond poorly to TKIs. While the numbers were too small to fully evaluate the association with the presence or absence of RETM918T, studies on TKI resistance have confirmed that RETM918T alteration is not near the TKI binding domain and this mutation would not be predicted to contribute to TKI resistance [46]. Equally important is the observation that intact ATF4 protein expression correlated with response to TKI therapy by both RECIST and serum calcitonin biomarkers. A stable disease response to TKIs is clinically advantageous in these advanced patients who are not placed on systemic therapy until tumor progression. While correlations are overall survival and TKI response were identified at both the genomic and protein levels for ATF4, protein evaluation identified a higher number of ATF4 altered cases. This higher number of tumors with low ATF4 protein levels is likely due to other mechanisms such as ATF4 protein degradation. This finding is advantageous as the immunohistochemical analysis is a rapid modality available in clinical laboratories and readily applicable to formalin-fixed paraffin-embedded archival clinical material over genomic allelic evaluation. ATF4 protein expression by immunohistochemistry has the potential to be integrated as part of clinical trials to allow for further validation of our findings as a predictive marker for response to TKIs.
Given the small number of patients included in the study, particularly when analyzing the response to TKI therapy, these findings should be considered exploratory in nature but essential to pursue because defining TKI’s resistance pathways allows the development of targeted cancer therapies. Building upon historic genomic observations, the recognition of ATF4 as a critical factor in MTC pathogenesis alongside RET has the promise of further patient prognostication and biomarker potential for personalizing patient care for MTC patients that can be therapeutically targeted.
Acknowledgments
We thank Sarah Bronson from the Research Medical Library for editing the manuscript. This work used the histology and sequencing core facilities, which receive funding from NCI Cancer Center Support Grant P30 CA016672 and was supported by the American Thyroid Association (Thyroid Cancer Survivors).
Disclosure of conflict of interest
None.
References
- 1.Cote GJ, Grubbs EG, Hofmann MC. Thyroid C-cell biology and oncogenic transformation. Recent Results Cancer Res. 2015;204:1–39. doi: 10.1007/978-3-319-22542-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dadu R, Bagheri-Yarmand R, Ringel MD, Grubbs EG, Zafereo M, Cote G, Gagel RF, Robinson BG, Shaw KR, Hu MI. Hereditary endocrine tumours: current state-of-the-art and research opportunities: the state of science in medullary thyroid carcinoma: current challenges and unmet needs. Endocr Relat Cancer. 2020;27:T27–T39. doi: 10.1530/ERC-20-0110. [DOI] [PubMed] [Google Scholar]
- 3.Modigliani E, Cohen R, Campos JM, Conte-Devolx B, Maes B, Boneu A, Schlumberger M, Bigorgne JC, Dumontier P, Leclerc L, Corcuff B, Guilhem I. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: results in 899 patients. The GETC Study Group. Groupe d’etude des tumeurs a calcitonine. Clin Endocrinol (Oxf) 1998;48:265–273. doi: 10.1046/j.1365-2265.1998.00392.x. [DOI] [PubMed] [Google Scholar]
