

Abstract
Genetic causes are thought to underlie about half of infertility cases, but understanding the genetic bases has been a major challenge. Modern genomics tools are empowering more sophisticated exploration of genetic causes of infertility through population, family-based, and individual studies. Nevertheless, potential therapies based on genetic diagnostics will be limited until there is certainty about causality of genetic variants identified in an individual. Genome modulation and editing technologies have revolutionized our ability to test functionally such variants, and also provide a potential means for clinical correction of infertility variants. This review addresses strategies being used to identify causative variants of infertility in people.
Keywords: Infertility, variants of uncertain significance (VUS), high-throughput sequencing (HTS), genome-wide association study (GWAS), CRISPR/Cas9 genome editing, precision medicine
Challenges for determining genetic causes of infertility in patients
Infertility affects approximately 15% of couples worldwide. About half of cases are estimated to be attributable to female factors alone, 20-30% to male factors alone, and the remaining 20-30% from a combination of male and female factors. Clinically, infertility is a highly heterogeneous disease with complex etiologies (Box 1), and genetic causes account for about half of cases. Several known causative genetic aberrations are used for clinical diagnosis of male infertility, such as Klinefelter Syndrome (XXY) and Y chromosome microdeletions. However, a large proportion of cases are grouped as idiopathic infertility, having unknown genetic or biological causes.
Box 1. Etiologies of Infertility.
Male infertility is mainly caused by decreased (oligozoospermia), absent (azoospermia), or functionally defective sperm. Azoospermia is caused either by spermatogenesis failure, called non-obstructive azoospermia (NOA), or by obstruction (OA) of the reproductive tract. NOA can be divided into three distinct types: Sertoli-cell-only syndrome, spermatogenic arrest at different stages of germ cell maturation, and hypospermatogenesis. The partial or complete obstruction in OA may occur at the rete testis, efferent ducts, epididymis, vas deferens, ampulla of the vas, and the ejaculatory duct. Sperm qualitative defects include globozoospermia (abnormal sperm head), multiple morphological abnormalities of sperm flagella (MMAF), primary ciliary dyskinesia (PCD), acephalia (absence of sperm head), and macrozoospermia (excessively large sperm head).
Female infertility can be caused by issues such as ovarian hyperstimulation syndrome, polycystic ovarian syndrome (PCOS), recurrent pregnancy loss, premature ovarian insufficiency (POI), hypogonadotropic hypogonadism, diminished ovarian reserve (DOR), failure in egg activation, and endometriosis. PCOS manifests clinically as obesity, infertility, impaired glucose tolerance, insulin resistance and an increased risk of endometrial cancer, metabolic syndrome, and cardiovascular disease. POI results in an early loss of ovarian function and leads most often to definitive infertility with adverse health outcomes. It is characterized by amenorrhea, increased follicle-stimulating hormone levels, and hypoestrogenism, leading to infertility before the age of 40 years. DOR is another cause of infertility that shares many features and etiologies with POI. Endometriosis is a benign, estrogen-dependent gynecological disease that is characterized by the growth of stromal and glandular tissue of endometrial origin outside the uterine cavity.
The genetic causes of idiopathic infertility can be rooted in de novo mutations, rare (including pedigree-specific), or segregating population variants. Variants can take the form of small deletions or insertions (indels), larger scale structural variants, copy number alterations, and single nucleotide polymorphisms (SNPs; see Glossary). In human populations, SNPs account for approximately 90% of genetic variants and contribute a major source of genetic heterogeneity. However, proving causality of a SNP can be exceedingly difficult. Advances in high-throughput sequencing (HTS) and bioinformatic methods are having a major impact with respect to characterizing variants present in human populations [1]. However, the vast majority are classified as “Variants of Uncertain Significance (VUS)” due to the lack of functional evidence to determine whether they are pathogenic or benign. Identifying the genetic causes of idiopathic infertility and interpreting VUS in essential fertility genes is challenging but profoundly important for clinical management and genetic counseling.
HTS has been making a large impact on the clinical identification of causative mutations responsible for rare diseases, because these are predominantly Mendelian in nature, and, since most are identified at an early age, parents and siblings are often available for genotyping [2]. More recently, the same methods are being applied towards infertility patients, but identification of causative mutations or variants faces several hurdles: it is often not certain that infertility in a patient is genetic in nature; the disease is often detected later in life; infertility is highly heterogeneous, likely reflecting the large number of genes known to be required for fertility; some cases may have complex genetic origins; and the affected patient is often the only family member manifesting the disease (referred to as the “n = 1” problem). Therefore, modalities beyond HTS are crucial for determining, with high confidence, the actual genetic causes of infertility in a given patient (see Clinician’s Corner). This review addresses genetic and genomic strategies to identify candidate infertility variants, and to validate them.
Clinician’s Corner.
• Can genomics help diagnose idiopathic infertility?
A person may be infertile for various reasons: anatomic defects (induced or innate); hormonal abnormalities; injury; environmental exposures; or complications from other medical situations. For idiopathic infertility, clinical presentation can limit the likely range of etiologies, and can lead to a suspected genetic basis. If there is no family history, that does not necessarily rule out a genetic cause, since spontaneous mutations or a inheritance of recessive alleles from both parents can be responsible. Though not yet standard-of-care, sequencing of patients has become quite inexpensive, and can help address the problem.
• What type of genomic sequencing is done?
The most inexpensive (<$1,000) is whole exome sequencing. This will reveal whether any genes harboring mutations that disrupt the amino acid sequence of proteins known to be important for fertility.
• How accurate is sequencing for diagnosis?
The actual method is highly accurate. The major issue has to do with making a conclusion as to whether any mutations/variants found are actually causative for the infertility. This review discusses how such conclusions can be made.
• Will the results be actionable?
Assuming that conventional Artificial Reproductive Technologies (ART) cannot be applied successfully, the likely answer – TODAY – is no. However, read on…
• Will it ever be possible to cure genetically-based infertility?
Depending on sex and the stage of germ cell development disrupted, this may be possible. For males, germ cell-intrinsic mutations that impact meiotic or postmeiotic cells can be repaired, via gene editing, in spermatogonial stem cells (SSCs). The technology of SSC culture and testicular transfer has been honed over 2 decades, so this is a promising solution. For females, postnatal diagnosis may be too late if the mutation affects oocyte development; key steps of meiosis occur during fetal life. However, the possibility of in vitro differentiation of stem cells, for example starting with reprogrammed somatic cells or even oogonial stem cells (with due recognition of the surrounding controversy), combined with genome editing, could be a solution. Of course, these strategies have major ethical and safety considerations.
Conventional genetic and genomic approaches to identify genes and variants causing idiopathic infertility
A crucial resources for identifying the genetic bases of infertility (and indeed, any disease) is to define the universe of genes that can cause it. The compendium of genes known to be vital for mammalian reproduction and fertility have been populated primarily by mutagenesis studies in mice [3]. This information is instrumental for informing the methods used most commonly by contemporary human geneticists for identifying infertility alleles or mutations in people, delineated below. The methods employed are determined by the goal of the study, whether it be to identify the cause of infertility in an individual patient, or to characterize variants or genes in the population that are responsible for a substantial fraction of infertility cases. By virtue of ever-increasing deployment of such methods, the number of identified infertility-causing variants has been rising dramatically, as reflected in the ClinVar database (Fig. 1).
Fig. 1. Infertility variants in the ClinVar database.
