

Abstract
A majority of breast cancer patients die of widespread aggressive multidrug-resistant tumors. Aspartate β-hydroxylase (ASPH) is an α-ketoglutarate-dependent dioxygenase and oncofetal antigen involved in embryogenesis. To illustrate if ASPH could be targeted for metastatic breast cancer, embedded and on-top three-dimensional (3-D) cultures, 3-D invasion, mammosphere formation, immunofluorescence, immunohistochemistry, Western blot, co-IP and microarray were conducted. In vitro metastasis was developed to imitate how cancer cells invade basement membrane at the primary site, transendothelially migrate, consequently colonize and outgrow at distant sites. Orthotopic and experimental pulmonary metastatic (tail vein injection) murine models were established using stable breast cancer cell lines. Cox proportional hazards regression models and Kaplan-Meier plots were applied to assess clinical outcome of breast cancer patients. In adult non-cancerous breast tissue, ASPH is undetectable. Pathologically, ASPH expression re-emerged at ductal carcinoma in situ (DCIS), and enhanced with disease progression, from early-stage invasive ductal carcinoma (IDC) to late-stage carcinoma. ASPH at moderate to high levels contribute to aggressive molecular subtypes, early relapse or more frequent progression and metastases, whereas substantially shortened overall survival and disease-free survival of breast cancer patients. Through direct physical interactions with A disintegrin and metalloproteinase domain-containing protein (ADAM)-12/ADAM-15, ASPH could activate SRC cascade, thus upregulating downstream components attributed to multifaceted metastasis. ASPH-SRC axis initiated pro-invasive invadopodium formation causing breakdown/disorganization of extracellular matrix (ECM), simultaneously potentiated epithelial-mesenchymal transition (EMT), induced cancer stem cell markers (CD44 and EpCAM), enhanced mammosphere formation and intensified 3-dimentional invasion. Oncogenic SRC upregulated matrix metallopeptidases (MMPs) were assembled by invadopodia, acting as executive effectors for multi-step metastasis. ASPH-SRC signal guided multi-organ metastases (to lungs, liver, bone, spleen, lymph nodes, mesentery or colon) in immunocompromised mice. Malignant phenotypes induced by ASPH-SRC axis were reversed by the third-generation small molecule inhibitor (SMI) specifically against β-hydroxylase activity of ASPH in pre-clinical models of metastatic breast cancer. Collectively, ASPH could activate ADAMs-SRC-MMPs cascades to promote breast cancer tumor progression and metastasis. ASPH could direct invadopodium construction as a biomechanical sensor and pro-metastatic outlet. ASPH-mediated cancer progression could be specifically/efficiently subverted by SMIs of β-hydroxylase activity. Therefore, ASPH emerges as a therapeutic target for breast cancer.
Keywords: Aspartate β-hydroxylase (ASPH), invadopodium, SRC, metastasis, breast cancer, small molecule inhibitor (SMI), extracellular matrix (ECM)
Introduction
As the most common malignancy in women, breast cancer ranks the 2nd leading cause of mortality, with approximately 279,100 new cases and 42,690 deaths in the United States in 2020 [1]. Breast cancer accounts for 30% of all newly diagnosed cancer population [1], and most patients die of metastatic recurrence after treated for early stage tumors. Here, we explored if ASPH, an oncofetal protein, could be a novel therapeutic target for fatal late-stage disease. Substrates of ASPH are aspartyl and asparaginyl residues in epidermal growth factor-like domains within multiple signal transduction molecules [2-7]. ASPH is highly expressed during early embryo development, acting as a critical initiator for cell motility and invasion in placenta [8]. After birth and throughout adulthood, ASPH is undetectable in normal tissues, only upregulated during tumorigenesis, which is essential to initiate, sustain and boost malignant phenotypes [9-11]. High expression of ASPH has been illustrated in malignancies arising from pancreas, liver (hepatocellular carcinoma [HCC]), bile duct (cholangiocarcinoma) [10], colon, rectum and lung [9-13]. Full-length or truncated homolog of ASPH confers poor prognosis of colon cancer [14], non-small cell lung cancer [15] and HCC [11]. Transcription of ASPH is upregulated by IGF1-initiated signal transduction through WNT/β-catenin, PI3K/AKT and RAS/MAPK/ERK pathways [16-19]. Subsequently, ASPH activates oncogenic cascades as serial fundamental processes attributed to aggressively malignant phenotypes in breast cancer [7]. Which signaling pathway is a downstream effector for ASPH?
Invadopodia, membrane protrusions rich in actin, dynamically degrade ECM and locally deposit proteases to intensify invasion. Invadopodia form in response to hypoxia, ECM mechanochemical features, cell contact-mediated signal transduction and soluble growth factors [20]. Invadopodia sense tumor microenvironment’s physical properties, which enables dissemination and metastasis. Invadopodia structures consist of MMPs family members, in particular MMP14, actin-regulators [e.g., Neural Wiskott-Aldrich syndrome protein (N-WASP), cortactin] and adaptors Tks4/Tks5 [21-23]. Integrated with localized MMPs activity, acidic stromal pH, invadopodia regulate tumor cell communication via modifying stroma-mediated premetastatic niche [20-23]. However, is invadopodium architecture as biomechanical sensor involved in ASPH-mediated breast cancer invasion and metastasis? What signaling pathway is responsible for its biopathology? Here, we carry out serial in vitro and in vivo experiments to address these outstanding questions.
Materials and methods
Cell lines
All cell lines were purchased from American Type Culture Collection and authenticated by short tandem repeat profile. Stable cell lines were established as previously described [24].
Plasmids and reagents
pRP-Hygro-CMV-ADAM12 and pRP-Hygro-CMV-ADAM12 were purchased from Vectorbuild. Dasatinib (CDS023389-25MG; SRC family tyrosine kinase inhibitor) and Wiskostatin (W2270-5MG; N-WASP inhibitor) were purchased from Sigma-Aldrich.
Microarray
The cDNA Microarray was performed with OneArray platform by PhalanxBio Inc. (San Diego, CA, USA). For reverse transcription, 2 μg of total RNA was extracted from each cancer cell line by using ABI High-Capacity cDNA reverse transcription Kits. Each sample was tested in triplicate on BIO-RAD CFX Connect real-time PCR machine. BIO-RAD CFX Manager Version 3.0 software was used for experimental setup and data analysis. Target gene qPCR data were normalized to GAPDH. Standard selection criteria to identify differentially expressed genes were as follows: (1) log2 |Fold change| ≥ 1 and P < 0.05; (2) log2 ratios = “NA” and the differences of intensity between the two samples ≥ 1000. Intensity data were pooled and calculated to identify differentially expressed genes based on the threshold of fold change and p-value. The correlation of expression profiles between samples was demonstrated by unsupervised hierarchical clustering analysis.
Western blot
Primary antibodies included: ASPH (homemade); MMP14 (13130), SRC (2109), SRCY416 (6943), SRCY527 (2105), FAK (13009), Tyr397 (8556), Tyr566/567 (3281), Tyr925 (3284) from Cell Signaling Technology (Danvers, MA); MMP1 (sc-58377), SNAIL1 (sc-393172), SNAIL2/SLUG (sc-166476), TWIST1 (sc-81417), ZEB1 (sc-81428), ZEB2 (sc-271984), ADAM12 (sc-25579, sc-293225) and ADAM15 (sc-16530, sc-365752) from Santa Cruz Biotechnology (Dallas, Texas).
