

Abstract
Skeletal muscle wasting and weakness caused by cancer and its treatments (known as “cachexia”) drastically impair quality of life and worsen survival outcomes in cancer patients. There are currently no approved treatments for cachexia. Hence, further investigation into the causes of cachexia induced by cancer and chemotherapy is warranted. Here, we sought to investigate skeletal muscle wasting, weakness and loss of motor unit function in mice bearing cancers or administered chemotherapeutics. Mice bearing colorectal cancers, including C26, MC38 and HCT116, and mice receiving the chemotherapeutics folfiri and cisplatin were assessed for in vivo and ex vivo muscle force, and for in vivo electrophysiological indices of motor unit connectivity, including compound muscle action potential and motor unit number estimation (MUNE). In vivo and ex vivo muscle force, as well as MUNE were reduced in C26, MC38, HCT116 hosts, and in mice receiving folfiri and cisplatin compared to their respective experimental controls. In addition, MUNE was correlated with muscle force and muscle mass in all experimental conditions, while assessment of neuromuscular junction (NMJ) protein expression and changes in presynaptic morphology suggested that cancer and chemotherapy significantly alter muscle innervation. The present results demonstrate that the loss of motor unit connectivity may contribute to skeletal muscle wasting and weakness that occur with cancer and chemotherapy.
Keywords: Cachexia, cancer, chemotherapy, muscle weakness, motor unit
Introduction
Despite decades of advances in treatment, cancer remains a critical clinical concern, with nearly 2 million new cases and over 600,000 deaths expected to occur in 2021 [1]. Upwards to 80% of these patients will experience cachexia, a multi-organ wasting syndrome, which is directly responsible for 30% of cancer-associated deaths [2,3]. In particular, skeletal muscle wasting and weakness, hallmarks of cachexia, lower treatment tolerance, impede the ability to perform daily activities of living and worsen survival in cancer patients [4-9]. Similar to cancer, anti-cancer chemotherapy regimens have robust off-target effects and are known to promote cachexia-like phenotypes. In fact, we have shown that chemotherapeutics directly promote body weight loss, skeletal muscle wasting and skeletal muscle weakness [10-13]. Unfortunately, there are currently no approved treatment options to combat cachexia in cancer patients.
To date, a majority of research has placed emphasis on directly improving muscle mass in order to improve muscle strength. However, evidence suggests that loss of muscle function may precede muscle wasting, highlighting the importance of investigating mechanisms mediating weakness [14,15]. Of note, an important determinant of muscle function is the number of motor units, which are functional units made up of a motor neuron and all the myofibers it innervates. Interestingly, loss of motor unit number, as suggested by motor unit number estimation (MUNE), has shown to precede loss of muscle strength and is correlated with muscle weakness and muscle atrophy in aging rodents [16]. Regarding cachexia, recent investigations interrogating indices of motor neuron function, neuromuscular junctions (NMJs) and denervation in cancer- and chemotherapy-induced muscle wasting have revealed conflicting results [17-19]. Moreover, investigations into changes in motor unit number in cachexia have not occurred, leaving a gap towards understanding the mechanisms that contribute to muscle weakness caused by cancer and chemotherapy.
To address this point, we utilized an established MUNE technique to gain insight into the number of motor neurons functionally connected to the triceps surae muscles of the mouse hindlimb [20,21]. Taking advantage of three colorectal cancer (CRC) cell lines (C26, MC38, HCT116) and two chemotherapeutic regimens (folfiri and cisplatin) known to induce cachexia, we assessed alterations of MUNE to investigate whether muscle wasting and weakness were associated with loss of motor unit number. Our present findings indicate that loss of motor unit number is associated with muscle wasting and muscle weakness caused by cancer and chemotherapy. Moreover, we demonstrate that cachexia induced by cancer and its treatments is accompanied by loss of NMJ-associated proteins and abnormal presynaptic morphology, further suggestive of altered innervation.
Materials and methods
Cell lines
Murine Colon-26 (C26), provided by Donna McCarthy (Ohio State University) and MC38, provided by Dr. Xiongbin Lu (Indiana University School of Medicine), cells were grown in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S), 1% sodium pyruvate and 2 mM L-glutamine. Human HCT116 cells (ATCC; Manassas, VA, USA; #CRL-247) were grown in McCoy’s medium containing 10% FBS, 1% glutamine, 1% sodium pyruvate, and 1% P/S. All cell lines were cultured and passaged in 5% CO2 at 37°C, trypsinized when sub-confluent, and prepared for animal injections in sterile saline.
