

Abstract
Tumor immunotherapy, especially T cell based therapy, is becoming the main force in clinical tumor therapies. Bispecific T cell engager (BiTE) uses the single chain variable fragments (scFv) of two antibodies to redirect T cells to kill target cells. BiTEs for hematologic tumors has been approved for clinical use, and BiTEs for solid tumors showed therapeutic effects in clinical trials. Oncolytic viruses (OVs) of the adenovirus expressing p53 and herpes simplex virus expressing GM-CSF was approved for clinical use in 2003 and 2015, respectively, while other OVs showed therapeutic effects in clinical trials. However, BiTE and Oncolytic virus (OV) have their own limitations. We propose that OV-BiTE has a synergistic effect on tumor immunotherapy. Feng Yu et al. designed the first OV-BiTE in 2014, which remarkably eradicated tumors in mice. Here we review the latest development of the structure, function, preclinical studies and/or clinical trials of BiTE and OV-BiTE and provide perspective views for optimizing the design of OV-BiTE. There is no doubt that OV-BiTE is becoming an exciting new platform for tumor immunotherapy and will enter clinical trial soon. Exploring the therapeutic effects and safety of OV-BiTE for synergistic tumor immunotherapy will bring new hope to tumor patients.
Keywords: Bispecific T cell engager, oncolytic virus, tumor, immunotherapy
Introduction
Live bacteria was used for immunostimulatory treatment of tumors in 1893 as the beginning of tumor immunotherapy although its clinical effect was not satisfactory [1]. As our understanding of the immune system becomes more comprehensive, tumor immunotherapy is attracting more and more attention. Recently tumor immunotherapy, in combination with others, such as surgery therapy, chemotherapy, and radiotherapy, become one of the best methods to treat tumors [2]. Bispecific T-cell engager (BiTE) represents a novel type of immunotherapy. BiTE is a combination of two single chain fragment variables (scFv) from different antibodies, one targeting CD3, and the other targeting the antigen on the surface of cells, which are connected by a short flexible linker [3]. BiTE does not require major histocompatibility complex I (MHC I) for antigen presentation to activate T cells, form artificial immune synapses, and release perforin and granzyme to exert cytotoxic effects. BiTE’s mode of action independent of MHC I antigen presentation has an important impact on its application as an irreplaceable treatment option. BiTE have been successfully used clinically to treat hematological and solid tumors and may have great potential to treat other non-tumor diseases.
Oncolytic viruses (OVs) are the viruses that preferentially replicate in and lyse cancer cells. Oncolytic virus (OV) represent a multimodal mechanism immunotherapy [4]. Most of the OV that can be used are genetically modified to make them less cytotoxic to non-cancer cells but increase their ability to target cancer cells. OV provoke pro-inflammatory and immunostimulatory environment by increasing antigen release, antigen recognition and subsequent immune activation to prevent immune escape of cancer cells [4-6]. H101 adenovirus type 5 with E1B-55 KD and partial E3 gene deletions was the first OV allowed to be marketed in China in 2003. The antitumor effect of H101 combined with chemotherapy is better than that of chemotherapy alone [7].
Specific attention should be paid to the synergistic effect of OV-BiTE because it should work better than either OV or BiTE alone and has the advantage compared with other combination therapies. Viruses co-evolve with their hosts symbiotically with complex strategies. The anti-tumor activity of OVs depends on their direct oncolytic effect and participation in innate and adaptive immune responses [8]. However, the high heterogeneity of malignant cells and the antiviral response induced by the interferon (IFN) signal transduction pathway makes the therapeutic effect of OV monotherapy limited. Therefore, a new strategy that can minimize the antiviral immune response and stimulate the anti-tumor immune response is needed [9]. The large and highly stable oncolytic DNA virus genome made them possible to insert large foreign genes without reducing their self-replication and oncolytic properties. Therefore, OVs can be used as a genetic engineering platform for expressing transgenes, such as BiTE, which enables successful combination of different immunotherapies as one therapeutic agent [10,11]. BiTE has its unique therapeutic advantages as a specific tumor immunotherapy without the requirement of MHC-1 antigen presentation. However, the small molecular weight and short half-life of BiTEs requires their continuous infusion, which may cause adverse reactions due to the non-tumor expression of tumor associated antigens, such as CEA and EGFR. When OVs are used as a transgene platform to express BiTEs, the selective replication of OVs in tumors can limit the expression of BiTEs in tumors, thus reduce the possibility of non-tumor side effects of BiTEs. Therefore OV-BiTE has a synergistic effect. In addition, OV-BiTE has the advantages of less time consuming, and low cost compared with chimeric antigen receptor T (Car-T) cells because it does not require genetic modification of T cells to obtain anti-tumor specificity.
Therefore, this article systemically reviews the latest development of the structure, function, preclinical studies and clinical trials of BiTE and OV-BiTE in tumor immunotherapy and immunotherapy of other diseases. This review will also provide perspective views for optimizing the design to get maximum advantages of OV-BiTE synergistic tumor immunotherapy. This review may have significant implication for future research and development of tumor immunotherapy.
The structure, function, preclinical studies and clinical trials of BiTE
The structure and function of BiTE
Antibodies are large Y-shaped proteins utilized by the immune system to recognize and neutralize foreign substances, such as germs, viruses, infected or mutated cells. The antigen binding sites of antibodies can recognize specific antigens, directly neutralize the antigens or mark the antigens for other immune responses. The smallest unit of an immunoglobulin molecule with antigen-binding activity is a variable fragment (Fv). The single chain Fv (scFv) refers to an engineered antibody fragment composed of a flexible peptide linker connecting the variable regions of the heavy chain (VH) and the light chain (VL). The length, composition and conformation of the flexible peptide linker is essential for the correct folding of the polypeptide chain without serious steric interference. The linker must contain hydrophilic sequences to avoid the insertion of peptides into or between variable domains during the entire protein folding process. The most used linker contains Gly (glycine or G) and Ser (serine or S) residues or interspersed with some charged residues (such as Glu and Lys) to improve solubility, such as the motif (G4S)n [12,13] and (G2S1) 4GG [14]. BiTE contains the scFv fragments of two distinct monoclonal antibodies connected with a flexible peptide linker, which allows the two scFv arms to rotate freely for binding to the targeting antigen and the CD3 molecule of T cells [13]. The most used linker for BiTE to connect two scFvs is GGGGS with some special linkers, such as SGSG [15] and ASTGS [16].
The traditional BiTE does not have an Fc domain, has a molecular weight of about 55 KD and is excreted through kidney, thus the half-life is as short as 1-4 hours [17]. The short half-life and inability to self-expand means that continuous systemic infusion is required, and the lack of effective biodistribution may be related to toxicity [18]. At present BiTE can also be engineered to better integrate with the human immune system. For example, adding Fc domain to BiTE will increase the molecular weight and prolong the half-life to ~7 days [19]. AMG757 is a BiTE with Fc domain. In non-human primate models, the half-life of AMG757 is estimated to be greater than 200 µh. In addition, the pharmacokinetic model predicts that its weekly dose in humans may be less [20].
BiTE binds to both malignant cells and T cells, and specifically guides the cytolytic T cells to malignant cells (Figure 1). After the cytolytic synapse is formed, T cells release perforin and granzyme B to lyse the malignant cell. The activation of T cells results in the transient release of cytokines to expand the immune response, leading to T cell proliferation and continuous killing of malignant cells [20]. The most common target of BiTE is tumor surface antigen of hematologic malignancies or solid tumors. There are also some studies that designed BiTE to target some tumor stroma components, such as CD206 and folate receptor β (FRβ) of tumor associated macrophages [21] and fibroblast activation protein (FAP) [10,22]. The side effects of systematically administrated BiTE targeting non-tumor cells are worthy of attention, while OV-BiTE seems to avoid such side effects [22]. The exciting therapeutic efficacy of BiTE has inspired some researchers to use this model to treat other diseases, such as human cytomegalovirus (HCMV) infection [23] and acquired immune deficiency syndrome (AIDS) [24].
Figure 1.
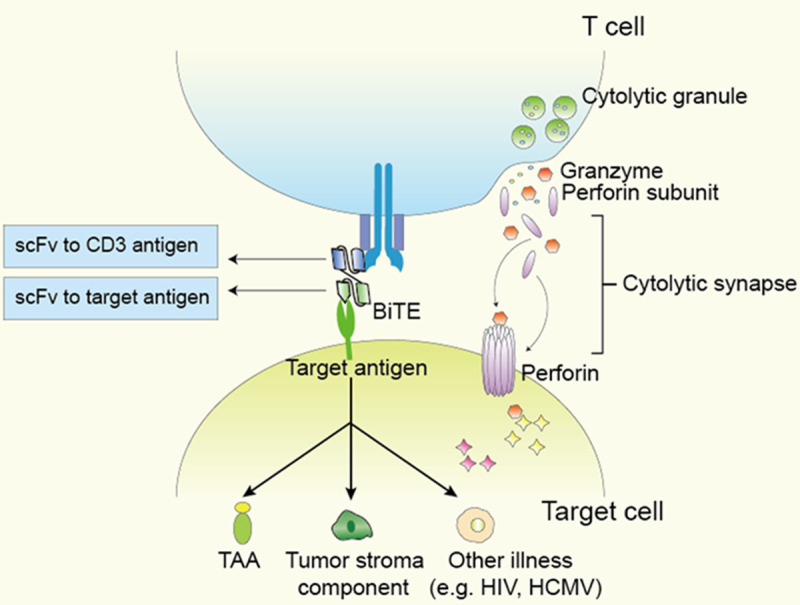
Structure and function of BiTE. BiTE simultaneously binds CD3 and target antigen to induce T cell activation and proliferation. Activated T cells release cytokine and cytolytic granules containing perforin and granzyme. Perforin forms a tube structure on target cell membrane, which allowed granzyme to enter target cells and induce programmed cell death of target cells. Currently designed BiTE can target tumor associated antigens (TAA), tumor stromal cells and non-tumor pathogens, such as human immunodeficiency virus (HIV) and human cytomegalovirus (HCMV).
Preclinical studies of BiTE
Next, we will elaborate on the relevant preclinical studies of BiTE against different target proteins of hematopoietic tumors, solid tumors, and non-tumor diseases (Table 1).
Table 1.
Preclinical studies of BiTE
| Indication | scFv1-target | scFv2-target | scFv1 linker | scFv2 linker | scFv1-scFv2 linker | Model | Model experiment results | Ref |
|---|---|---|---|---|---|---|---|---|
| AML | CD33 | CD3 | NR | NR | NR | Immunodeficient mice | Volume decreases by about 1000 mm3 | [27] |
| AML | CD123 | CD3 | NR | NR | NR | Xenograft mice model | Leukemia eradication | [28] |
| AML | CLEC12A | CD3 | NR | NR | NR | Human xenograft mouse model | Median survival increases 14.5 days | [29] |
| MM | BCMA | CD3ε | NR | NR | NR | L-363 xenograft model | Median survival increases 9 days | [30] |
| MM | A2/NY-ESO-1157 | CD3ε | NR | NR | SGSG | Tumor-bearing mice model | Tumor growth was significantly suppressed | [15] |
| Solid tumors | EpCAM (CD326) | CD3 | NR | NR | NR | 4T1 orthotopic breast cancer | Reduction of volume was 82% after treatment with 5 μg/kg | [40] |
| Solid tumors | mu-EpCAM (CD326) | CD3 | NR | NR | NR | NOD/SCID mouse models | Tumors eliminate | [32] |
| Solid tumors | CEA | CD3ε | (G4S) 3 | NR | NR | Xenograft mice model | Significantly inhibited tumor growth | [45] |
| HCC | Glypican 3 (GPC3) | CD3 | (G4S) 3 | (G4S) 3 | G4S | Mice model | Tumor weight decreases about 1 g | [48] |
| HCC | TfR | CD3 | (G4S) 3 | (G4S) 3 | ASTGS | Xenograft model | Volume decreases by about 1000 mm3 | [16] |
| SCLC | DLL3 | CD3 | NR | NR | NR | Orthotopic mice model | Tumor regression | [20] |
| GBM | EGFRvIII | CD3 | (G4S) 3 | (G4S)3 | G4S | Orthotopic xenograft models | Durable complete cure at rates up to 75% | [57] |
| Solid tumors | GD2 | CD3 | NR | NR | NR | Co-culture | Effectively killed GD2-positive neuroblastoma cell lines | [59] |
| Solid tumors | PSMA | CD3ε | NR | NR | NR | Immunodeficient NOD/SCID mice model | Complete tumor eradication | [65] |
| Solid tumors | CLDN6 | CD3 | (G4S) 3 | (G4S) 3 | (G4S) 3 | NSG mice xenograft tumors model | Nearly complete tumor eradication | [26] |
| Solid tumors | EphA2 | CD3 | (G4S) 3 | (G4S) 3 | NR | SCID xenograft model | 2 log or greater reduction in their tumor signal | [18] |
| Both hematopoietic tumors and solid tumors | CD206 | CD3ε | NR | NR | GGGGS | Malignant ascites fluids | Triggering robust T cell activation and cytotoxicity | [21] |
| Both hematopoietic tumors and solid tumors | FRβ | CD3ε | NR | NR | GGGGS | Malignant ascites fluids | Greatly diminished | [21] |
| Both hematopoietic tumors and solid tumors | ESK1 | CD3ε | NR | NR | NR | NSG mice model | Survival time extended by about one month | [71] |
| Both hematopoietic tumors and solid tumors | B7H6 | CD3ε | (G4S) 3 | (G4S) 3 | (G4S) 3 | Co-cultured | Produce IFN-γ, not pro-inflammatory monocytes | [70] |
| HCMV | gB | CD3 | NR | NR | NR | Co-cultured | Secrete IFN-γ and TNF | [23] |
| AIDS | gp120 | CD3ε | (G4S) 3 | (G4S) 3 | SG4S | Co-cultured | >90% HIV inhibition at 1 μg/ml | [24] |
NR, not reported; ALL, acute lymphoblastic leukemia; MM, multiple myeloma; SCLC, small cell lung cancer; AML, acute myelocytic leukemia; HCC, hepatocellular carcinoma; GBM, glioblastoma; AIDS, acquired immunodeficiency syndrome; HCMV, human cytomegalovirus.
