

Abstract

Iboga alkaloids are a group of monoterpenoid indole alkaloids with promising and intriguing biological activities. Ibogaine is the representative member of the series and has become widely known as a potent atypical psychedelic with promising effects to treat substance use disorder. Nowadays, an efficient and scalable enantioselective total synthesis of ibogaine and related iboga alkaloids is still lacking, so direct extraction from natural sources or semi-synthetic schemes are the methods of choice to obtain them in a preparative scale. In particular, ibogaine can be obtained either by a low yielding direct isolation from Tabernanthe iboga or using a semi-synthetic procedure from voacangine, an iboga alkaloid occurring in a higher yield in the root bark of Voacanga africana. In this work, we describe an optimized process to obtain voacangine from V. africana root bark as a precursor of the iboga scaffold. Using a direct acetone-based extraction procedure (0.5 kg of root bark), voacangine was isolated in ∼0.8% of root bark dried weight, while the major alkaloids isolated from the bark were identified as iboga-vobasinyl dimers (∼3.7%) such as voacamine and voacamidine. Since these alkaloids contain the voacangine moiety in their structure, the cleavage of the dimers was further optimized, affording an extra amount of voacangine in ∼50% isolated molar yield. In this manner, the total amount of voacangine obtained by application of the whole procedure to the plant material (extraction and dimer cleavage) could almost duplicate the content originally found in the root bark.
Introduction
Iboga alkaloids are a group of monoterpenoid indole alkaloids (MIA) chemically characterized by the presence of a fused isoquinuclidine-tetrahydroazepine ring system.1,2 They are usually found in plants of several genera of the Apocynaceae family such as Tabernanthe, Voacanga, Tabernaemontana, Catharanthus, Coryanthe, and Aspidosperma. Ibogaine (1), originally isolated in 1901 as the main alkaloid from the root bark of Tabernanthe iboga (Figure 1), is the representative member of the series and has become widely known as a potent and atypical psychedelic with promising effects to treat substance use disorder (SUD).3 As found in anecdotal reports and observational studies, ibogaine reduces withdrawal symptoms and cravings to drugs of abuse such as heroin and cocaine.4−7 These effects were also found in extensive preclinical work in rodents’ models for SUD (such as self-administration and withdrawal paradigms).8 As mentioned in a recent review,2 there are many unsolved problems regarding the chemistry and neuropharmacology of ibogaine, which are of high importance considering the increased interest of psychedelic compounds as promising experimental therapeutics in neuropsychiatric disorders. Our group has recently published new findings regarding the intriguing neuropharmacology of ibogaine: its ability to induce the expression of neurotrophic factors in the brain,9 its antidepressive-like effect in rodents models,10 and the characterization of its unusual oneirogenic psychedelic effects through electrophysiological studies in the rat brain.11,12 From a chemical standpoint, ibogaine and other structurally related iboga alkaloids still lack efficient and scalable enantioselective total syntheses despite the vast amount of outstanding synthetic studies found in the field.2 Thus, direct extraction from natural sources or semi-synthetic preparations are the methods of choice to obtain ibogaine and related analogues in a preparative scale.
Figure 1.