Animals
All animal work was approved by the Institutional Animal Care and Use Committee at the Indiana University School of Medicine and was in compliance with the National Institutes of Health Guidelines for Use and care of Laboratory Animals as well as the 1964 Declaration of Helsinki and all subsequent amendments. For the C26 experiments, 8-week-old CD2F1 male mice (Envigo, Indianapolis, IN, USA) were randomized into 2 groups: mice subcutaneously injected (intrascapularly) with 1.0 × 106 C26 cells in sterile saline (n = 8) and mice receiving isovolumetric subcutaneous injections of vehicle (n = 5) [22]. All mice were sacrificed 14 days following tumor implantation. For the MC38 experiments, 8-week-old C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were randomized into 2 groups: mice subcutaneously injected (intrascapularly) with 1.0 × 106 MC38 tumor cells in sterile saline (n = 8) and mice receiving an isovolumetric subcutaneous injection of vehicle (n = 5) [23]. All mice were sacrificed 28 days following tumor implantation. For the HCT116 experiments, 8-week-old male NOD-scid/IL2Rgnull (NSG) immunodeficient mice (In Vivo Therapeutics Core Facility, IU Simon Comprehensive Cancer Center, Indianapolis, IN) were used and housed in a pathogen-free facility at IU LARC and randomized into 2 groups: mice subcutaneously injected (intrascapularly) with 3.0 × 106 HCT116 tumor cells in sterile saline (n = 5) and mice receiving an isovolumetric subcutaneous injection of vehicle (n = 5) [24]. All mice were sacrificed 30 days following tumor implantation. For the folfiri experiments, 8-week-old CD2F1 male mice (Envigo, Indianapolis, IN, USA) were randomized into 2 groups: mice administered intraperitoneal injections of folfiri (30 mg/kg 5-fluorouracil, 90 mg/kg leucovorin, 24 mg/kg CPT-11; 2x/week) in sterile saline (n = 8) and mice treated with isovolumetric injections of vehicle (n = 5) for 5 weeks [11]. For the cisplatin experiments, 8-week-old C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were randomized into 2 groups: mice administered intraperitoneal injections of cisplatin (2.5 mg/kg; 9 total injections) in sterile saline (n = 5) and mice treated with isovolumetric injections of vehicle (n = 5) for 2 weeks [12]. Gastrocnemius muscles were harvested, weighed and snap frozen in liquid nitrogen, while extensor digitorum longus (EDL) muscles underwent ex vivo muscle contractility, or were processed for NMJ staining.
In vivo muscle contractility
All experimental animals were tested for muscle force by in vivo plantarflexion 2 days prior to euthanasia (Aurora Scientific, Aurora, ON, Canada), as performed previously [23,25]. In short, the foot of the right hindlimb was taped into a footplate force transducer and the knee secured at the femoral condyles. To stimulate the tibial nerve, two monopolar electrodes (Natus Neurology, Middleton, WI, USA) were inserted subcutaneously posterior and medial to the knee. The stimulus intensity needed to elicit maximal twitch force was determined and animals were subjected to a 100Hz stimulation (0.2 ms).
In vivo electrophysiology
Triceps surae muscles of all animals were subjected to electrophysiological functional assessment using the Sierra Summit 3-12 Channel EMG (Cadwell Laboratories Incorporated, Kennewick, WA, USA), as performed previously [20]. Two 28-gauge stimulating needle electrodes (Natus Neurology, Middleton, WI, USA) were used to stimulate the sciatic nerve of the left hindlimb, a duo shielded ring electrode (Natus Neurology, Middleton, WI, USA) was used for recording, and a ground electrode was placed over the animal’s tail. Baseline-to-peak and peak-to-peak compound muscle action potential (CMAP) responses were recorded (Figure 1A) utilizing supramaximal stimulations (constant current intensity: < 10 mA; pulse duration: 0.1 ms). In addition to CMAP responses, all animals were assessed for peak-to-peak single motor unit potential (SMUP) responses, using the incremental stimulation technique, as previously described [16,20] (Figure 1B, 1C). Briefly, the sciatic nerve was submaximally stimulated until stable, minimal all-or-none responses occurred. Ten successive SMUP increments were recorded and averaged. Baseline-to-peak CMAP amplitudes were used for comparison between experimental groups and MUNE was calculated using peak-to-peak CMAP and average SMUP, using the following equation: MUNE = CMAP amplitude (peak-to-peak)/average SMUP (peak-to-peak).
Figure 1.
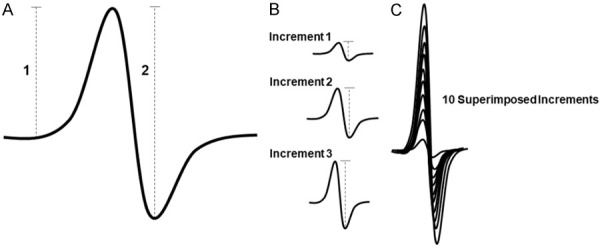
Overview of electrophysiological assessment. (A) Representative illustration of a compound muscle action potential (CMAP) response, showing the measured baseline-to-peak amplitude (1) and the measured peak-to-peak amplitude (2). (B) Representative illustration of three individual incremental single motor unit potential (SMUP) responses, showing the measured peak-to-peak amplitude. (C) Representative illustration of ten superimposed SMUP responses that would be averaged and used for motor unit number estimation (MUNE) calculation. Calculation of MUNE: MUNE = CMAP (peak-to-peak)/average SMUP (peak-to-peak).
Ex vivo muscle contractility
EDL muscles were subjected to whole-muscle contractility assessment as done previously [23]. The EDLs were quickly dissected and stainless-steel hooks were tied to both tendons using 4-0 silk sutures. The muscles were placed between a mounted force transducer (Aurora Scientific) and incubated in a stimulation bath containing Tyrode solution (121 mM NaCl, 5.0 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 0.4 mM NaH2PO4, 24 mM NaHCO3, 0.1 mM EDTA, and 5.5 mM glucose) supplemented with continuous O2/CO2 (95/5%). Following a 10-minute incubation period, the maximum twitch force was obtained by determining optimal muscle length (L0) and force-frequency relationships were assessed using a supramaximal incremental frequency stimulation sequence (10, 25, 40, 60, 80, 100, 125 and 150 Hz for 350 ms). Force data was collected and analyzed with the Dynamic Muscle Control/Data Acquisition and Dynamic Muscle Control Data Analysis programs (Aurora Scientific) and EDL muscle weight and L0 were used to determine specific force.