Hematopoietic tumors
Blinatumomab is the first BiTE approved by the food and drug administration (FDA) for the therapy of acute lymphoblastic leukemia (ALL) [25]. Blinatumomab connects CD3 scFv and CD19 scFv through a GS linker. Another blood disease, acute myeloid leukemia (AML), is currently undergoing BiTE research. The occurrence of immunodeficiency in AML patients makes other immunotherapies unsatisfactory. AML cells will lose MHC I expression, but the recognition and activation of T cells by BiTE does not require the antigen presentation of MHC I molecules [26]. Based on the effective experience of Blinatumomab treatment, there are currently three targets that were used to treat AML: CD33, CD123, CLEC12A. The CD33/CD3-BiTE named AMG330 can activate and expand the T cells of primary AML patient samples and can also effectively mediate the redirected lysis of AML blasts and normal myeloid cells [27]. CD123/CD3-BiTE was tested to treat AML patients by targeting primary AML cell lines in an antigen-dependent manner, redirecting bystander T cells to AML cells, producing cytokines and killing AML cells [28]. Similarly, CLEC12AxCD3 BiTE was found to kill AML cells in a CLEC12A antigen-dependent manner and are non-toxic to hematopoietic progenitor cells [29]. In addition, the three BiTE showed remarkable tumor growth inhibitory effects in the in vivo experimental model of mice. A mouse experiment showed that AMG330 can drive T cells to infiltrate tumor tissues, and its efficacy may not be affected by the initial state of T cell infiltration of the tumors [27]. Interestingly, T cells have been genetically modified to secret bispecific engager molecules (ENG-T cells) targeting CD123 (CD123-ENG T cells) or CLEC12A (CLEC12A-ENG T cells), which showed increased anti-tumor efficiency. To eliminate ENG-T cells, CD123-ENG T cells were further modified to contain the CD20 suicide gene (CD20.CD123-ENG T cells), which did not reduce the anti-AML activity, but allowed rituximab-mediated ENG-T cell elimination [28,29].
The survival rate of patients with multiple myeloma (MM) has been significantly improved in the past decade, but relapsed and refractory patients are resistant to present therapies and poor prognosis. BiTE targeting B cell maturation antigen (BCMA) and NY-ESO-1 may become a new strategy for the treatment of MM. The activity of BiTE against BCMA (BI-836909) is not affected by soluble BCMA, bone marrow stromal cells and proliferation inducing ligand (APRIL). BI-836909 induces T cell activation, proliferation, the release of cytokines and selective lysis of BCMA positive MM cells, [30]. BiTE targeting HLA-A2/NY-ESO-1157-165 complex (A2/NY-ESO-1157) and BI 836909 produced a certain anti-tumor response to myeloma cells in mouse xenograft experiments in vivo [15].
Solid tumors
BiTE not only has exciting effects in hematological malignancies, but also shows certain effects in solid tumors [31]. Epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein widely expressed in gastric cancer, lung cancer, ovarian cancer, prostate cancer, and other epithelial tumors, but not expressed in lymphoma, melanoma, and schwannoma. A BiTE targeting EpCAM was named MT110, which induced costimulation-independent polyclonal activation of CD8 and CD4 positive T cells. MT110 induces the specific lysis of EpCAM-positive tumor cells by redirecting unstimulated human peripheral blood T cells. MT110 was found to eliminate tumor tissues in both metastatic ovarian cancer and SW480 human colon cancer mouse models [32]. Following the activation by MT110, T cells lyse the target cells through pore formation, PARP lysis, caspase activation, and DNA fragmentation mediated by perforin and granzyme B. In addition, BiTE can also lengthen the contact time between malignant cells and lymphocytes [31]. Importantly, BiTE MT110 was shown kill tumor-initiating cells (TICs) or cancer stem cells (CSCs) [33]. A study showed that MT110 can lyse malignant cells derived from TICs of colorectal cancer. In the immunodeficiency mouse TIC xenograft tumor model, MT110 inhibited tumor growth by more than 5000-fold [34]. In a mouse model of primary pancreatic cancer, it was shown that MT110 can target and kill the highly carcinogenic CSC population of pancreatic cancer [35-39]. On the other hand, the expression of human and murine EpCAM protein in various tissues is similar in distribution and level. A BiTE (muS110) targeting mouse EpCAM and CD3 was studied. In the 4T1 orthotopic breast cancer model, the tumor volume of mice treated with 15 μg/kg and 5 μg/kg muS110 for 30 days decreased by 68% and 82%, respectively, and at the end of the experiment, one of the 6 mice in the 15 μg/kg treatment group and two of the 6 mice in the 5 μg/kg treatment group had tumors less than 0.1 cm3. However, treatment with 0.5 μg/kg and 1.5 μg/kg had no effect. In the CT-26 lung cancer mouse model, the number of lung colonies decreased with the increase of the therapeutic dose. The number of colonies was reduced by up to 63% with the injection dose of 12.5-400 μg/kg. The highest-dose group experienced short-term side effects, such as diarrhea, weight loss, and mild excitement/sedation. But the lower dose group did not have such side effects. These adverse effects may be mainly caused by acute T cell activation. Both mouse models described above showed significant anti-tumor activity when treated with muS110 at a dose as low as 5 μg/kg. The dose window of muS110 during the treatment of mice may be used for the prediction of MT110 in humans [31,40].
In multiple solid tumors (such as gastric cancer, colorectal cancer, breast cancer and non-small cell lung cancer) the common tumor biomarker glycoprotein carcinoembryonic antigen (CEA) is over-expressed [41-44]. A BiTE targeting CEA named AMG211 is a potentially effective treatment for solid tumors overexpressing CEA. The LS174T human colon cancer xenograft mouse model was transplanted with human CD3 positive T cells from healthy donors, and AMG211 was shown to inhibit tumor growth in a dose-dependent manner [45]. Another group showed that AMG211 binds to and kills CEA-positive cancer cells in a CEA-specific manner and in a positive correlation with the CEA antigen density, regardless of the mutation status [45]. The explants of tumors of colorectal cancer transferred from patients after chemotherapy can be lysed in vitro after injection of AMG211. Furthermore, AMG211 can be combined with immune checkpoint suppression therapy, which can produce stronger cytotoxicity to CEA-positive cancer cells [41]. Radioisotope labelled 89Zr-AMG211 was developed to show the systemic distribution and stability of AMG211 in vivo, and 89Zr-AMG211 was demonstrated to have a dose-dependent tumor uptake at 6 hours and can remain in the tumor tissue for at least 24 hours [46].
The incidence of hepatocellular carcinoma (HCC) has recently increased rapidly in Western and Asian countries. The most common and effective treatment option is surgery [47]. However, the 5-year survival rate of HCC patients is only about 10%, and the recurrence rate is very high [48]. It can be seen from the expression profile of HCC that the expression of glypican 3 (GPC3) is significantly increased [49]. During development, GPC3 is expressed in most tissues, but GPC3 expression in most adult tissues is inhibited by DNA methylation [48]. In a tumor xenograft mouse model, mice were inoculated with Huh-7 cells mixed with unstimulated fresh human PBMC. One hour after inoculation, mice were injected with 1 μg and 10 μg of anti-GPC3 BiTE for continuous treatment for 1-10 days. The results indicated that the tumors in mice injected with the doses of 1 μg and 10 μg of BiTE were effectively inhibited at the end of the study (22 days). The anti-tumor ability of GPC3/CD3 BiTE in vivo was shown to be target-dependent [48].
In addition, the transferrin receptor (TfR) is expressed at high levels on HCC and other tumor (precursor) cells, predicting poor prognosis and becoming a potential target for immunotherapy [50]. TfR-BiTE can activate CD8 and CD4 T cells and plays a crucial role in long-lasting cell-mediated immunity [16]. In the HCC HepG2 xenograft mouse model, low concentration of TfR-BiTE showed an inhibitory effect on tumor growth. On the 23rd day, the tumor volume of the TfR-BiTE group was significantly smaller than that of the other control groups of mice [16].
Small cell lung cancer (SCLC) is a common cause of death from cancer, and treatment options are limited with low more than 5 years survival rate [51,52]. Delta-like protein 3 (DLL3) has a higher expression level in SCLC tumors and a lower level in normal tissues, so it can be a potential target for SCLC immunotherapy [53,54]. AMG757 is a BiTE construct against DLL3 and fused with an Fc domain to extend the pharmacokinetic half-life. In in vitro experiments, low concentrations of AMG757 killed DLL3-positive SCLC (including SCLC cells with <1000 DLL3 molecules per cell) tumor cells by redirecting T cells. In the SCLC orthotopic model, the tumors were regressed after injection of low-dose AMG757. The half-life of AMG757 in non-human primate models is greater than 200 µh. In addition, the pharmacokinetic model predicts that it may be administered less frequently in humans per week. In non-clinical toxicology studies, AMG757 at a dose of 4.5 μmg/kg showed good tolerance [20].
Glioblastoma (GBM) has a median survival rate of only 15 months under the current treatment regimens [55]. Current treatment methods lack specificity and have limitations of the damage to the surrounding normal brain [56]. Immunotherapy specifically targeting malignant cells may be a potentially effective treatment option. The mutant form of EGF receptor (EGFR), EGFRvIII, is located at the center of the carcinogenic process. It is often expressed on the surface of glioblastoma and other common cancers but is not found in normal tissues [57,58]. Bryan D Choi et al. conducted preclinical evaluation of BiTE targeting EGFRvIII (bscEGFRvIIIxCD3). They reported that bscEGFRvIIIxCD3 mediates the effective antigen-specific lysis of EGFRvIII-expressing GBM by activating T cells at very low concentration (10 ng/mL) and the 2.5:1 ratio of effector to target in vitro. Mice with intracerebral tumors prolonged survival after intravenous injection of bscEGFRvIIIxCD3 (P<0.05) and achieved a complete and lasting cure of up to 75% [57].
The most common extracranial tumor in children is neuroblastoma. Stage 4 neuroblastoma in children of more than 18 months of age at diagnosis is uniformly aggressive and often recurrent following successful induction therapy [59]. Disialyl-ganglioside (GD2) is ubiquitously highly expressed on neuroblastoma cells [60,61]. Maxim Yankelevich et al. constructed a GD2×CD3 BiTE named 3F8BiAb and showed that 3F8BiAb can direct the cytotoxicity of redirected activated T cells (ATC) to GD2-positive neuroblastoma. The levels of chemokines RANTES, MIP-1α and MIP-1β, TNF-α and IFN-γ induced by 3F8BiAb are higher than those of ATC stimulated by cancer cells alone [59].
A large part of prostate cancer patients develops into advanced metastatic disease and become incurable [62,63]. About 20-40% of prostate cancer patients have biochemical recurrence within decade of treatment [64]. Antibodies that target prostate specific membrane antigen (PSMA) may become a supplement to prostate cancer treatment options [62]. A PSMA/CD3-BiTE named AMG212/BAY 2010112 was first constructed and found to effectively redirect T cells to PSMA-positive tumor cells, induce target cell-dependent activation of T cells and tumor cell lysis. When the concentration between 0.1 and 4 ng/mL, AMG212 lysed the human prostate cancer cell lines VCaP, C4-2, PC-3 huPSMA, MDA PCa 2b, 22Rv1 and LnCaP, but not the PSMA-negative human PCa cell lines. In the NOD/SCID mouse model, daily subcutaneous injection or intravenous injection of AMG212 can rapidly reduce tumors in mice and achieve complete remission. They also pointed out that subcutaneous injection of AMG212 may be more effective than intravenous administration [65]. Claudin 6 (CLDN6) is a meconium tight junction molecule, which is expressed only in the early developmental stage in human normal tissues and in human ovarian carcinoma and other tumors. Therefore, CLDN6 is a potential target for immunotherapy [66]. Christiane R Stadler et al. designed a CLDN6xCD3 BiTE named 6PHU3. Experiments have shown that 6PHU3 can significantly activate T cells. In the NSG mouse xenograft model, human PBMC was implanted and 6PHU3 was administrated. The tumors were infiltrated by CD8+ and CD4+ T cells, and finally eradicated. The survival period of mice treated with 6PHU3 was significantly prolonged [26].
Ephrin type-A receptor 2 (EphA2) receptor is a 130 kDa transmembrane glycoprotein expressed normal tissues with a high proportion of epithelial cell divisions [67] and overexpressed in solid tumors, such as ovarian cancer, prostate cancer, lung cancer and human glioma [68,69]. Kota Iwahori et al. designed and constructed EphA2-BiTE and showed that EphA2-BiTE induced immunostimulatory cytokines, redirected bystander T cells to target cells, and killed EphA2-positive tumor cells in vitro. In mouse models of lung cancer and human glioma, the tumor signal of mice treated with EphA2-BiTE was significantly lower than that of control mice [18].
