

Abstract
Niclosamide, an established anti-helminthic drug, has anticancer activity against various cancers including prostate cancer, but the underlying mechanisms have not yet been defined. We demonstrated the anticancer effects of niclosamide in castration-resistant prostate cancer (CRPC) cells, and elucidated the mechanism of action of niclosamide in CRPC. Niclosamide reduced cell proliferation and induced apoptosis of CRPC cells in vitro, and also reduced xenograft tumor growth in vivo. Niclosamide significantly increased the number of γH2AX- and 53BP1-positive cells. In RNA-sequencing, niclosamide induced extensive changes in gene expression including cell division, DNA replication, and DNA repair. Bioinformatics analysis using TCGA data set revealed that FOXM1 is an important target of niclosamide. In microarray assays, FOXM1 knockdown significantly inhibited several genes involved in DNA repair, and homologous recombination, in particular. Finally, FOXM1 strongly bound to EXO1 in CRPC cells, and FOXM1 knockdown significantly reduced EXO1-driven luciferase activity. Taken together, our results suggest that niclosamide exerts anticancer activity through inhibition of the FOXM1-mediated DNA damage response in CRPC.
Keywords: Castration-resistant prostate cancer, niclosamide, FOXM1, EXO1, DNA damage response
Introduction
Prostate cancer (PCa) is the most prevalent male cancer in 103 countries, and the main cause of cancer-related death in 29 countries [1]. Androgen deprivation therapy (ADT) is the frontline treatment option for advanced or recurrent PCa [2]. Most patients initially respond well to ADT, but unfortunately almost all cases eventually progress to fatal castration-resistant prostate cancer (CRPC), with a mean survival of 9-13 months [3]. Clinically available therapeutic options for CRPC have expanded rapidly over the past 10 years; however, some patients are primarily resistant to these novel agents, and others develop resistance to novel agents after treatment. In addition, CRPC is associated with significantly increased economic burden, averaging $417,862.92 per overall survival benefit of 20.1 months, or $20,789.20 per additional month of survival [4]. Therefore, the development of effective anticancer drugs for CRPC with lower overall costs remains a key goal.
Niclosamide is an FDA-approved old oral anti-helminthic drug that has been used worldwide to treat millions of patients with tapeworms [5]. Recent accumulating evidence indicates that niclosamide has broad clinical applications for the treatment of several diseases including cancers, bacterial and viral infections, and rheumatoid arthritis [6]. In CRPC, niclosamide has been considered as a promising anticancer agent that inhibits androgen receptor (AR) splicing variant 7 (AR-V7), which is one of the feature of fatal prognosis and the main resistance mechanism to abiraterone and enzalutamide [7]. Liu et al. reported that niclosamide has anticancer effects in PCa through downregulation of the protein expression of AR-V7 [8] and inhibition of the IL-6/STAT3/AR pathway [9]. However, additional mechanisms not involving the AR pathway have not yet been defined. Since CRPC is composed of cells that exhibit a range of AR expression levels [10], identifying a non-AR-dependent mechanism of action of niclosamide would be very valuable.
In this study, we describe for the first time that inhibition of the forkhead box M1 (FOXM1)-mediated DNA damage response by niclosamide inhibits proliferation and induces apoptosis of human CRPC in vitro and in vivo. These findings may have therapeutic value and result in significantly improved clinical outcomes for patients with CRPC.
Materials and methods
Reagents and cell cultures
Niclosamide and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. (St. Louis, MO). RWPE-1 (cells derived from histologically normal adult human prostate), DU145 (cells derived from AR-negative CRPC), VCaP (cells derived from AR-positive CRPC), and 22Rv1 (cells derived from AR-positive CRPC) cells were obtained from the American Type Culture Collection (Manassas, VA), and LNCaP (cells derived from hormone sensitive PCa) and PC-3 (cells derived from AR-negative CRPC) cells were obtained from Korean Cell Line Bank (Seoul, Republic of Korea). RWPE-1 cells were grown in keratinocyte serum-free medium containing 50 μg/mL bovine pituitary extract and 5 ng/mL epidermal growth factor. LNCaP, PC-3, and 22Rv1 cell lines were cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 units/mL penicillin, and 0.1 mg/mL streptomycin (Gibco). DU145 and VCaP cell lines were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% FBS, 100 units/mL penicillin, and 0.1 mg/mL streptomycin. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Cell viability assay
PC-3 (1×104/well) and 22Rv1 (2×104/well) cells were seeded in 96-well plates and after 24 hr of incubation, cells were incubated either with DMSO alone (0.1%) or with various concentrations of niclosamide (0.25-10 µM) for 48 hr in a CO2 incubator at 37°C. Cell viability was assessed using EZ-CYTOX (Daeil Lab Service Co. Ltd, Seoul, Republic of Korea) according to the manufacturer’s instructions.
Soft agar colony formation assay
Cells (5×103/well) were plated in the top agar layer in each well of a six-well culture plate with 0.4% top agar layer and 0.8% bottom agar layer (SeaPlaque Agarose, Lonza, Basel, Switzerland). The Medium, containing 2.5 µM of niclosamide or DMSO, was replaced every 3 days. After 4 weeks of growth, the colonies were fixed and stained with 0.1% crystal violet (Sigma-Aldrich). The number of colonies was counted.
Cell invasion assay
Cell invasion assays were carried out using 24-well transwell chambers with an 8 μm pore polycarbonate membrane insert (Corning Inc., Corning, NY). The inserts of the transwell chamber were coated with 25 µg/mL MatrigelTM (Corning). PC-3 cells were seeded into inserts at a density of 5×104 per insert in serum-free medium and then transferred to wells filled with the culture medium containing 10% FBS as a chemoattractant. The cells on the bottom of the membrane were fixed and stained using a 0.1% crystal violet solution (Sigma-Aldrich) and observed using a light microscope (Carl Zeiss Inc., Jena, Germany). The membranes were dissolved in 20% acetic acid and the solubilized color was measured at 570 nm.
Fluorescence-activated cell sorting (FACS)
PC-3 (2×105/well) and 22Rv1 (4×105/well) cells were seeded in 6-well plates and treated with 2.5 µM of niclosamide or DMSO control. At 24 or 48 hr post treatment, cells were harvested, and washed three times with phosphate-buffered saline (PBS). The cells were stained with Annexin V and propidium iodide (PI) according to the instructions of the FITC Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA) and analyzed using a FACS Canto II system (BD Biosciences).
Immunocytochemistry
Cells were grown on coverslips and incubated with 2.5 µM of niclosamide or DMSO control. After the 24 hr incubation, cells were fixed with 4% formaldehyde for 15 min, then permeabilized with 0.1% Triton-100 for 10 min. Samples were blocked and then incubated overnight with the primary anti-phospho-H2A histone family member X (Ser139; γH2AX; Millipore Sigma, Burlington, MA) and anti-p53-binding protein 1 (53BP1; NOVUS Biologicals, Centennial, CO). After washing with PBS, the slides were incubated with secondary antibody and DNA was stained with 4’,6-diamidino-2-phenylindole (DAPI). Confocal images were obtained using an LSM510 Meta confocal laser scanning microscope (Carl Zeiss Inc.).