Iboga alkaloids from natural sources. Isolation of ibogaine and voacangine from Tabernanthe iboga and Voacanga africana, respectively. (Plant’s drawings are courtesy of Juan Pablo Rodríguez)
Regarding extraction from natural sources, ibogaine direct isolation from T. iboga has become problematic because of the low content of the alkaloid in the root bark (∼0.3% isolated yield)13,14 and its high demand by the growing ibogaine medical subculture (global community of people who provide ibogaine therapy for SUD in medical, alternative, or underground settings).15 These facts have negatively impacted on the availability of the plant in West Africa because of an uncontrolled exploitation.16 In addition to these environmental concerns, T. iboga is also considered sacred in the traditional spiritual discipline of Bwiti, where it plays an important cultural role, especially in Gabon where the access to the plant has been threatened.16,17 As an alternative source, Voacanga africana a plant that has been cultivated in a larger scale in Africa, contains voacangine (2), another iboga alkaloid that is found in a high content in its stem and root barks.13,16 Voacangine allows for the commercial preparation of ibogaine in a semi-synthetic procedure that involves basic hydrolysis followed by acid decarboxylation.13 This process, although lengthy, affords a method for the easier purification and production of highly pure ibogaine, in contrast to the direct extraction from T. iboga where other structural related alkaloids (e.g., ibogamine and ibogaline) are present, making it difficult to achieve ibogaine of high purity. Although the qualitative composition of alkaloids in the stem and root barks of V. africana has been previously described,18−21 fewer studies show a quantitative analysis of its composition. An estimation of voacangine content using analytical methods afforded ∼1.7% of the dry weight in the root bark and ∼0.8% in the dry stem bark.17 On the other hand, only a few studies actually report isolated yields for voacangine (e.g., ∼0.5% of the dry weight of the stem bark) and detailed extraction procedures.13,14,22 Recently, voacangine was also detected in high amounts in some Tabernaemontana species, but again no isolated yields in a preparative scale were reported.17,23,24
So, it was decided to carry out an optimization study of the extraction of alkaloids from Voacanga africana root bark with the two-fold objective of using voacangine as a building block for our medicinal chemistry program (aimed at mapping the structure–activity relationship of iboga-inspired analogs) and obtaining ibogaine of pharmaceutical grade for future clinical and basic research projects. To this end, two extraction strategies were tested: a traditional acid/base method and a direct organic solvent-solid extraction. Voacangine (2) was isolated in ∼0.9% from both protocols (50–100 grams scale), while the major alkaloids of both extracts were identified as iboga-vobasine dimers such as voacamine (3) and voacamidine (4) (see Figure 2 for structures). Since these alkaloids present the voacangine moiety in their structure, a process for the cleavage of these dimers in acid media was further optimized to increase the isolated amount of voacangine available as a precursor to the iboga scaffold.
Figure 2.

Alkaloids isolated in this study from Voacanga africana root bark.
Results and Discussion
Isolation of Alkaloids Using Acid–Base vs Direct Organic Solvent Extraction
Initially, an acid–base extraction procedure inspired in the pioneer work by Jenks14,22 was used to isolate alkaloids from ground V. africana root bark (see the Supporting Information for experimental details and Figure S1). In short, 50 g of the ground of root bark was extracted with a 1% aqueous solution of HCl (6 × 250 mL) until no more voacangine was detected by HPLC-DAD. The combined filtered aqueous extracts were made alkaline by adding concentrated NH4OH, and the resulting brown precipitate was separated by centrifugation and dried. This solid was taken in acetone and filtered to discard root impurities. The solvent was evaporated in vacuo to afford a total alkaloid crude (∼3.7% of root bark dried weight), which was subjected to column chromatography to obtain voacangine (2) accounting for ∼0.85% of the root bark dried weight, voacristine (5) in ∼0.38%, which constitutes another interesting highly valuable building block containing the iboga moiety, and an inseparable mixture of three dimeric iboga-vobasine alkaloids (∼2.4% of root bark dried weight in a ratio of 3.5:1:0.3). Crystallization of the mixture rendered pure voacamine (3) as the major component, while further purification of the remaining alkaloids in the mother liquor allowed for identification of voacamidine (4) as the second major component. The minor alkaloid of the mixture remained unidentified (LC-MS analysis revealed the same molecular weight for the minor alkaloid, voacamine, and voacamidine, see the Supporting Information for details). Although these dimeric alkaloids were previously detected in V. africana,18,19,21,25−27 to our knowledge this is the first report to indicate quantitative data including isolated yields, showing them as the major alkaloids present in the root bark (∼65% of the total alkaloids found in the extract). All the alkaloids were fully characterized by 1H and 13C NMR using 2D NMR techniques as COSY, HSQC, and HMBC. A single-crystal X-ray diffraction analysis was carried out for voacamine, allowing for a stereochemistry confirmation which complements recently reported structural data for this alkaloid (Figure 3A).28 According to our second objective, voacangine was converted to ibogaine using a modification of previously published decarboxylation conditions29 (see the Supporting Information for details) and crystallized as its hydrochloride to afford a solid suitable for single-crystal X-ray diffraction analysis (Figure 3B).
Figure 3.

(A) ORTEP view of the voacamine molecule. The ellipsoids are represented with 30% probability. Crystallization water molecules were excluded for clarity. (B) ORTEP view of the ibogaine hydrochloride molecule. The ellipsoids are represented with 30% probability. Color code: H = gray, O = red, N = blue, Cl = green.