Both hematopoietic tumors and solid tumors
Here we discuss certain tumor antigen targets that are widely expressed in both hematopoietic tumors and solid tumors, including cancer cell antigens, such as B7H6 [70] and ESK1 [71], and tumor stroma antigens, such as FAP [22]. A major obstacle to the success of tumor immunotherapy is immunosuppressive stromal cells [72,73]. Anti-FAP×anti-CD3 BiTE targeting cancer-associated fibroblasts (CAF) can eliminate CAF in tumor stroma. In this way, a “cold tumor” in the immune concept becomes a “hot tumor”. In addition, tumor-associated macrophages (TAM) also play an important oncogenic role. Like other BiTE, CD206 and FRβ BiTE did not induce T cell-mediated macrophage killing through the death receptor pathway but through perforin. The cytotoxicity of macrophages repolarized the remaining incomplete cells due to the pro-inflammatory signals induced by BiTE [21].
In 2009, Wilm tumor gene 1 (WTI gene) was voted “highest priority antigen for cancer antigens” by the National Cancer Institute (NCI). Nearly all types of tumors have WT1 and can be treated with a part of its “WT1 peptide”. Tao Dao et al. described BiTE derived from the high-affinity TCR mimic antibody ESK1, which specifically binds to the WT1 epitope RMF. Although the expression density of peptide-MHC complex is extremely low, ESK1-BiTE still has a certain therapeutic effect on primary ALL, BV173 Ph ALL, SET-2 AML and JMN mesothelioma. In AML and JMN peritoneal mesothelioma mouse models, the tumors can be observed later in mice treated with ESK1-BiTE. The ESK1-BiTE group showed a significant inhibitory effect on mouse tumors compared with the control group. It is worth noting that no central nervous system sequelae of leukemia were observed in mice treated with ESK1-BiTE for 40 days, while sequelae were seen in almost all mice in the control group. In addition, research showed that ESK1-BiTE provided a further effective long-term and extensive reaction than BiTE that acted on other targets [71].
A group of ligands that are specifically expressed on a variety of cancers and have activating receptors NKp46, NKp44, NKp30 and NKG2D can be recognized by NK cells [74]. The broad tumor-targeting activity of NK cells causes these receptors attractive targets for immunotherapy [70]. One of the ligands of NKp30, B7H6, is expressed in about 20% of tumor cell lines and some primary tumors (such as leukemia and ovarian cancer), but it is not detected in 48 normal tissues. Ming-Ru Wu et al. developed a B7H6-BiTE [75] and showed in the co-culture experiment that B7H6 specific BiTE instructed T cells to mediate IFN-γ secretion and cytotoxicity. In the RMA lymphoma mouse model, B7H6-specific BiTE extremely improved the survival advantage of mice through IFN-γ effector and perforin mechanisms. The mice with prolonged survival were protected against RMA lymphoma rechallenge. A trend of decreasing tumor burden was also observed in mouse models of ovarian cancer and melanoma. The above experimental results indicated that B7H6-specific BiTE has certain therapeutic potential for various B7H6-positive hematomas and solid tumors [70].
Non-tumor diseases
Human cytomegalovirus (HCMV) infection is currently not curable even by a good treatment, and it will still be reactivated after allogeneic hematopoietic stem cell transplantation (HSCT) [76,77]. A team was inspired by the successful application of BiTE in tumors and designed BiTE for the treatment of HCMV. HCMV glycoprotein B (gB) is the most conserved glycoprotein encoded by HCMV and expressed on the surface of infected cells [23,78]. Co-culture experiments of fibroblasts infected with HCMV strain AD169 and anti-CD3 and anti-gB BiTE showed that gB-BiTE effectively induced T cells to secrete TNF and IFN-γ. It also showed that gB at low levels was sufficient for BiTE to effectively trigger T cells. Although BiTE did not lyse infected target cells, it inhibited HCMV replication by inducing cytokine production. Although evidence showed that HCMV-infected cells are effectively protected against T cell-mediated cytotoxicity, gB-BiTE restricted HCMV replication through non-cytolytic mechanisms [23].
The current gold standard for human immunodeficiency virus 1 (HIV-1) treatment is combined antiretroviral therapy (cART), which requires patients to take the drugs for life and strictly adhere to it [79]. cART can only prevent HIV-related immunodeficiency and reduce HIV-1 load below the detection limit and cannot cure patients [24]. Johannes Brozy et al. used scFv VRC01 or B12, the first two extracellular domains (1+2) of human CD4 independent or joined to the scFv of antibody 17b (CD4L17b) fused with anti-human CD3 scFv to construct an antibody targeting HIV-1 envelope protein gp120 (HIV gp120) [80]. These BiTE significantly inhibited HIV-1 replication in macrophages co-cultured with PBMCs and autologous CD8+ T cells, and induced T cells to redirect lysis of HIV gp120-transfected CHO cells. The BiTEs with the best inhibitory effects on replication are CD4(1+2) L17b BiTE and human CD4(1+2) BiTE [named CD(1+2) h BiTE]. The CD4(1+2) h BiTE increased HIV infection of human CD4-/CD8+ T cells. On the contrary, the neutralizing B12 and the VRC01 BiTE antibody and the CD4(1+2) L17b BiTE antibody, did not. The CD417b, B12, CD4(1+2) and BiTE were the most efficient ones, with >90% HIV suppression at 1 μg/ml; the CD417b BiTE antibody displayed >90% HIV suppression at 0.1 μg/mL. Significantly, T cell activation reversed the latency of HIV and facilitated viral replication in patients [81]. Hence, this BiTE antibody could cause cell death by activating replication in latently infected cells. HIV may eliminate the effects of BiTE through immune escape. Therefore, human CD4(1+2) BiTE, whose efficacy has nothing to do with HIV gp120, has theoretical advantages, if it is CD4-dependent, it can play a role [24]. They may be particularly valuable for cART patients to eliminate latent reservoirs. Combination therapy with compounds that reverse the incubation period, cART and BiTE antibodies that recognize HIV-gp120 may be a very attractive treatment strategy [24].
Clinical trials of BiTE
A growing number of BiTEs are entering clinical trials (Table 2). From the initial BiTE designed for blood diseases to clinical trials, now increasing BiTE for solid tumors are also applying for clinical medical experiments to test their effectiveness, safety, pharmacokinetics, etc. There have been articles that have summarized the clinical trials related to blinatumomab in detail [82]. 53 countries have approved Blinatumomab for the treatment of relapse/refractory (R/R) ALL. Blinatumomab also applies to the following categories: minimal residual disease (MRD) ALL patients and Philadelphia chromosome (Ph) + R/R ALL patients. Blinatumomab can be combined with other therapies to treat ALL, such as blinatumomab + tyrosine kinase inhibitors, blintumomab + immune checkpoint inhibitors, blinatumomab + chemotherapy, and blinatumomab + miniHCVD + inotuzumab [82]. We describe below the clinical trials related to BiTE other than blinatumomab.
Table 2.
Clinical trials of BiTE
| Drugs | Combination therapy | Target | Indication | Administration route | Case | Status | Adverse event | ID | Ref |
|---|---|---|---|---|---|---|---|---|---|
| MCLA 117 | No | CLEC12A | AML | NR | 50 | Phase 1 | NR | NCT03038230 | |
| AMG 330 | No | CD33 | AML | 0.5-960 µg/day cIV infusion in cycles from 14 to 28 days | 100 | Phase 1 | NR | NCT02520427 | |
| AMG673 | No | CD33 | AML | A short term intravenous (IV) infusion | 50 | Phase 1 | NR | NCT03224819 | |
| JNJ-63709178 | No | CD128 | AML | NR | 120 | Phase 1 | NR | NCT02715011 | |
| AMG 420 | No | BCMA | MM | Continuous intravenous infusion | 42 | Phase 1 | Infections, polyneuropathy, edema | NCT02514239 | [83] |
| MT110 | No | EpCAM | EpCAM-positive solid tumors | Continuous intravenous infusion | 65 | Phase 1 | Primarily diarrhea, elevated liver parameters, and elevated lipase | NCT00635596 | [84] |
| MEDI-565 | No | CEA | Gastrointestinal Adenocarcinomas | NR | 78 | Phase 1 | Diarrhea, CRS, increased alanine aminotransferase, hypertension, and hypoxia | NCT01284231 | [85] |
| AMG 211 | No | CEA | Gastrointestinal Adenocarcinomas | NR | 39 | Phase 1 | NR | NCT02291614 | |
| AMG 211 | No | CEA | Gastrointestinal Adenocarcinomas | NR | 9 | Phase 1 | NR | NCT02760199 | |
| AMG 757 | Pembrolizumab | DLL3 | SCLC | Intravenous infusion once every 2 weeks | 132 | Phase 1 | NR | NCT03319940 | |
| AMG 596 | No | EGFRvIII | Glioblastoma | NR | 200 | Phase 1 | NR | NCT03296696 | |
| BAY2010112 | No | PSMA | Prostate cancer | Be given daily as subcutaneous injection or as continuous intravenous infusion | 47 | Phase 1 | Influenza-like Symptoms, fatigue, decreased lymphocyte counts and infections (both in 44%) | NCT01723475 |
NR, not reported; MM, multiple myeloma; SCLC, small cell lung cancer; AML, acute myelocytic leukemia.
Acute myelocytic leukemia
Currently, BiTE that specifically targets CD33, CD123 and CLEC12A for the treatment of AML have entered phase I clinical trials. AMG330 (target CD33 x CD3 BiTE) is undergoing clinical evaluation in patients with relapsed/refractory AML (NCT02520427) to determine the biologically active dose/maximum tolerated dose of continuous infusion. The clinical trial is expected to end in May 2021. The Phase I clinical trial of the specific anti-CD33 BiTE fused to the Fc domain named AMG673 began in September 2017, and the results showed that the half-life of AMG673 can be extended to 7 days, and it can be administered at one time due to the inclusion of the Fc domain [19]. The study (NCT03224819) is a first-in-human, open-label, phase I, sequential dose escalation study. AMG673 will be evaluated as a short term intravenous (IV) infusion in adult subjects with relapsed/refractory AML. This clinical trial is expected to be completed in October 2022. CD123xCD3 BiTE, JNJ-63709178 (Jensen, USA) is conducting a phase I clinical trial for relapsed and refractory AML (NCT02715011). The anti-CLEC12AxCD3 BiTE named MCLA-117 is undergoing a phase I clinical trial (NCT03038230) to evaluate the effectiveness and preliminary safety in adult AML patients.
Multiple myeloma
The BiTE that specifically targets BCMA is named AMG420, previously BI836909, and has completed a first-in-human dose escalation trial (NCT02514239). The patients with refractory/relapsed MM in this trial received at most 10 cycles of AMG420 treatment after ≥ 2 lines of therapy, including immunomodulatory drugs (IMiDs) and proteasome inhibitors. 42 patients received AMG420 at a dose of 0.2-800 μg/d. Median exposure was 7 cycles and 1 cycle for responders. Patients with completion of 10 cycles (n = 3), discontinued for disease progression (n = 25), consent withdrawal (n = 1), death (n = 4), and adverse events (AEs; n = 7). In addition, two patients are still receiving treatment. Two patients died from AEs caused by aspergillosis/influenza and adenovirus-related hepatitis. Severe adverse events (n = 20; 48%) involved polyneuropathy (n = 2) and infections (n = 14); Grade 1 and grade 3 edema, and grade 2 and grade 3 polyneuropathy are serious AEs related to the treatment. There were no anti-AMG 420 antibody or grade ≥ 3 central nervous system (CNS) toxicity. Because one case of grade 3 polyneuropathy and one case of grade 3 cytokine release syndrome have been resolved, it is considered that 800 μg/d is intolerable. The total response rate was 31% (n = 13 of 42). The maximum tolerated dose (MTD) of this clinical trial was 400 μg/d, and its response rate was 70%, containing 50% complete remission with negative MRD [83].
EpCAM-positive solid tumors
The specific anti-EpCAM BiTE named Solitomab (AMG110, MT110) completed its phase I dose escalation clinical trial (NCT00635596) in 2015. 65 patients with solid tumors (including lung adenocarcinoma, small cell lung cancer, gastric cancer or adenocarcinoma of the gastro-esophageal junction, colorectal cancer, breast cancer, hormone-refractory prostate cancer, and ovarian cancer) were injected with solitomab. Six of the patients had diarrhea dose-limiting toxicity (DLT): four patients had relief of symptoms (grade 4, n = 1; grade 3, n = 3); one case (grade 3) was upgraded to grade 5 after Solitomab was discontinued; and the other (grade 3) death case (not related to the treatment) was still ongoing after Solitomab withdrawal. DLT occurred in 15 patients: 1 patient had supraventricular tachycardia (grade 3), 8 patients had transient abnormal liver parameters shortly at the beginning of the injection or after the dose were increased (grade 4, n = 4; grade 3, n = 4), all cases were resolved after Solitomab withdrawal. The maximum tolerated dose of Solitomab was 24 µg/day. From the results of clinical trials, AEs appeared in the first few days after administration. In addition, the test showed that step-by-step administration was more tolerable, which was consistent with the results of muS110 preclinical experiments [84].