Immunoprecipitation
Cells (80% of confluence) were transfected with 10 µg corresponding plasmids [22-24].
Mammosphere formation
As previously described [22-24], size and number of mammospheres were calculated in triplicate wells for each condition and repeated at least twice.
Invadopodia function/ECM degradation [22-24]
As previously described [22-24], activity of invadopodium was quantified according to gelatin degradation with NIH IMAGEJ in triplicate wells for each condition and repeated at least twice. Individual degraded area was normalized to nuclei number in the same field.
3D-embedded and 3D-on top cultures [22-24]
As previously described [22-24], cells growing on GFR Matrigel for 5-8 days were evaluated in triplicate wells for each condition and repeated in triplicate. Different compounds mixed with complete media were applied when plating and changed daily.
3-D (spheroid) invasion [22-24]
As previously described [22-24], in 3-5 days post-seeding (1×104 cells/ml), spheroids of 300-500 µm in diameter were expected. Tumor spheroid formation in response to individual compound treatment was visually confirmed and proceeded with 3-D invasion. Invasion was visualized microscopically and quantitated with NIH IMAGEJ in triplicate wells for each condition and repeated at least twice.
In vitro metastasis [22-24]
As previously described [22-24], after incubation for 3 days in inserted chambers, breast cancer cells having transendothelially migrated HUVECs were imaged and counted with fluorescence microscope. Consequently, breast cancer cells transmigrated into the lower chamber and were buried in Matrigel. After 7 days, mammospheres were detailed for size and number.
Orthotopic model of breast cancer [23]
This protocol was approved by IACUC of Rhode Island Hospital. NSG mice (Jackson Laboratory) (6-8-week-old female) were injected subcutaneously with stable MDA-MB-231 cells (1×106 in 100 µL of 1:1 Matrigel/Collagen I solution) into the right fourth mammary fat pad. Tumor length (L), width (W) and depth (D) was measured every 3-4 days for up to 28 days; and tumor volume (in mm3) was calculated as LWD×π/6.
The criteria for euthanasia included (1) tumor size ≥ 1 cm or tumor weight ≥ 10% of body weight; (2) development of ascites. Orthotopic tumors and micro-/macro-metastases in suspected involved organs/tissues were evaluated with necropsy and macroscope. Invasiveness/dissemination was quantified visually and histologically. IHC was performed on FFPE sections (4-µm thick) and evaluated by two pathologists independently. The mitotic index was determined by counting 1000 cells/field from 5 randomly selected fields. Microscopic images were analyzed with NIH ImageJ. To explore pharmacologic action of the 3rd generation SMI [2,17], all mice were randomly divided into 2 groups (n = 6/group) and treated for established tumors (~100 mm3) with either DMSO (control) or MO-I-1182 at a dose of 10 mg/kg, intraperitoneal injection (i.p.), every other day.
Experimental lung metastatic (tail vein injection) model [23]
This protocol was approved by IACUC of Harbin Medical University. Stable MDA-MB-231 cells (1×106 in 100 µl PBS) were injected into the tail vein of BALB/c athymic nude mice (4-6-week-old female). One day later, mice were administered with either 5% DMSO (control) or MO-I-1182 (10 mg/kg, i.p., every other day). D-luciferin (100 mg/kg, i.p.) was injected to identify pulmonary metastases with Berthold NIGHTOWL LB983 in vivo imaging system. Bioluminescent signals were photographed with different exposure time points (1-60 seconds). Acquired grayscale photographic and pseudo-colored luminescent images were automatically superimposed with Living Image software. Then, luciferase signal was matched with its anatomic location. After 28 days, all mice were sacrificed; involved organs were dissected and fixed for H&E and IHC staining. Pulmonary micro-/macro-metastases were quantified with noninvasive bioluminescence system or manually counted under dissecting microscope. ASPH-SRC network components were semi-quantified by IHC.
Patient selection and IHC [23]
This study was approved by IRBs of Rhode Island Hospital/Brown University, The First Affiliated Hospital of Harbin Medical University and Harbin Medical University Cancer Hospital, complying with respective regulations and guidelines. Tissue specimens were collected from 228 de-identified patients with breast cancer. As previously described [22-24], IHC was performed on 4-μm thick FFPE slides with antibodies to ASPH (FB50, homemade); MMP1 (ab38929), MMP14 (ab3644) from Abcam (Cambridge, MA); SRC Y416 (PAB25310) from Abnova (Taipei, Taiwan) and ADAM12 (14139-1-AP) from Proteintech (Rosemont, IL).
Statistical analysis
Data were analyzed with SPSS and GraphPad. Nonparametric parameters were evaluated with Kruskal-Wallis one-way ANOVA and Tamhane’s post hoc tests. Indices with normal distributions (presented as mean ± SD) were compared with one-way ANOVA and Bonferroni post hoc tests. Expression ASPH network in tumor tissue was calculated with IHC scoring system. Associations between molecules were explored with Spearman’s rank correlation coefficient (ρ) and Pearson’s correlation coefficient (r). Associations between ASPH levels with molecular subtypes and tumor characteristics were examined with χ2 or Fisher’s Exact tests. Overall survival (OS) was defined as the time from diagnosis to death or last follow-up. Progression-free survival (PFS) was defined as the time from surgery to clinically detectable relapse, progression or metastasis, death or last follow-up. Kaplan-Meier plots were applied to estimate median survival time (MST). Difference in MST was evaluated with log-rank test. A P < 0.05 (2-sided) was considered statistically significant.
Results
ASPH expression is enhanced with disease progression in breast cancer patients and drives malignant phenotypes dependent on β-hydroxylase activity
Differential expression of ASPH [23] between tumor and adjacent tissue was illustrated in breast cancer. Demographics and clinical indices of 228 patients were summarized in Table 1. Overall positive rate of ASPH reached 90.35%, categorized by negative, low, moderate or high expression as 9.65%, 16.23%, 40.79%, and 33.33%, respectively (Figure 1A, 1B). Notably, mean staining intensity score approached 2.0, where 0 = negative, 1 = low, 2 = moderate, 3 = high. ASPH was silenced in normal breast, inflammatory (mastitis) or benign diseases (fibroadenoma and intraductal papilloma) (Figure 1A, 1C), whereas upregulated at early-stage preinvasive breast neoplasm (DCIS), subsequently amplified with disease progression from IDC to advanced and spontaneously metastatic carcinoma (Figure 1A, 1D).
Table 1.
