

Abstract
Both in vivo and in vitro evidence has supported a key role of myeloid cells in immune suppression in melanoma and in promoting melanocytic metastases. Some single-nucleotide polymorphisms (SNPs) have been shown to predict cutaneous melanoma-specific survival (CMSS), but the association between genetic variation in myeloid cell-related genes and cutaneous melanoma (CM) patient survival remains unknown. Methods: we investigated associations between SNPs in myeloid cell-related pathway genes and CMSS in a discovery dataset of 850 CM patients and replicated the findings in another dataset of 409 CM patients. Results: we identified two SNPs (EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C) as independent prognostic factors for CMSS, with adjusted allelic hazards ratios of 1.56 (95% confidence interval =1.19-2.05, P=0.001) and 1.66 (1.22-2.26, P=0.001), respectively; so were their combined unfavorable alleles in a dose-response manner in both discovery and replication datasets (P trend<0.001 and 0.002, respectively). Additional functional analysis revealed that both EML1 rs10151787 G and HIST1H4E rs2069018 C alleles were associated with elevated mRNA expression levels in normal tissues. Conclusions: Our findings suggest that EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C are independent prognostic biomarkers for CMSS.
Keywords: Cutaneous melanoma, myeloid cell, single-nucleotide polymorphism, survival, prognostic factors
Introduction
Cutaneous melanoma (CM) is one of the most aggressive skin malignancies, with more than 100,000 new cases and 6,850 deaths estimated in 2020 in the United States [1]. Despite significant advances in the treatment of CM in the previous decade, CM-specific survival (CMSS) varies greatly and is hardly predicted accurately in patients with CM of any stage, partly due to the high metastatic potential of CM [2,3]. Although some clinical predictors for CM survival exist, identification of additional prognostic factors for metastatic CMSS could offer more precision in treatment decisions for CM patients.
Increasing evidence has shown that pre-metastatic tumor microenvironment, consisting mainly of myeloid cells, is critical for tumor cell recruitment and survival to facilitate metastasis [4-8]. Myeloid cells were recently found to suppress dendritic cell differentiation and CD8+ T cell proliferation, thus promoting the growth of B16F10 melanoma cells and lung metastasis [9]. Another study showed that myeloid cell accumulation in pre-metastatic tissues suppressed the anti-tumor cytotoxicity of tumor-specific CD8+ T cells in mouse models of melanoma and a significant increase in myeloid cells in the histopathology of metastasis-negative sentinel lymph nodes from melanoma patients [10]. Furthermore, specific myeloid cells were found to be expanded in melanoma patients treated with dendritic cell vaccines, causing enhanced PD-L1 expression as well as hindering CD4+ T cell proliferation [11]. Since these studies obviously support a prominent role of myeloid cells in driving melanoma cell metastases in both in vivo and in vitro models, we believe myeloid cells may serve as a prognostic marker for CM patient survival.
However, myeloid cell markers are seldom cell-type specific and vary considerably across different cancer tissues [12], making accurate detection of myeloid cells in CM patient tissue difficult [13]; therefore, there have been few studies on associations between myeloid cells and CM patient survival. Recent studies suggest that single-nucleotide polymorphisms (SNPs) may be useful biomarkers for tumor progression and patient survival [14,15], which implies that genetic variation could be a prognostic factor for CMSS. For example, studies have identified some specific SNPs as targeting molecules involved in melanoma pathogenesis, consequently controlling melanoma progress and patient outcomes [16-18].
Although several genome-wide association studies (GWASs) have identified some susceptibility loci for CM [19-21], few functional SNPs have been reported to be associated with CMSS at the GWAS level [22,23]. An alternative approach is a novel hypothesis-driven post-GWAS strategy that uses available genotyping data to identify functional genetic variants in the targeted biological pathway genes and reveal their associations with CMSS at a pathway level [24,25]. Therefore, to explore the value of myeloid cells in the prognosis of CMSS, we hypothesize that genetic variants of myeloid cell-related pathway genes are associated with CMSS, and we verified this hypothesis using two publically available CM GWAS datasets in the present study.
Materials and methods
Study populations
The discovery group derived from The University of Texas MD Anderson Cancer Center (MDACC) CM GWAS study that comprised genotyping and outcome data on 858 non-Hispanic white CM patients. The replication group used data on an additional 409 white participants from CM GWAS datasets in the Nurses’ Health Study (NHS) and Health Professionals Follow-up Study (HPFS). Participant selection and data collection for both discovery and replication groups have been published in detail elsewhere [20,26]. All the subjects in the present study provided a written informed consent under a protocol approved by the Institutional Review Boards of the MDACC, Brigham and Women’s Hospital, and the Harvard T.H. Chan School of Public Health, and those of participating registries as required.
Gene selection
Based on the Molecular Signatures Database of the Gene Set Enrichment Analysis (GSEA) website, we comprehensively extracted 280 myeloid cell-related pathway genes located only on the autosomes (Supplementary Table 1).
SNP genotyping
The genotyping of DNA samples in both the discovery MDACC dataset and the NHS/HPFS replication datasets are detailed in Supplementary Methods.
Statistical methods
The details of statistical analyses are presented in Supplementary Methods.
Results
Baseline characteristics of study populations
The baseline characteristics of CM patients from the MDACC and NHS/HPFS datasets are described in Supplementary Table 2. CM patients from the MDACC dataset were between 17 and 94 years of age with a mean age of 52.4 (± 14.4) years at diagnosis, of whom 82.6% had stage I/II CM; the median follow-up time was 81.1 months with 95 CM-related deaths. CM patients from the NHS/HPFS dataset were between 34 and 87 years of age with a mean age of 61.1 (± 10.8) years at diagnosis; the median follow-up time was 179.0 months, and the mortality rate was 11.7%. There were no associations between principal components in GWAS datasets and CM survival in these datasets; therefore, there was unnecessary to adjust for principal components.