- 4.Cote GJ, Evers C, Hu MI, Grubbs EG, Williams MD, Hai T, Duose DY, Houston MR, Bui JH, Mehrotra M, Waguespack SG, Busaidy NL, Cabanillas ME, Habra MA, Luthra R, Sherman SI. Prognostic significance of circulating RET M918T mutated tumor DNA in patients with advanced medullary thyroid carcinoma. J Clin Endocrinol Metab. 2017;102:3591–3599. doi: 10.1210/jc.2017-01039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu JY, Murphy WA Jr, Milton DR, Jimenez C, Rao SN, Habra MA, Waguespack SG, Dadu R, Gagel RF, Ying AK, Cabanillas ME, Weitzman SP, Busaidy NL, Sellin RV, Grubbs E, Sherman SI, Hu MI. Bone metastases and skeletal-related events in medullary thyroid carcinoma. J Clin Endocrinol Metab. 2016;101:4871–4877. doi: 10.1210/jc.2016-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moley JF. Medullary thyroid carcinoma: management of lymph node metastases. J Natl Compr Canc Netw. 2010;8:549–556. doi: 10.6004/jnccn.2010.0042. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi M. The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev. 2001;12:361–373. doi: 10.1016/s1359-6101(01)00012-0. [DOI] [PubMed] [Google Scholar]
- 8.van Weering DH, Bos JL. Signal transduction by the receptor tyrosine kinase ret. Recent Results Cancer Res. 1998;154:271–281. doi: 10.1007/978-3-642-46870-4_18. [DOI] [PubMed] [Google Scholar]
- 9.Plaza-Menacho I, Barnouin K, Goodman K, Martinez-Torres RJ, Borg A, Murray-Rust J, Mouilleron S, Knowles P, McDonald NQ. Oncogenic RET kinase domain mutations perturb the autophosphorylation trajectory by enhancing substrate presentation in trans. Mol Cell. 2014;53:738–751. doi: 10.1016/j.molcel.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bagheri-Yarmand R, Sinha KM, Gururaj AE, Ahmed Z, Rizvi YQ, Huang SC, Ladbury JE, Bogler O, Williams MD, Cote GJ, Gagel RF. A novel dual kinase function of the RET proto-oncogene negatively regulates activating transcription factor 4-mediated apoptosis. J Biol Chem. 2015;290:11749–11761. doi: 10.1074/jbc.M114.619833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bagheri-Yarmand R, Williams MD, Grubbs EG, Gagel RF. ATF4 targets RET for degradation and is a candidate tumor suppressor gene in medullary thyroid cancer. J Clin Endocrinol Metab. 2017;102:933–941. doi: 10.1210/jc.2016-2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagheri-Yarmand R, Sinha KM, Li L, Lu Y, Cote GJ, Sherman SI, Gagel RF. Combinations of tyrosine kinase inhibitor and ERAD inhibitor promote oxidative stress-induced apoptosis through ATF4 and KLF9 in medullary thyroid cancer. Mol Cancer Res. 2019;17:751–760. doi: 10.1158/1541-7786.MCR-18-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bagheri-Yarmand R, Dadu R, Ye L, Jebaraj YS, Martinez JA, Ma J, Tarapore RS, Allen JE, Sherman SI, Williams MD, Gagel RF. ONC201 shows anti-cancer activity against medullary thyroid cancer via transcriptional inhibition of RET, VEGFR2, and IGFBP2. Mol Cancer Ther. 2021;20:665–675. doi: 10.1158/1535-7163.MCT-20-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khosla S, Patel VM, Hay ID, Schaid DJ, Grant CS, van Heerden JA, Thibodeau SN. Loss of heterozygosity suggests multiple genetic alterations in pheochromocytomas and medullary thyroid carcinomas. J Clin Invest. 1991;87:1691–1699. doi: 10.1172/JCI115186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mulligan LM, Gardner E, Smith BA, Mathew CG, Ponder BA. Genetic events in tumour initiation and progression in multiple endocrine neoplasia type 2. Genes Chromosomes Cancer. 1993;6:166–177. doi: 10.1002/gcc.2870060307. [DOI] [PubMed] [Google Scholar]
- 17.Huang SC, Koch CA, Vortmeyer AO, Pack SD, Lichtenauer UD, Mannan P, Lubensky IA, Chrousos GP, Gagel RF, Pacak K, Zhuang Z. Duplication of the mutant RET allele in trisomy 10 or loss of the wild-type allele in multiple endocrine neoplasia type 2-associated pheochromocytomas. Cancer Res. 2000;60:6223–6226. [PubMed] [Google Scholar]