RNA sequencing
RNA was extracted from PC-3 cells incubated with DMSO or 1 µM of niclosamide. Libraries were prepared from total RNA using the NEBNext Ultra Directional RNA-Seq Kit (New England BioLabs, Inc., Rowley, MA). The isolation of mRNA was performed using the Poly(A) RNA Selection Kit (LEXOGEN, Inc., Vienna, Austria). The isolated mRNAs were used for cDNA synthesis according to the manufacturer’s instruction. Subsequently, libraries were checked using the Agilent 2100 bioanalyzer (DNA High Sensitivity Kit, Agilent Technologies, Amstelveen, Netherlands) to evaluate the mean fragment size. High-throughput sequencing was performed as paired-end 100 sequencing using NovaSeq 6000 (Illumina, Inc., San Diego, CA). The RNA-Seq reads were mapped to the reference genome using TopHat [11], and gene expression levels were estimated using FPKM (fragments per kb per million reads) values by Cufflinks [12]. The FPKM values were normalized based on the quantile normalization method using EdgeR within R [13]. Data mining and graphic visualization were performed using ExDEGA (E-biogen, Inc., Seoul, Republic of Korea). Gene ontology (GO) analyses were performed using DAVID to identify the biological processes influenced by niclosamide.
Analysis of TCGA dataset
We applied the TCGAbiolinks package to download all RNA-Seq data and clinical data from Genomic Data Commons (GDC) data portal on June, 2020. These databases (The Cancer Genome Atlas Prostate Adenocarcinoma; TCGA_PRAD), which were generated using the Illumina HiSeq RNASeq platform, included the mRNA sequencing data of 496 prostate cancer and 52 adjacent non-tumorous prostate tissue samples and the corresponding clinical data for these samples. The raw expression data for each GDC dataset were normalized and processed, and differentially expressed genes (DEGs) in normal and tumor tissue were identified with TCGAbiolinks pipeline [14]. Survival analyses based on expression of individual queried genes were performed using cBioPortal (http://www.cbioportal.org) and TCGA gene expression data set (TCGA, Firehose Legacy).
Western blot analyses
Cell lysates were prepared with RIPA lysis buffer (Cell Signaling Technology, Danvers, MA). Proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked and subsequently incubated overnight at 4°C with specific primary antibodies against FOXM1 (GeneTex, Inc., Irvine, CA) and β-actin (Santa Cruz Biotechnology, Inc., Dallas, TX). The membranes were washed and then incubated with a horseradish peroxidase-conjugated horse anti-rabbit or anti-mouse IgG (GenDEPOT, Barker, TX). The protein bands were visualized using an ECL Kit (DoGenBio, Seoul, Republic of Korea) and detected using X-ray film.
Small-interfering RNA (siRNA) transfection
SiRNAs targeting FOXM1 and exonuclease 1 (EXO1) were designed and synthesized by Bioneer Co., Ltd. (Daejeon, Republic of Korea). The sequences of the siRNAs are presented in Table S1. The siRNA transfections were performed using Lipofectamine® 2000 transfection reagent (Invitrogen). A non-targeting siRNA (siNC) was purchased from Bioneer Co., Ltd. and used as the negative control.
Quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted with TRIzol® reagent (Invitrogen Life Technologies; Thermo Fisher Scientific, Inc., Waltham, MA). Total RNA (1 µg) was reverse transcribed to cDNA using the PrimeScriptTM RT Reagent Kit (Takara Bio Inc., Shiga, Japan) and PCR was carried out with the TB GreenTM Premix Ex TaqTM II (Takara Bio Inc.). Relative gene expression was determined by normalizing to β-actin using the 2-ΔΔCT method. Primer pairs are presented in Table S1.
cDNA microarray and data analyses
RNA was extracted from two sample sets of PC-3 cells transfected with siNC or siFOXM1. Then, cDNA microarrays were performed on an Affymetrix GeneChip Human Gene 2.0 ST Array (Affymetrix, Santa Clara, CA), which covers 40,716 coding transcripts and 11,086 long intergenic non-coding RNA transcripts. DEGs between the negative control and FOXM1-knockdown groups were identified as those with at least a 1.5-fold change.
Dual luciferase assay
We amplified the -2451/-1952 and -1030/-481 regions of the human EXO1 promoter and -1279/-780 regions of the human BLM promoter from 22Rv1 genomic DNA by PCR using primers containing the restriction sites KpnI and HindIII, respectively. The cloning primers are listed in Table S1. After restriction digestion, the fragment was cloned in the pGL-4.10[luc2] vector (Promega, Madison, WI) to generate pGL4-EXO1 P1 (-2451/-1952), pGL4-EXO1 P2 (-1030/-481), and pGL4-BLM P (-1279/-780). All constructs were verified by DNA sequencing. These reporter constructs were co-transfected with siNC or siFOXM1 using Lipofectamine® 2000 transfection reagent (Invitrogen). Cells were co-transfected with pRL-TK (Promega) to normalize the transfection efficiency. Luciferase activity was measured after incubation for 48 hr using the Dual-Luciferase Reporter Assay System (Promega).
Chromatin immunoprecipitation (ChIP) assay
Cells were incubated with niclosamide or DMSO for 12 or 24 hr and fixed in 1% formaldehyde for 15 min to allow crosslinking and then quenched with glycine. Cells were then washed and collected, followed by lysis in SDS buffer. The lysates were sonicated to shear the cross-linked DNA. Sonicated chromatin-protein complexes were incubated overnight with Dynabeads Protein G (Invitrogen Life Technologies) pre-conjugated with anti-FOXM1 (G-5; Santa Cruz Biotechnology Inc.) or normal rabbit IgG (Novus Biologicals, Littleton, CO) antibodies. Immunoprecipitated DNA was reverse cross-linked, purified and analyzed by PCR analysis. The PCR products were resolved on agarose gel visualized by Gel DocTM XR+ System (Bio-Rad, Hercules, CA). ChIP primers are presented in Table S1.
In vivo study
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Catholic University of Korea (CUMC-2018-0050-02). Five-week-old male BALB/c nude mice (body weight 20-25 g) were obtained from Orient Bio (Gyenggi, South Korea). The mice (n=6) were injected subcutaneously in the flank with 4×106 22Rv1 cells in 100 µL PBS with 100 µL Matrigel (Corning). When tumor volumes reached approximately 200 mm3, each mouse was randomly assigned to one of the treatment groups: 150 µL of 1% DMSO and 1% Tween-80 in PBS (daily by oral gavage), 25 mg/kg of niclosamide (daily by oral gavage), or 50 mg/kg of niclosamide (daily by oral gavage). Body weight and tumor size were measured through the experimental period at 3-day intervals, and tumor volume was calculated as width2 × length × 0.5. The mice were sacrificed after 4 weeks of treatment and tumors were harvested for further assays.
Histologic analyses
For histological analyses, paraffin sections were stained with hematoxylin and eosin. The paraffin sections were immunostained with the following primary antibodies: rabbit Ki-67 (Abcam, Cambridge, United Kingdom), cleaved caspase-3 (Cell Signaling Technology), FOXM1 (G-5; Santa Cruz Biotechnology Inc.), and EXO1 (MyBioSource, Inc., San Diego, CA). And HRP-conjugated secondary antibody was used. The color reaction of the HRP-conjugated antibodies was completed using 3,3’-diaminobenzidine tetrahydrochloride (DAB) (Vector laboratories Inc., Burlingame, CA). Counter staining was performed using hematoxylin. Tissue sections were examined under a light microscope (Carl Zeiss Inc.).
Statistical analyses
The results of in vitro experiments are expressed as mean ± SD (standard deviation), whereas the results of in vivo experiments are expressed as mean ± SE (standard error). Differences between values were analyzed using Student’s t test or one-way analysis of variance (ANOVA) test in IBM SPSS version 24.0 software (SPSS, Inc., Chicago, IL). P-values < 0.05 were considered statistically significant.