Demographic and clinical characteristics of breast cancer patients (n = 228)
| Variables | No. of patients (%) |
|---|---|
| Age (years) | |
| ≤ 35 | 22 (9.6) |
| > 35 | 206 (90.4) |
| TNM stage | |
| 0 | 9 (3.9) |
| I | 7 (36.0) |
| II | 64 (47.4) |
| III | 16 (12.7) |
| Surgery | |
| Mastectomy | 211 (92.5) |
| BCS | 17 (7.5) |
| Tumor size (cm) | |
| < 2 | 72 (31.6) |
| 2-5 | 148 (64.9) |
| > 5 | 8 (3.5) |
| Lymph node involvement (n) | |
| 0 | 118 (51.8) |
| 1-3 | 62 (27.2) |
| 4-9 | 29 (12.7) |
| ≥ 10 | 19 (8.3) |
| Histological grade | |
| I-II (Well-Moderate differentiation) | 36 (15.8) |
| III-IV (Poor-Dedifferentiation) | 192 (84.2) |
| ER status | |
| (+) | 151 (66.2) |
| (-) | 77 (33.8) |
| PR status | |
| (+) | 132 (57.9) |
| (-) | 96 (42.1) |
| HER2 status | |
| (+) | 127 (55.7) |
| (-) | 101 (44.3) |
| Ki-67 index (%) | |
| ≤ 14 | 98 (43.0) |
| > 14 | 130 (57.0) |
| P53 status | |
| (+) | 78 (34.2) |
| (-) | 150 (65.8) |
| Pathology type | |
| DCIS | 15 (6.6) |
| IDC | 202 (88.6) |
| ILC | 7 (3.0) |
| Other | 4 (1.8) |
| Molecular subtype | |
| Luminal A | 71 (31.1) |
| Luminal B | 80 (35.1) |
| TNBC | 28 (12.3) |
| HER2 amplified | 49 (21.5) |
| Recurrence/Metastasis | |
| No | 192 (84.2) |
| Yes | 36 (15.8) |
| Chemotherapy | |
| No | 60 (26.3) |
| Yes | 168 (73.7) |
BCS, breast-conserving surgery; DCIS, ductal carcinoma in situ; IDC, invasive ductal carcinoma including Tubular, Medullary, Mucinous, Cribriform and Papillary subtypes; ILC, invasive lobular carcinoma.
Figure 1.
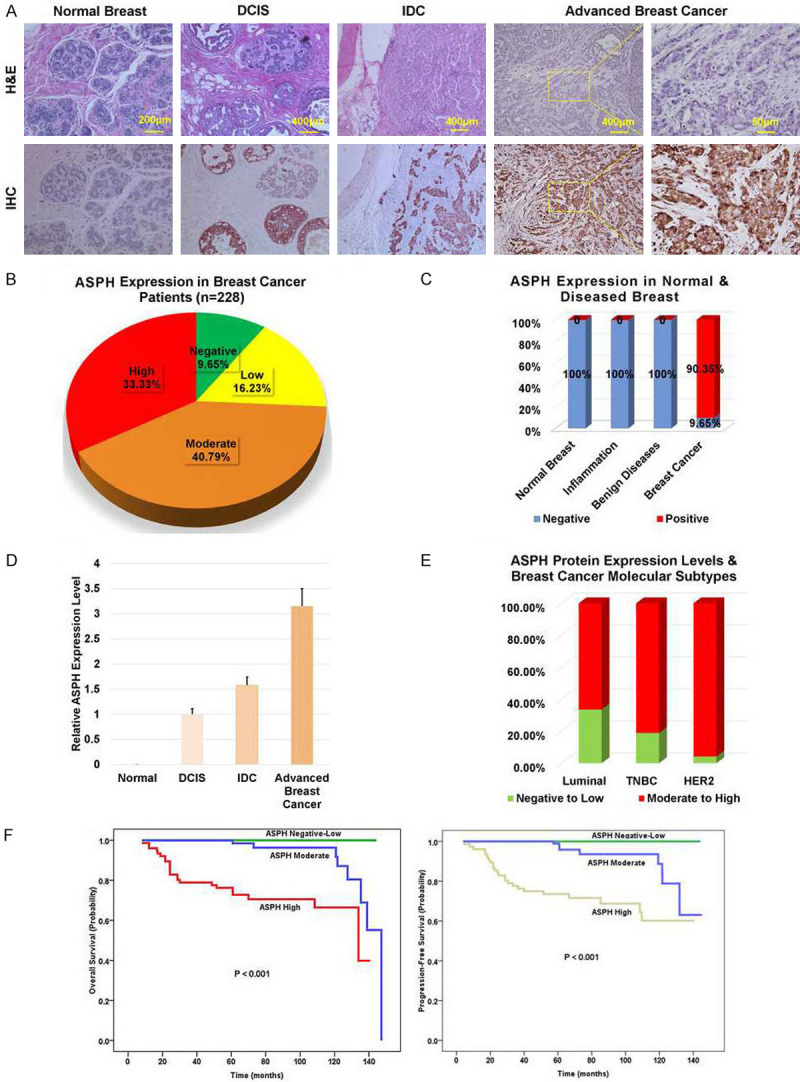
ASPH is upregulated with disease progression and correlates with worse clinical outcome in breast cancer patients. A. ASPH expression differentiated from normal breast (negative), to DCIS (low), IDC and advanced (moderate to high) breast cancer. DCIS: ductal carcinoma in situ; IDC: invasive ductal carcinoma. B, C. ASPH expression profile in normal adult breast, inflammatory breast disease (acute/chronic mastitis), benign breast tumor (fibrosis/fibroadenoma), and tumors derived from breast cancer patients (N = 228). D. Quantification of ASPH protein expression in representative different stages of breast tumors based on IHC staining scores. E, F. Moderate-high expression of ASPH correlated with more aggressive molecular subtypes (TNBC and HER2 amplified) (P < 0.001) and curtailed OS or PFS (log-rank tests, P < 0.001) of breast cancer patients. Kaplan-Meier method was used to assess differential effects of ASPH expression levels on median survival time. TNBC: triple-negative/basal-like.
To clarify whether ASPH could be a predictor for clinical outcome, survival cures were generated by Kaplan-Meier plots whereas hazard ratios estimated by Cox proportional regression models. Moderately to highly expressed ASPH were more frequently identified in aggressive molecular subtypes, i.e., triple negative/basal like breast cancer (TNBC) and HER2 amplification (Table 2; Figure 1E). When moderately or highly expressed (log-rank test, Ps < 0.001), ASPH represented increased incidence of recurrence/metastasis (Table 3) and curtailed OS or PFS (Figure 1F). Thus, ASPH is valuable for early diagnosis and prognosis of breast cancer.
Table 2.
Association of ASPH expression level with molecular subtypes of breast cancer patients (n = 228)
| ASPH expression | Molecular subtype | P | ||
|---|---|---|---|---|
| Luminal | TNBC | HER2 | 0.001 | |
| Negative to Low | 48 (33.6) | 7 (18.9) | 2 (4.2) | |
| Moderate to High | 95 (66.4) | 30 (81.1) | 46 (95.8) |
The p value (2-sided) from Fisher’s Exact. Luminal includes luminal A and B subtypes. TNBC, triple-negative/basal-like breast cancer; HER2, HER2 amplification.
Table 3.
Association of ASPH expression level with disease progression of breast cancer patients (n = 228)
| Recurrence/Metastasis | ASPH expression | P | ||
|---|---|---|---|---|
| Negative to Low | Moderate | High | < 0.001 | |
| No | 56 (98.2) | 80 (89.9) | 52 (63.4) | 0 |
| Yes | 1 (1.8) | 9 (10.1) | 30 (35.6) | 0 |
The p value (2-sided) from Fisher’s Exact.