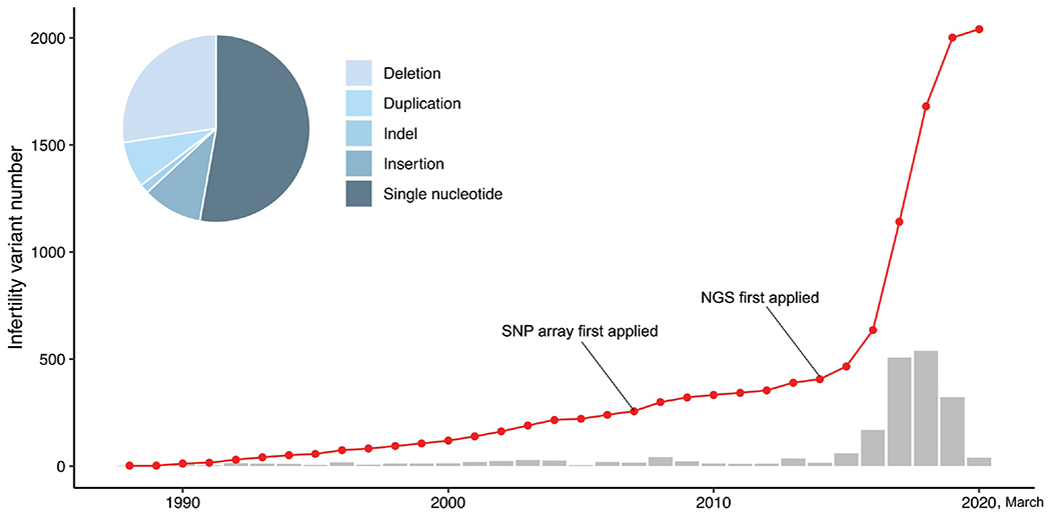
Red line, cumulative pathogenic variants in the database. Grey bars, number added annually. The pie chart shows the distribution of variant types. Graphs were made using R.
A dated but still common method for identifying genes that drive infertility, either in a given patient or in the general population, is candidate gene re-sequencing. This approach is applicable for addressing a specific subtype of infertility, such as nonobstructive azoospermia (NOA), or when an investigator is particularly interested in the role of a specific gene or class of genes known to cause that phenotype. Using the NOA example, there are some reports (e.g. [4,5]) of studies that involve re-sequencing of essential meiosis genes such as the meiosis-specific stromal antigen 3 (STAG3). Resequencing is readily accomplished by Sanger sequencing of PCR products. The experimental designs typically involve assessing whether there is an increased incidence of putatively deleterious variants (see below) in cases vs controls. This method is largely obsolete, since it is no longer cheaper or less labor intensive than some whole exome sequencing (WES), discussed below.
WES is presently the method of choice for rapid and cost-effective (currently costing under $500) genomic analysis as applied to diverse purposes in clinical and basic research. Regarding the former, it has gained increasing utilization for screening of individual patients, or larger cohorts of patients, that are infertile as a potential consequence of a Mendelian disorder. The exome contains ~85% of known disease variants [6], and compared to WGS, WES is cheaper, enabling higher read depth, and is simpler to analyze. This approach is being/has been used by various groups to identify mutations or population variants that may underlie primary/premature ovarian insufficiency (POI) [7–9] and NOA [4,10–12]. As with candidate gene resequencing, studies involving a substantial number of individuals utilize a case:control design to identify genes preferentially altered, and evidence for those candidate genes can be supported across multiple studies and by dint of large cohorts.
Finally, Genome-Wide Association (GWA), a mainstay approach in human genetics for identifying the locations of segregating variants (not de novo mutations) in the genome that affect a phenotype or predispose to a disease, is being used to address infertility genetics. Of course, this is not a diagnostic tool, and it can only identify phenotype-contributing alleles in haplotypes that are not very rare. However, once association to such loci are established, inferences to new patients can be drawn. Crucial to GWA study (GWAS) is defining the phenotype being studied quantitatively and qualitatively, and choosing a suitable study population [13]. The subjects are genotyped, then statistical analyses are performed to identify phenotype-associated genomic regions. For large cohorts, SNP arrays are a cost-effective genotyping platform, although WGS or WES provides data that essentially doubles as a SNP genotyping platform, in addition to providing information at non-polymorphic sites.
GWAS have been performed to identify infertility-causing loci with varying success (Fig. 2A). Variants associated with PCOS [14], early menopause and POI [15], endometriosis [16], azoospermia, and oligozoospermia [17–19] have been identified. SNPs most significantly associated with a trait by GWA are rarely causal or functional; they are simply marking genetic proximity, i.e., high linkage disequilibrium (LD), with the measured phenotype. Typically, GWA “hits” map predominantly to non-coding or intergenic regions [20], and the underlying causal sequence variants are more difficult to pinpoint and validate as compared to coding variants. Sequencing of the haplotypes, and finer mapping through larger cohorts and application of advanced statistical methods [21], can refine candidate genes to a manageable number, but proof of causality and mechanism remains elusive. By nature, GWAS are designed not to identify Mendelian mutations/variants, but rather quantitative trait loci (QTL), i.e., collections of alleles that on their own may not cause infertility, but rather have additive, opposing, or synergistic effects on a phenotype. Relevant to fertility, examples would be sperm count/quality, size of ovarian reserve, age to menopause, etc. Even if the precise identity, or mechanistic effects of, specific QTL may not be known, the identification of haplotypes impacting a fertility parameter will ultimately be useful for predictive medicine and for making conclusions as to the likelihood of genetic causes underlying a patient’s condition.
Fig. 2. Strategies to identify genetic variants causing human infertility.
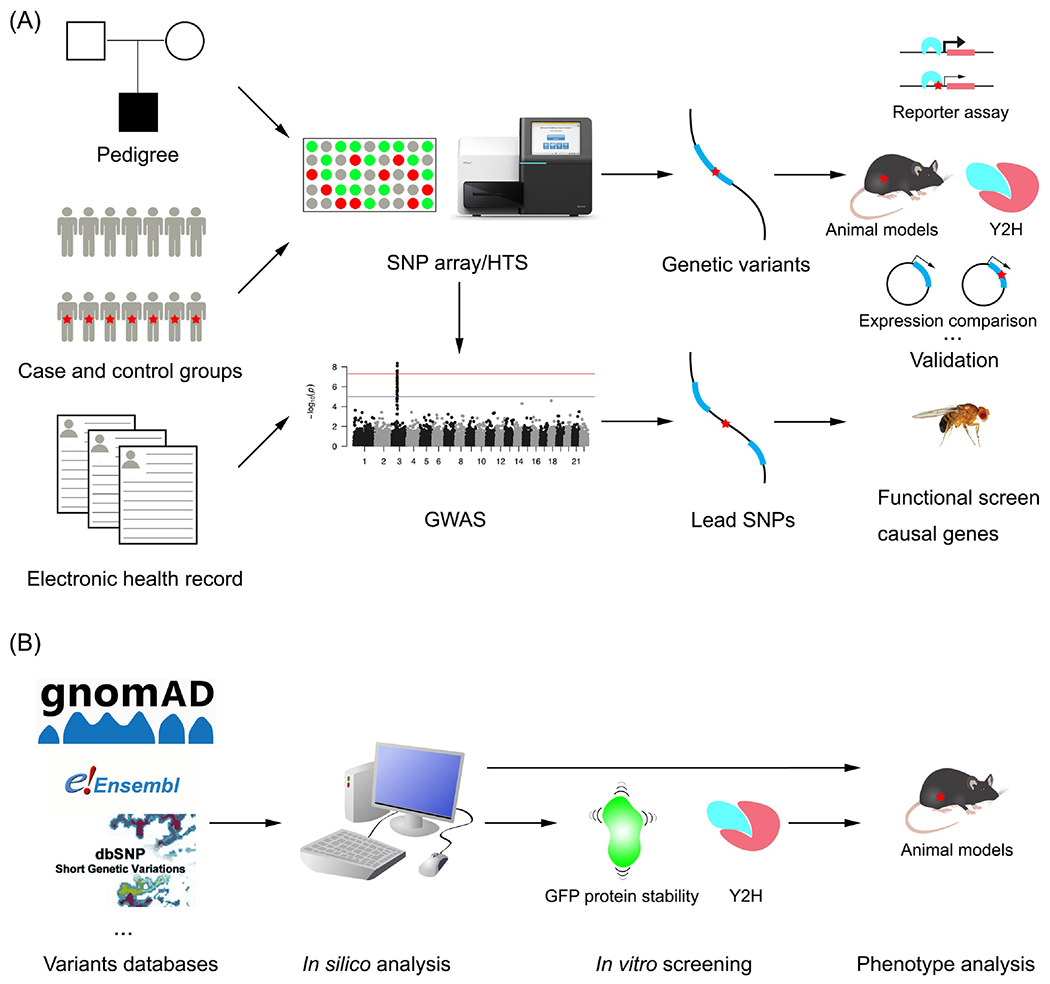
A. Conventional forward genetic approaches identify variants associated with disease, using SNP arrays or HTS. Follow-up experimental validation studies focus on potentially causative variants in the associated genetic intervals, and whether they affect expression of a nearby gene (regulatory variants) or coding regions of genes. B. In a reverse approach, the nsSNPs within genes known to be required for gametogenesis and fertility are retrieved from public variant databases. After in silico prediction, the deleterious nsSNPs are tested via in vitro screening and/or modeling in mice.