Results
Niclosamide induces apoptosis of human CRPC cells
To determine the anticancer effect of niclosamide of CRPC cells, PC-3 and 22Rv1 cells were incubated with various concentrations of niclosamide and the effects were assessed using cell viability and colony formation assays. Niclosamide significantly reduced the viability of PC-3 and 22Rv1 cells in a concentration-dependent manner (Figure 1A). The colony formation assay showed that 2.5 µM of niclosamide significantly reduced colony formation in both PC-3 and 22Rv1 cells (Figure 1B). To elucidate whether niclosamide induces apoptosis, we performed Annexin V-FITC/PI double-staining assays. As shown in Figure 1C, a niclosamide induced a considerable increase in apoptotic cells in a time-dependent manner. In LNCaP cells, niclosamide reduced cell viability in a concentration-dependent manner and increased apoptosis in a time-dependent manner (Figure S1).
Figure 1.
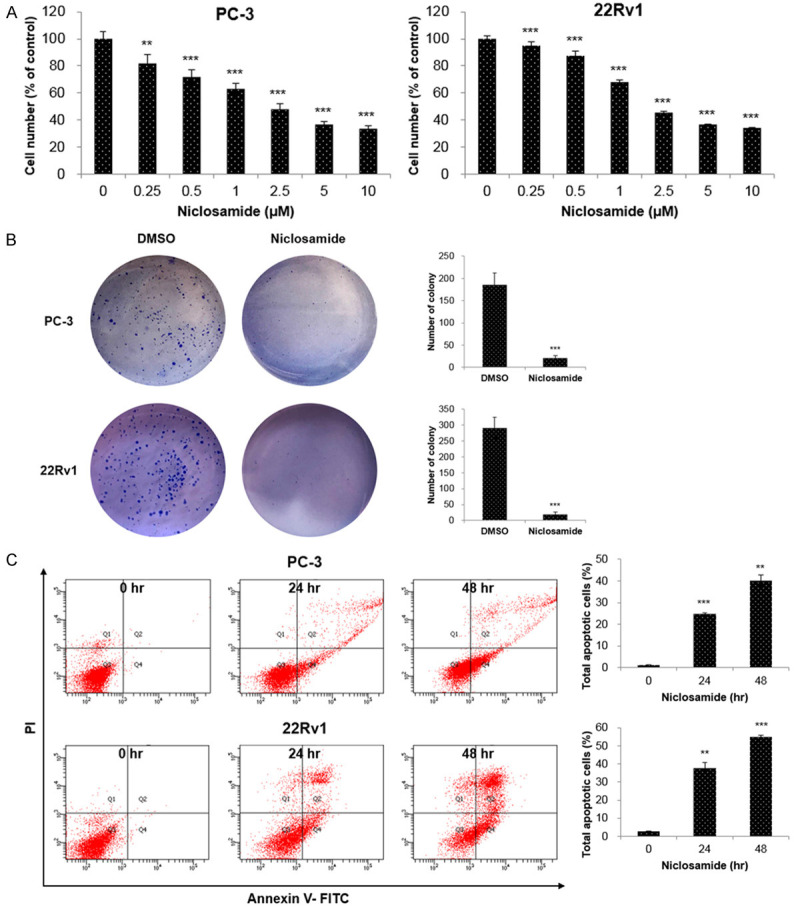
Niclosamide inhibits cell proliferation and induces apoptosis in CRPC cells. (A) PC-3 and 22Rv1 cells were treated with the indicated concentrations of niclosamide for 48 hr and cell viability was measured using the WST assay. (B) Colony formation assays were performed in PC-3 and 22Rv1 cells incubated with or without 2.5 µM niclosamide. The colonies were photographed and the number of colonies was counted. (C) PC-3 and 22Rv1 cells were incubated with 2.5 μM niclosamide for 24 and 48 hr. Apoptosis was determined by analyzing FITC Annexin V-PI staining. All data are presented as the mean 48 hr. Apoptosis was determined by analyzing: **P < 0.01, ***P < 0.001, two-tailed Student’s t test or one-way ANOVA test.
Niclosamide suppresses FOXM1-mediated DNA damage response
Because recent studies showed that consideration of DNA damage response pathways has the potential to improve diagnostic and therapeutic outcomes in PCa [15], we investigated whether niclosamide affects the DNA damage response. In light of these findings, we performed immunocytochemistry to assess the expression of γH2AX and 53BP1. In cells incubated with niclosamide, nuclear foci of γH2AX and 53BP1 increased compared to DMSO controls. Quantitative analyses revealed that the fluorescence intensity of γH2AX- and 53BP1-positive cells significantly increased in response to niclosamide (Figure 2A).
Figure 2.

Niclosamide induces DNA damage response in CRPC cells. (A) Nuclear γH2AX and 53BP1 foci were detected using immunofluorescence staining (γH2AX, green; 53BP1, red) and nuclei were counterstained with DAPI (blue). Scale bar, 10 µm. The fluorescence intensity of each group was analyzed using the ZEN 2012 Black Edition software. (B) Scatter plot of all expressed genes in PC-3 cells incubated with DMSO or niclosamide. Red dots indicated the up-regulated genes while blue dots indicated the down-regulated genes. The screening threshold is defined as a fold change > 2.0 and P < 0.05. (C) Dot plot of the significantly screened GO term of biological processes using DAVID. (D) Analysis of FOXM1 mRNA expression in tumor and adjacent normal tissues using the TCGA data set. (E) PC-3 and 22Rv1 cells were incubated with 2.5 μM niclosamide for 24 or 48 hr. FOXM1 mRNA expression was quantified using qRT-PCR. β-actin mRNA was used as an internal control to normalize the data. (F) Cell lysates were prepared after the same treatment as in (E). FOXM1 protein was detected by western blot analyses and β-actin was used as an internal control. Data in (A) and (E) are presented as the mean ± SD of two experiments performed in triplicate. **P < 0.01, ***P < 0.001, two-tailed Student’s t test or one-way ANOVA test.
To determine which genes were modulated by niclosamide, we performed RNA-sequencing. Niclosamide induced extensive changes in gene expression, up-regulating 904 genes and down-regulating 1,101 genes (fold change > 2, p-value < 0.05) in Figure 2B. The down-regulated genes were assessed using GO analyses and twenty GO terms (biological process) were significantly screened including cell division, DNA replication, and DNA repair (Figure 2C). Among the top 20 pathways, we focused on the DNA repair pathway. A total of 44 genes were identified from the DNA repair pathway, and we evaluated the clinical importance of each gene using the TCGA data set. Among these 44 genes in the DNA repair pathway, the expression of 10 genes including FOXM1 was significantly different in tumor tissue compared to adjacent normal tissue (Figures 2D, S3), and 3 genes (CDK1, P=0.027; FOXM1, P=0.011; EXO1, P < 0.001) were associated with poor overall survival in the TCGA data set (Table S2). Considering fold changes in DEGs, these results allowed us to postulate that FOXM1 is an important target of niclosamide. Finally, we performed qRT-PCR and western blot analyses, and found that niclosamide inhibited the expression of FOXM1 in PCa cell lines (Figures 2E, 2F and S2). These results suggest that niclosamide inhibits the DNA damage response in CRPC cells by reducing FOXM1 expression.