Gastrointestinal adenocarcinomas
The specific anti-CEA BiTE (AMG211/MEDI-565) ended its phase I clinical trial (NCT01284231) in January 2015. 4 out of 39 patients developed DLT (2 at 3 mg; 2 at 7.5 mg + dexamethasone): cytokine release syndrome (CRS), diarrhea, and hypoxia (n = 2). Six patients had serious grade 4 treatment-related AEs: vomiting, CRS (n = 1), diarrhea, hypoxia (n = 2), and fever. Five patients had level 3 treatment-related AEs: CRS, diarrhea, hypoxia (n = 2), elevated alanine aminotransferase, and hypertension (n = 1). Anti-drug antibodies appeared in 19 patients (48.7%). The results of this experiment showed that the maximum tolerated dose of MEDI-565 was 5 mg administered over 3 hours on days 1 through 5 every 28 days, with dexamethasone. The pharmacokinetics of MEDI-565 were linear. No objective reaction was observed [85]. Another phase I clinical trial (NCT02291614) evaluated the effect of AMG211 in patients with relapsed/refractory gastrointestinal adenocarcinoma. The experiment gave patients continuous intravenous administration to maintain the possibility of sustained exposure [44,46]. In addition, a clinical trial (NCT02760199) used the radionuclide Zirconium-89 (89Zr) labeled AMG211 to evaluate the distribution of 89Zr-AMG211 in normal organs and the uptake in metastatic and primary tumor lesions. 89Zr-AMG211 showed dose-dependent cancer uptake at 6 hours [46].
Small cell lung cancer
NCT03319940 is a dose evaluating phase I clinical trial to test the tolerability, pharmacokinetics, and safety of AMG757 (target DLL3 x CD3 BiTE). The study will evaluate AMG757 monotherapy and in combination with anti-PD1 therapy. The experiment is expected to be completed by June 2023.
Glioblastoma
Phase 1 study (NCT03296696) evaluating AMG596 (target EGFRvIII x CD3) for safety, tolerability, pharmacokinetics, and pharmacodynamics in glioblastoma patients will be completed in 2024.
Prostate cancer
A clinical trial of BAY2010112 (target PSMA x CD3) (NCT01723475) targets patients with castration-resistant prostate cancer. Serum PSA levels in three patients were reduced by >50% when administered 20-80 μg daily. Two of the patients had a long-term response, one patient was treated for 14 months and the other patient was for 19.4 months. The most common AEs above grade 3 were infection and decreased lymphocyte count (both 44%). The most common AEs of any grade were flu-like symptoms (fatigue in 50% of patients, algor in 69% of patients, and fever in 94%); no DLT was observed.
The structure, function, and preclinical studies of OV-BiTE
OVs are a type of virus that can preferentially infect and kill tumor cells. After the oncolytic virus enters the body, it may specifically infect tumor cells and multiply within them, and eventually kill tumor cells while releasing new virus particles to continue to specifically infect and kill tumor cells in other parts of the body [86]. OVs exert their “oncolytic effect” mainly through two aspects: on the one hand, they directly infect and kill tumor cells; on the other hand, they stimulate the host to produce an anti-tumor immune response [5]. Oncolytic viruses can be divided into two categories: one is naturally occurring, such as most oncolytic Newcastle disease virus and reovirus, etc.; the other is genetically modified oncolytic viruses, such as most oncolytic adenovirus, vaccinia virus and herpes simplex virus. At present, the research on OVs through genetic modification gives the potential to be more effective. Combining OV therapy with other immunomodulatory molecules is a very attractive immunotherapy. OV can encode and specifically express biological agents in tumor cells [87,88]. OV can use its own regulatory elements to create complex gene expression dynamics [69,89]. For example, the gene expression of vaccinia virus is subject to strict time regulation, and a single gene can be expressed individually or together with other genes at each stage of infection. Use temporal promoters can achieve ideal expression kinetics of immunostimulatory transgenes [11,90]. The herpes simplex virus (HSV) expressing human GM-CSF, called Imlygic, was approved for clinical therapy by the FDA [11,91-93]. Importantly, genetic engineering has successfully made OVs specifically express BiTE (OV-BiTE) from tumor cells. Feng Yu et al. designed the first OV-BiTE, vaccinia virus-BiTE in 2014 [94]. There are currently three types of viruses used as OV-BiTE vectors in the preclinical research stage (Figure 2 and Table 3): vaccinia virus (VV), adenovirus (AdV, EnAdenotucirev (EnAd) and ICOVIR-15K (ICO15K)) and measles virus (MV). No OV-BiTEs currently entered clinical trial yet.
Figure 2.
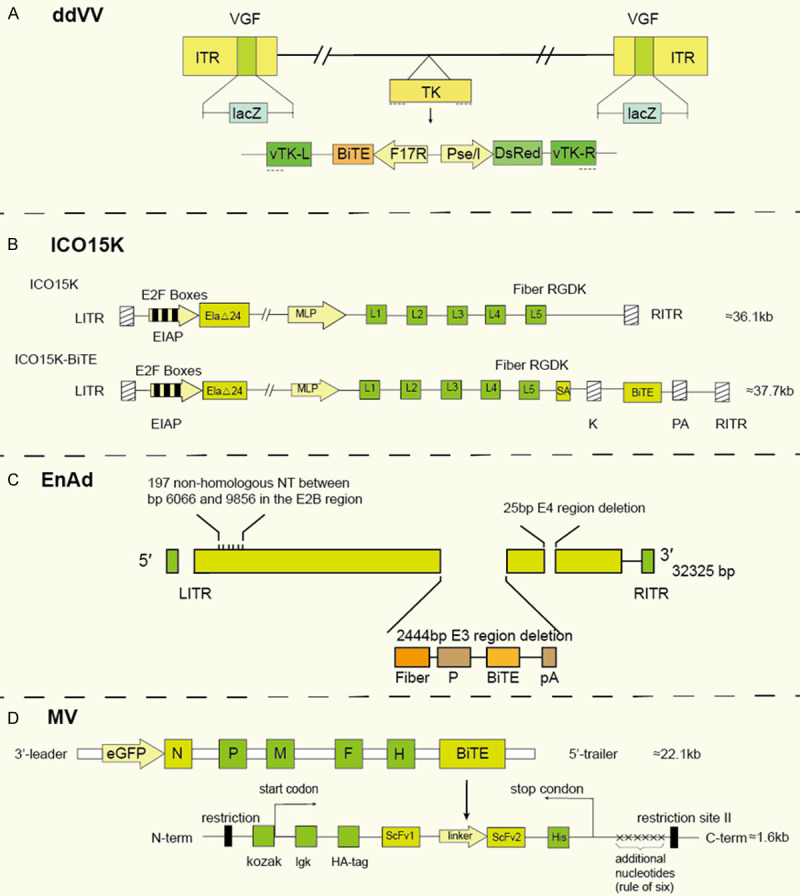
Structure of the genomes of OV-BiTE. A. vvDD (double-deleted VVs). The lacZ gene is inserted into its VGF site. vTK-L/R, the left and right segments of vaccinia TK gene; Pse/I, synthetic early/late promoter; F17R, late promoter. B. ICO15K (ICOVIR-15K). L/RITR, left/right inverted terminal repeats; SA, splicing acceptor; K, kozac sequence; C. EnAd (enadenotucirev). The Ad11p sequence between the base pair 6081 and 9322 in the E2B region is often replaced by the Ad3 sequence. In addition, the E3 region of EnAd is almost completely deleted, and the E4orf4 region of the virus has a 25 bp deletion. The BiTE transgenes were inserted in the downstream of a CMV promoter or splice acceptor site (P) followed by a polyadenylation sequence (pA). D. MV (measles virus). The transcription unit downstream of the leader sequence (ld) encodes enhanced green fluorescent protein (eGFP). F, H, L, M, N, and P respectively represent the coding genes for structural fusion protein, hemagglutinin protein, large protein (polymerase), matrix protein, nucleoprotein, P protein. BiTE was inserted into an ATU downstream of the H open reading frame. the Kozak sequence is located before the BiTE. To comply with the six-rule, other nucleotides are exemplarily included downstream of the coding region. Two restriction sites can be inserted into the corresponding ATU on both sides of the insertion box.
Table 3.
The structure of OV-BiTE
| OV | scFv1 | scFv2 | scFv1-linker | scFv2-linker | Linker | N-terminal | C-terminal | Virus insertion site | OV self-modification | Recombination method | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ddVVs | EphA2 | CD3 | NR | NR | NR | NR | NR | The TK gene, before the F17R late promoter | Deletions in the TK and VGF genes | pSEL shuttle plasmid recombination | [94] |
| ICO15K | EGFR | CD3 | (G4S1) 3 | (G2S1) 4GG | GGGS | Signal peptide | Flag tag | After the fiber gene under the control of the MLP | An RGDK motif replacing the KKTK domain | Recombination in bacteria | [14] |
| ICO15K | FAP | CD3β | (G4S1) 3 | (G2S1) 4GG | GGGGS | Signal peptide | Flag tag | After the fiber gene under the control of the MLP | An RGDK motif replacing the KKTK domain | Recombination in bacteria | [10] |
| EnAd | EpCAM | CD3ε | NR | NR | (G4S) 3 | SP, light chain immunoglobulin signal peptide | His, decahistidine affinity tag | After the fiber gene under the control of the MLP | Complete E3 region deletion and a samll E4 region deletion | Gibson assembly technology | [87] |
| EnAd | FAP | CD3ε | NR | NR | NR | Immunoglobulin signal sequence | Decahistidine tag | After the fiber gene under the control of the MLP | Complete E3 region deletion and a samll E5 region deletion | Gibson assembly technology | [22] |
| EnAd | FRβ | CD3ε | NR | NR | GGGGS | NR | NR | Downstream of the fibre gene under the control of CMV Promoter | Complete E3 region deletion and a samll E6 region deletion | Gibson assembly technology | [21] |
| MV | CEA or CD20 | CD3 | (G4S1) 3 | (G4S1) 3 | G4S | NR | His | in ATU downstream of the H ORF | NR | NR | [110] |
ddVVs, double-deleted VVs; ICO15K, ICOVIR-15K; EnAd, EnAdenotucirev; MV, Measles virus; NR, not report.
Structure and function of OV-BiTE
The site selection of BiTE insertion into an OV genome affects the curative effect of OV-BiTE therapy [95]. The insertion site of BiTE should limit its secretion to tumor cells, which can reduce its toxicity to non-tumor cells. In addition, the choice of insertion site also needs to consider whether it affects the replication and oncolytic properties of the OV itself (Figure 2 and Table 3).
Vaccinia virus-BiTE
Vaccinia virus (VV) can infect tumor cells, replicate in tumor cells, lyse tumor cells, and spread to other tumor cells, so VV is an attractive anti-cancer drug. VV has been shown to have certain anti-tumor efficacy in preclinical and clinical studies [96,97]. VV which is a big cytoplasmic DNA virus that can insert up to 25 kb of external DNA without deleting the viral DNA has unique advantages in gene therapy [98]. VV synthesizes viral RNA and DNA in the cytoplasm through viral factors, so its dependence on cytokines is lower than other OVs [99]. The vaccinia virus currently used as a gene therapy platform is a VV lacking VGF and thymidine kinase (TK) gene. The parental VGF deletion virus (VSC20) was created by inserting the lacZ gene into the VGF site of VV. The shuttle plasmid was used to plug EGFP into the TK locus of the VGF-deleted VV through homologous recombination, creating the double-deleted virus, vvDD-GFP (ddVV). The main advantage of ddVV is its significantly reduced toxicity in vivo, which has been evaluated in a nude mouse model [99].
Feng Yu et al. developed a T-cell engager-armed oncolytic ddVV (TEA-VV) encoding secretory BiTE targeting EphA2 (EphA2-TEA-VV) [94]. After VV infects tumor cells, the cascade viral late or early proteins are expressed under the control of late or early promoter, respectively. Usually, the virus activates the late promoter after replicating DNA in tumor cells, so the transgene is selectively expressed in tumor cells. Studies have shown that the late promoter F17R of VV is activated only after VV infects cancer cells. Therefore, Feng Yu et al. chose to express the transgene BiTE under the control of the F17R late promoter, which can enhance the selective expression of the transgene in tumors. In addition, the late expression of BiTE will also permit adequate viral replication before T cell activation and cancer cells lysis [94].
Adenoviru-BiTE
The killing effect of oncolytic adenovirus on tumor cells is based on multi-modal mechanisms, including direct oncolysis, immunogenic cell death-mediated anti-tumor immune response and bystander effect when equipped with therapeutic transgene. Therefore, oncolytic adenovirus is an exciting tumor immunotherapy agent [97]. The replication of oncolytic adenovirus in tumor cells is strictly regulated, so it is an attractive candidate for viral therapy. After transgene insertion, it is controlled by the main late promoter of adenovirus to ensure replication-dependent expression [100]. In addition, adenovirus shows low toxicity after systemic administration in tumor patients [14].
The currently used oncolytic adenoviruses are ICO15K and EnAdenotucirev (EnAd). ICO15K is an oncolytic adenovirus based on E1a-Δ24 that replaces the glycosaminoglycan binding domain of KKTK heparan sulfate in the fiber shaft with RGDK [101]. This modification achieves both tumor and liver targeting and shows low toxicity profiles. In addition, its E1a promoter has palindrome E2F binding sites [101]. The transgene BiTE was inserted under the control of the main late promoter (MLP) of ICO15K to promote the expression of the transgene in a replication-dependent mode without disturbing with viral oncolysis [10].
EnAdenotucirev (EnAd) is a chimera of group B type 3 and type 11 adenoviruses. EnAd almost deleted the entire 2444 bp E3 region and the smaller 25 bp E4orf4 region. In addition, the Ad11p sequence in the 6081 to 9322 base pairs in the E2B region was replaced with an Ad3 sequence [102]. Early clinical trials have proved that EnAd has good systemic pharmacokinetics [103]. Transgene BiTE was encoded within EnAd downstream of the fibre gene, employing a shuttle vector plugged into the virus backbone by Gibson assembly. BiTE is inserted downstream of the splice acceptor (SA) site of the MLP of EnAd. In the previous structure, if the virus infects cells successfully, BiTE will be expressed and secreted. However, transgenes inserted downstream of SA can only be expressed when MLP is activated in tumor cells that allow virus replication. In this way, BiTE may be restricted to be expressed and secreted only in tumor cells to reduce toxicity [87].