In aggregate, these various methods for both identifying fertility genes and population variants are constantly increasing our understanding of genetic architectures of infertility, thereby providing resources for basic and translational benefit. Nevertheless, there is a long way to go before we have a comprehensive understanding of how common, rare, or unique variants contribute to human infertility. The following sections address this problem, beginning with resources being developed for the overall community, followed by overview of methods for increasing the knowledgebase and for enabling genetic diagnosis of individual patients.
The knowledgebase of fertility genetics
There are multiple sources that curate genetic information, with varying emphases on clinical vs basic research data. For those concerned with human reproductive genetics and who are seeking to determine if particular sequence variants are disease related, the ClinVar database (Box 2) lists ~2,000 “pathogenic” infertility variants (predominantly single nucleotide mutations). However, most of these are associated with syndromic disorders such as cystic fibrosis or primary ciliary dyskinesia, and overall, these variants correspond to only ~150 distinct genes. OMIM (Online Mendelian Inheritance in Man) is a more gene: disease oriented resource better suited for, as the name implies, finding genes that, when mutated, cause a particular disease. It currently has 442 listings for infertility genes. On the basic research side, studies of model organisms (e.g., rodents, fruit flies, worms, etc.) have identified many hundreds of genes important for reproduction and fertility. The data from major model organisms (and humans), which includes substantial mechanistic information, can be mined at www.alliancegenome.org, a consortium of bioinformatics groups dedicated to specific organisms. Additionally, each maintains sites dedicated to those individual organisms such as the mouse (informatics.jax.org) or fruit flies (flybase.org).
Box 2. Genetic variant repositories.
Many databases have been founded to curate the rising number of genetic variants. Some of these are population-based repositories such as gnomAD (genome aggregation database) [1], 1000 Genomes Browser [98], dbSNP [99], ExAC (Exome aggregation consortium) [100] and Exome Variant Server (EVS). They provide variant information such as minor allele frequency in different populations, functional consequence, and nature of the variants. gnomAD is the successor to ExAC and version 3.0 aggregated 125,748 exomes and 15,708 genomes from human sequencing studies. It identified 443,769 high-confidence, predicted loss-of-function variants. The 1000 Genomes Project sequenced 2,504 individuals using WGS to catalogue variants and their frequencies genome-wide in 26 different population groups. dbSNP is a public archive for genetic variation within and across different species. It contains SNPs, short deletion and insertion polymorphisms (indels), microsatellite markers or short tandem repeats, multinucleotide polymorphisms, heterozygous sequences, and named variants. These databases are often integrated into overarching search engines, such as the Ensembl browser [101]. Geography of Genetic Variants (GGV) browser [102] and UCSC genome browser.
The others include the disease-specific variant atlases that have been reported in patients and obtained from bibliographic references or clinical laboratory submissions, such as ClinVar [104], ClinGen [25], OMIM (Online Mendelian Inheritance in Man) [105] and HGMD (human gene mutation database) [106]. ClinVar is a public archive of reported relationships among human variants and phenotypes along with supporting evidence. A key strength of this archive is the aggregation of data from multiple clinical laboratories, providing a growing record of support for each interpretation, in which the provenance for each interpretation is maintained. OMIM is a compendium of human genes and genetic phenotypes. HGMD constitutes a comprehensive, disease-oriented collection of published heritable mutations causing genetic diseases in people.
Existing knowledge of fertility genetics will continue to ramp over the next few years. In terms of basic knowledge, as most major model organisms are being subjected to whole genome functional analysis (see Outstanding questions). For example, over half of all mouse genes have been knocked out, and a group of international partners (the International Mouse Phenome Consortium) is systematically mutating all remaining genes (protein coding and non-coding) and conducting phenotypic analyses including fertility (www.mousephenotype.org). Thus, there will be information on the functions of all genes in next next few years, thus providing useful information for those conserved in humans. This information can be helpful in identifying candidates in regions identified by GWAS in people, or from genomic analysis (generally WES) of individual patients.
Outstanding questions.
• What are all the genes necessary for fertility of people?
We are still a long way from having this answer. Best estimates on the number of requisite genes come from studies of knockout mice, in which mutant models have been made for just over half of all genes. It is likely that ~1,000 genes can be mutated to cause fertility defects but be compatible with viability.
• What fraction of idiopathic infertility cases are genetic, non-genetic, or epigenetic in nature?
This will likely be a question that persists for many more years or decades. We are experiencing a remarkable increase in fertility parameters. For example, sperm number and quality is dropping precipitously in recent decades. Many suspect such issues are related to environmental effects, which may disrupt hormone signaling or have epigenetic consequences on gene expression. While we have the genomic technology to examine epigenetic alterations, it is more difficult to draw cause:effect relationships.
• For genetically-based infertilities, how many are of complex origin vs single gene origin?
Diseases caused by single gene mutations/variants are far easier to identify than those caused by interactions of multiple genetic variants. Because of the genetic complexity of infertility, such “complex” or quantitiative traits have been exceedingly difficult to dissect, even in laboratory models. However, traits with two or very few contributing loci are tractable.
Aside from GWAS directed at specific traits, increased usage and availability of healthcare system-based biobanks linked to electronic health records (EHRs) is adding to the compendium of human-specific reproductive genetics. For the subset of patients having genomic data associated with their EFIR, this provides new opportunities to discover genetic variants from large patient bases with the same phenotype definitions (Fig. 2A). Several initiatives, such as the Clinical Sequencing Exploratory/Evidence-Generating Research (CSER) Consortium, Clinical Genomic Resource (ClinGen), Implementing Genomics in Practice (IGNITE) network and the Electronic Medical Records and Genomics (eMERGE) Network [22–25] evaluate the use of genomic tests. The Geisinger MyCode Community Health Initiative (MyCode®) is being used to generate molecular data, including high-density genotype and exome sequence data, for diagnosis and lifetime patient health management. A recent study exploited EHRs to identify novel variants in 2,995 PCOS patients by GWA [26]. These patient data were extracted from 3 EHR collections: MyCode® [27], BioVU at Vanderbilt University [28], and the eMERGE [24]. The practice of routine patient sequencing, as exemplified by the individual eMERGE partners, presages the future standard of care for most for developed countries and their populations in the future. Until then, genome wide sequencing (WES or WGS) will remain a niche diagnostic that is performed on a limited basis by caregivers at locations with sufficient expertise, for patients with resources and for whom the diagnostic may be of potential benefit.
Genetic analysis of individual patients
In situations where an “idiopathic” diagnosis is reached following a comprehensive clinical workup of an infertile patient, including fundamental genetic tests such as those for karyotypic abnormalities or co-morbidities (such as cystic fibrosis or Klinefelter Syndrome, the latter of which is a major known genetic cause of NOA), advanced genetic analysis (typically WES) is increasingly becoming the next logical step. While the clinical portion of this “test” is as simple as a blood draw, the most problematic aspect lies in interpreting and understanding the data to draw conclusions as to possible genetic cause(s). At the root of this problem is the issue of genetic variation, whereas every individual carries many thousands of minor alleles (namely SNPs) of uncertain significance, including those in “fertility” genes. Aside from situations in which a patient inherits a known deleterious allele, or a private/rare allele that is obviously deleterious (say, a nonsense mutation), and the patient’s phenotype is congruous with a mutation in that gene, interpretation becomes more difficult and requires expert interpretation beyond the expertise of most clinicians. Of course, the potential character of a variant allele(s) (i.e., recessive, dominant, semidominant) must be considered, and crucially, the phasing (for autosomal genes) if two or more variants are identified in a gene by sequencing.