The cDNA Microarray data was interpretated with differentially up- and down-regulated genes in response to ASPH overexpression or knock-out in breast cancer cell lines. A subset of differential genes (277 in total) was selected for clustering analysis. An intensity filter was used to select genes where the difference between the maximum and minimum intensity values exceeds 4,000 among all microarrays. The most significantly differentially expressed molecules potentially regulated by ASPH had been enriched in signaling pathways/biological processes including Focal Adhesion, ECM-Receptor Interaction, Cytoskeleton (Actin Filament), Tyrosine Kinase, Metalloproteinase, EMT and Stemness. To validate that ASPH’s expression level or enzymatic activity could modify malignant properties of breast cancer cells through bioinformatically identified pathways and processes, 3-D invasion, mammosphere formation, and immunofluorescence were performed.
In serial breast cancer cell lines, ASPH expression was determined by Western blot (Figure 2A). ASPH was stably overexpressed using lentivirus expression system in MDA-MB-468 and MDA-MB-231. ASPH was knocked out (KO) using CRISPR-CAS9 system in T47D and BT474. ASPH enabled EMT by upregulating Vimentin (mesenchymal marker) (Figure 2B) or downregulating E-cadherin (epithelial marker) (Figure 2C, 2D), attributed to upregulation of transcription factor TWIST1 (Figure 2E, 2F). ASPH exacerbated ECM degradation (Figure 2G-J), 3-D invasion (Figure 3A-C), cancer stem cell marker upregulation and mammosphere formation (Figure 3D-G). Aggressive phenotypes were mitigated by H675Q mutant with 80% reduction in enzymatic activity compared to wide-type (WT)-ASPH [2]. To more accurately represent cytoarchitecture in vivo, embedded/on-top 3-D culture was employed. Exogenous ASPH distorted the morphology (Figure 3H-J) as described by on-top or embedded 3-D culture. These cells form dynamic/complicated tree-like (with a big trunk and small branches) or mesh-like networks, which was attenuated by H675Q mutant. ASPH mediated archetypal morphology could facilitate intracellular communication (tumor-tumor and tumor-microenvironment interactions).
Figure 2.

ASPH promotes EMT and ECM degradation. A. Expression profiling of ASPH in human breast cancer cell lines. B. Expression of mesenchymal marker Vimentin affected by WT- vs. mutant-ASPH. C, D. Expression of epithelial marker E-cadherin modified by WT- vs. mutant- or KO-ASPH. E, F. Expression profiling of major components attributed to EMT in breast cancer cell lines. G-J. WT-ASPH augmented ECM degradation, which was reversed by mutant- or KO-ASPH. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 3.

ASPH phenocopies aggressive behaviors. A. Scheme of 3-D invasion. B, C. 3-D invasion index intensified by WT-ASPH. D-G. Mammosphere formation regulated by WT- vs. mutant- or KO-ASPH. H-J. Morphological changes in on-top or embedded 3-D culture determined by WT- vs. mutant ASPH. *P < 0.05; **P < 0.01; ***P < 0.001.
To vividly imitate tumor-microenvironment interaction, close-to-truly simulate tumor characteristics (e.g., dormancy, hypoxia, anti-apoptosis) and practically estimate drug sensitivity vs. resistance, in vitro metastasis [22-24] was developed (Figure 4A). Notably, WT-ASPH aggravated in vitro metastasis, which was alleviated by H675Q mutant with reduced enzymatic activity (Figure 4B, 4C).
Figure 4.

Inhibitory effect of SMI on ASPH enzymatic activity. (A) Scheme of in vitro metastasis assay. (B, C) In vitro metastasis modulated by WT- vs. mutant-ASPH: (B) Transendothelial migration (intravasation/extravasation); (C) Invasion through basement membrane and subsequent mammosphere formation. (D, E) 14CO2/α-Ketoglutarate-Dependent Capture Assay. (F) Quantification of ASPH enzymatic activity in response to SMI. (G) Inhibitory effect of SMI (at high dose only) on cell viability. *P < 0.05; **P < 0.01; ***P < 0.001.
Because action mode of ASPH relies on β-hydroxylase activity, the 3rd generation candidate SMIs potentially blocking its catalytic site were characterized [17] (Figure 4D-F). The leading compound MO-I-1182 diminished cell viability only at a high dose (10 μM) but not at a low concentration (100 nM) as demonstrated by CCK-8 assay (Figure 4G). MO-I-1182 specifically reversed ASPH mediated ECM degradation (Figure 5A-C), 3-D invasion (Figure 5D, 5E), stemness (Figure 5F-I), EMT (Figure 6A-F), and in vitro metastasis (Figure 6G, 6H).
Figure 5.

Oncogenic properties of ASPH depend on its enzymatic activity. A-C. ECM degradation in response to SMI. D, E. 3-D invasion in response to SMI. F-I. Mammosphere formation in response to SMI. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 6.

SMI mitigates ASPH induced EMT, stemness and in vitro metastasis. A. Expression of epithelial marker E-cadherin in response to SMI. B. Expression of mesenchymal marker Vimentin in response to SMI. C, D. Expression of cancer stem cell marker EpCAM in response to SMI. E, F. Expression of cancer stem cell marker CD44 in response to SMI. G, H. In vitro metastasis in response to SMI. *P < 0.05; **P < 0.01; ***P < 0.001.
ASPH activates SRC pathway by directly interacting with ADAM12/15
The SRC-FAK cascade drives tumor growth and metastasis by regulating processes involved in control of focal adhesion, cytoskeleton dynamics and MMPs expression [25]. High-throughput yeast two-hybrid system was employed to identify potential interactors with ASPH. Notably, ADAMs family members participating in tumorigenesis were promising candidates [22,23]. SMART software was applied to predict binding sites to and hydroxylated sites by ASPH within ADAMs sequences. Among ADAMs members. ADAM12/ADAM15 were predicted to be potential substrates for ASPH. Thus, expression pattern of ADAM12/ADAM15 was explored (Figure 7A). As confirmed by co-IP and Western blot, ASPH physically interacted with ADAM12/ADAM15 (Figure 7B, 7C). Upon interaction with SH3 domain of SRC [26-28], ADAM12/ADAM15 subsequently activated SRC cascade (Figure 7D). Could SRC signaling be responsible for ASPH mediated tumor progression? ASPH did activate SRC signaling, as elucidated by upregulation of active SRC (phosphorylated at Y416). ASPH mediated SRC activation through ADAM12/ADAM15 was mitigated by both Dasatinib and SMI (Figure 7E-H).
Figure 7.