Measles virus-BiTE
Measles virus (MV) is a non-segmented, single-stranded, negative-stranded RNA virus of 16,000 nucleotides [104]. The attenuated measles virus vaccine strain Edmundston B (MV-Edm) is a naked-stranded RNA virus that has been evaluated in clinical trials [105]. Although MV-Edm has a significant attenuation effect in humans, its replication ability is stronger than that of unattenuated MV in a large amount of primate cell lines, thereby inducing cell fusion and forming characteristic multinucleate syncytia [106]. MV-Edm has been shown to have oncolytic activity in animal models of human ovarian cancer, malignant glioma, multiple myeloma, lymphoma, cutaneous T-cell lymphoma and fibrosarcoma [106,107]. MV-Edm has a large transgene capacity (over 5 kb), strict cytoplasmic replication allows it to have no risk of mutagenesis, effective spreading includes syncytial formation, natural oncotropism and high immunogenicity characteristics. All these are conducive for it to be an ideal oncolytic virus vector. A versatile reverse genetics system based on anti-viral genome cDNA allows transgenes to be inserted into other additional transcription units (ATUs) [108]. Study showed that the genome of MV-Edm requires each nucleocapsid protein to bind 6 nucleotides [108]. For insertion of transgenes into such viruses, the number of nucleotides of the complete genome should be divisible by six. This is also referred to as “rule of six”. If necessary, additional nucleotides can be added to the insert (downstream of the stop codon or upstream of the Kozak sequence) without introducing a stop codon prematurely or without introducing a frameshift. Avoidance of specific sequences similar to MV gene start (AGGRNCMARGW), RNA editing sequence (AAAAAGGG) and termination (RTTAWANAAAA) signals in the transgene have been reported [109]. Recombinant MV vectors were engineered to encode secretory BiTE (MV-BiTEs) in an ATU downstream of the H open reading frame. Furthermore, MV-BiTEs encoding enhanced GFP (eGFP) in an ATU upstream of the N ORF were generated to monitor viral spread and infection [110].
Preclinical studies of OV-BiTE
As proposed above that the OV-BiTE combination has a synergistic role on tumor immunotherapy, preclinical studies of OV-BiTE have showed exciting results (Table 4).
Table 4.
Preclinical studies of OV-BiTE
| Ovs | Insertion effect | Self-modification effect | Virus replication ability after inserting BiTE | Onocolytic properties after inserting BiTE, without T-cell | Experimental model | Anti-tumor efficacy | Ref |
|---|---|---|---|---|---|---|---|
| ddVVs | Enhance tumor selective expression of transgene and allow for sufficient viral replication | Selective tumor replication, significantly less pathogenic | No impair | No impair | Mice A549 xenograft tumor model | Volume decreases by about 2500 mm3 | [94] |
| ICO15K | Its expression in a replication-dependent manner without interfering with viral oncolysis | Enhances the systemic antitumor efficacy | NR | Reduced approximately 2-fold | SCID/beige mice bearing subcutaneous A549 tumors | Volume decreases by about 600 mm3 | [14] |
| ICO15K | Its expression in a replication-dependent manner without interfering with viral oncolysis | Enhances the systemic antitumor efficacy | No impair | No impair | Xenograft mouse models | Volume decreases by about 1300 mm3 | [10] |
| EnAd | Express BiTE only in cells supporting virus replication | Replicate more quickly and enhance potency | No impair | No impair | Liquid cancer biopsies model | Kill endogenous tumor cells | [87] |
| EnAd | Express BiTE only in cells supporting virus replication | Replicate more quickly and enhance potency | No impair | No impair | Malignant ascites model | Kill endogenous Fibroblasts | [22] |
| EnAd | NR | Replicate more quickly and enhance potency | NR | No impair | Malignant ascites model | Depletion of endogenous macrophages | [21] |
| MV | NR | NR | No impair | No impair | Xenograft mouse model | Significantly prolongs survival | [110] |
ddVVs, double-deleted VVs; ICO15K, ICOVIR-15K; EnAd, EnAdenotucirev; MV, Measles virus; NR, not report.
Vaccinia virus-BiTE
The study by Feng Yu et al. showed that EphA2-TEA-VV produced a similar amount of virus, proving that EphA2-TEA-VV did not reduce VV self-replication. In addition, in the absence of human T cells, the ability of EphA2-TEA-VV to lyse cancer cells was no different from that of GFP-VV, demonstrated that the expression of EphA2-TEs did not affect the oncolytic ability of the OV itself. EphA2-TEA-VVs activated human T cells and redirected human T cells to EphA2-positive lung cancer A549 cells. In addition, uninfected cancer cells are killed by bystander T cells induced by EphA2-TEA-VVs [94]. EphA2-TEA-VV expressed EphA2-TEA, which not only caused antigen-specific cancer lysis by binding to cancer cells and endogenous T cells, but also induced the secretion of pro-inflammatory cytokines to reverse the immunosuppressive environment. Although EphA2-TEA-VVs induced the secretion of IL-2 and IFN-γ, no obvious T cell proliferation was found in vivo and in vitro. Of note, after adding 100 μU/ml human IL-2 to the cell culture medium, EphA2-TEA-VVs induced obvious T cell proliferation. Therefore, EphA2-TEA-VV only activated T cells by binding to CD3 because the quantity of IL-2 produced by EphA2-TEA-VV activated T cells was not enough to induce T cell proliferation [94]. In the A549 mouse xenograft model, human PBMCs and EphA2-TEA-VVs completely relieved all treated animals. The “bystander effect” caused EphA2-TEA-VVs to kill uninfected and infected cancer cells through T cells after infecting EphA2-positive cancer cells. On day 41, the tumor volume of mice treated with EphA2-TEA-VV was approximately 0 mm3, that of GFP-VV control was approximately 700 mm3, PBS was approximately 2000 mm3, and A549 alone was approximately 2500 mm3 [94].
Another research team constructed mFAP-TEA-VV to better eliminate the physical barriers of OVs [111]. mFAP-TEA-VV destroyed FAP-positive stromal cells and caused a markedly increase in virus titer in the tumor. In the B16 melanoma mouse model, mFAP-TEA-VV markedly promoted CD4 and CD8 T cell activation and tumor infiltration than GM-CSF-VV and EphA2-TEA-VV controls. mFAP-TEA-VV was suitable for a wide range of solid tumors overexpressing FAP, removed the influence of tumor stroma on the spread of the virus, and highlighted the significance of targeting stroma for anti-tumor efficacy of immunotherapy.
Adenovirus-BiTE
The efficacy of Cetuximab for colorectal cancer has not reached a very satisfactory level, because some colorectal cancer patients develop drug resistance. One of the main resistance mechanisms is the mutation of downstream signaling genes such as PIK3CA, KRAS, PTEN and BRAF [112]. BiTE derived from Cetuximab overcomes drug resistance by redirecting T cells to kill KRAS and BRAF-mutated colorectal cancer cells [14]. Fajardo CA et al. constructed ICO15K-cBiTE using adenovirus ICO15K as a platform to express BiTE targeting EGFR. Both in vivo and in vitro experiments showed that ICO15K-cBiTE induced T cell-mediated killing of KRAS-mutated colon cancer HCT116 cells. Although ICO15K-cBiTE could be amplified, its oncolytic properties were about 2 times lower than that of unmodified ICO15K. Competing transcription and translation of cBiTE with viral genes may lead to the reduction of virus replication as well as cell toxicity. However, in the presence of T cells, the cBiTE still had obvious therapeutic advantages despite the low MOI in vitro. Of note, in vitro cytotoxicity loss of ICO15K-cBiTE did not translate into a loss of anti-tumor efficacy in vivo without PBMC. In ICO15K-cBiTE mouse model, the cBiTE-mediated tumor cells death did not harm the standing of the virus in the tumor [14]. ICO15K-cBiTE, ICO15K or PBS was injected intravenously in the SCID/beige mouse model of subcutaneous human colon cancer HCT116 xenograft tumor. On days 4, 8 and 11 after virus injection, pre-activated T cells were injected intravenously, and then IL2 was injected intraperitoneally. On day 22, the tumor volume of mice treated with ICO15K-cBiTE was approximately 550 mm3, ICO15K was approximately 800 mm3, and PBS was approximately 1250 mm3 [14]. Although TEA-VV induced PBMC-mediated bystander cell killing of tumor cells and T cell activation, T cell proliferation was not observed in the case of insufficient IL2. Adding enough IL2 to the co-culture induced T cell proliferation [94]. Without adding IL2 to the co-culture, ICO15K-BiTE induced significant proliferation of T cells in vitro [14]. Currently, the reason for the difference is not yet clear. It may be caused by different OV, different BiTE design and different antigen density.
The poor tumor targeting of adenovirus after systemic administration is supposed to be the main limitation of OV therapy for disseminated tumors. In response to this problem, Paula Barlabé et al. have indicated that menstrual blood-derived mesenchymal stem cells (MenSCs) loaded with adenovirus have increased anti-tumor efficacy. Although there was a significant improvement in efficacy, the anti-tumor efficacy gained was modest. Therefore, they combined ICOVIR15-cBiTE with MenSC. Experiments showed that the anti-tumor efficacy of this combination was stronger than MenSC loaded with unarmed ICOVIR15. MenSCs can produce cBiTE after virus infection, thus having higher anti-tumor efficacy in vivo and in vitro [113].
Sostoa JD et al. raised that ICO15K-cBiTE did not solve the problem of a tumor stroma, which can impede virus spread in the tumors. They genetically modified ICO15K to express FAP-targeted BiTE (FBiTE) and named it ICO15K-FBiTE. ICO15K-FBiTE not only mediated the redirection of T cell immune response to tumor stromal fibroblasts, but also mediated oncolysis to target other tumor cells. The replication ability of ICO15K-FBiTE was weaker than that of the parent virus (although not significantly), and it maintained oncolytic properties [10]. When FBiTE was secreted from infected tumor cells, it activated CD8 and CD4 T cells, causing FAP-positive cell cytotoxicity and FAP-negative cells to be lysed by bystanders. In addition, lysis of FAP-positive stromal cells increased the overall anti-tumor efficacy without causing enhanced toxicity. In the mouse model, the tumor of mice treated with ICO15K-FBiTE was significantly smaller than that of mice treated with ICO15K control virus or PBS. On day 45, the tumor volume of mice treated with ICO15K-FBiTE was approximately 450 mm3, ICO15K was approximately 750 mm3, and PBS was approximately 1750 mm3. Experiments showed that this therapy significantly improved the survival rate of mice treated with ICO15K-FBiTE [10].
Freedman JD et al. combined BiTE targeting EpCAM with adenovirus EnAd to construct EnAd-SA-EpCAM. Due to the powerful transgene packaging ability of the virus, BiTE did not affect the replication and oncolytic activity ability of EnAd without human T cells [87]. EnAd-SA-EpCAM activated CD8+ and CD4+ T cells to mediate cytotoxicity. The BiTE expressed and secreted by EnAd-SA-EpCAM can activate endogenous T cells in liquid cancer biopsy samples without additional stimulation and kill endogenous cancer cells. Importantly, this situation may also occur in the original fluid of the pleural effusion or peritoneal ascites microenvironment, as a substitute for the immunosuppressive microenvironment of solid tumors. EnAd-SA-EpCAM expanded the T cell population and reduced the count of cancer cells compared to EnAd. Experiments had shown that EnAd-SA-EpCAM can activate T cells and increased the expression of IL-13, IL-5, IL17F, IL17A and tumor necrosis factor (TNF) by about 10-fold, and the increase of interferon gamma by about 1,000-fold. However, the EnAd expressing the BiTE control group and the EnAd alone group only caused a 10-fold increase in gamma interferon [87].
Another team use EnAd to express and secrete BiTE targeting FAP. Experiments showed that EnAd-FAP-BiTE destroyed endogenous FAP fibroblasts and activates tumor-infiltreated T cells in biopsy samples of hematoma and solid tumors. In addition, many chemokines and pro-inflammatory cytokines were secreted, including IL17, IL2, IFNγ, TNFα and CXCL9. Importantly, this demonstrated that the patient’s own cancer-infiltrated T cells could be used for the treatment in the real environment of human advanced tumors. The T cells in the biopsy tissues of all patients showed PD1+ and probably anergic, were easily activated by BiTE and mediate cytotoxicity. In ascites, EnAd-SA-EpCAM led to up-regulation of pro-inflammatory cytokines, depletion of CAF-related immunosuppressive factors and increased gene expression of antigen presentation, transportation markers, and T cell function. The combination of the two therapies reversed the CAF-mediated immunosuppressive effect and killed tumor fibroblasts and tumor cells more effectively [21]. To avoid “off-target tumor” toxicity, Scott EM et al. also used EnAd as a transgene platform for expressing BiTE. It also showed that BiTE-armed EnAd targeting TAM provided a powerful treatment by combining the immunosuppression reversal with direct tumor cell cytotoxicity [21].
Measles virus-BiTE
Oncolyitc MV was used as a platform to combine with BiTE therapy, and its own replication and oncolytic ability was not impaired [110]. In the B16-CD20-CD46 (CD46, a MV receptor) mouse xenograft model, treatment efficacy did not depend on viral replication, but could not be realized by injecting local BiTEs only, suggesting immunostimulatory properties of MV was indispensable for effectiveness. In addition, analysis of BiTE mRNA and intratumoral MV-N mRNA levels showed that virus replication and virus-expressed BiTE were restricted in this model. Thus, stronger virus replication may be produced in more permissive tumors to increase the efficacy. Most of patients have been vaccinated against measles and have MV neutralizing antibodies in their bodies. The efficacy of MV-BiTE treatment in MV-immunized mice in the B16-CD20-CD46 model was not compromised. Significantly, intratumoral injection of MV-BiTE in mice might limit the accessibility of MV-BiTE neutralizing antibodies [110] MV-BiTE treatment of the B16-CD20-CD46 model caused protective immunity against the parent cell line B16 and tumor cells lacking the BiTE target antigens.