To simplify bioinformatic analysis of patient HTS data in a clinical setting, automated computational pipelines (such as C-PIPE [29]) have been developed to identify candidate causal variants in a patient and rank them on the bases of various factors such as the predicted consequence upon protein function, conservation of the affected amino acid(s), and frequency in a population [30,31]. ANNOVAR (ANNOtate VARiation) [32] is a basic and commonly-used tool to prioritize variants, gleaned from a variety of databases (Box 2), regarding probable impact on gene function. More sophisticated tools for in silico prediction of pathogenicity of coding variants include the widely-used SIFT [33] and PolyPhen2 [34] algorithms, which focus on particular metrics of analysis such as evolutionary conservation of amino acids or the predicted effects of variants on the physiochemical properties of a protein, and “ensemble” tools such as REVEL [35] and CADD [36] that weight predictions from other tools. The genetic Variant Impact Predictor Database (VlPdb) summarizes hundreds of prediction tools on its website (https://genomeinterpretation.org/vipdb), underscoring the importance of the issue and demand for solutions [37]. Finally, the PSAP (population sampling probability) approach prioritizes potentially causative variants obtained from WES data using a model-based likelihood framework, and is designed to address single cases (referred to as the “n = 1” problem) [38].
Unfortunately, these and other computational tools for in silico predictions are not flawless. For example, a human missense variant of MND1 (Meiotic nuclear divisions 1) that was predicted to be highly deleterious to protein function by several algorithms, did not impact fertility, fecundity or gametogenesis in a mouse model [39], though caveats with animal modeling are discussed later. The major challenge for a physician, genetic counsellor or researcher is to actually pinpoint, with high confidence, the actual causal lesion. Of course, this assumes that there is indeed a genetic cause, which, in the absence of compelling evidence, may not be the case; alternative etiologies may be related to environmental factors or even epigenetic mechanisms. Unless a suspect variant is a proven infertility allele, experiments are required to validate the candidate variant, which is currently unrealistic in a clinical context. Since this conundrum is representative of the overwhelming majority of cases, how can we move the field forward? Possible solutions are discussed in the following sections.
Shrinking the number of candidate VUS impacting infertility bioinformatically
As for most human diseases of genetic origin, a strong case can be made that comprehensive characterization of VUS would have the broadest impact on infertility genetics, because it would provide a definitive and permanent list of segregating populations variants to be consulted for diagnostic purposes. VUS fall into two broad categories: coding and non-coding. For several reasons, coding variants (predominantly nonsynonymous SNPs, or nsSNPs) are the most tractable to address. First, the universe of known “fertility genes,” as discussed earlier, is already very comprehensive. Second, the majority of mutations or variants known to cause Mendelian traits are in coding regions. Third, it is far simpler to predict consequences of coding than noncoding variants, and most functional prediction programs discussed above focus on coding SNPs and/or can call splice site alterations. Fourth, the consequences of intergenic (non-coding) SNPs are much more difficult to predict, and even if one was to alter function of a regulatory element, typically correspond to QTL as opposed to Mendelian loci [20]. Also, the genes regulated by elements such as enhancers can be difficult to define. Therefore, we will focus on coding SNPs in this section, but will address regulatory variants later.
The first step in defining the universe of potential infertility variants is to select candidate VUS for follow-up study (Fig. 2B). Obviously this should involve practical criteria for prioritization, namely that only genes known to be involved in fertility should be considered. This set of genes can then be cross listed with all known nsSNPs. The gnomAD database (Box 2) is a robust source of high-quality human genetic variants from different populations, reflecting over 200,000 whole exome and whole genome sequences [1]. Next, these SNPs should be evaluated and prioritized by in silico prediction packages as discussed earlier. Sites such as Ensembl and gnomAD have already pre-computed deleteriousness scores from some of these algorithms, simplifying bioinformatic analysis and rankings. Subsequent steps for prioritization can consider factors such as allele frequency. Common variants are unlikely to cause infertility in a Mendelian fashion, because selective forces wouldn’t allow them to persist at high levels in a population.
Missense variants can affect a protein in several ways, including tertiary structure, stability, efficiency of protein: macromolecule interaction, or post-translational modification (PTM). For example, two human RABL2A variants were found to destabilize the protein and cause male infertility and ciliopathy [40], and a SEPT12 variant was found to cause male subfertility by disrupting interaction with SEPT1 [41]. Aside from the deleteriousness prediction tools discussed earlier there are other in silico approaches are available to predict such outcomes. Some examples are: the neural network-based method DeepDDG, which predicts the consequences of variants/mutations upon protein stability [44]; SAAMBE-3D a machine learning algorithm that predicts the effects of amino acid substitutions (AAS) on PPIs [45]; and “Awesome” (A Website Exhibits SNP On Modification Event) that evaluates nsSNPs predicted to alter PTMs [46].
With time and more data, the universe of genes important for fertility will increase. Similarly, increased sequencing of people from diverse populations around the world will uncover more and rarer population variants. Finally, we can expect that functional assessment algorithms and modeling software will improve in accuracy, enabling better prioritization of VUS that are likely to impact fertility genes. Nevertheless, it remains essential to obtain functional evidence for these variants for eventual clinical utility. This is the focus of the remaining sections.
Functional assessment of putative infertility variants.
A hallmark of the genomics era is high-throughput biology. Strategies to assay roles of all human genes in a certain process can be performed in a single experiment via knockout, knockdown, or upregulation. Such experiments typically require cell culture platform, as well as an appropriate assay such as cell growth, expression of diagnostic markers, or even perturbations to the transcriptome of single cells [47–50]. Unfortunately, we have yet to develop such massively parallel reporter assays (MPRAs) relevant to fertility, because tenable platforms are not yet available. In particular, despite advances with in vitro germ cell development, there are currently no feasible ways to conduct MPRAs on effects of alleles – whether knockouts or VUS – in a manner that accurately recapitulates spermatogenesis or oogenesis. Until these culture systems are substantially improved in terms of developmental accuracy and efficiency, there are nevertheless alternative methods, albeit less efficient, that can be very informative. A few examples of such alternatives are discussed here.
Biochemical and in vitro approaches
There are several experimental methods for functional testing of variants in vitro and in vivo. One is biochemical, that is, to test if some activity or property of a protein is altered by a nsSNP. An advantage is that a relevant biological framework isn’t required. For proteins with a known activity, for example, kinases, sequence-specific DNA binding, or DNA repair function, or interaction with other proteins, relevant biochemical or in vitro assays in cells can be performed. For example, one study of patients with maturation arrest of oocytes found variants in TRIP13, encoding a gene important for recombination during meiosis [51,52], that affected protein abundance and binding to a partner protein using in transfection studies [53]. Pathogenic variants in Anti-Mullerian Flormone (AMFI), causing polycystic ovarian syndrome (PCOS) were validated in one study using luciferase assays detecting robustness of signaling to a BMP-response element target [54]. A third example used reporter assays to test a missense variant in the transcription factor NR5A1 (nuclear receptor subfamily 5 group A member 1) present in some POI patients. The variant reduce ability to stimulate transcription of target gene promoters [55].
Disease causing mutations in people preferentially map to the interfaces of protein-protein interactions (PPI), presumably disrupting association with partners [56]. For example, two variants of KLFIL10 (Kelch like family member 10) found in oligozoospermic patients impair homodimerization with the wide-type protein [57]. A high-throughput yeast two-hybrid screen for nsSNPs that disrupt PPIs, in conjunction with structural predictions and human cell assays, allowed identification of a deleterious allele of SEPT12 that proved to disrupt sperm function in CRISPR-edited mice [41].
