

Abstract
In this study, we intended to explore a novel combination treatment scheme for pancreatic cancer, using irreversible electroporation (IRE) and OX40 agonist. We further aimed to investigate the capacity and mechanism of this combination treatment using an in vivo mouse aggressive pancreatic cancer model. To this end, mice subcutaneously injected with KPC1199 pancreatic tumor cells were treated with IRE, followed by intraperitoneal injection of OX40 agonist. Tumor growth and animal survival were observed. Flow cytometry analysis, immunohistochemistry, and immunofluorescence were used to evaluate the immune cell populations within the tumors. The tumor-specific immunity was assessed using ELISpot assay. Besides, the cytokine patterns both in serum and tumors were identified using Luminex assay. After combination therapy with IRE and OX40 agonist, 80% of the mice completely eradicated the established subcutaneous tumors, during the 120 days observation period. Rechallenging these tumor-free mice at day 120 with KPC1199 tumor cells leads to complete resistance to tumor growth, suggesting that the combination therapy generated long-term-specific antitumor immune memory. Moreover, combination therapy significantly delayed the growth of contralateral untreated tumors, and significantly prolonged animal survival, suggesting that a potent systematic anti-tumor immunity was induced by combination therapy. Mechanically, combination therapy amplified antitumor immune response induced by IRE, as manifested by the increased quality and quantity of CD8+ T cells trigged by IRE. Together, these results provide strong evidence for the clinical assessment of the combination of IRE and OX40 agonist in patients with pancreatic cancer.
Keywords: Pancreatic cancer, OX40, irreversible electroporation, immunotherapy, combination treatment
Introduction
Pancreatic cancer (PC) remains one of the most deadly solid cancers in the world and one of the most difficult to diagnose and cure, making it the seventh leading cause of cancer-related deaths [1]. However, in all PC cases, more than 90% are pancreatic ductal adenocarcinoma (PDAC). Furthermore, the 5-year survival rate has been reported less than 9% for all stages among patients diagnosed with PDAC [2]. Complete surgical resection of the tumor remains a potentially curative treatment, but unfortunately, nearly 80% of patients with PDAC are diagnosed at locally advanced or distant metastasis stages, meaning that these patients miss the opportunity for radical surgical therapy. Moreover, after surgical resection, the recurrence rate remains high despite adjuvant chemotherapy [3-5]. Recently, the advances made in systemic chemotherapy and radiation therapy have contributed to modest improvements in patient survival [6-8]. Besides, immunotherapy, including immune checkpoint inhibitors (ICIs), chimeric antigen receptor T cells (CAR-T), or cancer vaccines, has demonstrated promising results for many cancers but did not include PC [9]. To achieve superior therapeutic effects against PC, new strategies are needed.
As a member of the tumor necrosis factor receptor (TNFR) superfamily, the costimulatory molecule OX40 is chiefly expressed in activated CD8+ and CD4+ T cells, which regulate multiple functions of T cells [10-12]. The proliferation, differentiation, survival, and effector function of T cells and the generation of memory T cells can be enhanced by OX40 costimulation [13-16]. Manipulation of OX40 signaling using recombinant soluble OX40 ligand (OX40L) or the anti-OX40 agonist monoclonal antibody (anti-OX40 mAb), either alone or in combination with other treatments, is an efficient approach to treating multiple preclinical tumor models, including PC [13,17-24]. However, in clinical settings, anti-OX40 mAb has shown limited efficacy against advanced solid tumors [25,26]. One of the potential reasons is the lack of OX40 expression at baseline, which might limit the anti-tumor efficiency of anti-OX40 mAb. Therefore, there is increasing interest in combining anti-OX40 mAb with additional strategies to enhance antigen availability or the expression of OX40 in T cells.
Currently, a novel ablation strategy, irreversible electroporation (IRE), has emerged as a non-thermal minimally invasive ablation technique against multiple solid tumors. Unlike conventional ablation methods, IRE relies on high-voltage electrical pulses across the target tissues aimed to kill tumor cells by producing irreversible nanopores in cellular membranes. These nanopores result in increased transmembrane transport and loss of homeostasis, while there is less damage to surrounding normal tissues, such as blood vessels, intestines, and bile ducts. These unique advantages make IRE an ideal choice for the treatment of PC, which is not suitable for traditional ablation methods [27,28]. In clinical settings, IRE has shown promising results in treating PC [29-33]. Prior clinical studies demonstrated that apart from the primary function of IRE to ablate PC, IRE has also shown immunomodulatory properties [34,35]. Besides, previous studies reported that IRE ablation might invoke a more robust immune response than conventional ablation methods, such as radiofrequency ablation or cryoablation [36-38]. However, micrometastases, recurrence after incomplete ablation, or residual microscopic tumor remain the problematic aspects of delivering curative treatment by IRE in clinical settings. This reality implies that the immunomodulatory effects induced by IRE therapy might be weak and transitory which are incapable of eradicating the residual tumor and preventing disease progression. Therefore, the immune response caused by IRE requires augmentation by other therapies to exert an effective anti-tumor immunity.
To this end, we hypothesized that IRE induces the exposure of tumor antigens that could serve as tumor vaccination in situ, which might further activate T cells in vivo and upregulate the expression of OX40 in T cells. Therefore, therapy using anti-OX40 mAb might augment and prolong the tumor-specific adaptive immune responses induced by IRE. Ultimately, these effects might stimulate the immune system to attack residual microscopic tumor or micrometastases, and systemic benefits for distant tumors might be obtained from such combination therapy. To test our hypothesis, this study assessed the efficacy and mechanisms of the combination of IRE therapy and anti-OX40 mAb in a PC model.
Materials and methods
Tumor cells and animals
The KPC1199 cell line is a mouse PC cell line, which was isolated from a spontaneous PDAC mouse model (LSL-KrasG12D, Pdxcre, LSL-TP53R172H) on a C57BL/6 background [39], and was kindly provided by Dr. Liwei Wang (Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China). Cells were cultured in DMEM medium (ThermoFisher, Waltham, MA, USA) supplemented with 1% penicillin-streptomycin (PS) solution (Gibco, New York, NY, USA), 10% fetal bovine serum (FBS) (ThermoFisher) at 37°C, and 5% CO2 in a humidified atmosphere. Male 6-8-week-old C57BL/6 mice were obtained from SLAC Laboratory Animal Co. Ltd. (Shanghai, China). And, all mice were housed under specific pathogen-free conditions. All our studies were performed in line with the Guidelines for the Care and Use of Laboratory Animals of Shanghai Jiao Tong University School of Medicine.
Establishment of subcutaneous PC mouse model and treatment protocol
For therapeutic efficacy studies, all-male wild-type (WT) C57BL/6 mice aged 6-8 weeks underwent a subcutaneous injection of 4×106 KPC1199 tumor cells into the right flank. This first injection into the right flank was done to simulate the primary tumor. To further assess the effects of IRE+anti-OX40 mAb combination therapy on distant metastatic tumors, 3 days later, the left flank was injected with 2×106 KPC1199 tumor cells, which represented distant metastatic tumors. Twelve days after injection of the primary tumor, when the tumor on the right flank reached a volume of approximately 200 mm3 or the length of the primary tumor had reached 6-9 mm, mice (n = 10 in each group) were randomly allotted into the following groups: control group (untreated group), IRE therapy group, anti-OX40 mAb group, and IRE followed by anti-OX40 mAb combination therapy group. For IRE therapy alone, mice were anesthetized using intraperitoneal (IP) injection of sodium pentobarbital (10 mg/mL, 50 mg/kg body weight) before the treatment. Subsequently, an insulating plate was used to fix the mouse, and then an ECM 830 Electroporation System with a pair of adjustable electrodes (BTX Harvard Apparatus, Holliston, MA, USA) was used to perform the IRE procedure. Adjustable electrodes with a maximum gap of 10 mm were inserted into the margin of the subcutaneous tumor, and the gap between electrodes was adjusted depending on the size of the tumor, enabling the entire tumor to receive electric pulses. And, the parameters of IRE treatment in our study were set as follows: voltage, 1250 V; pulse duration, 90 μs; pulse interval, 100 ms. For anti-OX40 mAb therapy alone or combination therapy mice, anti-OX40 mAb (5 mg/kg, Cat #BE0031-100, clone OX86, Bio X Cell, Lebanon, NH, USA) in 100 μL phosphate-buffered saline (PBS) was administered intraperitoneally starting on day 13 (24 h after IRE therapy) and continued every 2 days for a total of six doses. The mice were weighed every 4 days. During our study, tumors were measured every 2-4 days with calipers, and the volume of the tumor was calculated as length (mm) × width (mm)2/2. Animals were observed at least three times per week for survival studies. When animals were moribund or their tumor volume reached 1500 mm3, they were euthanized and considered to have reached the “survival” endpoint following Institutional Animal Care and Use Committee guidelines. For mechanism study, mice in each group were euthanized at specific time points according to the purpose of the experiment, and tumors, spleens, serum, or tumor-draining lymph nodes (TDLN) were isolated for further analysis.