Efficacy of the MV-hCD3xCEA MV-BiTE against genetic and functional heterogeneous cancer cells that closely mimic clinical situation was demonstrated in a spherical patient xenograft metastasis mouse model with human PBMC injection. MV-hCD3xCEA did not induce the selection of CEA-negative tumor cells, possibly because the persistence of injected PBMC was limited [110]. Significantly, the efficacy of MV-hCD3xCEA not irradiated with ultraviolet was equivalent to that of irradiated MV-BiTE, which indicated that the BiTE mediated immunotherapy effect was better than direct oncolysis by the oncolytic measles virus [110].
According to the target design, BiTEs used for arming OV can be divided into two types, targeting cancer-specific antigens or stromal cells. BiTE that specifically targets cancer-specific antigens when armed with OV, the secreted BiTE acts on both uninfected and infected cancer cells, so the production of virus-expressed BiTE in cancer cells and its utilization in the tumor microenvironment are reduced. To conquer this limitation, OV can be armed with BiTE targeting stromal cells, which can prevent the exhaustion of BiTE-expressing cancer cells and promote continuous BiTE production [10]. In addition, the BiTE-armed OV targeting stromal cells acts on both cancer cells and stromal cells, overcoming the physical barrier caused by the tumor matrix, and increasing the mode of action and curative effect.
Conclusion and perspectives
BiTE has good therapeutic efficiency but has limitations. First, we introduced the current preclinical and clinical studies of BiTE. The BiTE currently approved for clinical use is only suitable for hematologic tumors. BiTE for solid tumors is still in the clinical research stage, but they have shown exciting effects. In addition, some researchers have been inspired to design BiTE to treat diseases other than tumors, and the results of preclinical studies have shown that BiTE is an effective treatment option for CMV and HIV infections. BiTE will provide new treatment options for various diseases with suitable targets. Second, currently the most common BiTEs are designed to target tumor cell surface antigens but targeting tumor stromal cells seems to be a way to break through the barrier of tumor stromal. Third, in clinical studies of solid tumors, most BiTEs do not have combined drug experiments. In future experiments, more exploration of combined drug treatment may be a way to provide better efficacy. Fourth, on the other hand, BiTE itself is also evolving. There are engineered antibodies like BiTE, such as dual affinity retargeting (DART) and trispecific killer engagers (TriKEs).
OVs show obvious advantages in tumor therapy with multiple modes of anti-tumor activity and relative safety. Future research may enhance their anti-tumor activity by evading the antiviral immune responses. Current promising strategies include genetic modification of oncolytic viruses for membrane protein deglycosylation [114] and polymer coatings [115]. As a gene therapy platform, OVs should be further tested for their regulated transgene expression, tumor antigen targeting, pharmacokinetics, routes of administration, safety and commodity cost.
We propose that the OV-BiTE combination has a synergistic role on tumor immunotherapy (Figure 3). Although both BiTE therapy and OV therapy have shown effects, they still have certain limitations. Combining the two therapies miraculously compensated for each other’s shortcomings and enhanced the curative effect. The combination of OV as a gene therapy platform and BiTE therapy brings out the advantages of the two strategies and overcomes the multiple obstacles of the two single therapies. The spread of OV in cancer is limited by the interstitial barrier, which is another limitation except the antiviral immune response. The much smaller size of BiTE molecules compared to virus particles is good for distribution and penetration in tumors. Even if the spread of OV in the tumor is restricted, the overall bystander effect of OV can be improved [14]. OV therapy also helps BiTE overcome some of its limitations. BiTE has a short half-life in serum and requires continuous infusion for treatment, which is closely related to the side effects that occur during the treatment. OV-BiTE is an attractive solution to overcome the disadvantage of the short half-life of BiTE. OV can replicate preferentially in tumor cells and express BiTE in a large amount locally in tumors, which can avoid repeated injections of BiTE and reduce the adverse events of systemically administered BiTE [6]. The continuous secretion of BiTE only from infected tumor cells may also reduce the probability of its targeting to normal tissues and adverse events but increase its availability to tumor cells and T cells [14]. Furthermore, combination therapy will provide a “multimodal” therapy mechanism that simultaneously targets different types of cells in tumor tissue. When BiTE specifically targets FAP, FAP-BiTE is secreted from cancer cells to target tumor infiltrating lymphocytes (TIL), such as CAF and CAM, thus reversing the immune suppression [21]. The OV-BiTE combination provides direct OV mediated cancer cell lysis, direct BiTE mediated cytotoxicity, immune stimulation, and a reversal of local immunosuppression, thus transforming immunologically inactive “cold” tumors into “hot” tumors. Therefore, OV-BiTE combination provides greater immune infiltration and a more effective and comprehensive immunotherapy response (Figure 3) [21].
Figure 3.
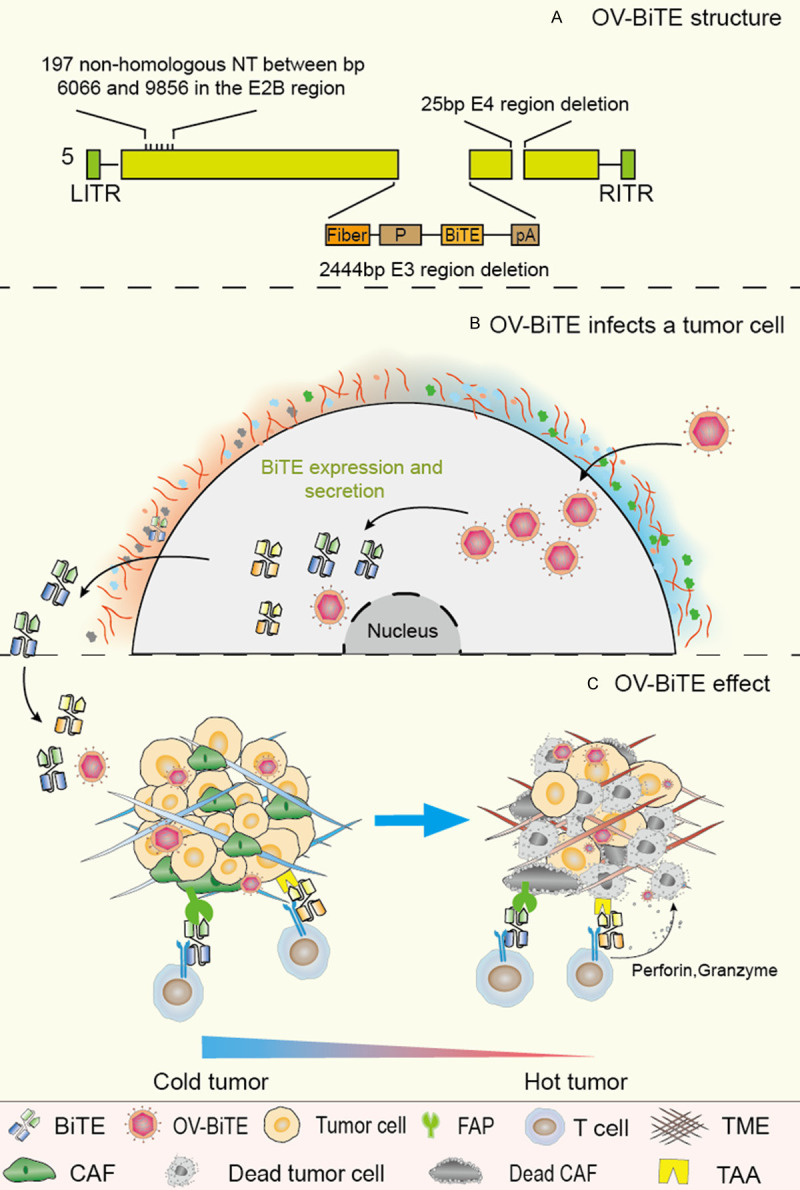
Mechanisms of action of OV-BiTE. A. One type of OV-BiTE, EnAd. B. The OV-BiTE infects a tumor cell, replicates in the tumor cell, and then expressed BiTE is secreted outside the tumor cell. C. OV-BiTEs lyse tumor cells directly, at the same time secret BiTEs targeting tumor cells and stromal cells (such as CAF), providing a new multimodal anti-tumor model. CAFs are targeted and killed, which reduces TME-mediated immunosuppression. This mode of action of OV-BiTE may transform an immune “cold” tumor into an immune “hot” tumor.
Research on OV-BiTE has made great progress but may be improved further. First, through reasonable engineering and modification, selective or restrictive BiTE expression and secretion can almost be achieved without affecting the replication and oncolytic ability of the OV itself. Second, OV selection should be made carefully to express BiTE for different diseases, which requires more comprehensive and detailed exploration. We can also screen or engineer better OVs. For example, the recently developed Tupaia paramyxovirus may represent a substitute for MV because of the absence of cross-neutralizing antibodies [110]. Third, with more and more in-depth research on the anti-tumor immune response, it is found that OV-BiTE may become an increasingly important platform for tumor immunotherapy. In this aspect, combining chemotherapy or other immunotherapy with OV-BiTE therapy may also have marked strengths. For example, a recent experiment showed that combining OV-BiTE with CAR-T cells can enhance the proliferation and activation of CAR-T cells in vitro and in vivo, thus improving T cell-mediated cytotoxicity [116]. Ribas et al. reported the significantly improved immune recognition of tumor when the oncolytic virus T-VEC was combined with an anti-PD1 antibody [117].
There is no doubt that the OV-BiTE is becoming a new platform for the exciting synergistic tumor immunotherapy. We believe that OV-BiTEs will enter clinical trials soon. Exploring the therapeutic effects and safety of OV-BiTE synergistic tumor immunotherapy will bring new hope to tumor patients.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81872412 to XHW, 81602303 to XY, 31700736 to WXW), the Natural Science Foundation of Hubei Province (2019CFB591 to Z.M.), Hubei Province Scientific and Technological Research Project (D20201306 to WXW), and Guangzhou Key Medical Discipline Construction Project (CSZ).
Disclosure of conflict of interest
None.