- 18.Koch CA, Brouwers FM, Vortmeyer AO, Tannapfel A, Libutti SK, Zhuang Z, Pacak K, Neumann HP, Paschke R. Somatic VHL gene alterations in MEN2-associated medullary thyroid carcinoma. BMC Cancer. 2006;6:131. doi: 10.1186/1471-2407-6-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koch CA, Huang SC, Moley JF, Azumi N, Chrousos GP, Gagel RF, Zhuang Z, Pacak K, Vortmeyer AO. Allelic imbalance of the mutant and wild-type RET allele in MEN 2A-associated medullary thyroid carcinoma. Oncogene. 2001;20:7809–7811. doi: 10.1038/sj.onc.1204991. [DOI] [PubMed] [Google Scholar]
- 20.Hemmer S, Wasenius VM, Knuutila S, Franssila K, Joensuu H. DNA copy number changes in thyroid carcinoma. Am J Pathol. 1999;154:1539–1547. doi: 10.1016/S0002-9440(10)65407-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frisk T, Zedenius J, Lundberg J, Wallin G, Kytola S, Larsson C. CGH alterations in medullary thyroid carcinomas in relation to the RET M918T mutation and clinical outcome. Int J Oncol. 2001;18:1219–1225. doi: 10.3892/ijo.18.6.1219. [DOI] [PubMed] [Google Scholar]
- 22.Marsh DJ, Theodosopoulos G, Martin-Schulte K, Richardson AL, Philips J, Roher HD, Delbridge L, Robinson BG. Genome-wide copy number imbalances identified in familial and sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab. 2003;88:1866–1872. doi: 10.1210/jc.2002-021155. [DOI] [PubMed] [Google Scholar]
- 23.Ye L, Santarpia L, Cote GJ, El-Naggar AK, Gagel RF. High resolution array-comparative genomic hybridization profiling reveals deoxyribonucleic acid copy number alterations associated with medullary thyroid carcinoma. J Clin Endocrinol Metab. 2008;93:4367–4372. doi: 10.1210/jc.2008-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castells A, Gusella JF, Ramesh V, Rustgi AK. A region of deletion on chromosome 22q13 is common to human breast and colorectal cancers. Cancer Res. 2000;60:2836–2839. [PubMed] [Google Scholar]
- 25.Poli-Frederico RC, Bergamo NA, Reis PP, Kowalski LP, Zielenska M, Squire JA, Rogatto SR. Chromosome 22q a frequent site of allele loss in head and neck carcinoma. Head Neck. 2000;22:585–590. doi: 10.1002/1097-0347(200009)22:6<585::aid-hed7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 26.Takahashi K, Kudo J, Ishibashi H, Hirata Y, Niho Y. Frequent loss of heterozygosity on chromosome 22 in hepatocellular carcinoma. Hepatology. 1993;17:794–799. [PubMed] [Google Scholar]
- 27.Miyakawa A, Wang XL, Nakanishi H, Imai FL, Shiiba M, Miya T, Imai Y, Tanzawa H. Allelic loss on chromosome 22 in oral cancer: possibility of the existence of a tumor suppressor gene on 22q13. Int J Oncol. 1998;13:705–709. doi: 10.3892/ijo.13.4.705. [DOI] [PubMed] [Google Scholar]
- 28.Lin H, Pizer ES, Morin PJ. A frequent deletion polymorphism on chromosome 22q13 identified by representational difference analysis of ovarian cancer. Genomics. 2000;69:391–394. doi: 10.1006/geno.2000.6357. [DOI] [PubMed] [Google Scholar]
- 29.Nishioka M, Kohno T, Tani M, Yanaihara N, Tomizawa Y, Otsuka A, Sasaki S, Kobayashi K, Niki T, Maeshima A, Sekido Y, Minna JD, Sone S, Yokota J. MYO18B, a candidate tumor suppressor gene at chromosome 22q12.1, deleted, mutated, and methylated in human lung cancer. Proc Natl Acad Sci U S A. 2002;99:12269–12274. doi: 10.1073/pnas.192445899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou CZ, Peng ZH, Zhang F, Qiu GQ, He L. Loss of heterozygosity on long arm of chromosome 22 in sporadic colorectal carcinoma. World J Gastroenterol. 2002;8:668–673. doi: 10.3748/wjg.v8.i4.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17:1471–1474. doi: 10.1245/s10434-010-0985-4. [DOI] [PubMed] [Google Scholar]