Associations between SNPs in myeloid cell-related pathway genes and CMSS in the MDACC and NHS/HPFS datasets
An overall flowchart of the present study is presented in Figure 1. We first explored the associations between all acquired SNPs (including 4,054 genotyped and 20,801 imputed SNPs) in 280 myeloid cell-related pathway genes with CMSS in the MDACC dataset and found that 1126 SNPs were associated with CMSS (P<0.05) in an additive model with multiple test correction. After replication in the NHS/HPFS dataset, only 22 SNPs remained significant. These 22 SNPs are located in seven genes, i.e., three SNPs in WNT2B (Wnt family member 2B), three SNPs in HIST1H4D (histone cluster 1), eight SNPs in HIST1H4E (H4 clustered histone 5), two SNPs in HIST1H4F (H4 clustered histone 6), one SNP in ARL11 (ADP ribosylation factor like GTPase 11), one SNP in EML1 (echinoderm microtubule-associated protein-like 1), and four SNPs in UBASH3B (ubiquitin-associated and SH3 domain containing B) (Supplementary Table 3).
Figure 1.
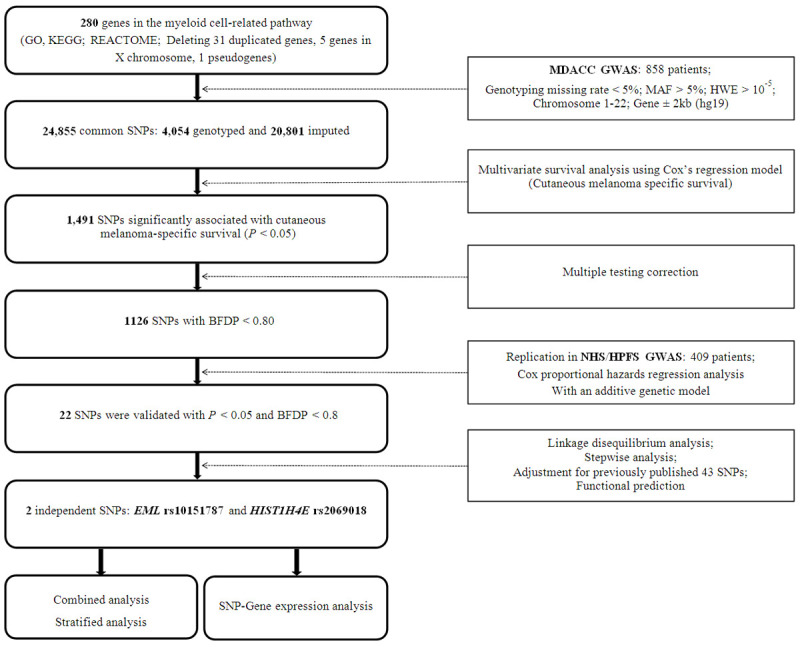
The flowchart of the study. Abbreviations: SNP, single-nucleotide polymorphism; MDACC, The University of Texas MD Anderson Cancer Center; GWAS, genome-wide association study; MAF, minor allele frequency; HWE, Hardy Weinberg equilibrium; BFDP, Bayesian false-discovery probability; NHS/HPFS, the Nurses’ Health Study/Health Professionals Follow-up Study.
Two SNPs independently predict CMSS
To detect SNPs that are independently associated with CMSS, we first performed stepwise multivariable Cox regression analyses to assess the effects of our 22 validated SNPs on CMSS in the MDACC dataset. Four SNPs (rs1175649 in WNT2B, rs2069018 in HIST1H4E, rs61959910 in ARL11, and rs10151787 in EML1) remained significantly associated with CMSS (P<0.05) in the presence of clinical covariates (i.e., age, sex, Breslow thickness of tumor, regional/distant metastasis, mitotic rate, and ulceration). We then expanded this survival model by including 43 previously reported SNPs in the MDACC GWAS dataset; finally we found that two SNPs (EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C) remained significantly associated with CMSS (P=0.018 and 0.001, respectively) (Table 1). The results of our meta-analysis of these two independent SNPs in each dataset are presented in Table 2, showing the absence of heterogeneity across these datasets.
Table 1.
Two independent SNPs identified by multivariate Cox proportional hazards regression analysis including the selected variables and previously published survival-associated SNPs in the MDACC dataset
| Variables1 | Category2 | Frequency | HR (95% CI)1 | P 1 | HR (95% CI)3 | P 3 |
|---|---|---|---|---|---|---|
| Age | ≤50/>50 | 371/487 | 1.02 (1.00-1.04) | 0.007 | 1.04 (1.02-1.06) | <.0001 |
| Sex | Female/Male | 362/496 | 1.49 (0.93-2.37) | 0.096 | 1.35 (0.79-2.29) | 0.269 |
| Regional/distant metastasis | No/Yes | 709/149 | 3.93 (2.54-6.06) | <0.0001 | 12.00 (6.63-21.72) | <.0001 |
| Breslow thickness (mm) | ≤1/>1 | 347/511 | 1.19 (1.12-1.26) | <0.0001 | 1.25 (1.15-1.35) | <.0001 |
| Ulceration | No/Yes | 681/155 | 3.04 (1.95-4.76) | <0.0001 | 5.08 (2.90-8.91) | <.0001 |
| Mitotic rate (mm2) | ≤1/>1 | 275/583 | 2.32 (1.13-4.75) | 0.022 | 2.30 (0.99-5.36) | 0.053 |
| EML1 rs10151787 A>G | AA/AG/GG | 509/311/38 | 1.56 (1.11-2.18) | 0.010 | 1.64 (1.09-2.48) | 0.018 |
| HIST1H4E rs2069018 T>C | TT/TC/CC | 636/204/18 | 1.51 (1.03-2.22) | 0.036 | 2.28 (1.39-3.73) | 0.001 |
Abbreviations: SNP, single-nucleotide polymorphism; MDACC, The University of Texas MD Anderson Cancer Center; HR, hazards ratio; CI, confidence interval.