References
- 1.Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335–3337. doi: 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Xiang Y, Xin VW, Wang XW, Peng XC, Liu XQ, Wang D, Li N, Cheng JT, Lyv YN, Cui SZ, Ma Z, Zhang Q, Xin HW. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13:107. doi: 10.1186/s13045-020-00939-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slaney CY, Wang P, Darcy PK, Kershaw MH. CARs versus BiTEs: a comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. 2018;8:924–934. doi: 10.1158/2159-8290.CD-18-0297. [DOI] [PubMed] [Google Scholar]
- 4.Raja J, Ludwig JM, Gettinger SN, Schalper KA, Kim HS. Oncolytic virus immunotherapy: future prospects for oncology. J Immunother Cancer. 2018;6:140. doi: 10.1186/s40425-018-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018;18:498–513. doi: 10.1038/s41577-018-0014-6. [DOI] [PubMed] [Google Scholar]
- 6.Wu ZJ, Tang FR, Ma ZW, Peng XC, Xiang Y, Zhang Y, Kang J, Ji J, Liu XQ, Wang XW, Xin HW, Ren BX. Oncolytic viruses for tumor precision imaging and radiotherapy. Hum Gene Ther. 2018;29:204–222. doi: 10.1089/hum.2017.189. [DOI] [PubMed] [Google Scholar]
- 7.Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007;28:326–333. doi: 10.1016/j.tips.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang B, Cheng P. Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy. Mol Cancer. 2020;19:158. doi: 10.1186/s12943-020-01275-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 10.de Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. 2019;7:19. doi: 10.1186/s40425-019-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu XQ, Xin HY, Lyu YN, Ma ZW, Peng XC, Xiang Y, Wang YY, Wu ZJ, Cheng JT, Ji JF, Zhong JX, Ren BX, Wang XW, Xin HW. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. 2018;25:1950–1962. doi: 10.1080/10717544.2018.1534895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NB, Hamid M. scFv antibody: principles and clinical application. Clin Dev Immunol. 2012;2012:980250. doi: 10.1155/2012/980250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Chen Y, Park J, Liu X, Hu Y, Wang T, McFarland K, Betenbaugh MJ. Design and production of bispecific antibodies. Antibodies (Basel) 2019;8:43. doi: 10.3390/antib8030043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, June CH, Alemany R. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res. 2017;77:2052–2063. doi: 10.1158/0008-5472.CAN-16-1708. [DOI] [PubMed] [Google Scholar]
- 15.Maruta M, Ochi T, Tanimoto K, Asai H, Saitou T, Fujiwara H, Imamura T, Takenaka K, Yasukawa M. Direct comparison of target-reactivity and cross-reactivity induced by CAR- and BiTE-redirected T cells for the development of antibody-based T-cell therapy. Sci Rep. 2019;9:13293. doi: 10.1038/s41598-019-49834-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu M, He Q, Guo Z, Zhou X, Li H, Zhao L, Tang H, Zhou X, Zhu H, Shen G, He Y, Lei P. Therapeutic bispecific T-cell engager antibody targeting the transferrin receptor. Front Immunol. 2019;10:1396. doi: 10.3389/fimmu.2019.01396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 18.Iwahori K, Kakarla S, Velasquez MP, Yu F, Yi Z, Gerken C, Song XT, Gottschalk S. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol Ther. 2015;23:171–178. doi: 10.1038/mt.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guy DG, Uy GL. Bispecific antibodies for the treatment of acute myeloid leukemia. Curr Hematol Malig Rep. 2018;13:417–425. doi: 10.1007/s11899-018-0472-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Owen DH, Giffin MJ, Bailis JM, Smit MD, Carbone DP, He K. DLL3: an emerging target in small cell lung cancer. J Hematol Oncol. 2019;12:61. doi: 10.1186/s13045-019-0745-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott EM, Jacobus EJ, Lyons B, Frost S, Freedman JD, Dyer A, Khalique H, Taverner WK, Carr A, Champion BR, Fisher KD, Seymour LW, Duffy MR. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J Immunother Cancer. 2019;7:320. doi: 10.1186/s40425-019-0807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, Campo L, Bryant RJ, Verrill C, Lambert A, Miller P, Champion BR, Seymour LW, Fisher KD. An oncolytic virus expressing a T-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018;78:6852–6865. doi: 10.1158/0008-5472.CAN-18-1750. [DOI] [PubMed] [Google Scholar]
- 23.Brey CU, Proff J, Teufert N, Salzer B, Brozy J, Munz M, Pendzialek J, Ensser A, Holter W, Lehner M. A gB/CD3 bispecific BiTE antibody construct for targeting Human Cytomegalovirus-infected cells. Sci Rep. 2018;8:17453. doi: 10.1038/s41598-018-36055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brozy J, Schlaepfer E, Mueller CKS, Rochat MA, Rampini SK, Myburgh R, Raum T, Kufer P, Baeuerle PA, Muenz M, Speck RF. Antiviral activity of HIV gp120-targeting bispecific T cell engager antibody constructs. J Virol. 2018;92:e00491-18. doi: 10.1128/JVI.00491-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S, Horst HA, Raff T, Viardot A, Schmid M, Stelljes M, Schaich M, Degenhard E, Kohne-Volland R, Bruggemann M, Ottmann O, Pfeifer H, Burmeister T, Nagorsen D, Schmidt M, Lutterbuese R, Reinhardt C, Baeuerle PA, Kneba M, Einsele H, Riethmuller G, Hoelzer D, Zugmaier G, Bargou RC. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011;29:2493–2498. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- 26.Stadler CR, Bahr-Mahmud H, Plum LM, Schmoldt K, Kolsch AC, Tureci O, Sahin U. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. Oncoimmunology. 2016;5:e1091555. doi: 10.1080/2162402X.2015.1091555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aigner M, Feulner J, Schaffer S, Kischel R, Kufer P, Schneider K, Henn A, Rattel B, Friedrich M, Baeuerle PA, Mackensen A, Krause SW. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27:1107–1115. doi: 10.1038/leu.2012.341. [DOI] [PubMed] [Google Scholar]
- 28.Bonifant CL, Szoor A, Torres D, Joseph N, Velasquez MP, Iwahori K, Gaikwad A, Nguyen P, Arber C, Song XT, Redell M, Gottschalk S. CD123-engager T cells as a novel immunotherapeutic for acute myeloid leukemia. Mol Ther. 2016;24:1615–1626. doi: 10.1038/mt.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krawczyk E, Zolov SN, Huang K, Bonifant CL. T-cell Activity against AML Improved by dual-targeted T cells stimulated through T-cell and IL7 receptors. Cancer Immunol Res. 2019;7:683–692. doi: 10.1158/2326-6066.CIR-18-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hipp S, Tai YT, Blanset D, Deegen P, Wahl J, Thomas O, Rattel B, Adam PJ, Anderson KC, Friedrich M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia. 2017;31:1743–1751. doi: 10.1038/leu.2016.388. [DOI] [PubMed] [Google Scholar]
- 31.Yu S, Li A, Liu Q, Yuan X, Xu H, Jiao D, Pestell RG, Han X, Wu K. Recent advances of bispecific antibodies in solid tumors. J Hematol Oncol. 2017;10:155. doi: 10.1186/s13045-017-0522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, Offner S, Locher M, Urbig T, Raum T, Kleindienst P, Wimberger P, Kimmig R, Fichtner I, Kufer P, Hofmeister R, da Silva AJ, Baeuerle PA. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43:1129–1143. doi: 10.1016/j.molimm.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 33.Xin HW, Ambe CM, Miller TC, Chen JQ, Wiegand GW, Anderson AJ, Ray S, Mullinax JE, Hari DM, Koizumi T, Godbout JD, Goldsmith PK, Stojadinovic A, Rudloff U, Thorgeirsson SS, Avital I. Liver label retaining cancer cells are relatively resistant to the reported anti-cancer stem cell drug metformin. J Cancer. 2016;7:1142–1151. doi: 10.7150/jca.10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrmann I, Baeuerle PA, Friedrich M, Murr A, Filusch S, Ruttinger D, Majdoub MW, Sharma S, Kufer P, Raum T, Munz M. Highly efficient elimination of colorectal tumor-initiating cells by an EpCAM/CD3-bispecific antibody engaging human T cells. PLoS One. 2010;5:e13474. doi: 10.1371/journal.pone.0013474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cioffi M, Dorado J, Baeuerle PA, Heeschen C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin Cancer Res. 2012;18:465–474. doi: 10.1158/1078-0432.CCR-11-1270. [DOI] [PubMed] [Google Scholar]
- 36.Cioffi M, Heeschen C. Immuno-targeting of pancreatic cancer stem cells: a new therapeutic strategy against a devastating disease? Oncoimmunology. 2012;1:560–562. doi: 10.4161/onci.19368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xin HW, Ambe CM, Hari DM, Wiegand GW, Miller TC, Chen JQ, Anderson AJ, Ray S, Mullinax JE, Koizumi T, Langan RC, Burka D, Herrmann MA, Goldsmith PK, Stojadinovic A, Rudloff U, Thorgeirsson SS, Avital I. Label-retaining liver cancer cells are relatively resistant to sorafenib. Gut. 2013;62:1777–1786. doi: 10.1136/gutjnl-2012-303261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xin HW, Ambe CM, Ray S, Kim BK, Koizumi T, Wiegand GW, Hari D, Mullinax JE, Jaiswal KR, Garfield SH, Stojadinovic A, Rudloff U, Thorgeirsson SS, Avital I. Wnt and the cancer niche: paracrine interactions with gastrointestinal cancer cells undergoing asymmetric cell division. J Cancer. 2013;4:447–457. doi: 10.7150/jca.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hari D, Xin HW, Jaiswal K, Wiegand G, Kim BK, Ambe C, Burka D, Koizumi T, Ray S, Garfield S, Thorgeirsson S, Avital I. Isolation of live label-retaining cells and cells undergoing asymmetric cell division via nonrandom chromosomal cosegregation from human cancers. Stem Cells Dev. 2011;20:1649–1658. doi: 10.1089/scd.2010.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amann M, Brischwein K, Lutterbuese P, Parr L, Petersen L, Lorenczewski G, Krinner E, Bruckmeier S, Lippold S, Kischel R, Lutterbuese R, Kufer P, Baeuerle PA, Schlereth B. Therapeutic window of MuS110, a single-chain antibody construct bispecific for murine EpCAM and murine CD3. Cancer Res. 2008;68:143–151. doi: 10.1158/0008-5472.CAN-07-2182. [DOI] [PubMed] [Google Scholar]
- 41.Oberst MD, Fuhrmann S, Mulgrew K, Amann M, Cheng L, Lutterbuese P, Richman L, Coats S, Baeuerle PA, Hammond SA. CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. MAbs. 2014;6:1571–1584. doi: 10.4161/19420862.2014.975660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han ZW, Lyv ZW, Cui B, Wang YY, Cheng JT, Zhang Y, Cai WQ, Zhou Y, Ma ZW, Wang XW, Peng XC, Cui SZ, Xiang Y, Yang M, Xin HW. The old CEACAMs find their new role in tumor immunotherapy. Invest New Drugs. 2020;38:1888–1898. doi: 10.1007/s10637-020-00955-w. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Yu C, Wu Y, Sun X, Su Q, You C, Xin H. CD44(+) fibroblasts increases breast cancer cell survival and drug resistance via IGF2BP3-CD44-IGF2 signalling. J Cell Mol Med. 2017;21:1979–1988. doi: 10.1111/jcmm.13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xin HW, Hari DM, Mullinax JE, Ambe CM, Koizumi T, Ray S, Anderson AJ, Wiegand GW, Garfield SH, Thorgeirsson SS, Avital I. Tumor-initiating label-retaining cancer cells in human gastrointestinal cancers undergo asymmetric cell division. Stem Cells. 2012;30:591–598. doi: 10.1002/stem.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lutterbuese R, Raum T, Kischel R, Lutterbuese P, Schlereth B, Schaller E, Mangold S, Rau D, Meier P, Kiener PA, Mulgrew K, Oberst MD, Hammond SA, Baeuerle PA, Kufer P. Potent control of tumor growth by CEA/CD3-bispecific single-chain antibody constructs that are not competitively inhibited by soluble CEA. J Immunother. 2009;32:341–352. doi: 10.1097/CJI.0b013e31819b7c70. [DOI] [PubMed] [Google Scholar]
- 46.Waaijer SJH, Warnders FJ, Stienen S, Friedrich M, Sternjak A, Cheung HK, van Scheltinga AGTT, Schröder CP, de Vries EGE, Lub-de Hooge MN. Molecular imaging of radiolabeled bispecific T-cell engager (89)Zr-AMG211 targeting CEA-positive tumors. Clin Cancer Res. 2018;24:4988–4996. doi: 10.1158/1078-0432.CCR-18-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 48.Bi Y, Jiang H, Wang P, Song B, Wang H, Kong X, Li Z. Treatment of hepatocellular carcinoma with a GPC3-targeted bispecific T cell engager. Oncotarget. 2017;8:52866–52876. doi: 10.18632/oncotarget.17905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamauchi N, Watanabe A, Hishinuma M, Ohashi K, Midorikawa Y, Morishita Y, Niki T, Shibahara J, Mori M, Makuuchi M, Hippo Y, Kodama T, Iwanari H, Aburatani H, Fukayama M. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Mod Pathol. 2005;18:1591–1598. doi: 10.1038/modpathol.3800436. [DOI] [PubMed] [Google Scholar]
- 50.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Hirsch FR, Suda K, Wiens J, Bunn PA Jr. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet. 2016;388:1012–1024. doi: 10.1016/S0140-6736(16)31473-8. [DOI] [PubMed] [Google Scholar]
- 52.Powell HA, Tata LJ, Baldwin DR, Potter VA, Stanley RA, Khakwani A, Hubbard RB. Treatment decisions and survival for people with small-cell lung cancer. Br J Cancer. 2014;110:908–915. doi: 10.1038/bjc.2013.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furuta M, Kikuchi H, Shoji T, Takashima Y, Kikuchi E, Kikuchi J, Kinoshita I, Dosaka-Akita H, Sakakibara-Konishi J. DLL3 regulates the migration and invasion of small cell lung cancer by modulating Snail. Cancer Sci. 2019;110:1599–1608. doi: 10.1111/cas.13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, Desai R, Escarpe PA, Hampl J, Laysang A, Liu D, Lopez-Molina J, Milton M, Park A, Pysz MA, Shao H, Slingerland B, Torgov M, Williams SA, Foord O, Howard P, Jassem J, Badzio A, Czapiewski P, Harpole DH, Dowlati A, Massion PP, Travis WD, Pietanza MC, Poirier JT, Rudin CM, Stull RA, Dylla SJ. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med. 2015;7:302ra136. doi: 10.1126/scitranslmed.aac9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 56.Imperato JP, Paleologos NA, Vick NA. Effects of treatment on long-term survivors with malignant astrocytomas. Ann Neurol. 1990;28:818–822. doi: 10.1002/ana.410280614. [DOI] [PubMed] [Google Scholar]