For genes that also function in cell types outside of the reproductive system or germline, testing of variants is more accessible to high throughput assays relevant to that gene. As an example, variants in the BRCA2 gene, which functions in homologous recombination repair, is intensely studied due to its role in breast cancer. This gene also function in meiosis [58], and mutations in it may cause POI [59]. A recent report describes a high throughput assay that enabled rapid screening of nearly 200 VUS in BRCA2 for their impact on DNA repair function [60].
While in vitro systems yield useful information on the impact of variants on protein function, and thus help prioritize or classify them with greater confidence than in silico predictions, the impact of such functional variants on reproductive organs or phenotype is lacking. The following sections discusses in vivo systems that can be used to address this issue.
In vivo models of genetic variants.
In vivo functional validation can utilize appropriate non-mammalian experimental models. For example, because the flagellar 9+2 microtubule doublet axoneme structure is widely conserved across species, variants identified in patients suffering from MMAF and PCD (Box1) have been tested in the flagellated protist Trypanosoma brucei [61]. Since many key genes in meiosis are conserved across eukaryotes, yeast can be used to screen functional consequences of nsSNPs, as demonstrated in the case of a putative NOA allele of ENTPD6 [62], and for alleles of mismatch repair genes that may contribute to Lynch Syndrome [63]. The fruit fly Drosophila melanogaster is another workhorse genetic model, and a strategy was developed using in vivo RNA interference and genome editing for functional screening of gene variants associated with NOA in Chinese men [64,65]. Of course, this strategy is limited to highly conserved genes, and caveats about phenotype translatability apply.
While non-mammalian systems can help identify and validate infertility-causing mutations, the most relevant experimental model is the mouse, where human alleles can be modeled in orthologous genes. This approach has been possible for many years using homologous recombination technology to “knock in” specific changes into a mouse gene in embryonic stem cells (ESCs), such as was done to validate an infertility-causing MIWI allele [66]. However, this methodology requires building complex targeting vectors and screening hundreds of ESC clones for proper genome modification, before making chimeric mice. Fortunately, the advent of CRISPR/Cas9 genome editing technology, which enables the precise replacement of mouse genomic sequences with orthologous human sequences directly in single cell embryos, has become routine. Introduction of subtle changes (such as a single nucleotide) is especially efficient, taking advantage of homology-directed repair (HDR) of a Cas9-induced double strand break (DSB) with a synthetic template DNA molecule (most commonly a single-stranded oligodeoxynucleotide, or ssODN) homologous to the DNA sequence flanking the DSB (Fig. 3A). There are now numerous examples in the literature where such modeling has been used to confirm or reject potential human infertility-causing mutations or variants, some of which are cited here [39–41,43,67–71].
Fig. 3. Methods for precision genome editing of mice.
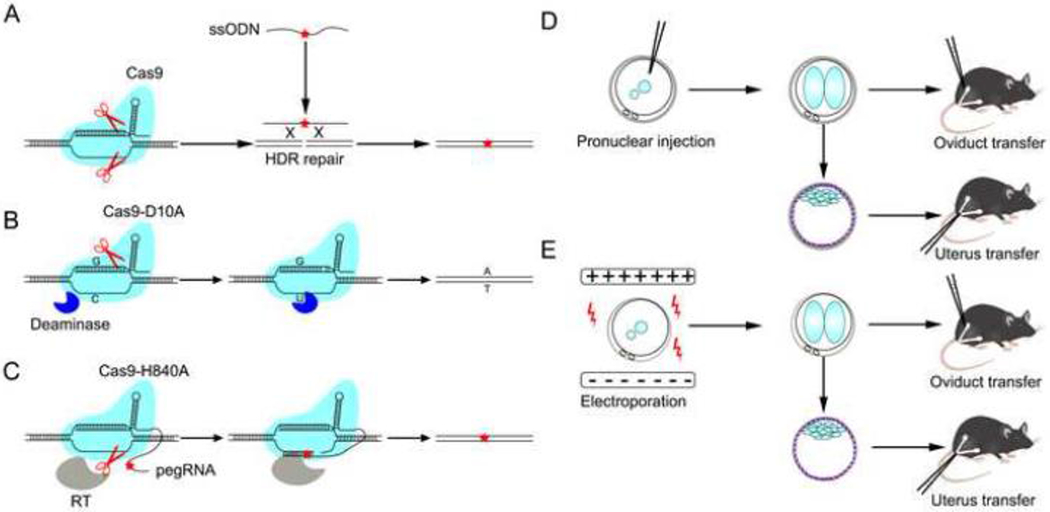
A. Conventional genome editing approach. A desired mutation (red star) can be incorporated into the genome when the host cell’s HDR-repair machinery repairs the DSB produced by Cas9. B. Base editing (BE) approach. A Cas9 that produces only single-strand breaks (nicks) works with a deaminase. The deaminase chemically modifies a specific cytidine base (C) to uracil (U). The nick is repaired, converting a guanine–uracil (G–U) intermediate to an adenine–thymine (A–T) base pair. C. Prime editing (PE) approach. A nick-producing Cas9 and a reverse transcriptase produce nicked DNA into which sequences corresponding to the guide RNA have been incorporated. The original DNA sequence is excised, then the nicked strand is repaired to produce a fully edited duplex. RT, Reverse transcriptase. D. Conventional pronuclear injection. The 2-cell stage zygotes are delivered to the oviduct, while blastocysts are delivered to the uterus. E. The CRISPR-EZ approach [107]. This method of electroporation embryos with CRISPR/Cas9 reagents eliminates the need for microinjection, which requires expensive intrumentation and substantial expertise.
The efficiency of CRISPR/Cas9 editing in mice has opened the door to larger, systematic identification of infertility alleles in humans. Because over half of mouse genes have already been mutated and the phenotypes described, there is substantial knowledge of the “reproductive genome” (reviewed in [3]). Therefore, nsSNPs in genes known to be required for gametogenesis and fertility, and which are selected as being potentially deleterious by computational predictions or other evidence, can be modeled in mice [43] and subjected to systematic phenotyping [72]. The authors and collaborators maintain a website (www.infertilitygenetics.org) overviewing progress on this ongoing project.
Although the generation and analysis of mutant mice can be time consuming due to the reproductive life cycle of the animal, several developments have made the process of making edited animals simpler and more efficient. Recently, promising alternatives to DSB-induced HDR have been developed that mitigate the non-homologous end-joining (NHEJ)-induced indels that occur when DSBs are introduced in the genome. One class includes base editors (BE) (Fig. 3B), enabling A·T>G·C and C·G>T·A changes [73,74]. However, recent studies found that the former type has DNA and RNA off-target effects [75–77]. Another promising strategy is “search-and replace” technology called prime editing (PE) [78] (Fig. 3C). Both systems were reported to function in mouse embryos [79,80]. However, more data on efficiency and fidelity is keenly anticipated.
The ability to model mutations in mice with reasonably high throughput is a powerful and biologically relevant approach. However, there are specific and general caveats. Most problematic are variants affecting amino acids that are not conserved in mice. In this case it might be possible to replace the entire mouse gene (or part of it) with the human counterpart, or to add the human allele as a transgene in the context of a mouse null for the orthologous allele, but this assumes that a divergent gene (and the relevant amino acids) is functionally interchangeable. Even for genes conserved at the sequence level, it isn’t a guarantee that deleterious variants in humans will actually be deleterious in mice (or vice versa). Non-human primates would be a more relevant system for modeling human variants via gene editing [81], though there are more logistical and regulatory obstacles in this regard.