Stepwise analysis included age, sex, regional/distant metastasis, Breslow thickness, ulceration, mitotic rate and 22 validated SNPs;
The “category/” was used as the reference;
Published SNPs were used for post-stepwise adjustment: rs1175649, rs1124379, rs10916352, rs6707820, rs6750552, rs6785564, rs2306574, rs11551405, rs1718404, rs12512631, rs788935, rs32579, rs3734398, rs7826362, rs10090371, rs7850212, rs3851552, rs10882807, rs61873997, rs35748949, rs11018104, rs7944031, rs11037684, rs508485, rs7933369, rs11225163, rs1990330, rs7953425, rs2342924, rs10846684, rs206118, rs10492396, rs3752447, rs2596191, rs782917, rs17204952, rs62068372, rs72635537, rs7253062, rs3918251, rs12663017, rs17676826, rs8012548.
Table 2.
Meta-analysis of the two independent SNPs in myeloid cell-related pathway genes
| SNP | Allele1 | Gene | Discovery-MDACC (n=858) | Replication-NHS/HPFS (n=409) | Combined-Meta-analysis (n=1267) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||||||
| EAF | HR (95% CI)2 | P 2 | BFDP3 | EAF | HR (95% CI)4 | P 4 | BFDP3 | P het | I2 | HR (95% CI)5 | P 5 | |||
| rs101517876 | A>G | EML1 | 0.23 | 1.51 (1.07-2.11) | 0.017 | 0.682 | 0.20 | 1.65 (1.04-2.61) | 0.033 | 0.769 | 0.761 | 0 | 1.56 (1.19-2.05) | 0.001 |
| rs20690187 | T>C | HIST1H4E | 0.14 | 1.64 (1.12-2.42) | 0.012 | 0.631 | 0.13 | 1.70 (1.02-2.84) | 0.042 | 0.798 | 0.913 | 0 | 1.66 (1.22-2.26) | 0.001 |
Abbreviations: SNP, single-nucleotide polymorphism; GWAS, genome-wide association study; MDACC, The University of Texas MD Anderson Cancer Center; NHS/HPFS, the Nurses’ Health Study/Health Professionals Follow-up Study; EAF, effect allele frequency; HR, hazards ratio; CI, confidence interval; BFDP, Bayesian false-discovery probability; Phet, P value for heterogeneity by Cochrane’s Q test.
Reference allele>effect allele;
Adjusted for age, sex, Breslow thickness, distant/regional metastasis, ulceration and mitotic rate in an additive genetic model;
BFDP was used for multiple test correction with detected a highest HR of 2.0 and a prior probability of 0.1;
Adjusted for age and sex in an additive genetic model;
Meta-analysis in a fix-effects model;
Imputed SNP in the MDACC GWAS dataset;
Genotyped SNP in the MDACC GWAS dataset.
Furthermore, as shown in Table 3, we noticed that the EML1 rs10151787 G allele and the HIST1H4E rs2069018 C allele were prognostic risk alleles for CMSS in the MDACC dataset (P trend=0.017 and 0.012, respectively) with similar results in both the NHS/HPFS dataset (P trend=0.033 and 0.042, respectively) and the combined MDACC and NHS/HPFS dataset (P trend=0.0004 and 0.006, respectively). All of the SNPs investigated in the present study are depicted in a Manhattan plot in Supplementary Figure 1, and regional association plots for these two independent SNPs are displayed in Supplementary Figure 2.
Table 3.