T cells depletion experiments
For the T cell depletion experiment, anti-mouse CD8 monoclonal antibody (Cat #BE0061, clone 2.43, Bio X Cell), anti-mouse CD4 monoclonal antibody (Cat #BE0003-1, clone GK1.5, Bio X Cell), or isotype control antibody (Cat #BE0094, Bio X Cell) (250 μg in 100 μL PBS) were delivered per mouse (n = 6 in each group) four times by IP injection, two times per week, beginning on day 11 after inoculation of the primary tumor (1 d before initiation of treatment). The extent of depletion was verified in spleens using flow cytometry analysis, and the depletion efficiency was found to be greater than 99% (Figure 5B).
Figure 5.
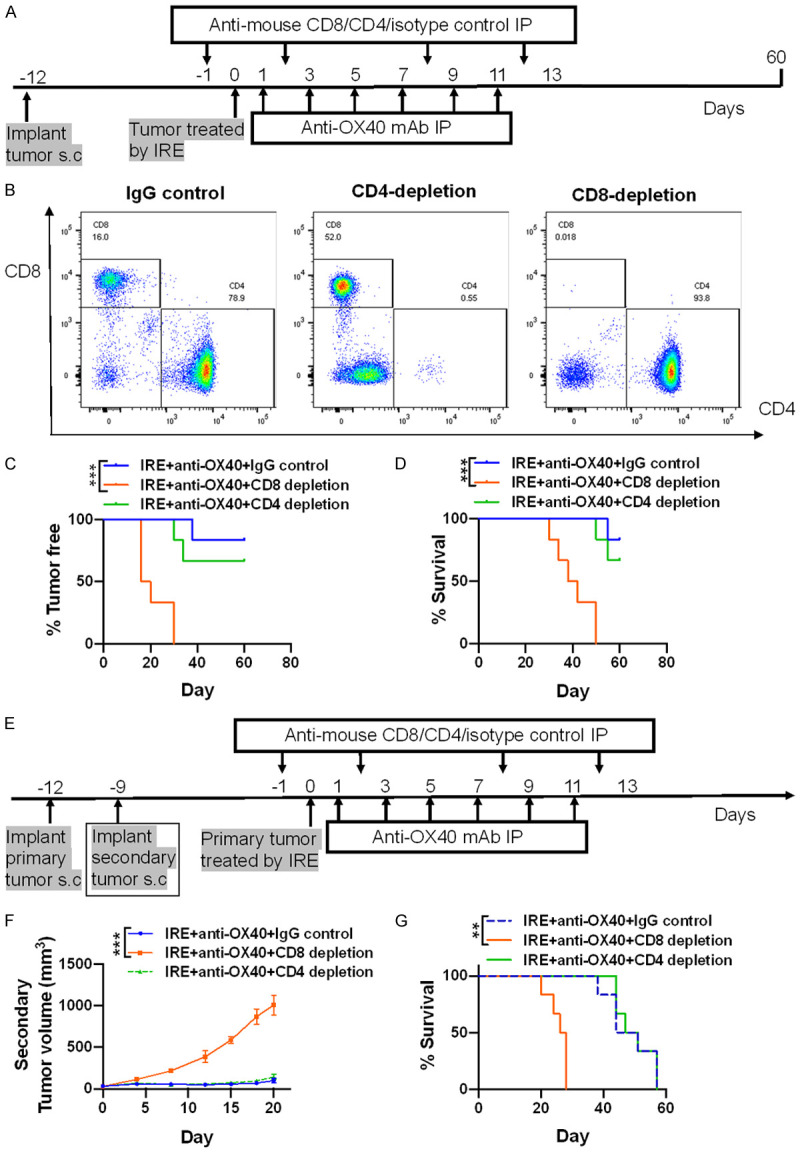
The anti-tumor activity of combination therapy is CD8+ T-cell dependent. A. Tumor cell implantation, immune cell depletion, and treatment strategy in unilateral subcutaneous mouse model. B. Successful depletion of CD4+ T and CD8+ T cells is confirmed using flow cytometry, and no cells are depleted after control IgG antibody injection. C and D. Tumor-free rates and survival rates after depletion of CD4+ or CD8+ T cells in unilateral subcutaneous tumor mice treated with combination therapy. N = 6 per group. E. Tumor cell implantation, immune cell depletion, and treatment strategy in bilateral subcutaneous tumor mouse model. F and G. Growth curves of the secondary untreated tumors and survival rates after depletion of CD4+ or CD8+ T cells in bilateral subcutaneous tumor mice treated with combination therapy. N = 6 per group. **P < 0.01, ***P < 0.001.
Tumor re-challenge experiments
To investigate long-term antitumor immunological memory, 120 days after initiation of treatment, cured mice in the IRE plus anti-OX40 mAb combination treatment group (n = 5) were re-challenged with 4×106 KPC1199 tumor cells in the contralateral (left) flank. Age-matched male WT C57BL/6 mice (n = 5) injected with the same numbers of KPC1199 tumor cells were used as control. Afterward, tumor growth was monitored over the next few weeks.
Interferon (IFN)-γ enzyme-linked immunospot (ELISpot) assays
For tumor-specific CD8+ T cell functional assays, 10 days after the initial therapy and 18 days after re-challenge, spleens were excised and CD8+ T cells were isolated using EasySep Mouse CD8α Positive Selection Kit (Cat #18953, Stemcell, Vancouver, Canada). Subsequently, irradiated (20 Gy) KPC1199 tumor cells were incubated with isolated CD8+ T cells (2×105) at the ratio of 1:10 for 48 h. Afterward, ELISpot assays to detect the IFN-γ cytokine spots were conducted according to the manufacturer’s protocol (Cat #551083, BD Pharmingen, San Diego, CA, USA). A CTL-ImmunoSpot® Plate Reader was used to scan the plates and CTL ImmunoSpot Software was used to analyze the data.
Flow cytometry
To obtain single-cell suspensions, TDLN or spleen samples isolated from the mice were first mechanically pushed through a 70 μm nylon sieve to prepare a single-cell suspension. For TDLN samples, the cells were followed by filtered through a 40 μm nylon mesh. For spleen samples, erythrocytes lysis was performed using red blood cell (RBC) lysis buffer (Beyotime, Shanghai, China) for 10 min at room temperature (RT), the cells were then washed (1500 rpm, 5 min) two times with fluorescence-activated cell sorting (FACS) buffer (1% FBS in PBS), followed by passed through a 40 μm nylon mesh. On day 10, tumors were harvested from different groups and minced using scissors, followed by digested for 30 min at 37°C in 6 mL RPMI 1640 (ThermoFisher) medium containing 2 mg/mL collagenase type IV (Cat #LS004188, Worthington, Lakewood, NJ, USA), 0.2 mg/mL hyaluronidase (Cat #H3506, Sigma, St. Louis, MO, USA), and 0.2 mg/mL DNase I (Cat #10104159001, Roche, Basel, Switzerland) under agitation, and then passed sequentially through a 70 μm cell filter and a 40-μm nylon mesh to obtain a single-cell suspension. Subsequently, cells were first Fc blocked with purified rat anti-mouse CD16/CD32 (Cat #553141, BD Pharmingen) in FACS buffer for 30 min at 4°C. Fixable Viability Stain 780 (1:200) (Cat #565388, BD Pharmingen) was then used to incubated with the cells in FACS buffer for 30 min at 4°C to discriminate dead cells. After washing in FACS buffer, surface marker staining was performed at 4°C for 30 with. Besides, the Foxp3/Transcription Factor Fixation/Permeabilization kit was used for intracellular staining, according to the producer’s instructions (Cat #00-5523-00, eBiosciences, San Diego, CA, USA). For T cells stimulation in vitro, 1×106 cells/mL were incubated in RPMI 1640 with 10% FBS and 2 μl/ml of Cell Stimulation Cocktail with Brefeldin A (Cat #423304, BioLegend, San Diego, CA, USA) for 5 h. The antibodies used are listed in Table S1. Data were acquired on a BD Biosciences Fortessa X20 flow cytometer and analyzed with FlowJo (10.0) software.
Immunohistochemistry (IHC)
Tumor samples were processed with 4% paraformaldehyde fixation and paraffin embedding and sectioned to a thickness of 5 μm. After deparaffinization and rehydration, all sections were treated with ethylenediaminetetraacetic acid (EDTA) (pH 9.0) for antigen retrieval, and then 3% H2O2 was used to deactivate endogenous peroxidase. Subsequently, anti-CD8 monoclonal antibody (Cat #98941, CST, Beverly, MA, USA), anti-CD3 monoclonal antibody (Cat #Ab16669, Abcam, Cambridge, MA, USA), and anti-CD4 monoclonal antibody (#Ab183685, Abcam) were used to incubated with sections at 4°C overnight. Afterward, horseradish peroxidase (HRP)-conjugated secondary antibodies were used to be stained with the sections. Subsequently, all sections were incubated with diaminobenzidine (DAB). Lastly, nuclei were stained with Mayer’s hematoxylin. To count positive cells, five random areas were observed under a fluorescence microscope (Nikon Eclipse Ci-S).