Regulatory variants
The discussion until this point has focused on variants affecting proteins, but most genetic variation lies within noncoding regions of the genome – the portion that contains regulatory sequences. Understanding how the genome is regulated has been a major emphasis of large-scale genomic efforts such as ENCODE project [20]. ENCODE’s focus on characterizing epigenetic features of regulatory sequences led to the realization that most QTL from GWAS studies map to regions containing regulatory features such as enhancers. However, identifying variants that may impact regulatory sequences in the germline is difficult both from the computational prediction and experimental angles. Modeling in non-primates is problematic because intergenic regulatory regions are typically not highly conserved. An ideal solution would be to perfect in vitro human gametogenesis. This would enable introduction of edits into regulatory sequences of stem cells, then measuring transcription of linked genes following differentiation to the desired cell state. As mentioned earlier, such methods suffer from low efficiency and scalability [82], but it remains very promising to test molecular endpoints such as gene expression. Alternatives for latter stages of germ cell development are spermatogonial stem cell (SSC) [83] and oogonial stem cell [84,85] lines, although there is controversy over the latter.
For intergenic variants that may alter regulation of key reproductive genes, the next steps might involve testing the impact of an individual SNP by mining eQTL (expression QTL) data, or, if biopsies from relevant tissues/cells can be obtained or modeled, testing alterations to chromatin state surrounding the variant (e.g., via ChIP-seq of ATAC-seq).
Treatment of genetically-caused infertility
The ultimate aim of identifying causative variants is to help clinicians diagnose and treat infertile patients, particularly for cases in which current ART (assisted reproductive technology) technologies such as IVF, ICSI, and microTESE (microscopic testicular sperm extraction) do not suffice. Opportunities for intervention with genomic-based technologies, such as gene therapy, will depend greatly upon the genes involved, the patient situation, and the stage of germ cell development being affected.
Options for permanently effective genetic interventions are most optimistic for men, due to the ongoing nature of spermatogenesis enabled by an SSC pool. The ability to isolate SSCs, then transfer them into the testis where they can proliferate and reestablish spermatogenesis, was first developed in mice, and later in primates [86,87]. Coupled with ongoing improvements to human SSC culture systems [88], there is potential for using gene therapy approaches in a patient’s SSCs, followed by autologous transfer of cell clones or cultures having the desired modification(s) or gene correction. Proof-in-principle for such an approach has been demonstrated in mice. In one experiment, SSCs isolated from a testis of a sterile c-Kit mutant mice were cultured in vitro, the mutation was corrected by CRISPR/Cas9-mediated FIDR, then the repaired cells were transferred back into the remaining testis restore spermatogenesis [89]. However, genetic manipulations to the germline are controversial, and there are legitimate safety concerns concerning off-target effects that may occur during genome editing, and which can be difficult to monitor unless spermatogonial clones can be readily edited, propagated, and deep sequenced to ensure fidelity of the genome. It is also conceivable that the long culture process may impart epigenetic alterations on SSCs that can affect resulting offspring. Nevertheless, assuming these technical and ethical concerns will be surmounted, the power and efficiency of genome editing can be applied conceivably not only for Mendelian traits, but also to situations where two or more variants, including those in multiple genes, are contributing to a trait. Multi-locus genome editing in mice was demonstrated several years ago [90].
There are also circumstances in which gene therapy can be performed without changing the genome. In particular, genetic defects in oocytes or early embryos can be compensated by introducing exogenous protein or synthetic mRNA (sometimes called complementary RNA, or cRNA). For example, defects in egg activation due to mutations in the sperm-borne protein phospholipase C zeta 1 (PLCZ1) can be rescued by recombinant protein or cRNA [91–93]. Proof-in-principle for the oocyte cRNA correction approach has also been reported for mutations in WEE2, CDC20 and TRIP13 [53,94,95]. However, for mutations in genes that are required for establishment of the ovarian reserve, which requires successful completion of key steps of Meiosis I including meiotic recombination, there are fewer options for intervention. One possibility is to derive patient-specific induced pluripotent stem cells (iPSCs), correct genetic lesions by genome editing, then differentiate them in vitro into oocytes. It may also be possible to reconstruct ovaries as has been done in rodents [96]. Promising results for implantation of in vitro-developed primordial germ cell like cells (PGCLCs) cells has been presented in rhesus macaques [97].
Concluding Remarks
Breathtaking advances in genome biology are enabling and revolutionizing advances in many areas of medicine such as cancer and regenerative medicine. Translation to the field of reproductive biology has lagged relative to genomic efforts applied to life-threatening diseases, and the extreme biological and genetic complexity of reproduction, including lack of effective and efficient in vitro systems that are so widely applied in the genomics arena, underlies this dichotomy. Nevertheless, there is much reason for optimism.
We can reasonably expect that, due to the radical decline in HTS costs and the increased number of individuals being trained in genome sciences and bioinformatics, that WES or WGS will become standard of care in the near future, possibly for newborns (see Clinician’s Corner and Highlights). With increasing functional information and intense focus on understanding human genetic variation, precision medicine will become more routine and practical, including for infertility patients. Sequencing at birth can predict genetic predisposition to potential fertility problems, potentially enabling earlier and more efficacious interventions for maladies such as POI, where steps might be taken to preserve oocytes or prevent loss prior to manifestation of the phenotype.
HIGHLIGHTS.
Infertility is an common disease of heterogeneous nature, and genetics is thought to play a role a large fraction of idiopathic cases. However, it has been difficult to identify the exact genetic lesions contributing to these cases in individual patients.
High-throughput, low cost genome sequencing technologies, coupled with increasing knowledge of genes required for fertility, are making it more practical to screen patients for potential causative genetic mutations. However, proving causality remains a difficult problem, but a pre-requisite to clinical treatment.
A variety of biochemical, molecular and genetic strategies are being used to test consequences of genetic variants found in people that may underlie disease, including infertility. CRISPR/Cas9 genome editing technology is extremely powerful in this respect, allowing modeling of such variants in relevant model organisms.
Regarding improvements to prediction or diagnosis of genetically-driven infertility phenotypes, a strong argument can be made that improvements to in vitro systems would be crucial for enabling a high-throughput platform not only for evaluating functions of VUS segregating in populations, but also as an advanced clinical diagnostic for individual patients. With certainty about the genetic etiology of a patient’s infertility, the next challenge lies with perfecting platforms (e.g., SSCs or iPSCs) for genetic correction or in vivo rescues, and addressing ethical boundaries. Finally, as with all other areas of medicine, the professionals in the reproductive medicine community must keep abreast of these developing technologies to provide patients with the most modern options.
Acknowledgement
JCS is supported by the following grants concerning infertility from the National Institutes of Health: R01HD082568 and P50HD0976723. X.D. was supported by a postdoctoral fellowship from the Empire State Stem Cell Fund through New York State Department of Health contract no. C30293GG.
GLOSSARY
- Electronic health record (EHR)
digitally stored patients’ medical history and data.
- Gene editing
a group of technologies that allow genetic material to be added, removed, or altered at particular locations in the genome.
- Genome-wide association study (GWAS)
an approach to identify inherited genetic variants statistically associated with risk of disease or a particular trait. It is accomplished by surveying the entire genome for genetic variants that occur more frequently in cases (for example, diseased individuals) than in controls (for example, healthy individuals).
- Homology-directed repair (HDR)
a mechanism to repair double-strand DNA breaks in chromosomes by homologous recombination with a highly-related stretch of DNA.
- In silico prediction
a group of methods predicting the biological consequences of genetic variants by computational tools.
- Linkage disequilibrium (LD)
a measure of non-random association between alleles at different loci at the same chromosome in a given population. SNPs are in LD when the frequency of association of their alleles is higher than expected under random Mendelian assortment. LD concerns patterns of correlations between SNPs.
- Multiple morphological abnormalities of the sperm flagella (MMAF)
a specific kind of asthenoteratozoospermia with severe flagellar morphological abnormalities such as absent, short, bent, coiled, and irregular flagella.
- Massively parallel reporter assay (MPRA)
a high-throughput technology that enables genome-scale functional screening of sequences and their variants for functional effects in a single experiment.
- Non-homologous end-joining (NHEJ)
a pathway to repair double strand breaks in DNA by directly ligation without the need for a homologous template.