Associations between the two identified independent SNPs in myeloid cell-related pathway genes and CMSS of patients in the MDACC dataset, the NHS/HPFS dataset and the combined dataset
| Genotype | MDACC (n=858) | NHS/HPFS (n=409) | MDACC + NHS/HPFS (n=1267) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||||
| Frequency | Multivariate analysis1 | Frequency | Multivariate analysis2 | Frequency | Multivariate analysis3 | |||||||
|
|
|
|
|
|
|
|||||||
| All | Death (%) | HR (95% CI) | P | All | Death (%) | HR (95% CI) | P | All | Death (%) | HR (95% CI) | P | |
| EML1 rs10151787 A>G | ||||||||||||
| AA | 509 | 44 (8.64) | 1.00 | 262 | 24 (9.16) | 1.00 | 771 | 68 (8.82) | 1.00 | |||
| AG | 311 | 45 (14.47) | 1.55 (1.01-2.38) | 0.045 | 134 | 22 (16.42) | 1.93 (1.08-3.45) | 0.026 | 445 | 67 (15.06) | 1.81 (1.29-2.53) | <0.001 |
| GG | 38 | 6 (15.79) | 2.13 (0.89-5.11) | 0.089 | 13 | 2 (15.38) | 1.81 (0.43-7.67) | 0.423 | 51 | 8 (15.69) | 2.03 (0.97-4.22) | 0.059 |
| Trend test | 0.017 | 0.033 | <0.001 | |||||||||
| HIST1H4E rs2069018 T>C | ||||||||||||
| TT | 636 | 64 (10.06) | 1.00 | 309 | 32 (10.36) | 1.00 | 945 | 96 (10.16) | 1.00 | |||
| TC | 204 | 28 (13.73) | 1.60 (1.01-2.52) | 0.045 | 92 | 13 (14.13) | 1.34 (0.71-2.57) | 0.369 | 296 | 41 (13.85) | 1.41 (0.98-2.03) | 0.066 |
| CC | 18 | 3 (16.67) | 3.03 (0.93-9.85) | 0.066 | 8 | 3 (37.50) | 5.08 (1.55-16.62) | 0.007 | 26 | 6 (23.08) | 2.97 (1.30-6.78) | 0.010 |
| Trend test | 0.012 | 0.042 | 0.006 | |||||||||
| Number of combined risk alleles4 | ||||||||||||
| 0 | 377 | 33 (8.75) | 1.00 | 193 | 13 (6.74) | 1.00 | 570 | 46 (8.07) | 1.00 | |||
| 1 | 359 | 36 (10.03) | 1.22 (0.75-1.99) | 0.431 | 167 | 26 (15.57) | 2.35 (1.20-4.57) | 0.012 | 526 | 62 (11.79) | 1.54 (1.05-2.25) | 0.027 |
| 2 | 100 | 23 (23.00) | 2.82 (1.63-4.87) | <0.001 | 46 | 8 (17.39) | 2.95 (1.22-7.16) | 0.016 | 146 | 31 (21.23) | 3.00 (1.90-4.73) | <.0001 |
| 3-4 | 22 | 3 (13.64) | 2.21 (0.67-7.31) | 0.195 | 3 | 1 (33.33) | 6.86 (0.89-53.00) | 0.065 | 25 | 4 (16.00) | 2.38 (0.85-6.64) | 0.098 |
| Trend test | <0.001 | 0.002 | <.0001 | |||||||||
| 0-1 | 736 | 69 (9.38) | 1.00 | 360 | 39 (10.83) | 1.00 | 1096 | 108 (9.85) | 1.00 | |||
| 2-4 | 122 | 26 (21.31) | 2.48 (1.56-3.95) | <0.001 | 49 | 9 (18.37) | 1.95 (0.94-4.06) | 0.073 | 171 | 35 (20.47) | 2.33 (1.59-3.41) | <.0001 |
Abbreviations: SNP, single-nucleotide polymorphism; CMSS, cutaneous melanoma-specific survival; MDACC, The University of Texas MD Anderson Cancer Center; NHS/HPFS, the Nurses’ Health Study/Health Professionals Follow-up Study; HR, hazards ratio; CI, confidence interval.
Adjusted for age, sex, Breslow thickness, distant/regional metastasis, ulceration and mitotic rate in Cox models of SNPs and CMSS in MDACC study;
Adjusted for age and sex in Harvard NHS/HPFS study;
Adjusted for age and sex in MDACC and Harvard NHS/HPFS study.
Risk alleles include EML1 rs10151787 G allele and HIST1H4E rs2069018 C allele.
Combined risk alleles of two independent CMSS-associated SNPs
To identify the collective effect of EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C on CMSS, we combined their risk alleles (i.e., EML1 rs10151787 G allele and HIST1H4E rs2069018 C allele) into one variable as a genetic score. Patients in each dataset were categorized into four groups (i.e., 0, 1, 2, and 3-4) according to their number of risk alleles (NRA), and the trend test in each dataset showed a significant risk-allele dose-response effect on CMSS. Specifically, higher NRA was associated with a worse survival in the MDACC dataset (P trend=0.0008), the NHS/HPFS dataset (P trend=0.002), and the combined MDACC and NHS/HPFS dataset (P trend<0.0001) after adjustment for available covariates (Table 3). Moreover, we also dichotomized all CM patients into two groups: 0-1 or 2-4 NRA. As shown in Table 3, the 2-4 NRA group had a significantly worse CMSS in the MDACC dataset (HR=2.48; 95% CI=1.56-3.95, P=0.0001), compared with the 0-1 NRA group. Similarly, the 2-4 NRA group showed a significantly worse CMSS in the combined MDACC and NHS/HPFS dataset (HR=2.33; 95% CI=1.59-3.41, P<0.0001). Furthermore, we constructed Kaplan-Meier (KM) survival curves to display the associations between NRA and CMSS (Figure 2A-C).
Figure 2.

Two independent SNPs in myeloid cell-related pathway genes predict cutaneous melanoma survival and eQTL analysis for them. Kaplan-Meier survival curves of combined risk alleles of EML1 rs10151787 and HIST1H4E rs2069018 on CMSS: dichotomized 0-1 risk allele group and 2-4 risk alleles group in the MDACC dataset (A), the NHS/HPFS dataset (B) and the combined MDACC and NHS/HPFS dataset (C). The correlation of rs10151787 genotypes and EML1 mRNA expression in skin tissues from the GTEx (D). The correlation of rs2069018 and HIST1H4E mRNA expression in whole blood samples from the GTEx (E). Abbreviations: SNP, single-nucleotide polymorphism; eQTL, expression quantitative trait loci; CMSS, cutaneous melanoma-specific survival; MDACC, The University of Texas MD Anderson Cancer Center; NHS, the Nurses’Health Study; HPFS, the Health Professionals Follow-up Study; GTEx, Genotype-Tissue Expression project.
Stratified analysis for the effect of NRA on the CMSS
To determine whether the impact of NRA on CMSS was modified by other available clinical covariates, we employed stratified analysis in both MDACC (clinical covariates including sex, age, ulceration, mitotic rate, distant/regional metastasis, ulceration, and Breslow thickness of tumor) and NHS/HPFS datasets (clinical variables including age and sex). In the MDACC dataset, compared with CM patients having 0-1 NRA, those with 2-4 NRA had a significantly poorer CMSS, except for the subgroup with stage III/IV, mitotic rate ≤1, and Breslow thickness ≤1 mm. No significant effects of interaction between risk alleles and each covariate on CMSS were found in these datasets (Supplementary Table 4).