Immunofluorescence (IF)
After incubation with the primary anti-CD4 monoclonal antibody (Cat #Ab183685, Abcam), anti-Foxp3 monoclonal antibody (Cat #12653, Cell Signaling Technology), anti-CD11b monoclonal antibody (Cat #Ab133357, Abcam), and anti-Gr-1 monoclonal antibody (Cat #GB11229, Servicebio, Wuhan, China,) at 4°C overnight, immunofluorescence for visualizing these markers was performed, on the basis of the tyramide signal amplification (TSA) methodology, for 10-15 min at RT. The nuclei were then stained with 4’,6-diamidino-2-phenylindole (DAPI). Afterward, a Leica SP5 confocal laser scanning microscope (Mannheim, Germany) was used to acquire images with high quality, and three channels were used sequentially: DAPI (405 nm, blue for nuclei), Cy3 laser (530 nm, red for Gr-1 or Foxp3), and FITC laser (488 nm, green for CD4 or CD11b). Images were first acquired separately and then merged. To find myeloid-derived suppressor cells (MDSCs) and regulatory T cells (T-regs), the cells showing co-localization of CD11b and Gr-1 were identified as MDSCs (CD11b+Gr-1+), and co-localization of CD4 and Foxp3 were identified as T-regs (CD4+Foxp3+). For quantification, an average of five fields per slide was used to count co-positive cells.
Tumor supernatant collection
On day 10, the untreated secondary tumors were removed, washed with PBS, weighed, followed by cut into small pieces using sterile scissors. Then, the tumor fragments were dissociated with a tissue homogenizer. Afterward, the tissue homogenate was lysed using a cell lysis buffer containing phosphatase inhibitor (Absin, Shanghai, China), protease inhibitor, and phenylmethylsulfonyl fluoride (PMSF). Tissues were lysed at 4°C for 20 min with gentle agitation. After centrifuged at 13,000 rpm for 10 min, the supernatant was collected carefully. Pierce BCA Protein Assays (Solarbio, Pequim, China) was performed on the supernatant following the manufacturer’s instructions. Total protein concentrations for each tumor sample were used for analyte normalization into pg/mg protein values.
Luminex assays
Intratumor and serum levels of interleukins (IL)-1α, IL-1β, CXCL1, G-CSF, GM-CSF, IL-13, IL-17A, CCL2 (MCP-1), CCL3 (MIP-1α), CCL4 (MIP-1β), CCL5 (RANTES), CCL11 (eotaxin), IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IFN-γ, and TNF-α were determined using a Bio-Plex Pro Mouse Cytokine 23-plex Assay kit (Cat #M60009RDPD, Bio-Rad, Hercules, CA, USA). Assays were performed according to the producer’s instructions. A Luminex X-200 System and xPONENT 3.1 software (Luminex, Austin, TX, USA) were used for machine operation, data collection, and analysis. The sensitivity of each cytokine test was pg/mL.
Statistical analysis
Unpaired t-tests were used to compare differences between individual groups. Kaplan-Meier curves and log-rank Mantel-Cox tests were used to analyze survival outcomes between groups. A two-way analysis of variance (ANOVA) was conducted to compare tumor growth curves between groups. The presented data are representative of three independent experiments. All data are presented as mean ± standard error of the mean (SEM). All statistical analyses were performed using GraphPad Prism 8.0 software. Statistical significance was set at P < 0.05.
Results
Efficacy of anti-OX40 mAb combined with IRE against subcutaneous pancreatic tumors
First, we evaluated the therapeutic potential of local IRE therapy followed by anti-OX40 mAb treatment of unilateral tumors. To this end, mice implanted subcutaneously on the right flank with KPC1199 tumor cells were randomized and treated with IRE therapy, anti-OX40 mAb monotherapy, or the combination of IRE and anti-OX40 mAb or left without treatment (the treatment protocol is shown in the methods), and animal survival and tumor growth rate were compared between those groups (Figure 1A). Our data showed that the combination therapy exerted the strongest anti-tumor effect during the observation period of 120 days. All 10 mice in the control group died within 36 days due to excessive tumor burden. Mice treated with anti-OX40 mAb monotherapy had a median tumor burden comparable to that in the control group. Furthermore, the survival time between the two groups was also shown no significant difference (22 vs. 24 days, P = 0.205) (Figure 1D). For either single IRE therapy or combination therapy, the growth of the treated tumors was immediately halted after IRE and completely unpalpable at 5-7 days post-IRE (Figure 1B). Unfortunately, in the IRE therapy alone group, tumor relapse was observed 17 days after IRE; consequently, these mice had a median survival of 51 days (Figure 1C, 1D). In sharp contrast, 8 of 10 (80%) of mice in the IRE plus anti-OX40 mAb combination therapy group had no tumor recurrence, with complete eradication of the established primary tumors, and survived to the end of the 120-day monitoring period (Figure 1C, 1D). In addition, combination therapy was well tolerated with no overt toxicities or adverse effects.
Figure 1.
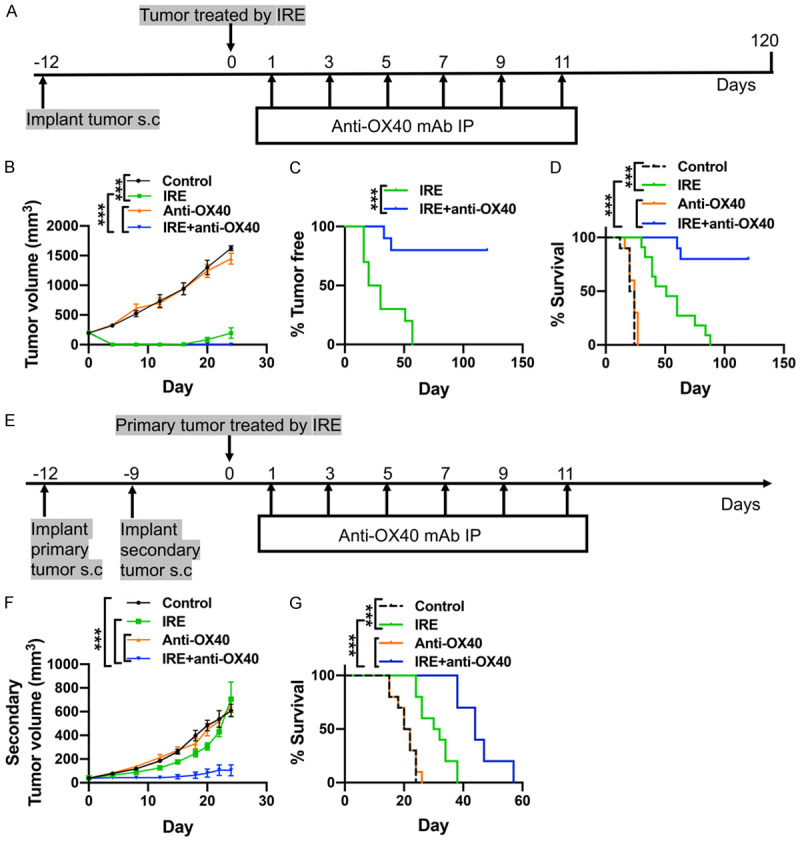
Efficacy of IRE and anti-OX40 mAb treatment of mouse subcutaneous pancreatic tumors. A. The schema for animal experiments using unilateral tumor mouse model. N = 10 per group. B. Volume changes in tumors in unilateral tumor mouse model following the indicated treatments. C. Tumor-free rates in IRE vs. IRE+Anti-OX40 groups after treatment initiation. D. Survival rates in unilateral tumor-bearing mice in the indicated treatment groups up to 120 days after initiating treatment. E. The schema for animal experiments using bilateral tumor mouse model. N = 10 per group. F. Volume changes of the untreated secondary tumors in bilateral tumor-bearing mice following the indicated treatments. G. Survival rates in bilateral tumor-bearing mice in the indicated treatment groups. N = 10 per group. ***P < 0.001. IRE, irreversible electroporation.
As seen in the potent efficiency of combination treatment in the control of primary subcutaneous tumors, we next determined whether anti-OX40 mAb combined with IRE treatment could induce systematic antitumor responses against untreated distant tumors. For this purpose, mice inoculated with KPC1199 tumor cells subcutaneously on both flanks were used. The primary tumor being selected for treatment and the secondary untreated tumor is considered a distant metastasis tumor (Figure 1E). Typically, compared with the control group, anti-OX40 mAb monotherapy did not slow down the growth rate of the secondary tumors. IRE therapy alone resulted in an insignificant reduction in the volume of the secondary tumors in a few weeks (Figure 1F). In contrast, treatment with IRE plus anti-OX40 mAb significantly slowed the growth of the secondary tumors (Figure 1F). In addition, the median survival time of the control, IRE therapy, anti-OX40 mAb monotherapy, and combination therapy groups were 21, 31, 21, and 44 days, respectively (P < 0.001) (Figure 1G).