- Primary cilia dyskinesia (PCD)
a family of autosomal-recession genetic disorders by affecting ciliary motility in several organ systems, resulting in recurrent respiratory infections, situs abnormalities and other forms of heterotaxy, abnormal sperm motility, as well as hydrocephalus.
- Quantitative trait locus (QTL)
a locus (section of DNA) contributing additively to variation of a trait that varies in a metric (for example, height or blood pressure).
- Single nucleotide polymorphism (SNP)
alternative nucleotide that is present at some frequency in one or more populations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
- 1000 Genomes Browser: https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/
- ClinGen: https://www.clinicalgenome.org/
- OMIM: https://www.omim.org/
- UCSC Genome Browser: http://qenome.ucsc.edu/
Competing interests
The authors declare no competing or financial interests.
References
- 1.Karczewski KJ et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581,434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Z et al. (2019) Toward Clinical Implementation of Next-Generation Sequencing-Based Genetic Testing in Rare Diseases: Where Are We? Trends Genet. 35, 852–867 [DOI] [PubMed] [Google Scholar]
- 3.Schimenti JC and Handel MA (2018) Unpackaging the genetics of mammalian fertility: strategies to identify the “reproductive genome”. Biol. Reprod 99, 1119–1128 [DOI] [PubMed] [Google Scholar]
- 4.van der Bijl N et al. (2019) Mutations in the stromal antigen 3 (STAG3) gene cause male infertility due to meiotic arrest. Hum. Reprod 34, 2112–2119 [DOI] [PubMed] [Google Scholar]
- 5.Riera-Escamilla A et al. (2019) Sequencing of a “mouse azoospermia” gene panel in azoospermic men: identification of RNF212 and STAG3 mutations as novel genetic causes of meiotic arrest. Hum. Reprod 34, 978–988 [DOI] [PubMed] [Google Scholar]
- 6.van Dijk EL et al. (2014) Ten years of next-generation sequencing technology. Trends Genet. 30, 418–426 [DOI] [PubMed] [Google Scholar]
- 7.Liu H et al. (2020) Whole-exome sequencing in patients with premature ovarian insufficiency: early detection and early intervention. J Ovarian Res 13, 114–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang R and Yu Q (2020) Novel variants in women with premature ovarian function decline identified via whole-exome sequencing. J. Assist. Reprod. Genet 37,2487–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaillard S et al. (2020) New insights into the genetic basis of premature ovarian insufficiency: Novel causative variants and candidate genes revealed by genomic sequencing. Maturitas 141, 9–19 [DOI] [PubMed] [Google Scholar]
- 10.Krausz C et al. (2020) Genetic dissection of spermatogenic arrest through exome analysis: clinical implications for the management of azoospermic men. Genet. Med DOI: 10.1038/s41436-020-0907-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fakhro KA et al. (2018) Point-of-care whole-exome sequencing of idiopathic male infertility. Genet. Med 20, 1365–1373 [DOI] [PubMed] [Google Scholar]
- 12.Chen S et al. (2020) Whole-exome sequencing of a large Chinese azoospermia and severe oligospermia cohort identifies novel infertility causative variants and genes. Hum. Mot. Genet 29, 2451–2459 [DOI] [PubMed] [Google Scholar]
- 13.Tam V et al. (2019) Benefits and limitations of genome-wide association studies. Nat. Rev. Genet 20, 467–484 [DOI] [PubMed] [Google Scholar]
- 14.Shi Y et al. (2012) Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat. Genet 44, 1020–1025 [DOI] [PubMed] [Google Scholar]
- 15.Perry JRB et al. (2013) A genome-wide association study of early menopause and the combined impact of identified variants. Hum. Mol. Genet 22, 1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uno S et al. (2010) A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat. Genet 42, 707–710 [DOI] [PubMed] [Google Scholar]
- 17.Zhao H et al. (2012) A genome-wide association study reveals that variants within the HLA region are associated with risk for nonobstructive azoospermia. Am. J. Hum. Genet 90, 900–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu Z et al. (2011) A genome-wide association study in Chinese men identifies three risk loci for non-obstructive azoospermia. Nat. Genet 44, 183–186 [DOI] [PubMed] [Google Scholar]
- 19.Aston KI and Carrell DT (2009) Genome-wide study of single-nucleotide polymorphisms associated with azoospermia and severe oligozoospermia. J Androl 30, 711–725 [DOI] [PubMed] [Google Scholar]
- 20.ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaid DJ et al. (2018) From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet 19, 491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green RC et al. (2016) Clinical Sequencing Exploratory Research Consortium: Accelerating Evidence-Based Practice of Genomic Medicine. Am. J. Hum. Genet 98, 1051–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weitzel KW et al. (2016) The IGNITE network: a model for genomic medicine implementation and research. BMC Med. Genomics 9, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottesman O et al. (2013) The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet. Med 15, 761–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rehm HL et al. (2015) ClinGen--the Clinical Genome Resource. N. Engl. J. Med 372, 2235–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y et al. (2020) A genome-wide association study of polycystic ovary syndrome identified from electronic health records. Am. J. Obstet. Gynecol DOI: 10.1016/j.ajog.2020.04.004 [DOI] [PubMed] [Google Scholar]
- 27.Carey DJ et al. (2016) The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet. Med 18, 906–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulley J et al. (2010) Principles of human subjects protections applied in an optout, de-identified biobank. Clin Transl Sci 3, 42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sadedin SP et al. (2015) Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med. 7, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pereira R et al. (2020) Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J Clin Med 9, 1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eilbeck K et al. (2017) Settling the score: variant prioritization and Mendelian disease. Nat. Rev. Genet 18, 599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H and Wang K (2015) Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc 10, 1556–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar P et al. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc 4, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 34.Adzhubei IA et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ioannidis NM et al. (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet 99, 877–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rentzsch P et al. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu Z et al. (2019) VIPdb, a genetic Variant Impact Predictor Database. Hum. Mutat 40, 1202–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilfert AB et al. (2016) Genome-wide significance testing of variation from single case exomes. Nat. Genet 48, 1455–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran TN et al. (2019) A predicted deleterious allele of the essential meiosis gene MND1, present in ~ 3% of East Asians, does not disrupt reproduction in mice. Mot. Hum. Reprod 25, 668–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding X et al. (2020) Variants in RABL2A causing male infertility and ciliopathy. Hum. Mol. Genet DOI: 10.1093/hmg/ddaa230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fragoza R et al. (2019) Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun 10, 4141–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh P et al. (2019) CDK2 kinase activity is a regulator of male germ cell fate. Development 146, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh P and Schimenti JC (2015) The genetics of human infertility by functional interrogation of SNPs in mice. Proc. Natl. Acad. Sci. USA 112, 10431–10436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao H et al. (2019) Deepddg: predicting the stability change of protein point mutations using neural networks. J. Chem. Inf. Model 59, 1508–1514 [DOI] [PubMed] [Google Scholar]
- 45.Pahari S et al. (2020) SAAMBE-3D: Predicting Effect of Mutations on Protein-Protein Interactions. Int. J. Mol. Sci 21, 2563–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y et al. (2019) AWESOME: a database of SNPs that affect protein post-translational modifications. Nucleic Acids Res. 47, D874–D880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.iu Y, Yu C, Daley TP, Wang F, Cao WS, Bhate S, et al. CRISPR Activation Screens Systematically Identify Factors that Drive Neuronal Fate and Reprogramming. Cell Stem Cell. 2018. November 1;23(5):758–771.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014. January 3;343(6166):84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell. 2016. December 15;167(7):1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korkmaz G et al. (2016) Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol 34, 192–198 [DOI] [PubMed] [Google Scholar]