ASPH activates SRC signaling pathway. A. Expression profiling of ADAM12/ADAM15 in breast cancer cell lines. B. Direct physical interaction of endogenous ASPH with endogenous ADAM12 or ADAM15 in T47D cells. C. A direct physical interaction of ASPH with ADAM12 or ADAM15 was detected by co-IP in HEK293 cells. D. ASPH or ADAM12 overexpression activated SRC. E. ADAM12/ADAM15 induced SRC activation was blocked by Dasatinib (BCR-ABL/SRC family tyrosine kinase inhibitor) in MDA-MB-231 cells. F-H. ASPH significantly enhanced activation of SRC signaling, which was reversed by the SMI and SRC inhibitor Dasatinib. I-L. ECM degradation in response to Dasatinib. *P < 0.05; **P < 0.01; ***P < 0.001.
Since SRC activity was inhibited by Dasatinib, ASPH induced ECM degradation (Figure 7I-L), 3-D invasion (Figure 8A, 8B), stemness (Figure 8C-F) and in vitro metastasis (Figure 8G, 8H) were compromised.
Figure 8.

Inhibition of SRC attenuates 3-D invasion, mammosphere formation and in vitro metastasis. A, B. 3-D invasion in response to Dasatinib. C-F. Mammosphere formation in response to Dasatinib. G, H. In vitro metastasis in response to Dasatinib. *P < 0.05; **P < 0.01; ***P < 0.001.
Invadopodium architecture responsible for degrading/remodeling ECM contributes to pro-oncogenic properties of ASPH
Invadopodia represent sites of attachment to and subsequent degradation of ECM. SRC mediated invadopodia were proposed to be required for cancer metastasis [21,29,30]. N-WASP could direct cytoskeleton to link invadopodia with ECM organization/remodeling; govern MMP14 trafficking into invadopodia [31] and organize actin polymerization at invadopodia [32]. As ASPH activates SRC signaling pathway, we hypothesized ASPH-SRC axis could integrate invadopodia dependent on N-WASP to initiate breast cancer invasiveness and metastasis.
As specific inhibition of N-WASP with Wiskostatin reduced the number and function of invadopodia, could ASPH mediated aggressive phenotypes be mitigated accordingly? Exogenous ASPH strengthened invadopodia formation and ECM degradation (Figure 9A-D), 3-D invasion (Figure 9E, 9F), stemness (Figure 10A-D) and in vitro metastasis (Figure 10E, 10F) were compromised by Wiskostatin. Interestingly, when treated at a high dosage (4 μM) with Wiskostatin, breast cancer cell viability was reduced (Figure 10G). The molecular mechanism is under further investigation.
Figure 9.

ASPH guides SRC signal mediated invadopodia function. A-D. Invadopodia formation and ECM degradation in response to Wiskostatin. E, F. 3-D invasion in response to Wiskostatin. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 10.

Invadopodia magnify stemness and in vitro metastasis. A-D. Mammosphere formation in response to Wiskostatin. E, F. In vitro metastasis in response to Wiskostatin. G. Inhibitory effect of Wiskostatin (at high dose [4 μM]) on cell viability. *P < 0.05; **P < 0.01; ***P < 0.001.
ASPH spurs multi-organ metastasis in pre-clinical animal models
Exogenous ASPH contributed to more aggressive xenografts as reflected by enlarged volume of primary tumor in NSG mice (Figure 11A-C). Those tumors exhibited increased mitosis index (Figure 11D, 11E), attributed to rapid tumor growth (Figure 11F). Exogenous ASPH dramatically reinforced widespread dissemination to lungs, liver, spleen, lymph nodes, mesentery or intestine (colon) (Figure 11G-J) in an orthotopic murine model.
Figure 11.

ASPH activates SRC signaling pathway to spur metastasis in orthotopic murine model. (A) Compared to empty vector, MDA-MB-231 cells stably expressing ASPH exhibited high tumorigenicity in orthotopic model (n = 5/group). Increased primary tumor volume demonstrated tumor development was strengthened by ASPH. Anti-tumor effect of MO-I-1182 was notable in tumors generated from MDA-MB-231 cells with exogenous ASPH. (B) Using fluorescent imaging system to detect potential pulmonary metastasis in mice from different groups of orthotopic model. (C) Histologic characteristics of the primary tumors from representative mice in different groups of orthotopic model. (D) Representative image for active mitosis. (E) Mitosis index of tumors in orthotopic model. (F) Correlation between mitosis index and tumor volume. (G) Histologic characteristics of multiple metastatic lesions (invasion into colon, mesenteric vasculature system, and lymph nodes) derived from representative mice in orthotopic model. These mice were injected with MDA-MB-231 cells stably overexpressing ASPH and treated with DMSO. (H, I) Representative gross appearance, quantification and histologic characteristics of (H, I Upper panel) pulmonary and (I) axillary lymph nodes metastatic lesions of representative mice from different groups in orthotopic model. Metastatic lesions were highlighted with yellow arrows. (I Lower panel) Metastatic lesions maintained high expression of ASPH. These mice were orthotopically injected with MDA-MB-231 stably expressing ASPH and treated with DMSO. (J) Histologic characteristics of pulmonary metastases in mice from different groups. (K, L) Expression profiling of key components in SRC signal pathway (activated SRC, ADAM12 and MMPs) was downregulated by the SMI. *P < 0.05; **P < 0.01.
As SMI destroyed pro-oncogenic/pro-metastatic properties of ASPH in vitro, we explored if it could abolish tumor development/progression in vivo. In response to MO-I-1182, both size/weight of primary tumor and number/size of metastatic lesions were significantly decreased in the orthotopic model (Figure 11A-F, 11H, 11J). Mechanistically, this SMI reversed exogenous ASPH-mediated SRC cascade activation in vivo as confirmed by downregulation of active SRC, ADAM12, MMP-1 and MMP-14 (Figure 11K, 11L).
Similarly, efficacy of SMI was demonstrated in tail vein injection model (Figure 12A, 12B). When treated with DMSO, marked metastases were developed in the lungs (Figure 12C-F), liver (Figure 12F) and bone (Figure 12G, 12H) attributed to exogeneous ASPH. Thus, ASPH propagated tumor development and progression. Accordingly, MO-I-1182 almost abrogated vasculature invasion and metastases in the lungs (Figure 12D, 12E). This SMI substantially suppressed exogenous ASPH-induced SRC cascade activation in vivo as confirmed by downregulation of active SRC, ADAM12, MMP-1 and MMP-14 (Figure 12I, 12J).
Figure 12.

Compared to empty vector, WT-ASPH significantly enhanced metastatic capability of MDA-MB-231 cells, which was efficiently reversed by the SMI in experimental pulmonary metastatic (tail vein injection) murine model. (A) Experimental design and (B) Therapeutic protocol for tail vein injection model (n = 5/group). (C) Using fluorescent imaging system to detect potential pulmonary metastasis in mice from different groups of tail vein injection model. (D) The number of macro-metastases in the lungs derived from mice in tail vein injection model. *P < 0.05; **P < 0.01. (E) Gross appearance of the lungs derived from representative mice in tail vein injection model. Metastatic lesions were highlighted with yellow arrows. (F) Gross appearance and histopathologic characteristics of (Upper) hepatic and (Bottom) pulmonary metastatic lesions of a representative mouse in ASPH+DMSO group of tail vein injection model. Noted the metastatic lesions also maintain high expression of ASPH. This animal was euthanized at 7th weeks. (G) Gross appearance of bone (spine) metastasis derived from a representative mouse in tail vein injection model. The mouse was tail vein injected with ASPH overexpressing MDA-MB-231 cells and treated with DMSO. (H) Histologic characteristics of bone and lung lesions in this specific mouse in (P). (I, J) Expression profiling of key components in SRC signaling pathway (p-SRC Y416, SRC regulator ADAM12 and downstream MMPs) was substantially downregulated by SMI.