Tissue preparation and staining of neuromuscular junctions (NMJs)
Non-contracted EDL muscles were immediately excised and processed for NMJ staining as described previously [26]. Briefly, EDLs were fixed in a 2% paraformaldehyde solution at room temperature, serially washed with phosphate buffered saline (PBS) and incubated overnight in a 30% Sucrose (PBS) solution at 4°C. Muscles were placed in OCT and frozen in liquid nitrogen-cooled isopentane and longitudinally sectioned (20 µm). Sections were blocked (PBS: 10% donkey serum, 0.1% Triton X-100) for 1 hour at room temperature and incubated overnight in SV2 (Developmental Studies Hybridoma Bank, Iowa City, IA) and neurofilament (NF-H, Aves Labs, Davis CA) at 4°C [27]. Sections were then serially washed with PBS and incubated with secondary antibodies directed to primary antibodies and anti-Bungarotoxin for 1 hour at room temperature. Samples were imaged using an Axio Observer.Z1 motorized microscope (Zeiss, Oberchoken, Germany).
Western blotting
Protein extracts were obtained from homogenizing whole gastrocnemius muscle and electrophoresed for western blotting, as performed previously [25]. Antibodies used were muscle-specific kinase (MuSK) (#ab92950), receptor-associated protein of the synapse (Rapsyn) (#ab156002) and downstream-of-kinase 7 (Dok7) (#ab75049) from Abcam; low-density receptor-related protein 4 (LRP4) (#AV46745) from MilliporeSigma. α-Tubulin (#12G10; Developmental Studies Hybridoma Bank, Iowa City, IA) was used as loading control.
Statistics
All statistical analyses were performed using GraphPad Prism 9.0.0 (GraphPad Software, San Diego, CA, USA). In general, Student’s t-tests were performed to determine significant differences between control and tumor-bearing (C26, MC38, HCT116) or chemotherapy-treated (folfiri, cisplatin) animals. If the variance between two groups was significantly different, a Mann-Whitney U test was used. A 2-way repeated-measures analysis of variance was performed, followed by Bonferroni’s post hoc comparisons for ex vivo muscle contractility of the EDL. For correlational analyses, Pearson correlation coefficients were calculated. Statistical significance was set at P ≤ 0.05, and the data are presented as means ± s.d.
Results
Cancer and chemotherapy promote muscle wasting and weakness
In line with our previous findings, animals bearing cancer or receiving chemotherapy underwent marked muscle atrophy [11,12,22-24]. In particular, mice bearing C26, MC38 and HCT116 CRC tumors had reduced gastrocnemius muscle weights compared to the respective control animals (C26: -21%, P < 0.001; MC38: -11%, P < 0.05; HCT116: -20%, P < 0.01) (Figure 2). Similarly, gastrocnemius weights in mice treated with chemotherapy were significantly reduced compared to the control animals (folfiri: -11%, P < 0.05; cisplatin: -16%, P < 0.001) (Figure 2). In order to validate our prior findings that cachectic mice also display muscle weakness, all experimental animals were assessed for plantarflexion force and whole-muscle contractility of the EDL muscle. In vivo force assessment across all experimental groups (Figure 3A) revealed marked reductions in skeletal muscle force with respect to the controls (C26: -17%, P < 0.05; MC38: -13%, P < 0.01; HCT116: -25%, P < 0.001; folfiri: -17%, P < 0.001; cisplatin: -20%, P < 0.01). Like in vivo force reductions, ex vivo contractility of the EDL revealed force reductions across all cancer-bearing and chemotherapy-treated mice. The C26 hosts saw an average force reduction of 20% beginning at 40 Hz (P < 0.05) and continuing through 150 Hz (P < 0.0001), whereas the MC38 bearers displayed an average force reduction of 15% beginning at 25 Hz (P < 0.05) and continuing through 150 Hz (P < 0.0001). Similarly, the HCT116 hosts exhibited a 14% reduction in average force beginning at 25 Hz (P < 0.05) and continuing through 150 Hz (P < 0.05). Among the chemotherapy-treated animals, the mice administered folfiri saw an average force reduction of 20% beginning at 25 Hz (P < 0.0001) and continuing through 150 Hz (P < 0.0001), whereas the animals exposed to cisplatin showed a 20% decrease in average force beginning at 25 Hz (P < 0.05) and continuing through 150 Hz (P < 0.001) (Figure 3B).
Figure 2.