Receiver operating characteristic (ROC) curves and time-dependent area under receiver curve (AUC) of the two independent SNPs for CMSS prediction
To further explore the predictive value of EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C, time-dependent AUC and ROC curves were designed for CM patients in the presence of available clinical covariates. In the MDACC dataset, although the time-dependent AUC in the model with clinical variables increased from 79.02% to 79.51% when risk alleles (i.e., EML1 rs10151787 G allele and HIST1H4E rs2069018 C allele) were added, the predictive performance of 5-year CMSS ROC curves did not significantly improve (P=0.752) (Supplementary Figure 3A, 3B). In the NHS/HPFS dataset, the predictive performance of 5-year CMSS ROC curves in the model with clinical covariables was dramatically improved by adding risk SNPs (P=1.46×10-4), and the time-dependent AUC increased from 54.05% to 73.27% (Supplementary Figure 3C, 3D). Finally, in the combined MDACC and NHS/HPFS datasets, the predictive performance of 5-year CMSS ROC curves in the model with common clinical variables (age, sex) did not significantly improve (P=0.119) (Supplementary Figure 3E, 3F).
Functional predictions of the two SNPs
To investigate specific biological functions of EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C associated with CMSS, we explored SNP-related genomics data using an online bioinformatics tool (HaploReg). We found that an A>G change in EML1 rs10151787 may disturb protein motifs. In addition, a T>C change in HIST1H4E rs2069018 was predicted to be located at a promoter histone marker region or DNAse region, which may also disturb protein motifs, while other identified significant SNPs showing a high linkage disequilibrium (LD) (r2 ≥ 0.8) with rs2069018 may be involved in regulating expression of histone in specific regions, DNase expression, and protein binding (Supplementary Table 3). Using data extracted from the Encyclopedia of DNA Elements (ENCODE) project, we found that rs10151787 was probably located on the H3K4Me1 motifs, while rs2069018 was highly probably located on the H3K27Ac, H3K4Me3, and H3K4Me1 motifs (Supplementary Figure 4). These findings suggest a strong possibility that EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C may disturb their gene’s expression through transcriptional regulation.
Two independent SNPs regulate their corresponding mRNA expression
To further investigate molecular mechanisms underlying the associations between two independent SNPs and CMSS, we explored correlations between risk alleles (i.e., EML1 rs10151787 G allele and HIST1H4E rs2069018 C allele) and their corresponding mRNA expression levels by the expression quantitative trait loci (eQTL) analysis. In the RNA-Seq data from the 1000 Genomes Project, the rs10151787 G allele showed no correlation with EML1 mRNA expression in any additive, dominant, or recessive model (Supplementary Figure 5A-C); no HIST1H4E mRNA expression data were available in the 1000 Genomes Project. Additionally, we also extracted data from the genotype-tissue expression (GTEx) Project, and the results showed that the rs10151787 G allele was significantly associated with increased EML mRNA expression in normal tissues from sun-exposed lower leg skin (P=0.0003) and unexposed suprapubic skin (P=0.03) (Figure 2D). Meanwhile, the rs2069018 C allele was significantly correlated with higher HIST1H4E mRNA expression levels in normal whole blood samples (P=0.022) (Figure 2E) but not in normal skin tissues (Supplementary Figure 5D).
Finally, we measured mRNA expression of EML and HIST1H4E in 104 primary CM tissues and 368 metastatic CM tissues available from the The Cancer Genome Atlas (TCGA) database. As shown in Supplementary Figure 6A and 6C, mRNA expression levels of EML and HIST1H4E were both significantly higher in metastatic CM tissues (P=0.003 and P=0.045, respectively) than in primary CM tissues. However, as displayed by KM survival curves in Supplementary Figure 6B and 6D, mRNA expression levels of both EML and HIST1H4E were not associated with CM survival in the TCGA database.
Discussion
In the present study, we investigated the associations between 24,855 SNPs of 280 myeloid cell-related pathway genes and CMSS using available genotyping and clinical outcome data from two previously reported CM GWAS datasets. We identified two SNPs (EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C) that were independently associated with CMSS. In addition, we found that the rs10151787 G allele was associated with significantly increased EML1 mRNA expression, while the rs2069018 C allele was associated with significantly increased HIST1H4E mRNA expression. Furthermore, our results revealed that mRNA expression levels of both EML1 and HIST1H4E were increased in CM metastatic tissues. In addition, these two SNPs were independent of other clinical characteristics of tumors including tumor thickness, presence of ulceration, and distant metastases in both the MDACC and the NHS/HPFS datasets. Therefore, the present study of 1267 Caucasian CM patients identified two SNPs as independently prognostic predictors for CMSS.
Metastasis is the major cause of mortality in CM patients, and tumor-infiltrating myeloid cells are the key component of the tumor microenvironment, likely involved in melanoma cell metastasis [9,10]. Notably, the presence of tumor-infiltrating myeloid cells in melanoma tumors has been reportedly to be correlated with melanoma patient survival [27], implying that functions of myeloid cells could be a prognostic marker for melanoma patient survival. However, myeloid cells are a highly diverse population, including various cellular subtypes and lacking cell-type specific markers [13,28,29]; hence, accurately assessing functions of myeloid cells in CM tumor tissue and further addressing their correlations with CM survival remains difficult. In the present study, we explored the prognostic value of genetic surrogates for functions of myeloid cells in CM by analyzing associations between SNPs in myeloid cell-related pathway genes and CMSS, instead of directly detecting the presence of myeloid cells in CM tissue. Given the accuracy of EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C in predicting CMSS in the present study, our results may provide easily detectable biomarkers for survival of CM patients, if validated by other investigators.