FOXM1 silencing modulates expression of DNA-repair-related genes
Little is known about the biological role of FOXM1 and relevant transcriptional targets in CRPC. FOXM1 expression was increased in PCa cells (LNCaP, DU-145, PC-3, VCaP and 22Rv1) compared to normal prostate epithelial cells (RWPE-1) (Figure S4A). To identify the biological role of FOXM1 in CRPC cells, we designed siRNA for FOXM1 to correspond to the nucleotides 112-130 (siFOXM1-1), 801-819 (siFOXM1-2) and 2073-2091 (siFOXM1-3) (Figure 3A). These three siRNAs were transfected into PC-3 and 22Rv1 cells for 48 hr. The expression of both FOXM1 RNA and protein was lower in siFOXM1-2-transfected cells than in cells transfected with the other siRNAs (Figure 3B, 3C). We selected siFOXM1-2 for further experiments. FOXM1 knockdown resulted in significantly reduced viability of CRPC cells in a time-dependent manner (Figure 3D).
Figure 3.
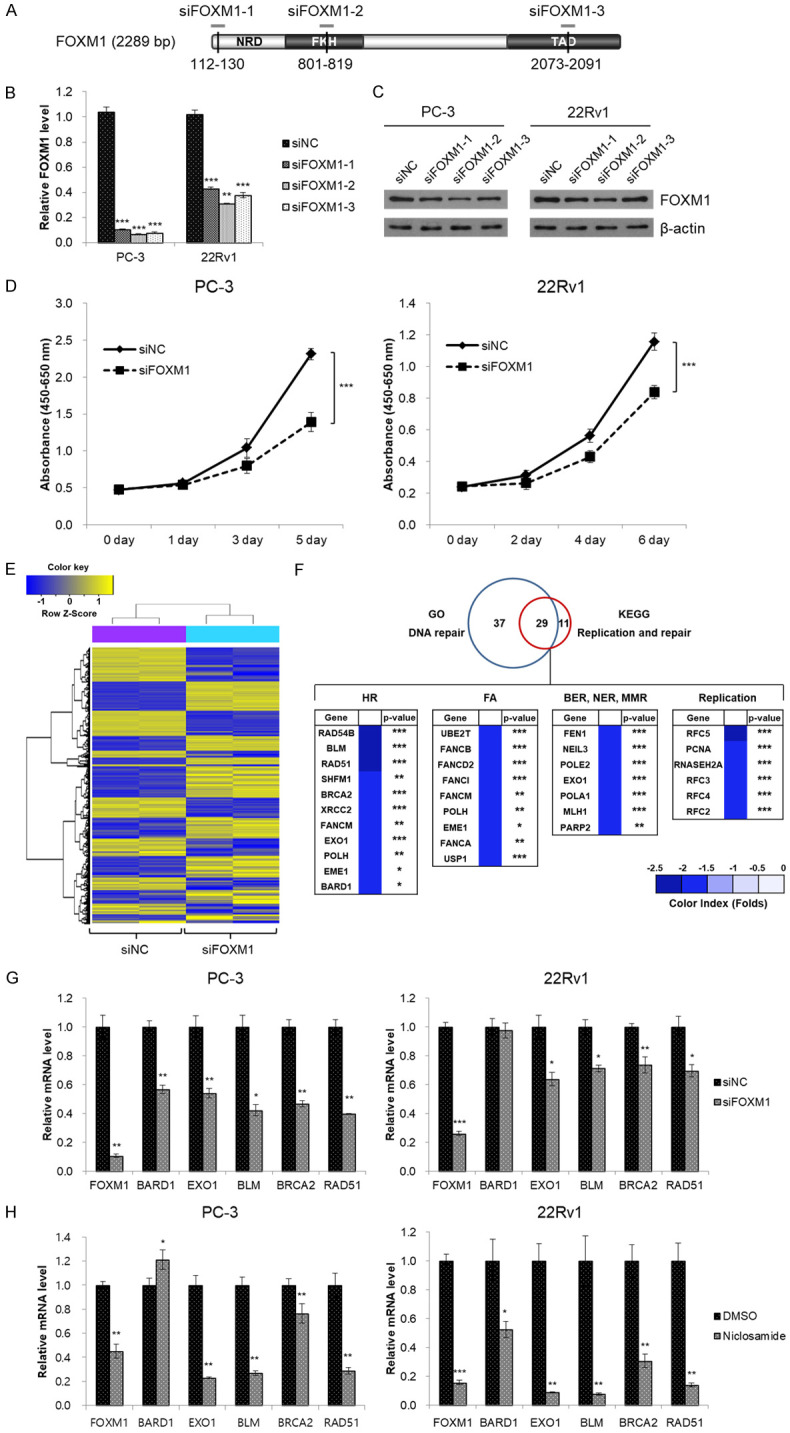
Knockdown of FOXM1 attenuates cell proliferation in CRPC cells and regulates expression of DNA-repair-related genes. (A) Schematic of FOXM1 mRNA including individual sites of three siRNA binding sites. (B, C) PC-3 and 22Rv1 cells were transiently transfected with three FOXM1 siRNAs (siFOXM1-1, siFOXM1-2 and siFOXM1-3, respectively) or negative control siRNA (siNC). After 48 hr of transfection, FOXM1 mRNA (B) and protein (C) expression was determined by qRT-PCR and western blot analyses, respectively. (D) PC-3 and 22Rv1 cells were transfected with siFOXM1 or siNC. Cell viability was measured using the WST assay at the indicated time point. (E) Heat map of differentially expressed genes (fold change > 1.5; P < 0.05) in PC-3 cells transfected with siFOXM1 or siNC. (F) Venn diagram of differentially expressed genes identified using gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. Overlap between the two circles represents 29 genes, DNA-repair-related genes. HR: homologous recombination; FA: Fanconi anemia; BER: base excision repair; NER: nucleotide excision repair; MMR: mismatch repair. (G, H) PC-3 and 22Rv1 cells were transfected with siFOXM1 for 48 hr (G) or were incubated with 2.5 μM niclosamide for 48 hr (H). mRNA expression was quantified using qRT-PCR. β-actin mRNA was used as an internal control to normalize the data. Data in (B, D, G, and H) are presented as the mean sed as an internal control to normalize the data. RP < 0.01, ***P < 0.001, two-tailed Student’s t test or one-way ANOVA test.
In order to identify the transcriptional targets of FOXM1 in CRPC cells, we performed microarray analyses after suppression of FOXM1 expression using siRNA. A total of 1,592 DEGs, including 824 up-regulated and 768 down-regulated DEGs, were identified after FOXM1 knockdown (P < 0.05, fold-change > 1.5; Figure 3E). Gene-enrichment and functional annotation analyses for DEGs were performed to obtain a functional interpretation of the gene expression changes using GO and KEGG pathway. The down-regulated DEGs were mainly involved in the DNA repair pathway, specifically in homologous recombination (HR; Figure 3F). After integrating information from the TCGA database and a PubMed search, five genes (BARD1, EXO1, BLM, BRCA2, and RAD51) were selected and validated in vitro. The qRT-PCR results showed that the expressions of EXO1, BLM, BRCA2, and RAD51 were reduced after FOXM1 knockdown in both PC-3 and 22Rv1 cells (Figure 3G). These results were consistent with those obtained after niclosamide treatment (Figure 3H). Among these four genes, we selected EXO1 and BLM as meaningful candidates for transcription targets of FOXM1 based on correlation coefficients and clinical significance determined by overall and disease-free survival data from the TCGA database (Table S3). In addition, expression of EXO1 and BLM RNA increased significantly in CRPC cell lines (Figure S4A). Like FOXM1, relative RNA expression of EXO1 and BLM was significantly reduced after niclosamide treatment in a concentration-dependent manner (Figure S4B).