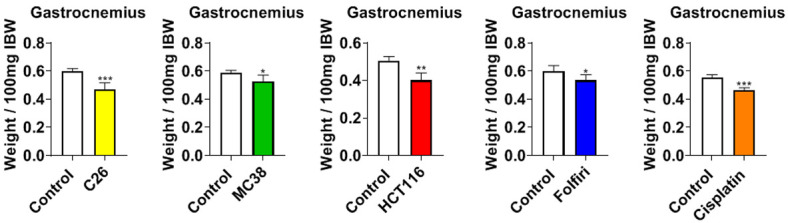
Cancer and chemotherapy promote muscle wasting. Gastrocnemius muscles normalized to initial body weight (IBW) of male mice (8 weeks) bearing subcutaneous C26 (1.0 × 106 cells/mouse in sterile saline), MC38 (1.0 × 106 cells/mouse in sterile saline) or HCT116 (3.0 × 106 cells/mouse in sterile saline) colorectal cancers and mice receiving intraperitoneal injections of folfiri or cisplatin. Control animals received equal amounts of empty vehicle for all experiments. Data are presented as mean ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Figure 3.

Cancer and chemotherapy induce muscle weakness. In vivo plantarflexion force assessment (A) reported as absolute force (expressed as mN·m) of male mice (8 weeks) bearing subcutaneous C26 (1.0 × 106 cells/mouse in sterile saline), MC38 (1.0 × 106 cells/mouse in sterile saline) or HCT116 (3.0 × 106 cells/mouse in sterile saline) colorectal cancers and mice receiving intraperitoneal injections of folfiri or cisplatin. Control animals received equal amounts of empty vehicle for all experiments. Assessment of ex vivo muscle contractility of EDL muscles (B), reported as specific ex vivo force (expressed as kN/m2). Data presented as mean ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. control.
Cancer and chemotherapy induce alterations in SMUP and MUNE
Given the prior evidence of impaired motor neuron function, denervation, and disrupted NMJs in cachexia [17-19], we sought to investigate functional indices of motor unit connectivity in mice exposed to cancer and chemotherapy. Interestingly, baseline-to-peak CMAP assessment revealed no statistically significant reductions in any experimental model of cancer- or chemotherapy-induced cachexia (Figure 4A; Table 1). However, investigation into SMUP revealed significant elevations in both cancer and chemotherapy experimental animals. SMUP values for C26 hosts (+114%; P < 0.05), MC38 hosts (+134%; P < 0.05) and HCT116 hosts (+215%; P < 0.01) were all increased compared to experimental controls (Figure 4B; Table 1). Similarly, mice receiving folfiri (+120%; P < 0.01) and cisplatin (+140%; P < 0.01) had significant elevations in SMUP compared to control mice (Figure 4B; Table 1). Meanwhile, MUNE estimations revealed reductions in C26 hosts (-50%; P < 0.01), MC38 hosts (-51%; P < 0.01), HCT116 hosts (-62%; P < 0.01), folfiri-treated mice (-42%; P < 0.01), and cisplatin-treated mice (-56%; P < 0.01) compared to control animals (Figure 4C; Table 1). Follow-up assessment demonstrated that MUNE was well correlated (Pearson r) with both gastrocnemius size (Figure 5A) and plantarflexion force (Figure 5B) across all experimental groups. Altogether, this suggests that MUNE is an additional useful tool when examining muscle wasting and weakness in mice bearing cancer or receiving chemotherapy.
Figure 4.
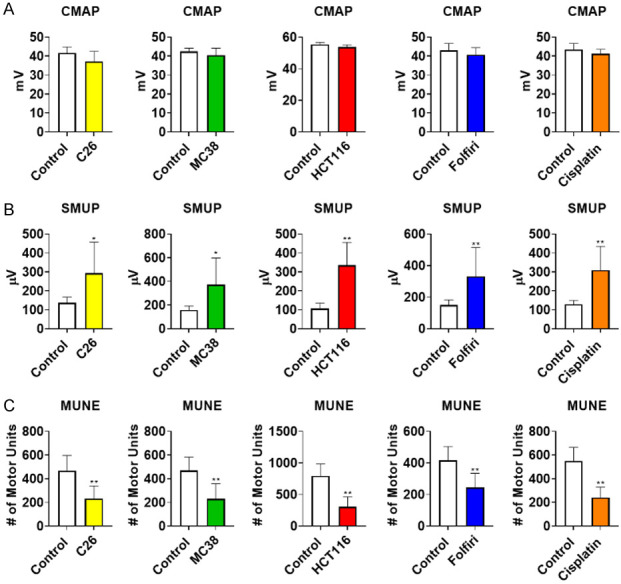
Cancer and Chemotherapy Induce Alterations in SMUP and MUNE. Compound muscle action potential (CMAP: millivolts (mV)) (A) of the triceps surae muscles of male mice (8 weeks) bearing subcutaneous C26 (1.0 × 106 cells/mouse in sterile saline), MC38 (1.0 × 106 cells/mouse in sterile saline) or HCT116 (3.0 × 106 cells/mouse in sterile saline) colorectal cancers and mice receiving intraperitoneal injections of folfiri or cisplatin. Control animals received equal amounts of empty vehicle for all experiments. Single motor unit potential (SMUP; microvolts (µV)) (B). Motor unit number estimation (MUNE) (C). Data presented as mean ± s.d. *P < 0.05, **P < 0.01 vs. control.
Table 1.