To date, however, no published studies have reported an association between EML1 or HIST1H4E and CM patient survival. EML1, also known as echinoderm microtubule-associated protein-like 1, has been identified as playing an important role in regulating molecular mechanisms underlying neuronal localization and normal cortical development [30]. Mutations in Eml1 have been shown to cause ectopic progenitors and neuronal heterotopia in both mouse models and human tissues [31]. Few studies have investigated the roles of EML1 in CM. One recent study suggested that EML1 fused with ABL1 might serve as a tyrosine kinase, because it induced myeloid cell transformation [32]. To our knowledge, the present study is the first to report an association between genetic variants of EML1 and CM survival. In addition, EML1 rs10151787 A>G showed a significant risk effect on CMSS and a significant association with elevated EML1 mRNA expression levels in normal skin tissues; furthermore, EML1 mRNA conspicuously accumulated in metastatic CM tissues, compared with primary CM tissues. These observations suggest that EML1 may serve as an oncogene in CM.
HIST1H4E, also called H4 clustered histone 5, is a member of the histone H4 family. Several histone variants have been confirmed as key epigenetic players implicated in the regulation of cancer progression [33]. Studies have linked changes in the global expression of some histones to melanoma metastasis [34,35]. However, no direct implication of HIST1H4E in melanoma has been reported. In the present study, we found that HIST1H4E rs2069018 T>C, a risk factor for CMSS, was associated with an elevated HIST1H4E mRNA expression in normal whole blood, while, HIST1H4E mRNA expression was significantly elevated in metastatic CM tissues, compared with primary CM tissues. Therefore, rs2069018 C allele-regulated HIST1H4E mRNA expression may be a molecular mechanism underlying the observed association between HIST1H4E rs2069018 and poor survival in CM patients.
In addition to these above-mentioned findings, these are some limitations in the present study. The CM patients were recruited only from Caucasian populations; thus, further validation of CM patient cohorts in different races/ethnicities should be conducted. Additionally, compared with the MDACC discovery dataset, fewer CM participants with a small number of clinical covariates were enrolled in the NHS/HPFS replication dataset, which could weaken statistical power in validating the effects of other SNPs identified in the discovery dataset. Finally, the sample sizes of CM patients from these two datasets were not large enough to perform the false discovery rate test; however, considering that up to 83% of selected SNPs in the present study were imputed, the Bayesian false-discovery probability test might be more appropriate for the highly correlated SNPs.
Overall, myeloid cells have been shown to be involved in melanoma progression, and pathologists have tried to determine whether the presence of myeloid cells predicts CM patient outcomes. Given the potential prognostic value of EML1 rs10151787 A>G and HIST1H4E rs2069018 T>C in myeloid cell-related pathway genes in predicting CMSS, as the AUC was not statistically significant, additional causal SNPs need to be identified in larger studies. These two SNPs may serve either as a new prognostic biomarker or as a precision clinical treatment indicator for CM patients and their caregivers, once these findings are validated in future studies.
Acknowledgements
The authors would like to thank Bingrong Zhou and Guiqing Lu for their technical assistance and all participants and staff members of the Nurses’ Health Study and Health Professionals Follow-Up Study for their valuable contributions as well as the following state cancer registries for their support: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors also thank the Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital, for its support as home of the NHS, Harvard Medical School. The authors assume full responsibility for analyses and interpretation of these data. We also thank the John Hopkins University Center for Inherited Disease Research for conducting high-throughput genotyping for our study. The results published herein are fully or partly based upon data from the TCGA pilot project established by the NCI and NHGRI. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov. We also thank all of the investigators and funding agencies that enabled the deposition of data in dbGaP that we used in this study (dbGaP Study Accession: phs000187.v1.p1). This work was supported by NIH/NCI R01 CA100264, 2P50CA093459, R01CA133996, R01 CA49449, P01 CA87969, UM1 CA186107, U01 CA167552, The University of Texas MD Anderson Cancer Center Various Donors Melanoma and Skin Cancers Priority Program Fund, the Miriam and Jim Mulva Research Fund, the McCarthy Skin Cancer Research Fund, and the Marit Peterson Fund for Melanoma Research. Qingyi Wei was partly supported by start-up funds from Duke Cancer Institute, Duke University Medical Center, and Duke Cancer Institute as part of the P30 Cancer Center Support Grant (Grant ID: NIH CA014236).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A, Ugurel S. Melanoma. Lancet. 2018;392:971–984. doi: 10.1016/S0140-6736(18)31559-9. [DOI] [PubMed] [Google Scholar]