In order to simplify the isolation procedure and to avoid the troublesome sequence of precipitation, filtration, and centrifugation steps, a direct extraction method with organic solvents was tried. Thus, a solvent screening using methanol, ethyl acetate, acetone, and hexane was carried out to extract 0.3 g of root bark in the presence of solid NaHCO3. The crude extracts were analyzed by NMR using an internal standard to estimate the amounts of voacangine and voacamine recovered by each solvent (see Supporting Information, Table S1). Acetone was selected since it recovered the highest amount of voacamine and voacangine, which accounted for ∼86% of the mass of the total extract. The process was scaled up to 100 g of root bark under ultrasound-assisted extraction conditions in order to maximize the performance.30 The combined acetone extracts were evaporated in vacuo to afford a crude material that was subjected to column chromatography to detect the same alkaloids as in the acid base extraction procedure. Average isolated yields and standard deviation (for three replicates) expressed as % of the root bark dried weight were: 1.1 ± 0.2 for voacangine, 0.46 ± 0.02 for voacristine, and 2.9 ± 0.2 for the mixture of dimers (voacamine, voacamidine, and the minor unidentified dimer) in a ratio of 1.9:1:0.5. Although the alkaloid isolated yields were just slightly higher than those for the acid–base extraction process, the ratio obtained for the dimers was different since more voacamidine was isolated in the acetone-based methodology.
Since the latter protocol is easier to perform (consisting in fewer practical steps), it was chosen for scaling up in a 10 L reactor using 0.5 kg of root bark. No sonication was possible in the reactor setup, so the temperature was set to 40 °C to facilitate the extraction process. The root bark was extracted in batch (5 × 4 L), and the combined acetone extracts were concentrated in vacuo to afford 40 g of crude extract, which was subjected to several chromatography columns (see the Supporting Information for experimental details). A mixture of 4.1 g of voacangine (0.82% of root bark dried weight), 2.2 g (0.45%) of voacristine, and 18.5 g (3.7%) of a mixture of voacamine, voacamidine, and the minor unidentified dimer in a relation of 1.7:1:0.4 was obtained. In addition, small amounts of 3-(2-oxopropyl)voacangine (6) (0.15 g, 0.03% of root bark dried weight) and tabernaricatine G (7) (0.07 g, 0.01%) were found. Although these alkaloids were previously described in Tabernaemontana specimens but not in V. africana,31,32 the possibility of their production as artifacts of the acetone extraction process cannot be ruled out.
Voacamine and Voacamidine Cleavage
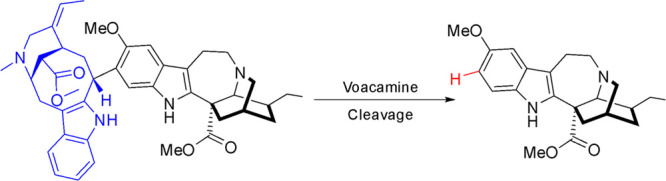
After identifying the dimers voacamine (3) and voacamidine (4) as the major alkaloids present in the root bark, we immediately envisioned the possibility of substituting the vobasine moiety in the dimers for a proton (using aromatic electrophilic substitution conditions) in order to produce higher quantities of voacangine. In the initial publication by Buchi regarding the structure elucidation for iboga-vobasine dimers,26 voacamine was treated with hydrogen chloride in deuterium oxide at reflux temperature to obtain voacangine-d3. Despite the lack of a reported molar yield, this precedent indicated the feasibility of the transformation. This condition was repeated using pure voacamine (0.02 M) in a 3:2 mixture of aqueous HCl and methanol (final concentration of HCl of 3 M). The reaction was completed after 6 h at reflux temperature to produce voacangine in 31% molar yield (no vobasine derived product was detected). In order to increase the yield in voacangine and reduce the reaction time, microwave-assisted heating was tried (Table 1). Upon irradiation at 100 W of potency the reaction was completed after only 9 min at 100 °C rendering the same molar yield of 30% for voacangine (entry 1). To avoid the potential decomposition of voacangine that could explain the low yield obtained, it was decided to run the reaction at lower temperatures. At 80 °C longer reaction times were required, which decreased the isolated molar yield of voacangine to 23% (entry 2), and at 60 °C no reaction was observed (entry 3). Higher concentrations of HCl produced decomposition of the starting material (entry 4).