Electrophysiological measures for all experimental interventions
| Group | CMAP (b-p; mV) | CMAP (p-p; mV) | SMUP (p-p; µV) | MUNE (#) |
|---|---|---|---|---|
| Control | 41.7 (3.1) | 60.8 (3.2) | 136.9 (30.2) | 466.8 (130.0) |
| C26 | 37.2 (5.3) | 55.0 (7.2) | 293.6 (163.8)* | 231.5 (108.5)** |
| Control | 42.4 (1.8) | 71.6 (4.9) | 158.8 (33.8) | 469.4 (113.8) |
| MC38 | 40.4 (3.8) | 63.3 (4.7)* | 372.2 (226.7)* | 230.1 (129.2)** |
| Control | 55.4 (1.3) | 80.92 (7.0) | 106.6 (28.9) | 795.6 (191.8) |
| HCT116 | 53.8 (1.3) | 88.4 (6.0) | 335.8 (120.4)** | 304.2 (158.6)** |
| Control | 43.1 (3.5) | 60.6 (2.4) | 150.7 (32.4) | 416.6 (86.6) |
| Folfiri | 40.9 (3.6) | 68.3 (9.9)* | 331.7 (182.6)** | 243.6 (90.7)** |
| Control | 43.4 (3.3) | 69.4 (7.7) | 129.3 (19.9) | 549.7 (115.0) |
| Cisplatin | 41.2 (2.4) | 66.4 (4.8) | 309.8 (124.9)** | 241.7 (87.0)** |
Baseline-to-peak (b-p); peak-to-peak (p-p).
P < 0.05 compared to experimental controls;
P < 0.01 compared to experimental controls.
Figure 5.

Loss of MUNE is associated with muscle wasting and weakness caused by cancer and chemotherapy. Gastrocnemius (GSN) normalized to initial body weight (IBW) correlated with motor unit number estimation (MUNE) (A) of male mice (8 weeks) bearing subcutaneous C26 (1.0 × 106 cells/mouse in sterile saline), MC38 (1.0 × 106 cells/mouse in sterile saline) or HCT116 (3.0 × 106 cells/mouse in sterile saline) colorectal cancers and mice receiving intraperitoneal injections of folfiri or cisplatin. Control animals received equal amounts of empty vehicle for all experiments. In vivo plantarflexion force (expressed as mN·m) correlated with MUNE (B). Data presented as mean ± s.d. *P < 0.05, **P < 0.01, ***P < 0.001 for pearson r value.
Cancer and chemotherapy disrupt NMJ innervation
Given that our in vivo measurements suggested that muscle wasting and weakness were accompanied by a functional loss of motor unit innervation, we decided to molecularly investigate proteins associated with NMJs. In particular, we assessed MuSK, LRP4, Dok7 and rapsyn, all proteins crucial to the formation and stability of NMJs [28]. As HCT116 hosts and cisplatin-treated mice demonstrated the strongest MUNE correlations for both muscle mass and weakness amongst their respective interventions, these experimental groups were selected for molecular analysis. Western blot analysis revealed that HCT116 hosts had reduced protein levels of MuSK (-15%, P < 0.01) and Rapsyn (-28%, P < 0.05) compared to control animals, while LRP4 and Dok7 were unchanged (Figure 6A). Interestingly, chemotherapy also appeared to cause changes in NMJ-associated proteins as cisplatin-treated mice exhibited reductions in MuSK (-29%, P < 0.05), LRP4 (-26%, P < 0.05) and Dok7 (-27%, P < 0.05) compared to control, while Rapsyn was unchanged (Figure 6A). In addition to measuring NMJ-associated proteins, we also wanted to gain insight into whether cancer and chemotherapy alter innervation of motor endplates. Interestingly, staining of EDL muscle longitudinal sections demonstrated that both HCT116 hosts and cisplatin-treated mice had loss of the presynaptic axonal terminal of the NMJ, suggesting possible denervation in these cachectic mice (Figure 6B).
Figure 6.
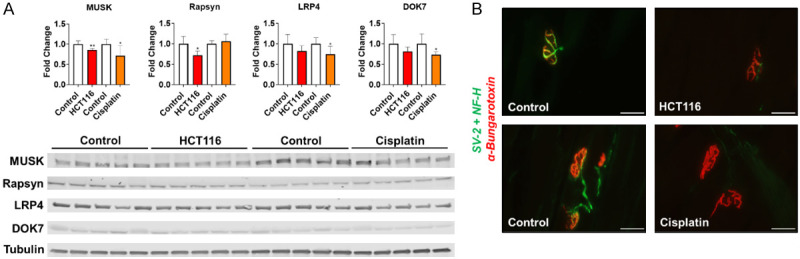
Cancer and Chemotherapy promote loss of NMJ-associated proteins. (A) Representative western blotting and quantification (expressed as fold change vs. control) for MuSK, Rapsyn, LRP4, Dok7 and Tubulin from gastrocnemius muscles of male mice (8 weeks) bearing subcutaneous HCT116 (3.0 × 106 cells/mouse in sterile saline) colorectal cancer and mice receiving intraperitoneal injections of cisplatin. Control animals received equal amounts of empty vehicle for all experiments. 40 × images of SV-2, NF-H and alpha-Bungarotoxin staining of EDL muscles (B). Scale bars: 10 µm. Data presented as mean ± s.d. *P < 0.05, **P < 0.01 vs. control.