- 51.Roig I et al. (2010) Mouse TRIP13/PCH2 is required for recombination and normal higher-order chromosome structure during meiosis. PLoS Genet. 6, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li XC and Schimenti JC (2007) Mouse pachytene checkpoint 2 (Trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet. 3, 1365–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Z et al. (2020) Bi-allelic Missense Pathogenic Variants in TRIP13 Cause Female Infertility Characterized by Oocyte Maturation Arrest. Am. J. Hum. Genet 107, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorsic LK et al. (2017) Pathogenic Anti-Müllerian Hormone Variants in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab 102, 2862–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jaillard S et al. (2020) Analysis of NR5A1 in 142 patients with premature ovarian insufficiency, diminished ovarian reserve, or unexplained infertility. Maturitas 131, 78–86 [DOI] [PubMed] [Google Scholar]
- 56.Wei X et al. (2014) A massively parallel pipeline to clone DNA variants and examine molecular phenotypes of human disease mutations. PLoS Genet. 10, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yatsenko AN et al. (2006) Non-invasive genetic diagnosis of male infertility using spermatozoal RNA: KLHL10 mutations in oligozoospermic patients impair homodimerization. Hum. Mol. Genet 15, 3411–3419 [DOI] [PubMed] [Google Scholar]
- 58.Sharan SK et al. (2004) BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development 131, 131–142 [DOI] [PubMed] [Google Scholar]
- 59.Miao Y et al. (2019) BRCA2 deficiency is a potential driver for human primary ovarian insufficiency. Cell Death Dis. 10, 474–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ikegami M et al. (2020) High-throughput functional evaluation of BRCA2 variants of unknown significance. Nat. Commun 11, 2573–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kherraf Z-E et al. (2018) A Homozygous Ancestral SVA-Insertion-Mediated Deletion in WDR66 Induces Multiple Morphological Abnormalities of the Sperm Flagellum and Male Infertility. Am. J. Hum. Genet 103, 400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Q et al. (2015) Yeast model identifies ENTPD6 as a potential nonobstructive azoospermia pathogenic gene. Sci. Rep 5, 11762–11774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martinez SL and Kolodner RD (2010) Functional analysis of human mismatch repair gene mutations identifies weak alleles and polymorphisms capable of polygenic interactions. Proc. Natl. Acad. Sci. USA 107, 5070–5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu H et al. (2016) Major spliceosome defects cause male infertility and are associated with nonobstructive azoospermia in humans. Proc. Natl. Acad. Sci. USA 113, 4134–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu J et al. (2015) Identification of seven genes essential for male fertility through a genome-wide association study of non-obstructive azoospermia and RNA interference-mediated large-scale functional screening in Drosophila. Hum. Mol. Genet 24, 1493–1503 [DOI] [PubMed] [Google Scholar]
- 66.Gou L-T et al. (2017) Ubiquitination-Deficient Mutations in Human Piwi Cause Male Infertility by Impairing Histone-to-Protamine Exchange during Spermiogenesis. Cell 169, 1090–1104.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tran TN and Schimenti JC (2019) A segregating human allele of SPO11 modeled in mice disrupts timing and amounts of meiotic recombination, causing oligospermia and a decreased ovarian reserve†. Biol. Reprod 101, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tran TN and Schimenti JC (2018) A putative human infertility allele of the meiotic recombinase DMC1 does not affect fertility in mice. Hum. Mol. Genet 27, 3911–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang B et al. (2020) A DNAH17 missense variant causes flagella destabilization and asthenozoospermia. J. Exp. Med 217, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Felipe-Medina N et al. (2020) A missense in HSF2BP causing primary ovarian insufficiency affects meiotic recombination by its novel interactor C19ORF57/BRME1. Elife 9, 1–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Imtiaz A et al. (2018) CDC14A phosphatase is essential for hearing and male fertility in mouse and human. Hum. Mol. Genet 27, 780–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Houston BJ et al. (2020) A framework for high-resolution phenotyping of candidate male infertility mutants: from human to mouse. Hum. Genet DOI: 10.1007/s00439-020-02159-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gaudelli NM et al. (2017) Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Komor AC et al. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou C et al. (2019) Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 571, 275–278 [DOI] [PubMed] [Google Scholar]
- 76.Zuo E et al. (2019) Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364, 289–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grünewald J et al. (2019) Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Anzalone AV et al. (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y et al. (2020) Efficient generation of mouse models with the prime editing system. Cell Discov. 6, 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Q et al. (2018) CRISPR-Cas9-mediated base-editing screening in mice identifies DND1 amino acids that are critical for primordial germ cell development. Nat. Cell Biol 20, 1315–1325 [DOI] [PubMed] [Google Scholar]
- 81.Kang Y et al. (2019) CRISPR/Cas9-mediated genome editing in nonhuman primates. Dis. Model. Mech 12, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saitou M and Miyauchi H (2016) Gametogenesis from Pluripotent Stem Cells. Cell Stem Cell 18, 721–735 [DOI] [PubMed] [Google Scholar]
- 83.Tan K et al. (2020) Transcriptome profiling reveals signaling conditions dictating human spermatogonia fate in vitro. Proc. Natl. Acad. Sci. USA 117, 17832–17841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.White YAR et al. (2012) Oocyte formation by mitotically active germ cells purified from ovaries of reproductive-age women. Nat. Med 18, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ding X et al. (2016) Human GV oocytes generated by mitotically active germ cells obtained from follicular aspirates. Sci. Rep 6, 28218–28234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hermann BP et al. (2012) Spermatogonial stem cell transplantation into rhesus testes regenerates spermatogenesis producing functional sperm. Cell Stem Cell 11, 715–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brinster RL and Avarbock MR (1994) Germline transmission of donor haplotype following spermatogonial transplantation. Proc. Natl. Acad. Sci. USA 91, 11303–11307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.David S and Orwig KE (2020) Spermatogonial stem cell culture in oncofertility. Urol Clin North Am 47, 227–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li X et al. (2019) Restore natural fertility of Kitw/Kitwv mouse with nonobstructive azoospermia through gene editing on SSCs mediated by CRISPR-Cas9. Stem Cell Res Ther 10, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang H et al. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yamaguchi T et al. (2017) The establishment of appropriate methods for egg-activation by human PLCZ1 RNA injection into human oocyte. Cell Calcium 65, 22–30 [DOI] [PubMed] [Google Scholar]
- 92.Sanusi R et al. (2015) Rescue of failed oocyte activation after ICSI in a mouse model of male factor infertility by recombinant phospholipase Cζ. Mol. Hum. Reprod 21, 783–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hirose N et al. (2020) Birth of offspring from spermatid or somatic cell by coinjection of PLCζ-cRNA. Reproduction 160, 319–330 [DOI] [PubMed] [Google Scholar]
- 94.Sang Q et al. (2018) Homozygous mutations in WEE2 cause fertilization failure and female infertility. Am. J. Hum. Genet 102, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao L et al. (2020) Biallelic mutations in CDC20 cause female infertility characterized by abnormalities in oocyte maturation and early embryonic development. Protein Cell DOI: 10.1007/s13238-020-00756-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eppig JJ and Wigglesworth K (2000) Development of mouse and rat oocytes in chimeric reaggregated ovaries after interspecific exchange of somatic and germ cell components. Biol. Reprod 63, 1014–1023 [DOI] [PubMed] [Google Scholar]
- 97.Sosa E et al. (2018) Differentiation of primate primordial germ cell-like cells following transplantation into the adult gonadal niche. Nat. Commun 9, 5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.1000 Genomes Project Consortium et al. (2010) A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smigielski EM et al. (2000) dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 28, 352–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lek M et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yates AD et al. (2020) Ensembl 2020. Nucleic Acids Res. 48, D682–D688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marcus JH and Novembre J (2017) Visualizing the geography of genetic variants. Bioinformatics 33, 594–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kent WJ et al. (2002) The human genome browser at UCSC. Genome Res. 12, 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Landrum MJ et al. (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McKusick VA (2007) Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet 80, 588–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stenson PD et al. (2017) The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet 136, 665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Modzelewski AJ et al. (2018) Efficient mouse genome engineering by CRISPR-EZ technology. Nat. Protoc 13, 1253–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]