- 57.Choi BD, Kuan CT, Cai M, Archer GE, Mitchell DA, Gedeon PC, Sanchez-Perez L, Pastan I, Bigner DD, Sampson JH. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc Natl Acad Sci U S A. 2013;110:270–275. doi: 10.1073/pnas.1219817110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi BD, Archer GE, Mitchell DA, Heimberger AB, McLendon RE, Bigner DD, Sampson JH. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009;19:713–723. doi: 10.1111/j.1750-3639.2009.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yankelevich M, Kondadasula SV, Thakur A, Buck S, Cheung NK, Lum LG. Anti-CD3 x anti-GD2 bispecific antibody redirects T-cell cytolytic activity to neuroblastoma targets. Pediatr Blood Cancer. 2012;59:1198–1205. doi: 10.1002/pbc.24237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Modak S, Cheung NK. Disialoganglioside directed immunotherapy of neuroblastoma. Cancer Invest. 2007;25:67–77. doi: 10.1080/07357900601130763. [DOI] [PubMed] [Google Scholar]
- 61.Wu ZL, Schwartz E, Seeger R, Ladisch S. Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res. 1986;46:440–443. [PubMed] [Google Scholar]
- 62.Elsasser-Beile U, Buhler P, Wolf P. Targeted therapies for prostate cancer against the prostate specific membrane antigen. Curr Drug Targets. 2009;10:118–125. doi: 10.2174/138945009787354601. [DOI] [PubMed] [Google Scholar]
- 63.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 64.Ward JF, Moul JW. Rising prostate-specific antigen after primary prostate cancer therapy. Nat Clin Pract Urol. 2005;2:174–182. doi: 10.1038/ncpuro0145. [DOI] [PubMed] [Google Scholar]
- 65.Friedrich M, Raum T, Lutterbuese R, Voelkel M, Deegen P, Rau D, Kischel R, Hoffmann P, Brandl C, Schuhmacher J, Mueller P, Finnern R, Fuergut M, Zopf D, Slootstra JW, Baeuerle PA, Rattel B, Kufer P. Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol Cancer Ther. 2012;11:2664–2673. doi: 10.1158/1535-7163.MCT-12-0042. [DOI] [PubMed] [Google Scholar]
- 66.Micke P, Mattsson JS, Edlund K, Lohr M, Jirstrom K, Berglund A, Botling J, Rahnenfuehrer J, Marincevic M, Ponten F, Ekman S, Hengstler J, Woll S, Sahin U, Tureci O. Aberrantly activated claudin 6 and 18.2 as potential therapy targets in non-small-cell lung cancer. Int J Cancer. 2014;135:2206–2214. doi: 10.1002/ijc.28857. [DOI] [PubMed] [Google Scholar]
- 67.Hafner C, Schmitz G, Meyer S, Bataille F, Hau P, Langmann T, Dietmaier W, Landthaler M, Vogt T. Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin Chem. 2004;50:490–499. doi: 10.1373/clinchem.2003.026849. [DOI] [PubMed] [Google Scholar]
- 68.Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. 2008;6:1795–1806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiang Y, Chen YJ, Yan YB, Liu Y, Qiu J, Tan RQ, Tian Q, Guan L, Niu SS, Xin HW. MiR-186 bidirectionally regulates cisplatin sensitivity of ovarian cancer cells via suppressing targets PIK3R3 and PTEN and upregulating APAF1 expression. J Cancer. 2020;11:3446–3453. doi: 10.7150/jca.41135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu MR, Zhang T, Gacerez AT, Coupet TA, DeMars LR, Sentman CL. B7H6-specific bispecific T cell engagers lead to tumor elimination and host antitumor immunity. J Immunol. 2015;194:5305–5311. doi: 10.4049/jimmunol.1402517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, Xiang J, Yan S, de Morais Guerreiro MD, Veomett N, Dubrovsky L, Curcio M, Doubrovina E, Ponomarev V, Liu C, O’Reilly RJ, Scheinberg DA. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015;33:1079–1086. doi: 10.1038/nbt.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cheng JT, Wang L, Wang H, Tang FR, Cai WQ, Sethi G, Xin HW, Ma Z. Insights into biological role of LncRNAs in epithelial-mesenchymal transition. Cells. 2019;8:1178. doi: 10.3390/cells8101178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kruse PH, Matta J, Ugolini S, Vivier E. Natural cytotoxicity receptors and their ligands. Immunol Cell Biol. 2014;92:221–229. doi: 10.1038/icb.2013.98. [DOI] [PubMed] [Google Scholar]
- 75.Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C, Moretta A, West R, Xu W, Vivier E, Levin SD. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med. 2009;206:1495–1503. doi: 10.1084/jem.20090681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Teira P, Battiwalla M, Ramanathan M, Barrett AJ, Ahn KW, Chen M, Green JS, Saad A, Antin JH, Savani BN, Lazarus HM, Seftel M, Saber W, Marks D, Aljurf M, Norkin M, Wingard JR, Lindemans CA, Boeckh M, Riches ML, Auletta JJ. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood. 2016;127:2427–2438. doi: 10.1182/blood-2015-11-679639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Green ML, Leisenring WM, Xie H, Walter RB, Mielcarek M, Sandmaier BM, Riddell SR, Boeckh M. CMV reactivation after allogeneic HCT and relapse risk: evidence for early protection in acute myeloid leukemia. Blood. 2013;122:1316–1324. doi: 10.1182/blood-2013-02-487074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meng W, Tang A, Ye X, Gui X, Li L, Fan X, Schultz RD, Freed DC, Ha S, Wang D, Zhang N, Fu TM, An Z. Targeting human-cytomegalovirus-infected cells by redirecting T cells using an anti-CD3/anti-glycoprotein B bispecific antibody. Antimicrob Agents Chemother. 2018;62:e01719-17. doi: 10.1128/AAC.01719-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shasha D, Walker BD. Lessons to be learned from natural control of HIV - future directions, therapeutic, and preventive implications. Front Immunol. 2013;4:162. doi: 10.3389/fimmu.2013.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao J, Song Y, Liu D. Recent advances on blinatumomab for acute lymphoblastic leukemia. Exp Hematol Oncol. 2019;8:28. doi: 10.1186/s40164-019-0152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, Kronke J, Facon T, Salnikov AV, Lesley R, Beutner K, Kalabus J, Rasmussen E, Riemann K, Minella AC, Munzert G, Einsele H. Anti-B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J. Clin. Oncol. 2020;38:775–783. doi: 10.1200/JCO.19.02657. [DOI] [PubMed] [Google Scholar]
- 84.Kebenko M, Goebeler ME, Wolf M, Hasenburg A, Seggewiss-Bernhardt R, Ritter B, Rautenberg B, Atanackovic D, Kratzer A, Rottman JB, Friedrich M, Vieser E, Elm S, Patzak I, Wessiepe D, Stienen S, Fiedler W. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE(R)) antibody construct, in patients with refractory solid tumors. Oncoimmunology. 2018;7:e1450710. doi: 10.1080/2162402X.2018.1450710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pishvaian M, Morse MA, McDevitt J, Norton JD, Ren S, Robbie GJ, Ryan PC, Soukharev S, Bao H, Denlinger CS. Phase 1 dose escalation study of MEDI-565, a bispecific T-cell engager that targets human carcinoembryonic antigen, in patients with advanced gastrointestinal adenocarcinomas. Clin Colorectal Cancer. 2016;15:345–351. doi: 10.1016/j.clcc.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 86.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Freedman JD, Hagel J, Scott EM, Psallidas I, Gupta A, Spiers L, Miller P, Kanellakis N, Ashfield R, Fisher KD, Duffy MR, Seymour LW. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol Med. 2017;9:1067–1087. doi: 10.15252/emmm.201707567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi F, Xin VW, Liu XQ, Wang YY, Zhang Y, Cheng JT, Cai WQ, Xiang Y, Peng XC, Wang X, Xin HW. Identification of 22 novel motifs of the cell entry fusion glycoprotein b of oncolytic herpes simplex viruses: sequence analysis and literature review. Front Oncol. 2020;10:1386. doi: 10.3389/fonc.2020.01386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang D, Wang XW, Peng XC, Xiang Y, Song SB, Wang YY, Chen L, Xin VW, Lyu YN, Ji J, Ma ZW, Li CB, Xin HW. CRISPR/Cas9 genome editing technology significantly accelerated herpes simplex virus research. Cancer Gene Ther. 2018;25:93–105. doi: 10.1038/s41417-018-0016-3. [DOI] [PubMed] [Google Scholar]
- 90.Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer. 2018;18:419–432. doi: 10.1038/s41568-018-0009-4. [DOI] [PubMed] [Google Scholar]
- 91.Grigg C, Blake Z, Gartrell R, Sacher A, Taback B, Saenger Y. Talimogene laherparepvec (T-Vec) for the treatment of melanoma and other cancers. Semin Oncol. 2016;43:638–646. doi: 10.1053/j.seminoncol.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 92.Cheng JT, Wang YY, Zhu LZ, Zhang Y, Cai WQ, Han ZW, Zhou Y, Wang XW, Peng XC, Xiang Y, Yang HY, Cui SZ, Ma Z, Liu BR, Xin HW. Novel transcription regulatory sequences and factors of the immune evasion protein ICP47 (US12) of herpes simplex viruses. Virol J. 2020;17:101. doi: 10.1186/s12985-020-01365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang Y, Xin Q, Zhang JY, Wang YY, Cheng JT, Cai WQ, Han ZW, Zhou Y, Cui SZ, Peng XC, Wang XW, Ma Z, Xiang Y, Su XL, Xin HW. Transcriptional regulation of latency-associated transcripts (LATs) of herpes simplex viruses. J Cancer. 2020;11:3387–3399. doi: 10.7150/jca.40186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu F, Wang X, Guo ZS, Bartlett DL, Gottschalk SM, Song XT. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther. 2014;22:102–111. doi: 10.1038/mt.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang YY, Lyu YN, Xin HY, Cheng JT, Liu XQ, Wang XW, Peng XC, Xiang Y, Xin VW, Lu CB, Ren BX, Liang YF, Ji JF, Ma Z, Cui SZ, Xin HW. Identification of putative UL54 (ICP27) transcription regulatory sequences binding to Oct-1, v-Myb, Pax-6 and hairy in herpes simplex viruses. J Cancer. 2019;10:430–440. doi: 10.7150/jca.29787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 97.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Smith GL, Moss B. Infectious poxvirus vectors have capacity for at least 25 000 base pairs of foreign DNA. Gene. 1983;25:21–28. doi: 10.1016/0378-1119(83)90163-4. [DOI] [PubMed] [Google Scholar]
- 99.McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, Moss B, Bartlett DL. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- 100.Nettelbeck DM. Cellular genetic tools to control oncolytic adenoviruses for virotherapy of cancer. J Mol Med (Berl) 2008;86:363–377. doi: 10.1007/s00109-007-0291-1. [DOI] [PubMed] [Google Scholar]
- 101.Rojas JJ, Gimenez-Alejandre M, Gil-Hoyos R, Cascallo M, Alemany R. Improved systemic antitumor therapy with oncolytic adenoviruses by replacing the fiber shaft HSG-binding domain with RGD. Gene Ther. 2012;19:453–457. doi: 10.1038/gt.2011.106. [DOI] [PubMed] [Google Scholar]
- 102.Kuhn I, Harden P, Bauzon M, Chartier C, Nye J, Thorne S, Reid T, Ni S, Lieber A, Fisher K, Seymour L, Rubanyi GM, Harkins RN, Hermiston TW. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS One. 2008;3:e2409. doi: 10.1371/journal.pone.0002409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Seymour LW, Fisher KD. Oncolytic viruses: finally delivering. Br J Cancer. 2016;114:357–361. doi: 10.1038/bjc.2015.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou D, Zhao ZY. Advances in measles virus for cancer therapy. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2015;44:458–464. doi: 10.3785/j.issn.1008-9292.2015.07.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Robinson S, Galanis E. Potential and clinical translation of oncolytic measles viruses. Expert Opin Biol Ther. 2017;17:353–363. doi: 10.1080/14712598.2017.1288713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Peng KW, Ahmann GJ, Pham L, Greipp PR, Cattaneo R, Russell SJ. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98:2002–2007. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 107.Allen C, Opyrchal M, Aderca I, Schroeder MA, Sarkaria JN, Domingo E, Federspiel MJ, Galanis E. Oncolytic measles virus strains have significant antitumor activity against glioma stem cells. Gene Ther. 2013;20:444–449. doi: 10.1038/gt.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Heidbuechel JPW, Engeland CE. Paramyxoviruses for tumor-targeted immunomodulation: design and evaluation ex vivo. J Vis Exp. 2019 doi: 10.3791/58651. [DOI] [PubMed] [Google Scholar]
- 109.Parks CL, Lerch RA, Walpita P, Wang HP, Sidhu MS, Udem SA. Analysis of the noncoding regions of measles virus strains in the Edmonston vaccine lineage. J Virol. 2001;75:921–933. doi: 10.1128/JVI.75.2.921-933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Speck T, Heidbuechel JPW, Veinalde R, Jaeger D, von Kalle C, Ball CR, Ungerechts G, Engeland CE. Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin Cancer Res. 2018;24:2128–2137. doi: 10.1158/1078-0432.CCR-17-2651. [DOI] [PubMed] [Google Scholar]
- 111.Yu F, Hong B, Song XT. A T-cell engager-armed oncolytic vaccinia virus to target the tumor stroma. Cancer Translational Medicine. 2017;3:122. [Google Scholar]
- 112.Cai WQ, Zeng LS, Wang LF, Wang YY, Cheng JT, Zhang Y, Han ZW, Zhou Y, Huang SL, Wang XW, Peng XC, Xiang Y, Ma Z, Cui SZ, Xin HW. The latest battles between EGFR monoclonal antibodies and resistant tumor cells. Front Oncol. 2020;10:1249. doi: 10.3389/fonc.2020.01249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barlabe P, Sostoa J, Fajardo CA, Alemany R, Moreno R. Enhanced antitumor efficacy of an oncolytic adenovirus armed with an EGFR-targeted BiTE using menstrual blood-derived mesenchymal stem cells as carriers. Cancer Gene Ther. 2020;27:383–388. doi: 10.1038/s41417-019-0110-1. [DOI] [PubMed] [Google Scholar]
- 114.Rojas JJ, Sampath P, Bonilla B, Ashley A, Hou W, Byrd D, Thorne SH. Manipulating TLR Signaling increases the anti-tumor T cell response induced by viral cancer therapies. Cell Rep. 2016;15:264–273. doi: 10.1016/j.celrep.2016.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Carlisle R, Choi J, Bazan-Peregrino M, Laga R, Subr V, Kostka L, Ulbrich K, Coussios CC, Seymour LW. Enhanced tumor uptake and penetration of virotherapy using polymer stealthing and focused ultrasound. J Natl Cancer Inst. 2013;105:1701–1710. doi: 10.1093/jnci/djt305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Correnti CE, Laszlo GS, de van der Schueren WJ, Godwin CD, Bandaranayake A, Busch MA, Gudgeon CJ, Bates OM, Olson JM, Mehlin C, Walter RB. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia. 2018;32:1239–1243. doi: 10.1038/s41375-018-0014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, Kirkwood JM, Gajewski TF, Chen L, Gorski KS, Anderson AA, Diede SJ, Lassman ME, Gansert J, Hodi FS, Long GV. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170:1109–1119. e1110. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]