These results indicated that anti-OX40 mAb synergized with IRE to induce a potent antitumor effect that controlled both the primary and the untreated secondary tumors.
Combination treatment changes immune cells infiltration into the primary tumors
To examine how combination treatment affected the primary tumor immune microenvironment (TIME), mice in all groups were sacrificed 10 days after initiation of therapy for analysis of the tumor-infiltrating immune cells by the means of IHC and IF. Our data showed that anti-OX40 mAb therapy alone had no significant impact on the population of CD3+, CD4+, CD8+ T cells, and MDSCs in the primary tumors. IRE alone induced a significant increase in CD3+ and CD8+ T cells and also induced a marked decrease in MDSCs compared with controls. In addition, combination therapy of IRE and anti-OX40 mAb led to further increase of CD3+ and CD8+ T cells and decreased MDSCs (Figure 2A, 2C, 2D). The numbers of CD4+FoxP3+ T-regs varied among the tumors with no significant difference for each therapy (Figure 2E). All these effects on immune cell infiltration into the primary tumors induced by combination therapy may lead to the eradication of the primary tumors.
Figure 2.

Immune cell infiltration in the primary tumor 10 days after initiation of treatments. (A-C) Representative images of immunohistochemistry and corresponding quantifications for CD3 (A), CD4 (B), and CD8 (C). (D, E) Representative images of immunofluorescence for MDSCs (D), and T-regs (E) and corresponding quantifications. MDSCs: CD11b+Gr1+; T-regs: CD4+Foxp3+. Scale bars = 50 µm. N = 5 per group. *P < 0.05 vs. control group, **P < 0.01 vs. control group; #P < 0.05 vs. IRE group. IRE, irreversible electroporation.
IRE combined with anti-OX40 mAb transforms the immune landscape of TIME in the distant untreated tumors
To further investigate the synergistic antitumor capacity induced by IRE and anti-OX40 mAb combination therapy, immune cells in the distant untreated tumors were analyzed 10 days after treatment initiation. As shown in Figure 3A, the results from flow cytometry showed that both treatments with IRE alone and anti-OX40 mAb alone failed to significantly affect all immune cell populations in the second tumors. However, IRE combined with anti-OX40 mAb induced a significant increase in CD8+, IFN-γ+ CD8+, and ki67+CD8+ T cells and a significant decrease in MDSCs in second tumors. Meanwhile, just as we saw in the primary tumors, no therapy had a significant effect on the infiltration of Tregs in the second tumors (Figure 3A). To further investigate whether T cells in the tumors were functional, a 23-plex bead-based assay was conducted to determine the levels of different cytokines and chemokines in the secondary tumors on day 10. As expected, the intratumoral levels of IFN-γ, IL-12p70, and IL-2 were significantly increased in the combination-treated group, while the levels of IL-10 were significantly decreased (Figure 3B). Besides, combination treatment significantly raised the intratumoral levels of CCL-2 and CCL-3 (Figure 3C).
Figure 3.
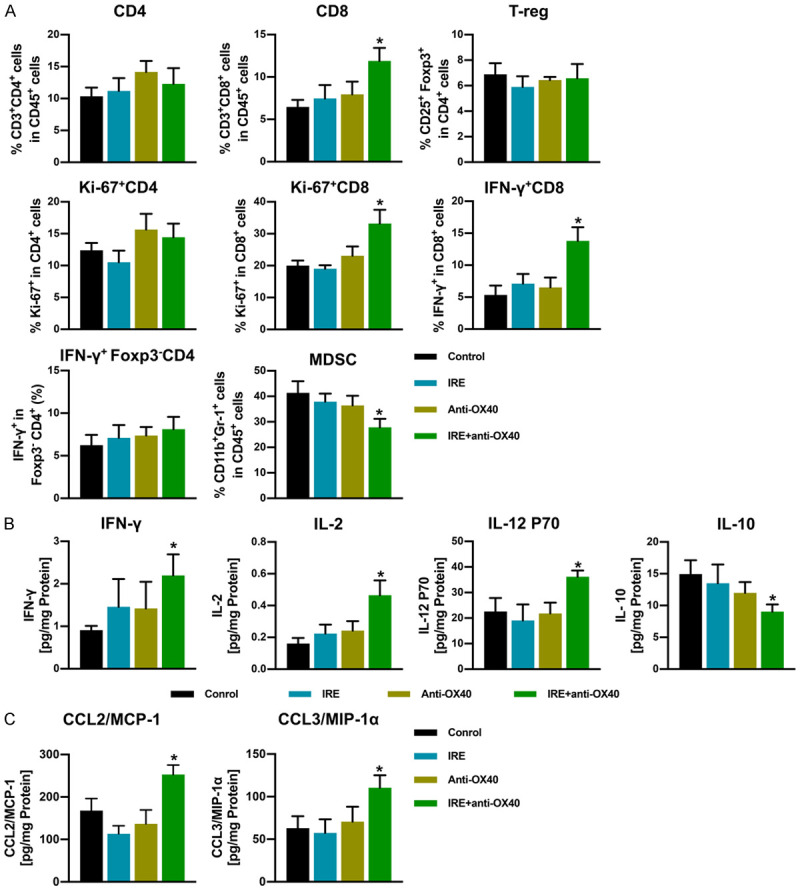
Immune cell infiltration and levels of cytokines or chemokines in the secondary tumor 10 days after initiation of treatments. A. Proportions of tumor-infiltrating CD3+CD8+ T cells, CD3+CD4+ T cells, ki-67+CD8+ cells, ki-67+CD4+ cells, IFN-γ+CD8+ cells, IFN-γ+Foxp3-CD4+ cells, T-regs (CD4+CD25+Foxp3+), and MDSCs (Gr-1+CD11b+) assessed using flow cytometry, n = 5 per group. B and C. Levels of cytokines or chemokines in the secondary tumor were measured using a Mouse Cytokine 23-plex Luminex Assay. Data values (pg/mL) are normalized to total protein content and are presented in pg/mg protein. Cytokines or chemokines that did not show any significant difference in all groups were not included in this figure. n = 5 per group. *P < 0.05 vs. control group.
These results further indicate that IRE combined with anti-OX40 mAb treatment synergistically shifts the intratumoral immune landscape toward a highly effective antitumoral state, which might play a pivotal role in suppresses the growth of the secondary untreated tumors.
Systemic antitumor immune response induced by IRE and anti-OX40 mAb combination therapy
We next asked if the anti-tumor activity observed in combination therapy reflected activation of systemic antitumor immune responses. To address this question, serum cytokine levels were measured 10 days after treatment initiation, using a mouse-specific 23 cytokine Luminex assay. Our data showed that anti-OX40 mAb monotherapy significantly elevated the levels of IL-3 and IL-12p70, while IRE therapy alone significantly increased the serum levels of TNF-α, IL-1α, IL-2, IL-3, IL-12p70, and IFN-γ (Figure 4A). Furthermore, higher levels of all six of these cytokines were detected in the combination therapy group than in the IRE group, especially the level of IFN-γ (Figure 4A), which further indicated the synergistic antitumor effect when IRE therapy was combined with anti-OX40 mAb. Apart from these six cytokines, combination therapy also leads to significantly higher levels of IL-9 and IL-17A in the serum (Figure 4A). To further evaluate the systematic antitumor immune effect, we next carried out ELISpot assays for IFN-γ secretion upon ex vivo restimulation of T cells with irradiated KPC1199 tumor cells. To this end, irradiated KPC1199 tumor cells were cocultured with CD8+ T cells isolated from spleens in vitro for 48 h. In agreement with the higher levels of intratumoral CD8+ T cells expressing IFN-γ, combination therapy markedly raised the number of tumor-specific IFN-γ-secreting CD8+ T cells in spleens when compared with other groups, although IRE alone also significantly raised the number of IFN-γ-secreting CD8+ T cells when compared with the control group or anti-OX40 mAb monotherapy group (Figure 4C).
Figure 4.