Discussion
Cancer remains an elusive global health concern, which will take the lives of over 600,000 individuals in the US alone this year [1]. One of the most debilitating complications of cancer is the progressive decline in skeletal muscle mass and strength, which impedes quality of life and is ultimately directly responsible for approximately 30% of cancer-related deaths [2,3]. We and others have demonstrated that commonly used chemotherapeutic drugs independently promote skeletal muscle wasting and weakness [11-13,29,30]. This represents a crucial area of cancer research, especially keeping in mind that no approved treatments for the skeletal muscle wasting and weakness that occur in cancer patients are currently available. Thus, it is essential to better understand the underlying causes of skeletal muscle wasting and weakness in a setting of chemotherapy treatment to improve overall quality of life and survival in cancer patients.
Though assessment of skeletal muscle mass is certainly an important measure for assessing cachexia in cancer patients and in experimental models of cachexia, it could be argued that assessment of skeletal muscle weakness is of greater importance. In fact, recent work has identified that muscle weakness and fatigue cannot be fully explained by reductions in muscle mass and that muscle weakness may precede the presence of muscle wasting [14,15,31]. Moreover, this is of particular interest given that skeletal muscle weakness and fatigue often persists in cancer survivors for several months, or even years following cancer remission [7,32]. Thus, in the present study we wanted to assess skeletal muscle weakness at both in vivo and ex vivo levels in several models of experimental cachexia. Our present study recapitulates and builds upon our prior findings that growth of CRC tumors (C26, MC38, HCT116) and administration of the chemotherapeutics folfiri and cisplatin promotes skeletal muscle weakness at both in vivo (plantarflexion torque) and ex vivo (EDL contractility) levels [11,12,23,24,33].
Currently, the mechanisms that underly muscle weakness caused by cancer and chemotherapy are not fully understood. Some of the more interrogated mediators include inflammation, mitochondrial dysfunction, and metabolic regulators, yet emerging evidence implicates that cancer and chemotherapy may cause deleterious alterations to motor neurons and NMJs, and possibly induce denervation [17-19]. Importantly, a key factor of muscle strength is the total number of functional motor units (i.e., motor neuron and its innervated myofibers). Despite the current suggestion that the neuromuscular system may be altered by cancer and chemotherapy, to our knowledge investigation into motor unit number has not been carried out in experimental cachexia.
Here, using an established incremental stimulation technique we have demonstrated that cancer and chemotherapy not only cause muscle weakness, but that they also cause a functional loss of motor unit number (MUNE). Indeed, all three CRC tumors, as well as folfiri and cisplatin, resulted in a reduced MUNE value compared to control animals. Moreover, further analysis revealed that MUNE was correlated with muscle size and in vivo muscle force production, while NMJ staining revealed a loss of presynaptic axonal terminal components. Taken together, our data suggests that loss of motor unit connectivity in cachectic mice may be in part responsible for the muscle wasting and weakness observed.
Our observations are consistent with recently published work examining functional electrophysiological indices in cachectic mice. Indeed, recent work from Brown et al. demonstrated that the commonly used Lewis lung carcinoma (LLC) model of cachexia led to reduced axon diameter and conduction velocity of the sciatic nerve, suggesting impaired motor neuron function as a possible cause of muscle weakness [17]. Moreover, Daou et al. demonstrated upregulated indices of denervation in mice bearing C26 tumors [18], whereas recent work from Huertas et al. revealed alterations in NMJ-associated proteins in rats exposed to the chemotherapeutic doxorubicin [19]. However, motor unit number was not assessed in any of these studies and a number of discrepancies warrant further interrogations. For example, Brown et al. and Daou et al. reported no significant changes in gene expression of the pivotal scaffold protein and NMJ organizer, MuSK, in gastrocnemius muscles of female LLC tumor hosts, or rectus abdominus muscles of female C26 tumor hosts, respectively [17,18]. On the contrary, Huertas et al. demonstrated significant upregulation of MuSK at both gene and protein levels in female rats treated with doxorubicin [19]. This is in contrast to our present findings demonstrating that MuSK levels were reduced in both male HCT116 tumor hosts and male mice treated with cisplatin. In addition, our present results demonstrate that mice treated with cisplatin also had reduced protein expression of LRP4, a membrane protein and Dok7, a cytoplasmic protein, both of which form complexes with MuSK and aid in NMJ function. Meanwhile, prior evidence demonstrated that doxorubicin treated rats did not present changes in either of these proteins [19].
These discrepancies could be explained by several factors including the sex used (male vs. female), the choice and duration of chemotherapeutic treatments (doxorubicin: 48 hrs vs. cisplatin: 2 weeks) or the tumor model (C26: 3 weeks vs. LLC: 3 weeks vs. HCT116: 30 days) used, the skeletal muscle assessed (rectus abdominus vs. soleus vs. gastrocnemius), and the model species (rat vs. mouse). It should be noted that despite the molecular signature differences between our current work and prior published studies, all of the mice bearing CRC or receiving chemotherapy in the current study displayed similar skeletal muscle wasting, weakness and functional declines in MUNE, regardless of study duration (C26: 2 weeks; MC38: 4 weeks; HCT116: 30 days; folfiri: 5 weeks; cisplatin: 2 weeks).
Despite demonstrating a functional loss of MUNE across multiple experimental models of CRC and chemotherapy-induced cachexia, this study is not without limitations. In particular, the current study was constrained to male mice. We identified molecular differences of the current study compared to previous published work in female mice, and though there are several other variables (tumor model, chemotherapy, muscle examined) to consider, whether parallel reductions in MUNE occur in female mice exposed to cancer or chemotherapy remains to be elucidated. In addition, in this study we only performed electrophysiological assessment at a single time point. Future studies should employ electrophysiological time course assessments to gain insight on whether changes in MUNE precede functional skeletal muscle weakness.