- 32.Luo J, Xia Y, Yin Y, Luo J, Liu M, Zhang H, Zhang C, Zhao Y, Yang L, Kong L. ATF4 destabilizes RET through nonclassical GRP78 inhibition to enhance chemosensitivity to bortezomib in human osteosarcoma. Theranostics. 2019;9:6334–6353. doi: 10.7150/thno.36818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrilli AM, Fernandez-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. 2016;35:537–548. doi: 10.1038/onc.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S, Jung K, Loening S, Dorken B, Daniel PT. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 2006;13:619–627. doi: 10.1038/sj.cdd.4401782. [DOI] [PubMed] [Google Scholar]
- 35.Sheikh HA, Tometsko M, Niehouse L, Aldeeb D, Swalsky P, Finkelstein S, Barnes EL, Hunt JL. Molecular genotyping of medullary thyroid carcinoma can predict tumor recurrence. Am J Surg Pathol. 2004;28:101–106. doi: 10.1097/00000478-200401000-00012. [DOI] [PubMed] [Google Scholar]
- 36.Flicker K, Ulz P, Hoger H, Zeitlhofer P, Haas OA, Behmel A, Buchinger W, Scheuba C, Niederle B, Pfragner R, Speicher MR. High-resolution analysis of alterations in medullary thyroid carcinoma genomes. Int J Cancer. 2012;131:E66–73. doi: 10.1002/ijc.26494. [DOI] [PubMed] [Google Scholar]
- 37.Takai S, Tateishi H, Nishisho I, Miki T, Motomura K, Miyauchi A, Kato M, Ikeuchi T, Yamamoto K, Okazaki M, et al. Loss of genes on chromosome 22 in medullary thyroid carcinoma and pheochromocytoma. Jpn J Cancer Res. 1987;78:894–898. [PubMed] [Google Scholar]
- 38.Tanaka N, Nishisho I, Yamamoto M, Miya A, Shin E, Karakawa K, Fujita S, Kobayashi T, Rouleau GA, Mori T, et al. Loss of heterozygosity on the long arm of chromosome 22 in pheochromocytoma. Genes Chromosomes Cancer. 1992;5:399–403. doi: 10.1002/gcc.2870050416. [DOI] [PubMed] [Google Scholar]
- 39.Wells SA Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab B, Vasselli JR, Read J, Langmuir P, Ryan AJ, Schlumberger MJ. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J. Clin. Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dadu R, Hu MN, Grubbs EG, Gagel RF. Use of tyrosine kinase inhibitors for treatment of medullary thyroid carcinoma. Recent Results Cancer Res. 2015;204:227–249. doi: 10.1007/978-3-319-22542-5_11. [DOI] [PubMed] [Google Scholar]
- 41.Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, Wirth LJ, Stock S, Smith S, Lauriault V, Corsi-Travali S, Henry D, Burkard M, Hamor R, Bouhana K, Winski S, Wallace RD, Hartley D, Rhodes S, Reddy M, Brandhuber BJ, Andrews S, Rothenberg SM, Drilon A. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol. 2018;29:1869–1876. doi: 10.1093/annonc/mdy137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, Hu W, Cao Q, Sheets MP, Wilson D, Wilson KJ, DiPietro L, Fleming P, Palmer M, Hu MI, Wirth L, Brose MS, Ou SI, Taylor M, Garralda E, Miller S, Wolf B, Lengauer C, Guzi T, Evans EK. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 2018;8:836–849. doi: 10.1158/2159-8290.CD-18-0338. [DOI] [PubMed] [Google Scholar]
- 43.Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab. 2017;28:794–806. doi: 10.1016/j.tem.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, Auwerx J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol. 2017;216:2027–2045. doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu X, Shen T, Mooers BHM, Hilberg F, Wu J. Drug resistance profiles of mutations in the RET kinase domain. Br J Pharmacol. 2018;175:3504–3515. doi: 10.1111/bph.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]