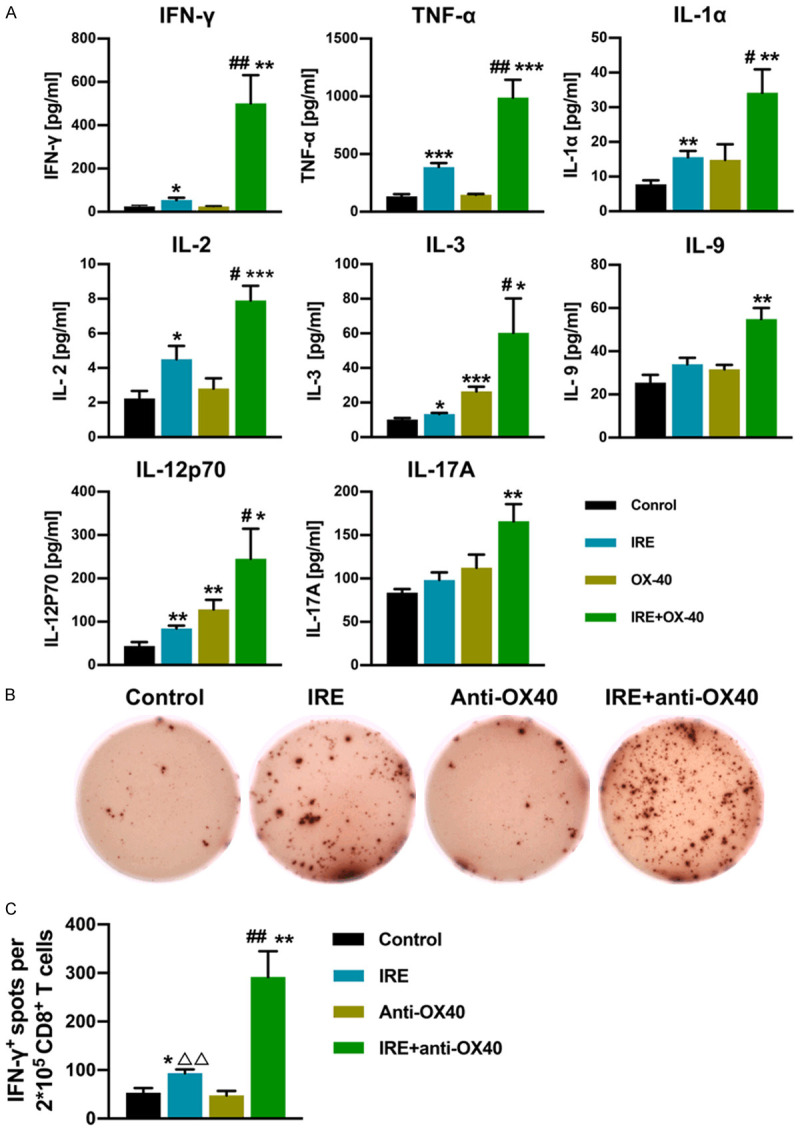
Levels of cytokines in serum and numbers of IFN-γ secreting CD8+ T cells in spleen at 10 days after initiation of treatments. (A) The concentration of serum cytokines in each group, assessed using Luminex assay. Cytokines that did not show any significant difference in all groups were not included in this figure. N = 5 per group. (B, C) IFN-γ secreting CD8+ T cells are analyzed using ELISpot. Representative ELISpot images from each group for (B), and the ELISpot count in each group for (C). N = 5 per group. *P < 0.05 vs. control group, **P < 0.01 vs. control group, ***P < 0.001 vs. control group, #P < 0.05 vs. IRE group, ##P < 0.01 vs. IRE group, ΔΔP < 0.01 vs. Anti-OX40 group. IRE, irreversible electroporation.
These data suggest that anti-OX40 mAb administration after IRE significantly amplified the systematic antitumor immune response triggered by IRE, synergistically inducing an effective tumor-specific systemic immune response, which might result in tumor eradication and prolonged animal survival.
Anti-tumor capacity of combination therapy is CD8+ T cell dependent
To evaluate the role of CD8+ and CD4+ T cells in the combination treatment, we assessed the antitumor capacity of combination therapy on established KPC1199 tumors in mice depleted of CD8+ or CD4+ T cells, both in unilateral and bilateral subcutaneous tumor mice groups (Figure 5A, 5E). Our data showed that in mice with unilateral subcutaneous tumors, the therapeutic effect provided by combination therapy was completely abrogated when mice were depleted of CD8+ T cells, which lead to local recurrence in all animals (Figure 5C). Furthermore, during the 60-day observation period, all these mice died, indicating that CD8+ T cells were responsible for the recurrence of the primary tumors (Figure 5D). However, the elimination of CD4+ T cells showed no significant impact on the recurrence rate of the ablated tumor and animal survival (Figure 5C, 5D). Next, we used mice bearing bilateral subcutaneous tumors to further confirm our findings. Similar to what we have observed in mice bearing unilateral subcutaneous tumors, neutralization of CD8+ T cells eliminated the anti-tumor capacity provided by combination therapy, as seen in Figure 5F, 5G. In contrast, elimination of CD4+ T cells was shown no significant impact on the delayed growth of the contralateral tumor and the survival benefit obtained from combination therapy.
These results indicate that CD8+ T cells have an indispensable function in the antitumor capacity of combination therapy in our models.
Combination therapy elicits a potent tumor-specific immunologic memory
To investigate whether long-term antitumor immunological memory was evoked in cured mice treated by combination therapy, a tumor rechallenge study was performed. Before rechallenge, three out of the eight long-term surviving cured mice were euthanized, then their spleens were obtained for further flow cytometry detection. The increased frequency of CD8+ effector memory T cells (CD3+CD8+CD44+CD62-) and CD8+ central memory T cells (CD3+CD8+CD44+CD62+) in splenocytes before rechallenge indicated the existence of immune memory in cured mice, induced by IRE plus anti-OX40 mAb combination therapy (Figure 6A, 6B). Afterward, five out of the eight cured mice in the combination therapy group were rechallenged with KPC1199 tumor cells (4×106) injected in the opposite flank at day 120, and age-matched healthy mice were used as controls. Surprisingly, all cured mice did not develop visible or palpable tumors after a few weeks. In sharp contrast, control mice developed palpable tumors (5/5) after one week, indicative of immunological memory (Figure 6C). To further investigate whether combination therapy was induced a tumor-specific memory response, re-challenged mice were euthanized at 18 days after re-challenge, serum and spleens were harvested, and then Luminex assays and ELISpot were conducted. The CD8+ T cells isolated from the spleens were stimulated in vitro by irradiated KPC1199 tumor cells, as quantified by the ELISpot assay. We found that the cured mice had almost 3 times more IFN-γ-secreting CD8+ T cells than the age-matched control mice (Figure 5D, 5E). In addition, the serum levels of IFN-γ, TNFα, IL-1α, IL-2, IL-3, IL-5, IL-12p70, and IL-13 were significantly increased in the cured mice (Figure 5F), indicating a strong immune response triggered by the tumor rechallenge.
Figure 6.
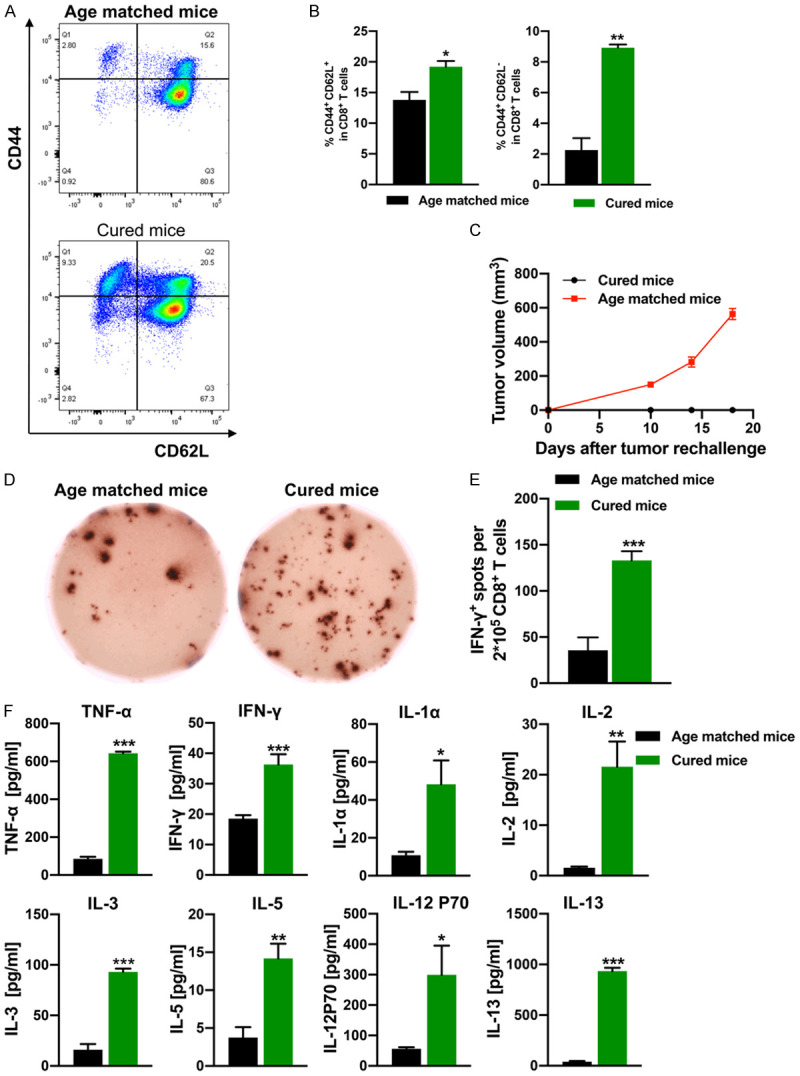
Immunological memory induced by combination therapy. A. Representative flow cytometry plots of splenic CD8+ central memory T cells (CD8+CD44+CD62L+) and CD8+ effector memory T cells (CD8+CD44+CD62L-) from cured mice isolated before rechallenge. B. Percentage of splenic CD8+ central memory T cells and CD8+ effector memory T cells from cured mice isolated before rechallenge. N = 3 per group. C. Growth curves of rechallenging tumors in the cured mice treated with IRE+anti-OX40 mAb. Age-matched healthy mice are used as controls (n = 5 per group). D. Representative images of IFN-γ spots from the ELISpot assay. E. Quantification of IFN-γ spots from the ELISpot assay. N = 5 per group. F. Cytokine levels in serum from mice isolated 18 days after cured mice were rechallenged with KPC1199 tumor cells. Cytokines that did not show any significant difference in all groups were not included in this figure. n = 5 per group. *P < 0.05, **P < 0.01, ***P < 0.001. IRE, irreversible electroporation.