ASPH-ADAMs-SRC axis is activated in breast cancer patients
In patients harboring ASPH negative tumors, major components of SRC signaling were negative or inactivated (Figure 13A-E). Accordingly, ADAM12-SRC-MMPs signaling was activated in ASPH positive breast cancer patients; where ASPH level was positively related to active SRC, ADAM12, and MMP1/MMP14, respectively; ADAM12 level was positively associated with that of active SRC (Figure 13F-J).
Figure 13.

ASPH activates SRC signaling in breast cancer patients. A, B. Expression pattern of ASPH-ADAM12-SRC axis in breast cancer patients. Inactivation vs. activation of SRC (phosphorylated at Tyr416) and consistent downregulation vs. upregulation of ADAM12 or MMPs in ASPH negative vs. ASPH positive tumor cells. C-E. Expression profile of ASPH, activated SRC (phosphorylated at Tyr416/418), ADAM12 and MMPs in tumor cells compared to adjacent non-malignant tissues of breast cancer patients (P < 0.001, 2-sided paired t test). ADAM12-SRC-MMPs pathway was consistently upregulated/activated or downregulated/inactivated in ASPH positive vs. negative breast cancer patients. F-J. ASPH positively correlated with active SRC, ADAM12, MMPs; ADAM12 positively correlated with active SRC expression in breast cancer patients. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Mechanistically, ASPH drives breast cancer progression partially rely on its physical interaction with ADAM12/15 to activate SRC cascade, which initiates invadopodium architecture to degrade and remodel ECM, empowers stemness, EMT, invasion, intravasation/extravasation and eventual metastases to multi-organs. Specific SMI blocks ASPH’s catalytic site, reduces its β-hydroxylase activity by 80-95% and thus inactivates SRC cascade. Consequently, multiple-step metastasis mediated by ASPH-ADAMs-SRC-MMPs axis is switched off and pro-invasive process guided by invadopodium is terminated [22] (Figure 14).
Figure 14.
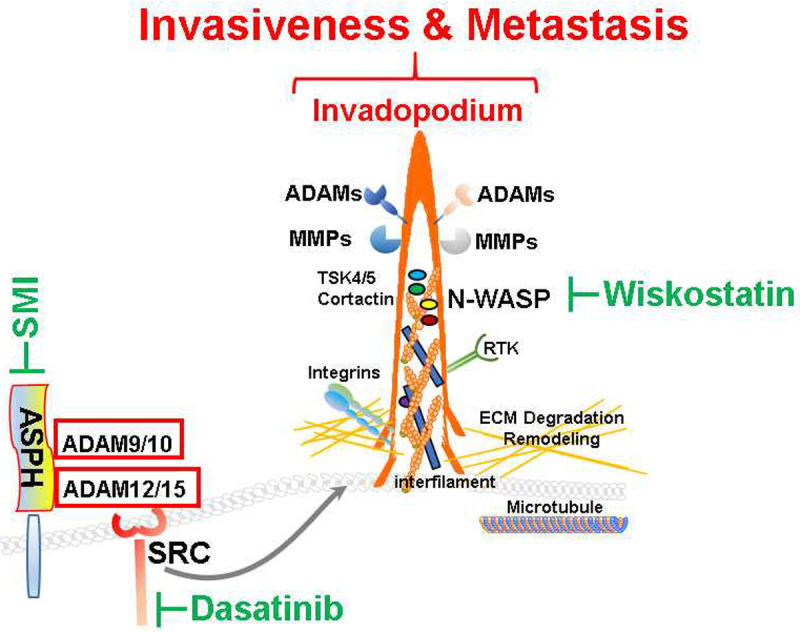
Hypothesized molecular mechanisms underlying prooncogenic properties of ASPH-SRC axis. ASPH promotes breast cancer progression through activating SRC signaling pathway, thus initiating invadopodia function/ECM degradation, and consequently forming metastatic colonization at distant organs.
ASPH reshapes breast cancer cells to construct invadopodia, invasive actin-rich protrusions as biomechanical sensors. Invadopodia guide remarkable morphology reorganization and avid ECM collapse. Inhibiting invadopodia structural protein N-WASP activity with Wiskostatin disables ECM breakdown. ASPH activates non-receptor protein tyrosine kinase SRC pathway through directly interacting with ADAM12/15 [26-28]. Subsequently, SRC escalates enzymatic activity of ADAMs [33-35]. As a result, SRC augments angiogenesis, invadopodia formation [30,32] and metastasis. We infer ASPH stabilizes ADAMs to act as pivotal building blocks for invadopodium architecture [32]. SRC disrupts endocytosis of MMP14, thus upregulating cell-surface expression of MMP14 and aggregating ECM disorgnaiztion [36]. Activation of SRC/EGFR-dependent ERK/AP1 signaling pathway promotes TNFα-induced MMP1 expression [37]. MMPs are immediate effectors for ECM degradation to initiate multi-step metastasis of breast cancer.
ASPH capacitates widespread dissemination of breast cancer to multi-organs in orthotopic and tail vein injection murine models. ASPH accelerated primary tumor growth and distant metastases are specifically/efficiently decimated by the 3rd generation SMI. Collectively, ASPH-ADAMs-SRC-MMPs axis triggers invadopodia as biomechanical sensors, giving rise to MMPs mediated ECM degradation and remodeling. These aggressive phenotypes are reversed by the SMI specifically against ASPH’s β-hydroxylase activity, whereas are lessened to different extents by inhibitors of SRC and N-WASP. This study reveals a fundamental role of ASPH network in propagating aggressive/metastatic capability of breast cancer cells.
ASPH is positively expressed in 90.7% of breast cancer patients, with 58.7% at a moderate-high level, whereas undetectable in inflammatory or benign breast diseases. ASPH is silenced in adult normal breast, upregulated at early stage preinvasive breast neoplasm (DISC) and markedly overexpressed in advanced or metastatic breast cancer. Moderately to highly expressed ASPH contributes to more aggressive molecular subtypes, early recurrence, higher incidence of local invasiveness and distant metastasis, and poorer survival of breast cancer patients. Thus, ASPH has substantial potential for early diagnosis and prognosis for breast cancer.
Metastasis [22-24] results from a series of complicated events: invading locally through basement membrane and remodeling ECM at the primary site, intravasating into and surviving in hematogenous/lymphatic circulation, arrested at a capillary bed in a distant organ, extravasating out of blood vessels/lymphatics, surviving/proliferating in a hostile environment, and ultimately forming metastatic colonization/outgrowth [38]. Invadopodia act as biomechanical media for ASPH mediated pro-oncogenic/pro-metastatic effects. ASPH is vital in multifaceted events of breast cancer metastasis. Through masking its catalytic site, the 3rd generation SMI specifically/substantially reduces ASPH’s β-hydroxylase activity [2-4,17,39] and consequently disables SRC cascade. Accordingly, pro-invasive role of invadopodia in breast cancer tumor progression initiated by ASPH-ADAMs-SRC-MMPs axis is eradicated and pro-oncogenic/pro-metastatic actions are collapsed. This study in combination with previous results [22-24,40-43] have distinguished ASPH as a potential therapeutic target for breast cancer.