Overall, our study demonstrates that mice bearing CRC or receiving chemotherapy experience muscle wasting and weakness that are associated with decreased MUNE, suggestive of a loss of motor unit connectivity. In addition, we show reductions of NMJ-associated proteins and a loss of pre-synaptic components in mice exposed to CRC and chemotherapy. Our data suggests that a functional loss of motor units contributes to the observed muscle wasting and weakness in experimental models of cachexia. Future studies should employ assessment of motor unit number when examining treatments for skeletal muscle wasting and weakness in models of chemotherapy- and cancer-induced cachexia.
Acknowledgements
This study was supported by the Department of Surgery and the Department of Otolaryngology-Head & Neck Surgery at Indiana University, and by grants from the Showalter Research Trust, the V Foundation for Cancer Research (V2017-021), and the American Cancer Society (Research Scholar Grant 132013-RSG-18-010-01-CCG) to A.B. J.R.H. was supported by a T32 Institutional Training Grant from NIH (AR065971). The #12G10 anti-Tubulin monoclonal antibody (developed by Frankel J. and Nelsen E.M. at the University of Iowa) and the SV2 anti-synaptic vesicle monoclonal antibody (developed by Buckley K.M. at Harvard Medical School) were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Bi-ology, Iowa City, IA.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 2012;16:153–166. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 3.Melstrom LG, Melstrom KA Jr, Ding XZ, Adrian TE. Mechanisms of skeletal muscle degradation and its therapy in cancer cachexia. Histol Histopathol. 2007;22:805–814. doi: 10.14670/HH-22.805. [DOI] [PubMed] [Google Scholar]
- 4.Dewys WD, Begg C, Lavin PT, Band PR, Bennett JM, Bertino JR, Cohen MH, Douglass HO Jr, Engstrom PF, Ezdinli EZ, Horton J, Johnson GJ, Moertel CG, Oken MM, Perlia C, Rosenbaum C, Silverstein MN, Skeel RT, Sponzo RW, Tormey DC. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am J Med. 1980;69:491–497. doi: 10.1016/s0149-2918(05)80001-3. [DOI] [PubMed] [Google Scholar]
- 5.Hayes S, Battistutta D, Newman B. Objective and subjective upper body function six months following diagnosis of breast cancer. Breast Cancer Res Treat. 2005;94:1–10. doi: 10.1007/s10549-005-5991-z. [DOI] [PubMed] [Google Scholar]
- 6.Luctkar-Flude M, Groll D, Woodend K, Tranmer J. Fatigue and physical activity in older patients with cancer: a six-month follow-up study. Oncol Nurs Forum. 2009;36:194–202. doi: 10.1188/09.ONF.194-202. [DOI] [PubMed] [Google Scholar]
- 7.Mustian KM, Peppone LJ, Palesh OG, Janelsins MC, Mohile SG, Purnell JQ, Darling TV. Exercise and cancer-related fatigue. US Oncol. 2009;5:20–23. [PMC free article] [PubMed] [Google Scholar]
- 8.Sun L, Quan XQ, Yu S. An epidemiological survey of cachexia in advanced cancer patients and analysis on its diagnostic and treatment status. Nutr Cancer. 2015;67:1056–1062. doi: 10.1080/01635581.2015.1073753. [DOI] [PubMed] [Google Scholar]
- 9.von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers-update 2014. J Cachexia Sarcopenia Muscle. 2014;5:261–263. doi: 10.1007/s13539-014-0164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, Bonetto A. Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front Physiol. 2016;7:472. doi: 10.3389/fphys.2016.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barreto R, Waning DL, Gao H, Liu Y, Zimmers TA, Bonetto A. Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget. 2016;7:43442–43460. doi: 10.18632/oncotarget.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Essex AL, Pin F, Huot JR, Bonewald LF, Plotkin LI, Bonetto A. Bisphosphonate treatment ameliorates chemotherapy-induced bone and muscle abnormalities in young mice. Front Endocrinol (Lausanne) 2019;10:809. doi: 10.3389/fendo.2019.00809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huot JR, Essex AL, Gutierrez M, Barreto R, Wang M, Waning DL, Plotkin LI, Bonetto A. Chronic treatment with multi-kinase inhibitors causes differential toxicities on skeletal and cardiac muscles. Cancers (Basel) 2019;11:571. doi: 10.3390/cancers11040571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson KM, Cole CL, Dunne RF. From bench to bedside: updates in basic science, translational and clinical research on muscle fatigue in cancer cachexia. Curr Opin Clin Nutr Metab Care. 2021;24:216–222. doi: 10.1097/MCO.0000000000000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VanderVeen BN, Hardee JP, Fix DK, Carson JA. Skeletal muscle function during the progression of cancer cachexia in the male Apc(Min/+) mouse. J Appl Physiol (1985) 2018;124:684–695. doi: 10.1152/japplphysiol.00897.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheth KA, Iyer CC, Wier CG, Crum AE, Bratasz A, Kolb SJ, Clark BC, Burghes AHM, Arnold WD. Muscle strength and size are associated with motor unit connectivity in aged mice. Neurobiol Aging. 