Taken together, these results indicate that anti-OX40 mAb plus IRE combination therapy could induce a potent specific immunological memory for tumor rejection.
IRE induces the expression of OX40 on CD8+ T cells in vivo
To further explore the synergistic mechanism by which anti-OX40 mAb treatment combined with IRE induced robust anti-tumor immunity, we analyzed the expression of OX40 on CD8+ and CD4+ T cells both in spleen and TDLN at 24 hours (h), 48 hours, 7 days, and 10 days after IRE. Our data showed that at 24 h and 48 h after IRE, in the spleen, the percentages of OX40+CD8+ T cells were significantly elevated, and modestly increased 7 days after IRE. While on day 10, the percentages of OX40+CD8+ T cells were returned to baseline, which was almost the same as that in the control mice (Figure 7B). However, in the spleen, when compared with the control group, the percentages of OX40+CD4+ T cells were not significantly different at all time points (Figure 7C). The same trend was observed for TDLN (Figure 7E, 7F). These results indicate that IRE therapy might play a pivotal role in the expansion of OX40+CD8+ T cells in vivo, which might be responsible for the synergic anti-tumor effects when anti-OX40 mAb combined with IRE therapy.
Figure 7.

Percentages of OX40+ T lymphocytes in spleen and TDLN in mice in the IRE and control groups 24 h, 48 h, 7 days, and 10 days post-IRE, analyzed using flow cytometry. A. Representative flow cytometry plots showing OX40+CD8+ T cells in spleen. B and C. Percentages of OX40+CD8+ and OX40+CD4+ T cells in spleen. D. Representative flow cytometry plots showing OX40+CD8+ T cells in TDLN. E and F. Percentages of OX40+CD8+ and OX40+CD4+ T cells in TDLN. n = 5 per group. *P < 0.05, **P < 0.01. IRE, irreversible electroporation; TDLN, tumor-draining lymph nodes.
Discussion
In this study, we assessed the therapeutic capacity and anti-tumor immunity triggered by the combination therapy of IRE and anti-OX40 mAb in a subcutaneous PC mouse model. Our results showed that anti-OX40 mAb synergized with IRE elicited a potent anti-tumor effect against established tumors, with a cure rate of 80% in mice bearing unilateral tumors during our 120-day observation period, and significantly prolonged animal survival and delayed distant tumor growth of mice bearing bilateral tumors. Also, we demonstrated that CD8+ T cells were necessary for the potent antitumor efficacy provided by combination therapy in our system. Besides, tumor-free cured mice capable of rejecting tumor re-challenge, which indicated that combination treatment also induced an anti-tumor immune memory. By this mechanism, IRE significantly up-regulates the expression of OX40 on CD8+ T cells, which might provide a rationale for combination treatment. To our knowledge, this is the first preclinical work investigating the effects of IRE followed by anti-OX40 mAb combination therapy for the treatment of PC. Our finding that IRE therapy cooperates with anti-OX40 mAb eradicates primary tumors and significantly delays the growth of secondary untreated tumors adds to earlier reports proving the usefulness of anti-OX40 mAb combined with surgery, chemotherapy, vaccination, and radiation therapy to treat other tumors [22,40-42].
In our study, we observed significantly increased CD8+ T cells infiltrating both the primary tumors and distant tumors, with the help of combination therapy. Consistent with the increased level of CD8+ T cells in the TIME, these cells also exhibited both functional phenotype (IFN-γ+CD8+) and proliferative phenotype (ki67+CD8+) in distant tumors. Several studies have reported that the agonistic action of OX40 may inhibit the expression of Foxp3 and further inhibit the suppressive function of T-regs, leading to enhanced anti-tumor activity [43-45]. However, in our model, the frequency of T-regs in the TIME treated by each therapy varied in both primary and distant tumors. Moreover, we observed a significant reduction in MDSCs within both primary and distant tumors treated with combination therapy. MDSCs can inhibit the function of T cells through diversity mechanisms, and play a vital role in tumor metastasis, recurrence, and resistance to chemotherapy, immunotherapy, and targeted therapy [46]. Furthermore, in patients with solid tumors, higher MDSCs infiltration is related to poor prognosis [47]. In addition to the decreased MDSCs infiltration in tumors treated with combination therapy, we also observed a marked reduction of intra-tumor IL-10 levels in the distant tumors. Furthermore, combination therapy also significantly raised the intra-tumoral secretion of cytokines, including IL-12p70, IL-2, and, notably, IFN-γ. These inflammatory phenomena are indicative of there might exist of extensive immune activation within the TIME. Besides, combination therapy significantly elevated intra-tumoral secretion of chemokines, including CCL-2 and CCL-3, which might play a role in promoting T cell and natural killer cell infiltration into the tumor [48]. These findings suggest that in addition to straightly promoting the proliferation of CD8+ T cells in the secondary tumor, combination therapy might also recruit more immune cells by changing the spectrum of cytokines and chemokines in the tumor to exert a more powerful anti-tumor effect. Additionally, combination therapy led to a dramatic increase in the level of IFN-γ in serum and the number of IFN-γ-secreting CD8+ T cells in the spleen, suggesting that systematic antitumor immune responses were successfully primed.
Our findings in this mouse model are of particular importance. Currently, OX40 agonists are studied clinically in the treatment of multiple solid tumors, but these studies utilizing humanized OX40 antibodies reported inefficient or limited effects either with therapy alone or combined with ICIs [49,50]. However, the use of novel combination therapies, such as interventional therapy, may help improve anti-tumor efficiency. Despite the expression of OX40 in resting CD8+ T cells is not high, our data indicate that IRE therapy significantly elevated the expression of OX40 in CD8+ T cells both in the spleen and TDLN. However, it is unclear why in our model, the upregulation of OX40 favored CD8+ over CD4+ T cells after IRE, since the expression of OX40 has been reported on both activated CD8+ and CD4+ T cells [10]. Furthermore, our data showed that OX40 was mainly expressed in CD4+ T cells at baseline, suggesting that CD4+ T cells should be responsible for the antitumor efficacy of combination therapy. However, contrary to our expectations, in our system, a T cell depletion study indicated that CD8+ T cells were necessary for the antitumor efficacy of combination therapy. A possible explanation could be that the anti-tumor immune efficiency produced by IRE is mainly dependent on CD8+ T cells [51,52]. When the OX40 agonist was added, this anti-tumor effect produced by IRE was augmented, which might lead to the dispensable role of CD4+ T cells in our combination therapy system. The exact mechanism by which this occurs, however, remains to be determined.
A critical question is how ablation of a local tumor by IRE followed by systematic administration of anti-OX40 mAb induced a potent antitumor immune response in our model. We propose the following hypotheses: First, in destroying tumor cells, IRE can induce tumor immunogenic cell death (ICD) and produce tumor-associated antigens (TAAs) in situ and release different types of damage-associated molecular patterns (DAMPs) which can prime CD8+ T cells by activating dendritic cells in the TDLN. Afterward, the activated tumor-specific CD8+ T cells can infiltrate the distant tumor and move back to the primary tumor under certain conditions. Meanwhile, the immunosuppressive TIME is destroyed by IRE in the primary tumor, which might contribute to the expansion of tumor-specific CD8+ T cells [51,53]. However, without other interventions, the immune response induced by IRE alone might be weak or not strong enough to inhibit metastasis and recurrence in both PC patients and tumor cell-bearing mice [53,54]. Finally, anti-OX40 mAb might further augment the immune response induced by IRE, eliminate residual tumor cells and micrometastasis, and generate a long-term anti-tumor immune memory. To validate these hypotheses, more data are needed to determine exact processes that happen after combination therapy, and other mechanisms of how IRE combined with OX40 mAb induce this potent anti-tumor immunity need to be fully elucidated in the future.