Acknowledgements
Structural characterization, synthesis and purification of small molecule inhibitors (SMIs) against ASPH [2, 17] were performed by Dr. Mark Olsen at the Department of Pharmaceutical Sciences, College of Pharmacy-Glendale, Midwestern University, Glendale (Arizona 85308, USA). Mass Spectrometry/Proteomics service was provided by Laboratory for Molecular Biology and Cytometry Research at the University of Oklahoma Health Sciences Center. This study was supported by National Institutes of Health (NIH) (CA123544, P30GM110759) and Institutional Funds of the United States (to J.R.W. and X.Q.D.); National Natural Science Foundation China (81573001, 81773295 to X.S.C.; 81772588 to L.X.L.); National Key Program for Science and Technology Research and Development of China (2016YFC0106503, 2016YFC0106500, 2016YFC0905902 to L.X.L.). This funding source had no role in experimental design and execution, data analyses and interpretation, or decision to submit results.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Dong X, Lin Q, Aihara A, Li Y, Huang CK, Chung W, Tang Q, Chen X, Carlson R, Nadolny C, Gabriel G, Olsen M, Wands JR. Aspartate beta-hydroxylase expression promotes a malignant pancreatic cellular phenotype. Oncotarget. 2015;6:1231–1248. doi: 10.18632/oncotarget.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iwagami Y, Huang CK, Olsen MJ, Thomas JM, Jang G, Kim M, Lin Q, Carlson RI, Wagner CE, Dong X, Wands JR. Aspartate beta-hydroxylase modulates cellular senescence through glycogen synthase kinase 3beta in hepatocellular carcinoma. Hepatology. 2016;63:1213–1226. doi: 10.1002/hep.28411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomimaru Y, Mishra S, Safran H, Charpentier KP, Martin W, De Groot AS, Gregory SH, Wands JR. Aspartate-beta-hydroxylase induces epitope-specific T cell responses in hepatocellular carcinoma. Vaccine. 2015;33:1256–1266. doi: 10.1016/j.vaccine.2015.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawton M, Tong M, Gundogan F, Wands JR, de la Monte SM. Aspartyl-(asparaginyl) beta-hydroxylase, hypoxia-inducible factor-alpha and Notch cross-talk in regulating neuronal motility. Oxid Med Cell Longev. 2010;3:347–356. doi: 10.4161/oxim.3.5.13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de la Monte SM, Tamaki S, Cantarini MC, Ince N, Wiedmann M, Carter JJ, Lahousse SA, Califano S, Maeda T, Ueno T, D’Errico A, Trevisani F, Wands JR. Aspartyl-(asparaginyl)-beta-hydroxylase regulates hepatocellular carcinoma invasiveness. J Hepatol. 2006;44:971–983. doi: 10.1016/j.jhep.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 7.Cantarini MC, de la Monte SM, Pang M, Tong M, D’Errico A, Trevisani F, Wands JR. Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms. Hepatology. 2006;44:446–457. doi: 10.1002/hep.21272. [DOI] [PubMed] [Google Scholar]
- 8.Gundogan F, Elwood G, Greco D, Rubin LP, Pinar H, Carlson RI, Wands JR, de la Monte SM. Role of aspartyl-(asparaginyl) beta-hydroxylase in placental implantation: relevance to early pregnancy loss. Hum Pathol. 2007;38:50–59. doi: 10.1016/j.humpath.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Ince N, de la Monte SM, Wands JR. Overexpression of human aspartyl (asparaginyl) beta-hydroxylase is associated with malignant transformation. Cancer Res. 2000;60:1261–1266. [PubMed] [Google Scholar]
- 10.Lavaissiere L, Jia S, Nishiyama M, de la Monte S, Stern AM, Wands JR, Friedman PA. Overexpression of human aspartyl(asparaginyl)beta-hydroxylase in hepatocellular carcinoma and cholangiocarcinoma. J Clin Invest. 1996;98:1313–1323. doi: 10.1172/JCI118918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Liu J, Yan ZL, Li J, Shi LH, Cong WM, Xia Y, Zou QF, Xi T, Shen F, Wang HY, Wu MC. Overexpression of aspartyl-(asparaginyl)-beta-hydroxylase in hepatocellular carcinoma is associated with worse surgical outcome. Hepatology. 2010;52:164–173. doi: 10.1002/hep.23650. [DOI] [PubMed] [Google Scholar]
- 12.Maeda T, Sepe P, Lahousse S, Tamaki S, Enjoji M, Wands JR, de la Monte SM. Antisense oligodeoxynucleotides directed against aspartyl (asparaginyl) beta-hydroxylase suppress migration of cholangiocarcinoma cells. J Hepatol. 2003;38:615–622. doi: 10.1016/s0168-8278(03)00052-7. [DOI] [PubMed] [Google Scholar]
- 13.Sepe PS, Lahousse SA, Gemelli B, Chang H, Maeda T, Wands JR, de la Monte SM. Role of the aspartyl-asparaginyl-beta-hydroxylase gene in neuroblastoma cell motility. Lab Invest. 2002;82:881–891. doi: 10.1097/01.lab.0000020406.91689.7f. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, de la Monte SM, Sabo E, Kethu S, Tavares R, Branda M, Simao L, Wands JR, Resnick MB. Prognostic value of humbug gene overexpression in stage II colon cancer. Hum Pathol. 2007;38:17–25. doi: 10.1016/j.humpath.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Luu M, Sabo E, de la Monte SM, Greaves W, Wang J, Tavares R, Simao L, Wands JR, Resnick MB, Wang L. Prognostic value of aspartyl (asparaginyl)-beta-hydroxylase/humbug expression in non-small cell lung carcinoma. Hum Pathol. 2009;40:639–644. doi: 10.1016/j.humpath.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung W, Kim M, de la Monte S, Longato L, Carlson R, Slagle BL, Dong X, Wands JR. Activation of signal transduction pathways during hepatic oncogenesis. Cancer Lett. 2016;370:1–9. doi: 10.1016/j.canlet.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aihara A, Huang CK, Olsen MJ, Lin Q, Chung W, Tang Q, Dong X, Wands JR. A cell-surface beta-hydroxylase is a biomarker and therapeutic target for hepatocellular carcinoma. Hepatology. 2014;60:1302–1313. doi: 10.1002/hep.27275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wands JR, Kim M. WNT/beta-catenin signaling and hepatocellular carcinoma. Hepatology. 2014;60:452–454. doi: 10.1002/hep.27081. [DOI] [PubMed] [Google Scholar]
- 19.Tomimaru Y, Koga H, Yano H, de la Monte S, Wands JR, Kim M. Upregulation of T-cell factor-4 isoform-responsive target genes in hepatocellular carcinoma. Liver Int. 2013;33:1100–1112. doi: 10.1111/liv.12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eddy RJ, Weidmann MD, Sharma VP, Condeelis JS. Tumor cell invadopodia: invasive protrusions that orchestrate metastasis. Trends Cell Biol. 2017;27:595–607. doi: 10.1016/j.tcb.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, Hague MN, Ablack A, Carmine-Simmen K, McPherson VA, Postenka CO, Turley EA, Courtneidge SA, Chambers AF, Lewis JD. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014;8:1558–1570. doi: 10.1016/j.celrep.2014.07.050. [DOI] [PubMed] [Google Scholar]