FOXM1 directly binds to the promoter region of EXO1
To elucidate whether FOXM1 regulates promoter activity of EXO1 and BLM in CRPC cells, we looked for potential FOXM1 binding sites, including TAAACA within the EXO1 and BLM promoter region. We found two potential FOXM1-binding sequences in the EXO1 promoter region and one potential FOXM1-binding sequence in the BLM promoter region. We made a promoter luciferase construct containing FOXM1-binding sequences, which were pGL4-EXO1 P1 (-2451/-1952), pGL4-EXO1 P2 (-1030/-481), and pGL4-BLM P (-1279/-780) (Figure 4A). The EXO1 and BLM promoter luciferase constructs were co-transfected with siFOXM1 in PC-3 cells. As shown in Figure 4B, EXO1 luciferase activity was reduced in FOXM1 knockdown cells compared to the control cells, while BLM1 luciferase activity did not change. To address direct binding of FOXM1 to the endogenous EXO1 promoter region, we performed ChIP assays with 22Rv1 cells. The results showed that FOXM1 binds to the predicted binding site of FOXM1 in the EXO1 promoter region and that binding is reduced after niclosamide treatment in a time-dependent manner (Figure 4C).
Figure 4.
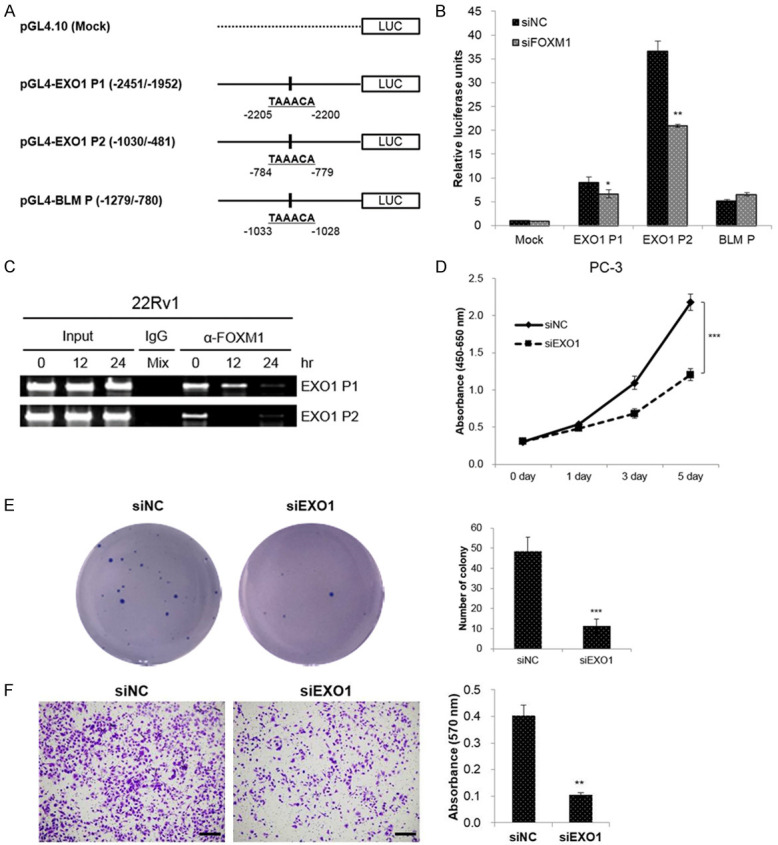
EXO1 is a direct transcriptional target of FOXM1. (A) Schematic structure of the promoter region of EXO1 and BLM, and the generated promoter reporter vectors. The nucleotide sequences in the right panel represent the predicted binding sequences. (B) PC-3 cells were co-transfected with the indicated luciferase reporter plasmids, and siFOXM1 or siNC. Luciferase activity was analyzed 48 hr later, and was normalized to renilla luciferase levels at each transfection. (C) 22Rv1 cells were treated with niclosamide (1 μM) for indicated period. The cells were fixed with formaldehyde and then immunoprecipitated with anti-FOXM1 antibody. Input loading controls was prepared prior to immunoprecipitation step. The immunoprecipitated DNA was amplified by PCR, using primers amplifying the FOXM1 binding site containing region, and resolved in 2% agarose gel. Normal rabbit IgG antibody was used as a control. (D) PC-3 cells were transfected with siEXO1 or siNC. Cell viability was measured using the WST assay at the indicated time. (E) Colony formation assays were performed in PC-3 cells transfected with siEXO1 or siNC. The colonies were photographed and the number of colonies was counted. (F) Transwell assays were performed to evaluate the influence of EXO1 knockdown on the invasive abilities of the PC-3 cells. Scale bar, 200 µm. The number of cells was calculated after crystal violet staining. Data in (B, D-F) are presented as the mean ± SD of two experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student’s t test.
To explore the biologic function of EXO1 in CRPC cells, several experiments were performed after EXO1 knockdown with siRNAs. Both cell viability and colony formation assays demonstrated that the proliferation of CRPC cells was significantly reduced compared to that in siNC cells (Figure 4D, 4E). Furthermore, transwell assays demonstrated that the invasive capability of CRPC cells was reduced after EXO1 knockdown (Figure 4F).
Niclosamide inhibits tumor growth in a CRPC xenograft mouse model
In order to confirm whether the anticancer effects of niclosamide observed in in vitro experiments are relevant in vivo, male BALB/c nude mice with 22Rv1 xenograft tumors were treated with vehicle, 25 mg/kg or 50 mg/kg of niclosamide. During the experimental period, niclosamide was well tolerated without any evidence of toxic reactions and weight changes were not significantly different among the three groups. Niclosamide significantly inhibited tumor growth and reduced final tumor weight in 22Rv1 xenograft tumors in a dose-dependent manner (Figure 5A, 5B).
Figure 5.