It should be noted that our research has some limitations. First, during IRE therapy, although we used adjustable electrodes to encompass the tumor, we could not simulate the clinically safe ablation margin in the mouse subcutaneous tumor model; therefore, the “residual tumor” after ablation might not have been completely avoided. However, this situation exactly simulates the residual microscopic tumor that occurs after IRE therapy in a clinical situation, this might make our research more meaningful. Second, subcutaneous tumor models do not fully recapitulate the complex tumor microenvironment of PC in patients, and ongoing research will evaluate the efficacy of combination therapy in orthotopic pancreatic cancer models. Third, the infiltration of tumor-associated macrophages was not examined in our study, and whether combination therapy affects these immune cell components requires study in the future. Finally, in our study, based on previous studies, we designed a preliminary administration regimen of anti-OX40 mAb in combination therapy [21,22,42]. However, there might be an optimal combination schedule as to when anti-OX40 mAb should be administered and what doses should be given to elicit powerful anti-tumor efficiency. This remains to be determined in a subsequent experiment.
In summary, our finding that OX40 agonist synergizes with IRE against pancreatic tumors in a mouse model is novel and implies that combining IRE with OX40 agonist might be a potentially helpful scheme for treating pancreatic cancer clinically. Our data also offer a basis for further study of the mechanism of this therapeutic strategy.
Acknowledgements
This work was supported by Shanghai Municipal Key Clinical Specialty (No. shslczdzk06002).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol. 2019;10:10–27. doi: 10.14740/wjon1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas RM, Truty MJ, Nogueras-Gonzalez GM, Fleming JB, Vauthey JN, Pisters PW, Lee JE, Rice DC, Hofstetter WL, Wolff RA, Varadhachary GR, Wang H, Katz MH. Selective reoperation for locally recurrent or metastatic pancreatic ductal adenocarcinoma following primary pancreatic resection. J Gastrointest Surg. 2012;16:1696–1704. doi: 10.1007/s11605-012-1912-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol. 2015;12:319–334. doi: 10.1038/nrclinonc.2015.53. [DOI] [PubMed] [Google Scholar]
- 5.Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63:318–348. doi: 10.3322/caac.21190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahadevan A, Miksad R, Goldstein M, Sullivan R, Bullock A, Buchbinder E, Pleskow D, Sawhney M, Kent T, Vollmer C, Callery M. Induction gemcitabine and stereotactic body radiotherapy for locally advanced nonmetastatic pancreas cancer. Int J Radiat Oncol Biol Phys. 2011;81:e615–622. doi: 10.1016/j.ijrobp.2011.04.045. [DOI] [PubMed] [Google Scholar]
- 7.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, Péré-Vergé D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 9.Sahin IH, Askan G, Hu ZI, O’Reilly EM. Immunotherapy in pancreatic ductal adenocarcinoma: an emerging entity? Ann Oncol. 2017;28:2950–2961. doi: 10.1093/annonc/mdx503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 11.Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50–66. doi: 10.1016/j.ejca.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 12.Jensen SM, Maston LD, Gough MJ, Ruby CE, Redmond WL, Crittenden M, Li Y, Puri S, Poehlein CH, Morris N, Kovacsovics-Bankowski M, Moudgil T, Twitty C, Walker EB, Hu HM, Urba WJ, Weinberg AD, Curti B, Fox BA. Signaling through OX40 enhances antitumor immunity. Semin Oncol. 2010;37:524–532. doi: 10.1053/j.seminoncol.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song A, Song J, Tang X, Croft M. Cooperation between CD4 and CD8 T cells for anti-tumor activity is enhanced by OX40 signals. Eur J Immunol. 2007;37:1224–1232. doi: 10.1002/eji.200636957. [DOI] [PubMed] [Google Scholar]
- 14.Serghides L, Bukczynski J, Wen T, Wang C, Routy JP, Boulassel MR, Sekaly RP, Ostrowski M, Bernard NF, Watts TH. Evaluation of OX40 ligand as a costimulator of human antiviral memory CD8 T cell responses: comparison with B7.1 and 4-1BBL. J Immunol. 2005;175:6368–6377. doi: 10.4049/jimmunol.175.10.6368. [DOI] [PubMed] [Google Scholar]
- 15.Ruby CE, Redmond WL, Haley D, Weinberg AD. Anti-OX40 stimulation in vivo enhances CD8+ memory T cell survival and significantly increases recall responses. Eur J Immunol. 2007;37:157–166. doi: 10.1002/eji.200636428. [DOI] [PubMed] [Google Scholar]
- 16.Yu Q, Yue FY, Gu XX, Schwartz H, Kovacs CM, Ostrowski MA. OX40 ligation of CD4+ T cells enhances virus-specific CD8+ T cell memory responses independently of IL-2 and CD4+ T regulatory cell inhibition. J Immunol. 2006;176:2486–2495. doi: 10.4049/jimmunol.176.4.2486. [DOI] [PubMed] [Google Scholar]
- 17.Morris A, Vetto JT, Ramstad T, Funatake CJ, Choolun E, Entwisle C, Weinberg AD. Induction of anti-mammary cancer immunity by engaging the OX-40 receptor in vivo. Breast Cancer Res Treat. 2001;67:71–80. doi: 10.1023/a:1010649303056. [DOI] [PubMed] [Google Scholar]
- 18.Pan PY, Zang Y, Weber K, Meseck ML, Chen SH. OX40 ligation enhances primary and memory cytotoxic T lymphocyte responses in an immunotherapy for hepatic colon metastases. Mol Ther. 2002;6:528–536. doi: 10.1006/mthe.2002.0699. [DOI] [PubMed] [Google Scholar]
- 19.Sadun RE, Hsu WE, Zhang N, Nien YC, Bergfeld SA, Sabzevari H, Lutsiak ME, Khawli L, Hu P, Epstein AL. Fc-mOX40L fusion protein produces complete remission and enhanced survival in 2 murine tumor models. J Immunother. 2008;31:235–245. doi: 10.1097/CJI.0b013e31816a88e0. [DOI] [PubMed] [Google Scholar]
- 20.Foote JB, Kok M, Leatherman JM, Armstrong TD, Marcinkowski BC, Ojalvo LS, Kanne DB, Jaffee EM, Dubensky TW Jr, Emens LA. A STING agonist given with OX40 receptor and PD-L1 modulators primes immunity and reduces tumor growth in tolerized mice. Cancer Immunol Res. 2017;5:468–479. doi: 10.1158/2326-6066.CIR-16-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jahan N, Talat H, Alonso A, Saha D, Curry WT. Triple combination immunotherapy with GVAX, anti-PD-1 monoclonal antibody, and agonist anti-OX40 monoclonal antibody is highly effective against murine intracranial glioma. Oncoimmunology. 2019;8:e1577108. doi: 10.1080/2162402X.2019.1577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jahan N, Talat H, Curry WT. Agonist OX40 immunotherapy improves survival in glioma-bearing mice and is complementary with vaccination with irradiated GM-CSF-expressing tumor cells. Neuro Oncol. 2018;20:44–54. doi: 10.1093/neuonc/nox125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma Y, Li J, Wang H, Chiu Y, Kingsley CV, Fry D, Delaney SN, Wei SC, Zhang J, Maitra A, Yee C. Combination of PD-1 inhibitor and OX40 agonist induces tumor rejection and immune memory in mouse models of pancreatic cancer. Gastroenterology. 2020;159:306–319. e312. doi: 10.1053/j.gastro.2020.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68:5206–5215. doi: 10.1158/0008-5472.CAN-07-6484. [DOI] [PubMed] [Google Scholar]
- 25.Diab A, El-Khoueiry A, Eskens FA, Ros W, Thompson JA, Konto C, Bermingham C, Joh T, Liao K, Ganguly B, Hamid O. A first-in-human (FIH) study of PF-04518600 (PF-8600) OX40 agonist in adult patients (pts) with select advanced malignancies. Ann Oncol. 2016;27:vi361. [Google Scholar]