- 22.Ogawa K, Lin Q, Li L, Bai X, Chen X, Chen H, Kong R, Wang Y, Zhu H, He F, Xu Q, Liu L, Li M, Zhang S, Nagaoka K, Carlson R, Safran H, Charpentier K, Sun B, Wands J, Dong X. Aspartate beta-hydroxylase promotes pancreatic ductal adenocarcinoma metastasis through activation of SRC signaling pathway. J Hematol Oncol. 2019;12:144. doi: 10.1186/s13045-019-0837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin Q, Chen X, Meng F, Ogawa K, Li M, Song R, Zhang S, Zhang Z, Kong X, Xu Q, He F, Bai X, Sun B, Hung MC, Liu L, Wands J, Dong X. ASPH-notch axis guided exosomal delivery of prometastatic secretome renders breast cancer multi-organ metastasis. Mol Cancer. 2019;18:156. doi: 10.1186/s12943-019-1077-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogawa K, Lin Q, Li L, Bai X, Chen X, Chen H, Kong R, Wang Y, Zhu H, He F, Xu Q, Liu L, Li M, Zhang S, Nagaoka K, Carlson R, Safran H, Charpentier K, Sun B, Wands J, Dong X. Prometastatic secretome trafficking via exosomes initiates pancreatic cancer pulmonary metastasis. Cancer Lett. 2020;481:63–75. doi: 10.1016/j.canlet.2020.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang Q, Cao Y, Zolkiewska A. Metalloprotease-disintegrin ADAM 12 binds to the SH3 domain of Src and activates Src tyrosine kinase in C2C12 cells. Biochem J. 2000;352 Pt 3:883–892. [PMC free article] [PubMed] [Google Scholar]
- 27.Poghosyan Z, Robbins SM, Houslay MD, Webster A, Murphy G, Edwards DR. Phosphorylation-dependent interactions between ADAM15 cytoplasmic domain and Src family protein-tyrosine kinases. J Biol Chem. 2002;277:4999–5007. doi: 10.1074/jbc.M107430200. [DOI] [PubMed] [Google Scholar]
- 28.Sun C, Wu MH, Guo M, Day ML, Lee ES, Yuan SY. ADAM15 regulates endothelial permeability and neutrophil migration via Src/ERK1/2 signalling. Cardiovasc Res. 2010;87:348–355. doi: 10.1093/cvr/cvq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ngan E, Stoletov K, Smith HW, Common J, Muller WJ, Lewis JD, Siegel PM. LPP is a Src substrate required for invadopodia formation and efficient breast cancer lung metastasis. Nat Commun. 2017;8:15059. doi: 10.1038/ncomms15059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paz H, Pathak N, Yang J. Invading one step at a time: the role of invadopodia in tumor metastasis. Oncogene. 2014;33:4193–4202. doi: 10.1038/onc.2013.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, Zech T, McDonald L, Gonzalez EG, Li A, Macpherson I, Schwarz JP, Spence H, Futo K, Timpson P, Nixon C, Ma Y, Anton IM, Visegrady B, Insall RH, Oien K, Blyth K, Norman JC, Machesky LM. N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. J Cell Biol. 2012;199:527–544. doi: 10.1083/jcb.201203025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stautz D, Sanjay A, Hansen MT, Albrechtsen R, Wewer UM, Kveiborg M. ADAM12 localizes with c-Src to actin-rich structures at the cell periphery and regulates Src kinase activity. Exp Cell Res. 2010;316:55–67. doi: 10.1016/j.yexcr.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 34.Maretzky T, Le Gall SM, Worpenberg-Pietruk S, Eder J, Overall CM, Huang XY, Poghosyan Z, Edwards DR, Blobel CP. Src stimulates fibroblast growth factor receptor-2 shedding by an ADAM15 splice variant linked to breast cancer. Cancer Res. 2009;69:4573–4576. doi: 10.1158/0008-5472.CAN-08-4766. [DOI] [PubMed] [Google Scholar]
- 35.Abram CL, Seals DF, Pass I, Salinsky D, Maurer L, Roth TM, Courtneidge SA. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src-transformed cells. J Biol Chem. 2003;278:16844–16851. doi: 10.1074/jbc.M300267200. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Gan B, Yoo Y, Guan JL. FAK-mediated src phosphorylation of endophilin A2 inhibits endocytosis of MT1-MMP and promotes ECM degradation. Dev Cell. 2005;9:185–196. doi: 10.1016/j.devcel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Lee CS, Bae IH, Han J, Choi GY, Hwang KH, Kim DH, Yeom MH, Park YH, Park M. Compound K inhibits MMP-1 expression through suppression of c-Src-dependent ERK activation in TNF-alpha-stimulated dermal fibroblast. Exp Dermatol. 2014;23:819–824. doi: 10.1111/exd.12536. [DOI] [PubMed] [Google Scholar]
- 38.Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169–175. doi: 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- 39.Huang CK, Iwagami Y, Aihara A, Chung W, de la Monte S, Thomas JM, Olsen M, Carlson R, Yu T, Dong X, Wands J. Anti-tumor effects of second generation beta-hydroxylase inhibitors on cholangiocarcinoma development and progression. PLoS One. 2016;11:e0150336. doi: 10.1371/journal.pone.0150336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeda T, Taguchi K, Aishima S, Shimada M, Hintz D, Larusso N, Gores G, Tsuneyoshi M, Sugimachi K, Wands JR, de la Monte SM. Clinicopathological correlates of aspartyl (asparaginyl) beta-hydroxylase over-expression in cholangiocarcinoma. Cancer Detect Prev. 2004;28:313–318. doi: 10.1016/j.cdp.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Noda T, Shimoda M, Ortiz V, Sirica AE, Wands JR. Immunization with aspartate-beta-hydroxylase-loaded dendritic cells produces antitumor effects in a rat model of intrahepatic cholangiocarcinoma. Hepatology. 2012;55:86–97. doi: 10.1002/hep.24629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palumbo KS, Wands JR, Safran H, King T, Carlson RI, de la Monte SM. Human aspartyl (asparaginyl) beta-hydroxylase monoclonal antibodies: potential biomarkers for pancreatic carcinoma. Pancreas. 2002;25:39–44. doi: 10.1097/00006676-200207000-00010. [DOI] [PubMed] [Google Scholar]
- 43.Shimoda M, Tomimaru Y, Charpentier KP, Safran H, Carlson RI, Wands J. Tumor progression-related transmembrane protein aspartate-beta-hydroxylase is a target for immunotherapy of hepatocellular carcinoma. J Hepatol. 2012;56:1129–1135. doi: 10.1016/j.jhep.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]