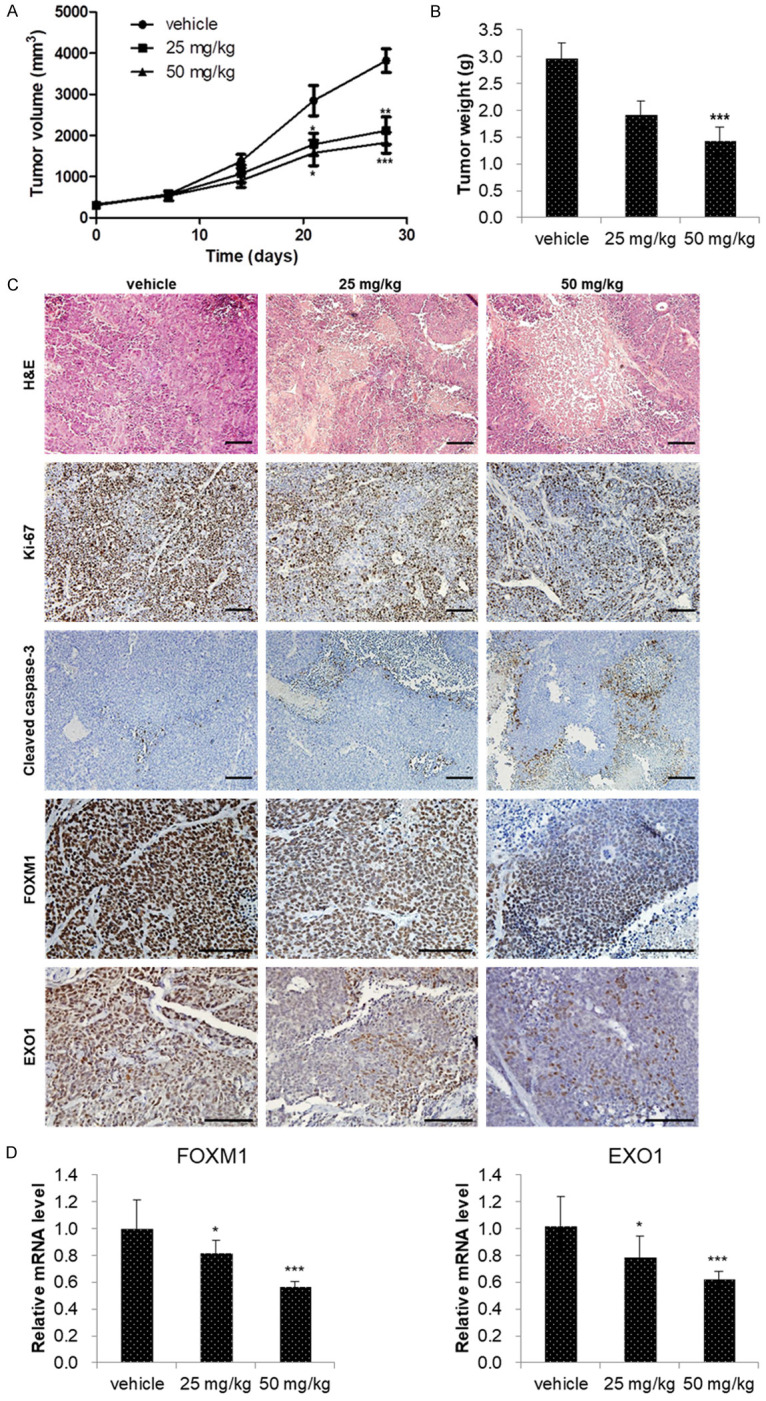
Niclosamide inhibited CRPC tumor growth in vivo. (A) Mice bearing 22Rv1 xenografts were treated with vehicle (1% DMSO and 1% Tween-80 in PBS), 25 mg/kg or 50 mg/kg of niclosamide for 4 weeks. Tumor volumes were measured twice weekly and the observed mean tumor volume are presented as the mean ± SE (n=6). (B) Mean tumor weights were measured after dissection at 28 days after implantation and data are presented as the mean ± SE (n=6). (C) The tumors were excised after 4 weeks of niclosamide treatment and processed for hematoxylin and eosin (H&E) and immunohistochemistry (IHC) staining. Images of H&E-stained paraffin-embedded tumor sections were captured using a microscope. Tumor tissues were processed for IHC staining for Ki-67, cleaved caspase-3, FOXM1, and EXO1 antibody (brown staining). Scale bar, 100 µm. (D) FOXM1 and EXO1 mRNA expression was determined using qRT-PCR. β-actin mRNA was used as an internal control to normalize the data. Data are presented as the mean ± SD (n=6). *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student’s t test or one-way ANOVA test.
We performed immunohistochemical staining to assess the expression of Ki-67, cleaved caspase-3, FOXM1, and EXO1 in xenograft tumors. In order to confirm the results of our in vitro experiments, we evaluated the expression of Ki-67 and cleaved caspase-3. In the niclosamide treatment group, expression of Ki-67 decreased and expression of cleaved caspase-3 increased compared to the control group. FOXM1 and EXO1 expressions in the niclosamide treatment group ranged from undetectable to low, whereas expression in the control group was much higher (Figure 5C). Finally, qRT-PCR revealed that niclosamide treatment significantly reduced the expression of FOXM1 and EXO1 RNA in xenograft tumors (Figure 5D).
Discussion
In recent years, niclosamide has emerged as a potential therapeutic agent for many solid cancers [6]. Our findings reveal that niclosamide, which is already known to have pharmacological properties, may serve as a novel therapeutic agent for CRPC because it inhibits proliferation and induces apoptosis of human CRPC cells in vitro and in vivo. Furthermore, our findings demonstrate that niclosamide exerts its anticancer effects through inhibition of the FOXM1-mediated DNA damage response. Our data suggest that niclosamide inhibits cell survival and efficient repair of DNA double-strand breaks (DSBs) in PCa cells. Niclosamide induced: (1) increased γH2AX and 53BP1 foci, suggesting increased DNA DSBs, and (2) reduced EXO1, BLM, BRCA2, and RAD51, suggesting suppressed HR, due to (3) inhibition of FOXM1 transcription factor. Ultimately, niclo-samide resulted in impaired DNA damage repair in PCa cells (Figure 6).
Figure 6.
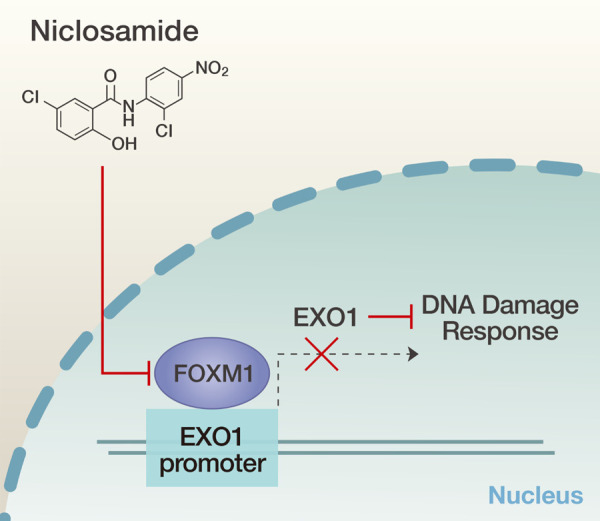
Schematic illustration of the suggested mechanism of niclosamide in CRPC cells.
Niclosamide is an anti-helminthic drug that has been shown to exhibit anticancer effects against several solid cancers including breast cancer, colorectal cancer, and liver cancer via various mechanisms of action [6]. Our understanding of the mechanistic basis of niclosamide treatment in PCa is limited, and has mainly been focused on the AR signaling pathway. Liu et al. demonstrated that niclosamide significantly reduced AR-V7 protein expression through a proteasome-dependent pathway [8]. Subsequently, they reported that niclosamide inhibited IL6-Stat3-AR pathway to overcome enzalutamide resistance in CRPC [9]. These previous studies suggested the great potential of niclosamide as a treatment option for CRPC patients who are AR-V7-positive. However, niclosamide also exhibited anti-cancer effects in AR-V7-negative LNCaP cells in our study (Figure S1). Thus, we hoped that the elucidation of a mechanism of action other than the AR signaling pathway would allow us to extend the indications of niclosamide to AR-V7-negative patients.
DNA damage response is defined as the coordinated cellular mechanisms responsible for preventing the accumulation of DNA damage and maintaining genomic stability [16]. Substantial evidence suggests that DNA damage response is important in PCa initiation, development, and progression [17,18]. In PCa, genetic alterations in the DNA damage response pathway are relatively common, having been reported in 22.7% of mCRPC patients [19]. In this regard, PARP inhibitors represent an emerging therapeutic option to target DNA damage response pathways in several solid cancers including PCa [20,21]. DNA damage can be repaired by various DNA damage response pathways, including HR, base excision repair (BER), and nucleotide excision repair (NER) [16]. Among these different pathways, we focused on HR because mutations of HR genes are known to be significant factors underlying cancer predisposition. We investigated the effects of niclosamide on the relative mRNA levels involved in HR pathways. Our results demonstrated that niclosamide significantly reduced the expression levels of BRCA2, RAD51, FOXM1, EXO1, and BLM which are important in HR pathways. The roles of RAD 51 or BRCA proteins have been widely studied, but the roles of FOXM1 or EXO1 in PCa remain poorly understood.