- 3.Enninga EAL, Moser JC, Weaver AL, Markovic SN, Brewer JD, Leontovich AA, Hieken TJ, Shuster L, Kottschade LA, Olariu A, Mansfield AS, Dronca RS. Survival of cutaneous melanoma based on sex, age, and stage in the United States, 1992-2011. Cancer Med. 2017;6:2203–2212. doi: 10.1002/cam4.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jian J, Pang Y, Yan HH, Min Y, Achyut BR, Hollander MC, Lin PC, Liang X, Yang L. Platelet factor 4 is produced by subsets of myeloid cells in premetastatic lung and inhibits tumor metastasis. Oncotarget. 2017;8:27725–27739. doi: 10.18632/oncotarget.9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim SY, Gordon-Weeks A, Allen D, Kersemans V, Beech J, Smart S, Muschel RJ. Cd11b(+) myeloid cells support hepatic metastasis through down-regulation of angiopoietin-like 7 in cancer cells. Hepatology. 2015;62:521–533. doi: 10.1002/hep.27838. [DOI] [PubMed] [Google Scholar]
- 6.Hirai H, Fujishita T, Kurimoto K, Miyachi H, Kitano S, Inamoto S, Itatani Y, Saitou M, Maekawa T, Taketo MM. CCR1-mediated accumulation of myeloid cells in the liver microenvironment promoting mouse colon cancer metastasis. Clin Exp Metastasis. 2014;31:977–989. doi: 10.1007/s10585-014-9684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitamura T, Fujishita T, Loetscher P, Revesz L, Hashida H, Kizaka-Kondoh S, Aoki M, Taketo MM. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc Natl Acad Sci U S A. 2010;107:13063–13068. doi: 10.1073/pnas.1002372107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itatani Y, Kawada K, Fujishita T, Kakizaki F, Hirai H, Matsumoto T, Iwamoto M, Inamoto S, Hatano E, Hasegawa S, Maekawa T, Uemoto S, Sakai Y, Taketo MM. Loss of SMAD4 from colorectal cancer cells promotes CCL15 expression to recruit CCR1+ myeloid cells and facilitate liver metastasis. Gastroenterology. 2013;145:1064–1075. e1011. doi: 10.1053/j.gastro.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 9.Papaspyridonos M, Matei I, Huang Y, do Rosario Andre M, Brazier-Mitouart H, Waite JC, Chan AS, Kalter J, Ramos I, Wu Q, Williams C, Wolchok JD, Chapman PB, Peinado H, Anandasabapathy N, Ocean AJ, Kaplan RN, Greenfield JP, Bromberg J, Skokos D, Lyden D. Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat Commun. 2015;6:6840. doi: 10.1038/ncomms7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Zhang C, Li W, Deng J, Herrmann A, Priceman SJ, Liang W, Shen S, Pal SK, Hoon DSB, Yu H. CD8+ T-cell immunosurveillance constrains lymphoid premetastatic myeloid cell accumulation. Eur J Immunol. 2015;45:71–81. doi: 10.1002/eji.201444467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bakdash G, Buschow SI, Gorris MA, Halilovic A, Hato SV, Skold AE, Schreibelt G, Sittig SP, Torensma R, Duiveman-de Boer T, Schroder C, Smits EL, Figdor CG, de Vries IJ. Expansion of a BDCA1+CD14+ myeloid cell population in melanoma patients may attenuate the efficacy of dendritic cell vaccines. Cancer Res. 2016;76:4332–4346. doi: 10.1158/0008-5472.CAN-15-1695. [DOI] [PubMed] [Google Scholar]
- 12.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 13.Gunther P, Schultze JL. Mind the map: technology shapes the myeloid cell space. Front Immunol. 2019;10:2287. doi: 10.3389/fimmu.2019.02287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van den Broeck T, Joniau S, Clinckemalie L, Helsen C, Prekovic S, Spans L, Tosco L, Van Poppel H, Claessens F. The role of single nucleotide polymorphisms in predicting prostate cancer risk and therapeutic decision making. Biomed Res Int. 2014;2014:627510. doi: 10.1155/2014/627510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zienolddiny S, Skaug V. Single nucleotide polymorphisms as susceptibility, prognostic, and therapeutic markers of nonsmall cell lung cancer. Lung Cancer (Auckl) 2012;3:1–14. doi: 10.2147/LCTT.S13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glatthaar H, Katto J, Vogt T, Mahlknecht U. Estrogen receptor alpha (ESR1) single-nucleotide polymorphisms (SNPs) affect malignant melanoma susceptibility and disease course. Genet Epigenet. 2016;8:1–6. doi: 10.4137/GEG.S31264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Figl A, Scherer D, Nagore E, Bermejo JL, Dickes E, Thirumaran RK, Gast A, Hemminki K, Kumar R, Schadendorf D. Single nucleotide polymorphisms in DNA repair genes XRCC1 and APEX1 in progression and survival of primary cutaneous melanoma patients. Mutat Res. 2009;661:78–84. doi: 10.1016/j.mrfmmm.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 18.Onken MD, Worley LA, Person E, Char DH, Bowcock AM, Harbour JW. Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma. Clin Cancer Res. 2007;13:2923–2927. doi: 10.1158/1078-0432.CCR-06-2383. [DOI] [PubMed] [Google Scholar]