- 26.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, Walker J, Gonzalez I, Meeuwsen T, Fox BA, Moudgil T, Miller W, Haley D, Coffey T, Fisher B, Delanty-Miller L, Rymarchyk N, Kelly T, Crocenzi T, Bernstein E, Sanborn R, Urba WJ, Weinberg AD. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73:7189–7198. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Sakere B, André F, Bernat C, Connault E, Opolon P, Davalos RV, Rubinsky B, Mir LM. Tumor ablation with irreversible electroporation. PLoS One. 2007;2:e1135. doi: 10.1371/journal.pone.0001135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.José A, Sobrevals L, Ivorra A, Fillat C. Irreversible electroporation shows efficacy against pancreatic carcinoma without systemic toxicity in mouse models. Cancer Lett. 2012;317:16–23. doi: 10.1016/j.canlet.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Martin RC 2nd, Kwon D, Chalikonda S, Sellers M, Kotz E, Scoggins C, McMasters KM, Watkins K. Treatment of 200 locally advanced (stage III) pancreatic adenocarcinoma patients with irreversible electroporation: safety and efficacy. Ann Surg. 2015;262:486–494. doi: 10.1097/SLA.0000000000001441. [DOI] [PubMed] [Google Scholar]
- 30.Narayanan G, Hosein PJ, Beulaygue IC, Froud T, Scheffer HJ, Venkat SR, Echenique AM, Hevert EC, Livingstone AS, Rocha-Lima CM, Merchan JR, Levi JU, Yrizarry JM, Lencioni R. Percutaneous image-guided irreversible electroporation for the treatment of unresectable, locally advanced pancreatic adenocarcinoma. J Vasc Interv Radiol. 2017;28:342–348. doi: 10.1016/j.jvir.2016.10.023. [DOI] [PubMed] [Google Scholar]
- 31.Scheffer HJ, Nielsen K, de Jong MC, van Tilborg AA, Vieveen JM, Bouwman AR, Meijer S, van Kuijk C, van den Tol PM, Meijerink MR. Irreversible electroporation for nonthermal tumor ablation in the clinical setting: a systematic review of safety and efficacy. J Vasc Interv Radiol. 2014;25:997–1011. doi: 10.1016/j.jvir.2014.01.028. [DOI] [PubMed] [Google Scholar]
- 32.Ruarus AH, Vroomen LGPH, Geboers B, van Veldhuisen E, Puijk RS, Nieuwenhuizen S, Besselink MG, Zonderhuis BM, Kazemier G, de Gruijl TD, van Lienden KP, de Vries JJJ, Scheffer HJ, Meijerink MR. Percutaneous irreversible electroporation in locally advanced and recurrent pancreatic cancer (PANFIRE-2): a multicenter, prospective, single-arm, phase II study. Radiology. 2020;294:212–220. doi: 10.1148/radiol.2019191109. [DOI] [PubMed] [Google Scholar]
- 33.Scheffer HJ, Vroomen LG, de Jong MC, Melenhorst MC, Zonderhuis BM, Daams F, Vogel JA, Besselink MG, van Kuijk C, Witvliet J, de van der Schueren MA, de Gruijl TD, Stam AG, van den Tol PM, van Delft F, Kazemier G, Meijerink MR. Ablation of locally advanced pancreatic cancer with percutaneous irreversible electroporation: results of the phase I/II PANFIRE study. Radiology. 2017;282:585–597. doi: 10.1148/radiol.2016152835. [DOI] [PubMed] [Google Scholar]
- 34.He C, Wang J, Sun S, Zhang Y, Li S. Immunomodulatory effect after irreversible electroporation in patients with locally advanced pancreatic cancer. J Oncol. 2019;2019:9346017. doi: 10.1155/2019/9346017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pandit H, Hong YK, Li Y, Rostas J, Pulliam Z, Li SP, Martin RCG. Evaluating the regulatory immunomodulation effect of irreversible electroporation (IRE) in pancreatic adenocarcinoma. Ann Surg Oncol. 2019;26:800–806. doi: 10.1245/s10434-018-07144-3. [DOI] [PubMed] [Google Scholar]
- 36.Bulvik BE, Rozenblum N, Gourevich S, Ahmed M, Andriyanov AV, Galun E, Goldberg SN. Irreversible electroporation versus radiofrequency ablation: a comparison of local and systemic effects in a small-animal model. Radiology. 2016;280:413–424. doi: 10.1148/radiol.2015151166. [DOI] [PubMed] [Google Scholar]
- 37.Shao Q, O’Flanagan S, Lam T, Roy P, Pelaez F, Burbach BJ, Azarin SM, Shimizu Y, Bischof JC. Engineering T cell response to cancer antigens by choice of focal therapeutic conditions. Int J Hyperthermia. 2019;36:130–138. doi: 10.1080/02656736.2018.1539253. [DOI] [PubMed] [Google Scholar]
- 38.White SB, Zhang Z, Chen J, Gogineni VR, Larson AC. Early immunologic response of irreversible electroporation versus cryoablation in a rodent model of pancreatic cancer. J Vasc Interv Radiol. 2018;29:1764–1769. doi: 10.1016/j.jvir.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 39.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 40.Gough MJ, Crittenden MR, Sarff M, Pang P, Seung SK, Vetto JT, Hu HM, Redmond WL, Holland J, Weinberg AD. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother. 2010;33:798–809. doi: 10.1097/CJI.0b013e3181ee7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, Weinberg AD, Wolchok JD, Houghton AN. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J Exp Med. 2009;206:1103–1116. doi: 10.1084/jem.20082205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niknam S, Barsoumian HB, Schoenhals JE, Jackson HL, Yanamandra N, Caetano MS, Li A, Younes AI, Cadena A, Cushman TR, Chang JY, Nguyen QN, Gomez DR, Diab A, Heymach JV, Hwu P, Cortez MA, Welsh JW. Radiation followed by OX40 stimulation drives local and abscopal antitumor effects in an anti-PD1-resistant lung tumor model. Clin Cancer Res. 2018;24:5735–5743. doi: 10.1158/1078-0432.CCR-17-3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, Xiao X, Lan P, Li J, Dou Y, Chen W, Ishii N, Chen S, Xia B, Chen K, Taparowsky E, Li XC. OX40 costimulation inhibits Foxp3 expression and treg induction via BATF3-dependent and independent mechanisms. Cell Rep. 2018;24:607–618. doi: 10.1016/j.celrep.2018.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitamura N, Murata S, Ueki T, Mekata E, Reilly RT, Jaffee EM, Tani T. OX40 costimulation can abrogate Foxp3+ regulatory T cell-mediated suppression of antitumor immunity. Int J Cancer. 2009;125:630–638. doi: 10.1002/ijc.24435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5:3–8. doi: 10.1158/2326-6066.CIR-16-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The role of myeloid-derived suppressor cells in patients with solid tumors: a meta-analysis. PLoS One. 2016;11:e0164514. doi: 10.1371/journal.pone.0164514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakasone Y, Fujimoto M, Matsushita T, Hamaguchi Y, Huu DL, Yanaba M, Sato S, Takehara K, Hasegawa M. Host-derived MCP-1 and MIP-1α regulate protective anti-tumor immunity to localized and metastatic B16 melanoma. Am J Pathol. 2012;180:365–374. doi: 10.1016/j.ajpath.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 49.Gutierrez M, Moreno V, Heinhuis KM, Olszanski AJ, Spreafico A, Ong M, Chu Q, Carvajal RD, Trigo J, Ochoa de Olza M, Provencio M, De Vos FY, De Braud F, Leong S, Lathers D, Wang R, Ravindran P, Feng Y, Aanur P, Melero I. OX40 agonist BMS-986178 alone or in combination with nivolumab and/or ipilimumab in patients with advanced solid tumors. Clin Cancer Res. 2021;27:460–472. doi: 10.1158/1078-0432.CCR-20-1830. [DOI] [PubMed] [Google Scholar]
- 50.Glisson BS, Leidner RS, Ferris RL, Powderly J, Rizvi NA, Keam B, Schneider R, Goel S, Ohr JP, Burton J, Zheng Y, Eck S, Gribbin M, Streicher K, Townsley DM, Patel SP. Safety and clinical activity of MEDI0562, a humanized OX40 agonist monoclonal antibody, in adult patients with advanced solid tumors. Clin Cancer Res. 2020;26:5358–5367. doi: 10.1158/1078-0432.CCR-19-3070. [DOI] [PubMed] [Google Scholar]
- 51.Zhao J, Wen X, Tian L, Li T, Xu C, Wen X, Melancon MP, Gupta S, Shen B, Peng W, Li C. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat Commun. 2019;10:899. doi: 10.1038/s41467-019-08782-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai Z, Wang Z, Lei K, Liao J, Peng Z, Lin M, Liang P, Yu J, Peng S, Chen S, Kuang M. Irreversible electroporation induces CD8(+) T cell immune response against post-ablation hepatocellular carcinoma growth. Cancer Lett. 2021;503:1–10. doi: 10.1016/j.canlet.2021.01.001. [DOI] [PubMed] [Google Scholar]
- 53.Narayanan JSS, Ray P, Hayashi T, Whisenant TC, Vicente D, Carson DA, Miller AM, Schoenberger SP, White RR. Irreversible electroporation combined with checkpoint blockade and TLR7 stimulation induces antitumor immunity in a murine pancreatic cancer model. Cancer Immunol Res. 2019;7:1714–1726. doi: 10.1158/2326-6066.CIR-19-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin RC 2nd, McFarland K, Ellis S, Velanovich V. Irreversible electroporation in locally advanced pancreatic cancer: potential improved overall survival. Ann Surg Oncol. 2013;20(Suppl 3):S443–9. doi: 10.1245/s10434-012-2736-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.