FOXM1 promotes the development and progression of PCa [22,23] and is upregulated in PCa tissues [24]. In a previous study, we found that elevated FOXM1 expression was associated with aggressiveness and poor prognosis in patients with PCa [25]. FOXM1 directly binds to the FOXM1 binding motif of the cell division cycle 6 (CDC6) promoter and PSA promoter/enhancer regions to regulate progression of PCa [26,27]. Interestingly, binding occurs regardless of the presence of androgen. Therefore, we speculate that FOXM1 is an important novel androgen-independent target in CRPC.
FOXM1 is a transcriptional regulator that directly or indirectly regulates HR-related genes. FOXM1 binds directly to the promoter to regulate transcription of Nijmegen breakage syndrome protein 1 (NBS1), BRCA2, RAD51, BRCA1-interacting protein 1 (BRIP1), and EXO1, and consequently promotes HR repair [28-32]. FOXM1 can also indirectly enhance HR repair by promoting transcription of S-phase kinase-associated protein 2 (Skp2), because Skp2 interacts with NBS1 and triggers K63-linked ubiquitination of NBS1 [33,34]. In particular, the relationship between FOXM1 and DNA repair genes has been scarcely studied in PCa. Mazzu et al. reported that FOXM1 directly regulates transcription of ribonucleotide reductase small subunit M2 (RRM2) [35] in PCa. RRM2 is one of the ribonucleotide reductases, which are essential for DNA synthesis and DNA repair but are not HR-related genes.
To the best of our knowledge, for the first time, we have demonstrated that FOXM1 and its target gene EXO1 are major factors influencing PCa. Even in other malignancies, little is known about the relationship between FOXM1 and EXO1. Zhou et al. reported that FOXM1 directly binds to the promoter region of EXO1 and facilitates DNA repair to protect ovarian cancer cells from cisplatin-mediated apoptosis [32]. Quist et al. demonstrated that a four-gene decision tree signature consisting of TP53BP2, RSU1, FOXM1, and EXO1 identified a subgroup of breast cancers sensitive to platinum-based chemotherapy [36]. These results indicate that inhibition of the FOXM1/EXO1 axis could be used as a therapeutic and diagnostic target in several malignancies, including PCa.
In conclusion, we found that the antihelminthic drug niclosamide inhibits proliferation and induces apoptosis of human CRPC cells in vitro and in vivo by inhibition of the FOXM1-mediated DNA damage response. Furthermore, we demonstrated that FOXM1, the therapeutic target of niclosamide, directly regulates EXO1 expression to promote the DNA repair pathway. This is the first study to reposition niclosamide for treatment of CRPC by regulating DNA repair pathways, and the first to confirm the significance and relationship of FOXM1 and its target gene EXO1 in PCa.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. 2017R1D1A1B03030051).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, Dandona L, Fleming T, Forouzanfar MH, Hancock J, Hay RJ, Hunter-Merrill R, Huynh C, Hosgood HD, Johnson CO, Jonas JB, Khubchandani J, Kumar GA, Kutz M, Lan Q, Larson HJ, Liang X, Lim SS, Lopez AD, MacIntyre MF, Marczak L, Marquez N, Mokdad AH, Pinho C, Pourmalek F, Salomon JA, Sanabria JR, Sandar L, Sartorius B, Schwartz SM, Shackelford KA, Shibuya K, Stanaway J, Steiner C, Sun J, Takahashi K, Vollset SE, Vos T, Wagner JA, Wang H, Westerman R, Zeeb H, Zoeckler L, Abd-Allah F, Ahmed MB, Alabed S, Alam NK, Aldhahri SF, Alem G, Alemayohu MA, Ali R, Al-Raddadi R, Amare A, Amoako Y, Artaman A, Asayesh H, Atnafu N, Awasthi A, Saleem HB, Barac A, Bedi N, Bensenor I, Berhane A, Bernabe E, Betsu B, Binagwaho A, Boneya D, Campos-Nonato I, Castaneda-Orjuela C, Catala-Lopez F, Chiang P, Chibueze C, Chitheer A, Choi JY, Cowie B, Damtew S, das Neves J, Dey S, Dharmaratne S, Dhillon P, Ding E, Driscoll T, Ekwueme D, Endries AY, Farvid M, Farzadfar F, Fernandes J, Fischer F, TT GH, Gebru A, Gopalani S, Hailu A, Horino M, Horita N, Husseini A, Huybrechts I, Inoue M, Islami F, Jakovljevic M, James S, Javanbakht M, Jee SH, Kasaeian A, Kedir MS, Khader YS, Khang YH, Kim D, Leigh J, Linn S, Lunevicius R, El Razek HMA, Malekzadeh R, Malta DC, Marcenes W, Markos D, Melaku YA, Meles KG, Mendoza W, Mengiste DT, Meretoja TJ, Miller TR, Mohammad KA, Mohammadi A, Mohammed S, Moradi-Lakeh M, Nagel G, Nand D, Le Nguyen Q, Nolte S, Ogbo FA, Oladimeji KE, Oren E, Pa M, Park EK, Pereira DM, Plass D, Qorbani M, Radfar A, Rafay A, Rahman M, Rana SM, Soreide K, Satpathy M, Sawhney M, Sepanlou SG, Shaikh MA, She J, Shiue I, Shore HR, Shrime MG, So S, Soneji S, Stathopoulou V, Stroumpoulis K, Sufiyan MB, Sykes BL, Tabares-Seisdedos R, Tadese F, Tedla BA, Tessema GA, Thakur JS, Tran BX, Ukwaja KN, Uzochukwu BSC, Vlassov VV, Weiderpass E, Wubshet Terefe M, Yebyo HG, Yimam HH, Yonemoto N, Younis MZ, Yu C, Zaidi Z, Zaki MES, Zenebe ZM, Murray CJL, Naghavi M. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524–548. doi: 10.1001/jamaoncol.2016.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pagliarulo V, Bracarda S, Eisenberger MA, Mottet N, Schroder FH, Sternberg CN, Studer UE. Contemporary role of androgen deprivation therapy for prostate cancer. Eur Urol. 2012;61:11–25. doi: 10.1016/j.eururo.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65:1180–1192. doi: 10.1111/j.1742-1241.2011.02799.x. [DOI] [PubMed] [Google Scholar]
- 4.Pollard ME, Moskowitz AJ, Diefenbach MA, Hall SJ. Cost-effectiveness analysis of treatments for metastatic castration resistant prostate cancer. Asian J Urol. 2017;4:37–43. doi: 10.1016/j.ajur.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearson RD, Hewlett EL. Niclosamide therapy for tapeworm infections. Ann Intern Med. 1985;102:550–551. doi: 10.7326/0003-4819-102-4-550. [DOI] [PubMed] [Google Scholar]
- 6.Chen W, Mook RA Jr, Premont RT, Wang J. Niclosamide: beyond an antihelminthic drug. Cell Signal. 2018;41:89–96. doi: 10.1016/j.cellsig.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sobhani N, Generali D, D’Angelo A, Aieta M, Roviello G. Current status of androgen receptor-splice variant 7 inhibitor niclosamide in castrate-resistant prostate-cancer. Invest New Drugs. 2018;36:1133–1137. doi: 10.1007/s10637-018-0653-2. [DOI] [PubMed] [Google Scholar]
- 8.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2014;20:3198–3210. doi: 10.1158/1078-0432.CCR-13-3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu C, Lou W, Armstrong C, Zhu Y, Evans CP, Gao AC. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate. 2015;75:1341–1353. doi: 10.1002/pros.23015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crnalic S, Hornberg E, Wikstrom P, Lerner UH, Tieva A, Svensson O, Widmark A, Bergh A. Nuclear androgen receptor staining in bone metastases is related to a poor outcome in prostate cancer patients. Endocr Relat Cancer. 2010;17:885–895. doi: 10.1677/ERC-10-0059. [DOI] [PubMed] [Google Scholar]