- 19.Barrett JH, Iles MM, Harland M, Taylor JC, Aitken JF, Andresen PA, Akslen LA, Armstrong BK, Avril MF, Azizi E, Bakker B, Bergman W, Bianchi-Scarra G, Bressac-de Paillerets B, Calista D, Cannon-Albright LA, Corda E, Cust AE, Debniak T, Duffy D, Dunning AM, Easton DF, Friedman E, Galan P, Ghiorzo P, Giles GG, Hansson J, Hocevar M, Hoiom V, Hopper JL, Ingvar C, Janssen B, Jenkins MA, Jonsson G, Kefford RF, Landi G, Landi MT, Lang J, Lubinski J, Mackie R, Malvehy J, Martin NG, Molven A, Montgomery GW, van Nieuwpoort FA, Novakovic S, Olsson H, Pastorino L, Puig S, Puig-Butille JA, Randerson-Moor J, Snowden H, Tuominen R, Van Belle P, van der Stoep N, Whiteman DC, Zelenika D, Han J, Fang S, Lee JE, Wei Q, Lathrop GM, Gillanders EM, Brown KM, Goldstein AM, Kanetsky PA, Mann GJ, Macgregor S, Elder DE, Amos CI, Hayward NK, Gruis NA, Demenais F, Bishop JA, Bishop DT GenoMEL Consortium. Genome-wide association study identifies three new melanoma susceptibility loci. Nat Genet. 2011;43:1108–1113. doi: 10.1038/ng.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amos CI, Wang LE, Lee JE, Gershenwald JE, Chen WV, Fang S, Kosoy R, Zhang M, Qureshi AA, Vattathil S, Schacherer CW, Gardner JM, Wang Y, Bishop DT, Barrett JH, Geno MELI, MacGregor S, Hayward NK, Martin NG, Duffy DL, Investigators QM, Mann GJ, Cust A, Hopper J, Investigators A, Brown KM, Grimm EA, Xu Y, Han Y, Jing K, McHugh C, Laurie CC, Doheny KF, Pugh EW, Seldin MF, Han J, Wei Q. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum Mol Genet. 2011;20:5012–5023. doi: 10.1093/hmg/ddr415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bishop DT, Demenais F, Iles MM, Harland M, Taylor JC, Corda E, Randerson-Moor J, Aitken JF, Avril MF, Azizi E, Bakker B, Bianchi-Scarra G, Bressac-de Paillerets B, Calista D, Cannon-Albright LA, Chin AWT, Debniak T, Galore-Haskel G, Ghiorzo P, Gut I, Hansson J, Hocevar M, Hoiom V, Hopper JL, Ingvar C, Kanetsky PA, Kefford RF, Landi MT, Lang J, Lubinski J, Mackie R, Malvehy J, Mann GJ, Martin NG, Montgomery GW, van Nieuwpoort FA, Novakovic S, Olsson H, Puig S, Weiss M, van Workum W, Zelenika D, Brown KM, Goldstein AM, Gillanders EM, Boland A, Galan P, Elder DE, Gruis NA, Hayward NK, Lathrop GM, Barrett JH, Bishop JA. Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet. 2009;41:920–925. doi: 10.1038/ng.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Tucker NR, Rizki G, Mills R, Krijger PH, de Wit E, Subramanian V, Bartell E, Nguyen XX, Ye J, Leyton-Mange J, Dolmatova EV, van der Harst P, de Laat W, Ellinor PT, Newton-Cheh C, Milan DJ, Kellis M, Boyer LA. Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. Elife. 2016;5:e10557. doi: 10.7554/eLife.10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hart AB, Kranzler HR. Alcohol dependence genetics: lessons learned from genome-wide association studies (GWAS) and post-GWAS analyses. Alcohol Clin Exp Res. 2015;39:1312–1327. doi: 10.1111/acer.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallagher MD, Chen-Plotkin AS. The post-GWAS era: from association to function. Am J Hum Genet. 2018;102:717–730. doi: 10.1016/j.ajhg.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Bailey SD, Lupien M. Laying a solid foundation for Manhattan--‘setting the functional basis for the post-GWAS era’. Trends Genet. 2014;30:140–149. doi: 10.1016/j.tig.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song F, Qureshi AA, Zhang J, Amos CI, Lee JE, Wei Q, Han J. Exonuclease 1 (EXO1) gene variation and melanoma risk. DNA Repair (Amst) 2012;11:304–309. doi: 10.1016/j.dnarep.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen TO, Schmidt H, Moller HJ, Donskov F, Hoyer M, Sjoegren P, Christensen IJ, Steiniche T. Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer. 2012;118:2476–2485. doi: 10.1002/cncr.26511. [DOI] [PubMed] [Google Scholar]
- 28.Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, Tay A, Lum J, Kairi M, Hwang YY, Becht E, Cheng Y, Chevrier M, Larbi A, Newell EW, Amit I, Chen J, Ginhoux F. Plasmacytoid dendritic cells develop from Ly6D(+) lymphoid progenitors distinct from the myeloid lineage. Nat Immunol. 2019;20:852–864. doi: 10.1038/s41590-019-0420-3. [DOI] [PubMed] [Google Scholar]
- 29.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyman KA, Chetkovich DM. Cortical compass: EML1 helps point the way in neuronal migration. Epilepsy Curr. 2015;15:43–44. doi: 10.5698/1535-7597-15.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kielar M, Tuy FP, Bizzotto S, Lebrand C, de Juan Romero C, Poirier K, Oegema R, Mancini GM, Bahi-Buisson N, Olaso R, Le Moing AG, Boutourlinsky K, Boucher D, Carpentier W, Berquin P, Deleuze JF, Belvindrah R, Borrell V, Welker E, Chelly J, Croquelois A, Francis F. Mutations in Eml1 lead to ectopic progenitors and neuronal heterotopia in mouse and human. Nat Neurosci. 2014;17:923–933. doi: 10.1038/nn.3729. [DOI] [PubMed] [Google Scholar]
- 32.Vanden Bempt M, Mentens N, Vandenberghe P, Cools J, De Keersmaecker K. EML1-ABL1 is activated by coiled-coil-mediated oligomerization and induces T-cell acute lymphoblastic leukemia or myeloproliferative disease in a mouse bone marrow transplant model. Hemasphere. 2018;2:e32. doi: 10.1097/HS9.0000000000000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boulard M, Bouvet P, Kundu TK, Dimitrov S. Histone variant nucleosomes: structure, function and implication in disease. Subcell Biochem. 2007;41:71–89. [PubMed] [Google Scholar]
- 34.Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, Polsky D, Osman I, Garcia BA, Hernando E, Bernstein E. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105–1109. doi: 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Konstantinov NK, Ulff-Moller CJ, Dimitrov S. Histone variants and melanoma: facts and hypotheses. Pigment Cell Melanoma Res. 2016;29:426–433. doi: 10.1111/pcmr.12467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.