2018;67:128–136. doi: 10.1016/j.neurobiolaging.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown JL, Lawrence MM, Ahn B, Kneis P, Piekarz KM, Qaisar R, Ranjit R, Bian J, Pharaoh G, Brown C, Peelor FF 3rd, Kinter MT, Miller BF, Richardson A, Van Remmen H. Cancer cachexia in a mouse model of oxidative stress. J Cachexia Sarcopenia Muscle. 2020;11:1688–1704. doi: 10.1002/jcsm.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daou N, Hassani M, Matos E, De Castro GS, Costa RGF, Seelaender M, Moresi V, Rocchi M, Adamo S, Li Z, Agbulut O, Coletti D. Displaced myonuclei in cancer cachexia suggest altered innervation. Int J Mol Sci. 2020;21:1092. doi: 10.3390/ijms21031092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huertas AM, Morton AB, Hinkey JM, Ichinoseki-Sekine N, Smuder AJ. Modification of neuromuscular junction protein expression by exercise and doxorubicin. Med Sci Sports Exerc. 2020;52:1477–1484. doi: 10.1249/MSS.0000000000002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arnold WD, Sheth KA, Wier CG, Kissel JT, Burghes AH, Kolb SJ. Electrophysiological motor unit number estimation (MUNE) measuring compound muscle action potential (CMAP) in mouse hindlimb muscles. J Vis Exp. 2015:52899. doi: 10.3791/52899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McComas AJ, Fawcett PR, Campbell MJ, Sica RE. Electrophysiological estimation of the number of motor units within a human muscle. J Neurol Neurosurg Psychiatry. 1971;34:121–131. doi: 10.1136/jnnp.34.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huot JR, Novinger LJ, Pin F, Narasimhan A, Zimmers TA, O’Connell TM, Bonetto A. Formation of colorectal liver metastases induces musculoskeletal and metabolic abnormalities consistent with exacerbated cachexia. JCI Insight. 2020;5:e136687. doi: 10.1172/jci.insight.136687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huot JR, Pin F, Essex AL, Bonetto A. MC38 tumors induce musculoskeletal defects in colorectal cancer. Int J Mol Sci. 2021;22:1486. doi: 10.3390/ijms22031486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huot JR, Novinger LJ, Pin F, Bonetto A. HCT116 colorectal liver metastases exacerbate muscle wasting in a mouse model for the study of colorectal cancer cachexia. Dis Model Mech. 2020;13:dmm043166. doi: 10.1242/dmm.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huot JR, Pin F, Narasimhan A, Novinger LJ, Keith AS, Zimmers TA, Willis MS, Bonetto A. ACVR2B antagonism as a countermeasure to multi-organ perturbations in metastatic colorectal cancer cachexia. J Cachexia Sarcopenia Muscle. 2020;11:1779–1798. doi: 10.1002/jcsm.12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tse N, Morsch M, Ghazanfari N, Cole L, Visvanathan A, Leamey C, Phillips WD. The neuromuscular junction: measuring synapse size, fragmentation and changes in synaptic protein density using confocal fluorescence microscopy. J Vis Exp. 2014:52220. doi: 10.3791/52220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blasco A, Gras S, Modol-Caballero G, Tarabal O, Casanovas A, Piedrafita L, Barranco A, Das T, Pereira SL, Navarro X, Rueda R, Esquerda JE, Caldero J. Motoneuron deafferentation and gliosis occur in association with neuromuscular regressive changes during ageing in mice. J Cachexia Sarcopenia Muscle. 2020;11:1628–1660. doi: 10.1002/jcsm.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zong Y, Jin R. Structural mechanisms of the agrin-LRP4-MuSK signaling pathway in neuromuscular junction differentiation. Cell Mol Life Sci. 2013;70:3077–3088. doi: 10.1007/s00018-012-1209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hain BA, Xu H, Wilcox JR, Mutua D, Waning DL. Chemotherapy-induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun. 2019;2:e00075. [PMC free article] [PubMed] [Google Scholar]
- 30.Hulmi JJ, Nissinen TA, Rasanen M, Degerman J, Lautaoja JH, Hemanthakumar KA, Backman JT, Ritvos O, Silvennoinen M, Kivela R. Prevention of chemotherapy-induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J Cachexia Sarcopenia Muscle. 2018;9:417–432. doi: 10.1002/jcsm.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanderveen BN, Fix DK, Counts BR, Carson JA. The effect of wheel exercise on functional indices of cachexia in tumor-bearing mice. Med Sci Sports Exerc. 2020;52:2320–2330. doi: 10.1249/MSS.0000000000002393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meeske K, Smith AW, Alfano CM, McGregor BA, McTiernan A, Baumgartner KB, Malone KE, Reeve BB, Ballard-Barbash R, Bernstein L. Fatigue in breast cancer survivors two to five years post diagnosis: a HEAL study report. Qual Life Res. 2007;16:947–960. doi: 10.1007/s11136-007-9215-3. [DOI] [PubMed] [Google Scholar]
- 33.Bonetto A, Rupert JE, Barreto R, Zimmers TA. The colon-26 carcinoma tumor-bearing mouse as a model for the study of cancer cachexia. J Vis Exp. 2016:54893. doi: 10.3791/54893. [DOI] [PMC free article] [PubMed] [Google Scholar]