- 11.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silva TC, Colaprico A, Olsen C, D’Angelo F, Bontempi G, Ceccarelli M, Noushmehr H. TCGA workflow: analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Res. 2016;5:1542. doi: 10.12688/f1000research.8923.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, van Gent DC, Incrocci L, van Weerden WM, Nonnekens J. Correction to: role of the DNA damage response in prostate cancer formation, progression and treatment. Prostate Cancer Prostatic Dis. 2020;23:194. doi: 10.1038/s41391-019-0153-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karanika S, Karantanos T, Li L, Corn PG, Thompson TC. DNA damage response and prostate cancer: defects, regulation and therapeutic implications. Oncogene. 2015;34:2815–2822. doi: 10.1038/onc.2014.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tapia-Laliena MA, Korzeniewski N, Hohenfellner M, Duensing S. High-risk prostate cancer: a disease of genomic instability. Urol Oncol. 2014;32:1101–1107. doi: 10.1016/j.urolonc.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, Stephens PJ, Mosquera JM, Cronin MT, Rubin MA. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920–926. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, Kunju LP, Hussain M, Feng FY, Tomlins SA, Cooney KA, Smith DC, Brennan C, Siddiqui J, Mehra R, Chen Y, Rathkopf DE, Morris MJ, Solomon SB, Durack JC, Reuter VE, Gopalan A, Gao J, Loda M, Lis RT, Bowden M, Balk SP, Gaviola G, Sougnez C, Gupta M, Yu EY, Mostaghel EA, Cheng HH, Mulcahy H, True LD, Plymate SR, Dvinge H, Ferraldeschi R, Flohr P, Miranda S, Zafeiriou Z, Tunariu N, Mateo J, Perez-Lopez R, Demichelis F, Robinson BD, Schiffman M, Nanus DM, Tagawa ST, Sigaras A, Eng KW, Elemento O, Sboner A, Heath EI, Scher HI, Pienta KJ, Kantoff P, de Bono JS, Rubin MA, Nelson PS, Garraway LA, Sawyers CL, Chinnaiyan AM. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, Seed G, Jain S, Ralph C, Protheroe A, Hussain S, Jones R, Elliott T, McGovern U, Bianchini D, Goodall J, Zafeiriou Z, Williamson CT, Ferraldeschi R, Riisnaes R, Ebbs B, Fowler G, Roda D, Yuan W, Wu YM, Cao X, Brough R, Pemberton H, A’Hern R, Swain A, Kunju LP, Eeles R, Attard G, Lord CJ, Ashworth A, Rubin MA, Knudsen KE, Feng FY, Chinnaiyan AM, Hall E, de Bono JS. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalin TV, Wang IC, Ackerson TJ, Major ML, Detrisac CJ, Kalinichenko VV, Lyubimov A, Costa RH. Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006;66:1712–1720. doi: 10.1158/0008-5472.CAN-05-3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Y, Balli D, Ustiyan V, Fulford L, Hiller A, Misetic V, Zhang Y, Paluch AM, Waltz SE, Kasper S, Kalin TV. Foxm1 expression in prostate epithelial cells is essential for prostate carcinogenesis. J Biol Chem. 2013;288:22527–22541. doi: 10.1074/jbc.M113.455089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon FA. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim MY, Jung AR, Kim GE, Yang J, Ha US, Hong SH, Choi YJ, Moon MH, Kim SW, Lee JY, Park YH. High FOXM1 expression is a prognostic marker for poor clinical outcomes in prostate cancer. J Cancer. 2019;10:749–756. doi: 10.7150/jca.28099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Gong Z, Sun L, Li X. FOXM1 and androgen receptor co-regulate CDC6 gene transcription and DNA replication in prostate cancer cells. Biochim Biophys Acta. 2014;1839:297–305. doi: 10.1016/j.bbagrm.2014.02.016. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Liu Y, Yuan B, Yin L, Peng Y, Yu X, Zhou W, Gong Z, Liu J, He L, Li X. FOXM1 promotes the progression of prostate cancer by regulating PSA gene transcription. Oncotarget. 2017;8:17027–17037. doi: 10.18632/oncotarget.15224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khongkow P, Karunarathna U, Khongkow M, Gong C, Gomes AR, Yague E, Monteiro LJ, Kongsema M, Zona S, Man EP, Tsang JW, Coombes RC, Wu KJ, Khoo US, Medema RH, Freire R, Lam EW. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene. 2014;33:4144–4155. doi: 10.1038/onc.2013.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monteiro LJ, Khongkow P, Kongsema M, Morris JR, Man C, Weekes D, Koo CY, Gomes AR, Pinto PH, Varghese V, Kenny LM, Charles Coombes R, Freire R, Medema RH, Lam EW. The Forkhead Box M1 protein regulates BRIP1 expression and DNA damage repair in epirubicin treatment. Oncogene. 2013;32:4634–4645. doi: 10.1038/onc.2012.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan Y, Raychaudhuri P, Costa RH. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol Cell Biol. 2007;27:1007–1016. doi: 10.1128/MCB.01068-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang N, Wu X, Yang L, Xiao F, Zhang H, Zhou A, Huang Z, Huang S. FoxM1 inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA-repair gene Rad51. Clin Cancer Res. 2012;18:5961–5971. doi: 10.1158/1078-0432.CCR-12-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou J, Wang Y, Wang Y, Yin X, He Y, Chen L, Wang W, Liu T, Di W. FOXM1 modulates cisplatin sensitivity by regulating EXO1 in ovarian cancer. PLoS One. 2014;9:e96989. doi: 10.1371/journal.pone.0096989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu J, Zhang X, Zhang L, Wu CY, Rezaeian AH, Chan CH, Li JM, Wang J, Gao Y, Han F, Jeong YS, Yuan X, Khanna KK, Jin J, Zeng YX, Lin HK. Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol Cell. 2012;46:351–361. doi: 10.1016/j.molcel.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazzu YZ, Armenia J, Chakraborty G, Yoshikawa Y, Coggins SA, Nandakumar S, Gerke TA, Pomerantz MM, Qiu X, Zhao H, Atiq M, Khan N, Komura K, Lee GM, Fine SW, Bell C, O’Connor E, Long HW, Freedman ML, Kim B, Kantoff PW. A novel mechanism driving poor-prognosis prostate cancer: overexpression of the DNA repair gene, ribonucleotide reductase small subunit M2 (RRM2) Clin Cancer Res. 2019;25:4480–4492. doi: 10.1158/1078-0432.CCR-18-4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quist J, Mirza H, Cheang MCU, Telli ML, O’Shaughnessy JA, Lord CJ, Tutt ANJ, Grigoriadis A. A four-gene decision tree signature classification of triple-negative breast cancer: implications for targeted therapeutics. Mol Cancer Ther. 2019;18:204–212. doi: 10.1158/1535-7163.MCT-18-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.