Table 1. Conditions for Voacamine Cleavage Using Microwave-Assisted Heatingc.
| entry | conditions | solvent | temperature (°C) | time (min) | voacangine (%) |
|---|---|---|---|---|---|
| 1 | HCl 3 M | H2O | 100 | 9 | 30 |
| 2 | HCl 3 M | H2O | 80 | 30 | 23a |
| 3 | HCl 3 M | H2O | 60 | 60 | no reaction |
| 4 | HCl 6 M | H2O | 100 | 15 | decomposition |
| 5 | HCl 3 M | H2O/MeOH (4:1) | 100 | 15 | 33 |
| 6 | HCl 3 M | H2O/MeOH (1:4) | 100 | 25 | no reaction |
| 7 | TFA 1.5 M | DMF | 100 | 15 | no reaction |
| 8 | HCl 3 M, NaN3(2.0 equiv.) | H2O | 100 | 10 | 30b |
| 9 | HCl 3 M, PrSH (10 equiv.) | H2O | 100 | 10 | 38b |
Conversion was not complete, prolonged heating time (90 min) rendered 11% of voacangine without voacamine consumption.
Conversion was not complete, prolonged heating times also reduced the yield of voacangine.
In all cases, reactions were run using 100 W and heated until no more voacamine was detected by TLC analysis.
The addition of small amounts of methanol to increase the solubility of voacamine in the reaction media made the reaction slower without significant improvement in the yield (entry 5), and using methanol as the main solvent produced no reaction (entry 6), indicating the importance of the presence of water in the reaction media. This was confirmed by running the reaction in an organic solvent as DMF with trifluoroacetic acid, resulting in no reaction (entry 7). Considering a standard SEAr mechanism for the dimer cleavage, a nucleophile (as water) is needed to trap the resulting voabasinyl cation. In order to provide stronger nucleophiles the transformation was carried out using sodium azide or propylthiol as additives (entries 8 and 9). No significant differences in the voacangine yield was found for both conditions, although a small increase was observed using 1-propanethiol.
Given the limited capacity of the microwave oven (1 mL, 0.05 M), it was decided to run the reaction with conventional heating using a round-bottom pressure flask in order to use higher amounts of voacamine and to increase the reaction temperature beyond the boiling point of the reaction mixture (110 °C) (Table 2). Thus, aqueous 3 M HCl required 5 h to completely consume voacamine and rendered the same 30% molar yield as before. Alternative acids (such as trichloroacetic, trifluoroacetic, and aqueous HBr, entries 2–4) and solvents (e.g., dioxane, entry 5) were assayed without good results.
Table 2. Conditions for Vocamine Cleavage Using Conventional Heating in a Round-Bottom Pressure Flaskd.
| entry | acid | solvent | additive | time (h) | voacangine (%) |
|---|---|---|---|---|---|
| 1 | HCl 3 M | H2O | 5 | 30 | |
| 2 | TCA 3 M | H2O | 7 | no reaction | |
| 3 | TFA 3 M | H2O | 7 | no reaction | |
| 4 | HBr 3 M | H2O/acetic acid | 2 | decomposition | |
| 5 | HCl 3 M | dioxane | 7 | decomposition | |
| 6 | HCl 3 M | H2O | TIS (50 equiv) | 3 | 41 |
| 7 | HCl 3 M | H2O | TIS-SH (50 equiv) | 2 | 44 |
| 8 | HCl 3 M | H2O | TIS (3.0 equiv.) | 4 | 39a |
| 9 | HCl 3 M | H2O | TIS (3.0 equiv.) | 2 | 51b |
| 10 | HCl 3 M | H2O | TIS (3.0 equiv.) | 1 | 48c |
17% of norvoacangine and 20% of ibogaine were also obtained.
7% of norvoacangine.
8% of norvoacangine.
In all cases, reactions were run at 110 °C using a voacamine concentration of 0.015 M, until no more starting material was detected by TLC analysis, except entry 10 where the reaction was stopped after 1 h. All reactions were carried out using 34 mg of voacamine except entries 8, 9, and 10 where 500 mg of voacamine was used.
The addition of cation scavengers as triisopropylsilane (TIS) and triisopropilsilanethiol (TIS-SH) in a molar excess (50 equivalents) allowed for an increase in reaction yields (∼40%) with significant reduction in the reaction time (entries 6 and 7). The reaction at a 500 mg scale of voacamine was tested using TIPS (3.0 equivalents, entry 8) to afford 39% molar yield for voacangine and other byproducts as norvoacangine (8) in 17% and ibogaine in 20% yield. Since the latter products were probably produced from voacangine, an optimization of the reaction time was undertaken in order to select an optimum reaction time to obtain the maximum concentration of voacangine. Figure 4 shows the time profiles for voacamine consumption and vocangine/norvoacangine formation in the first 2 h of reaction, determined by HPLC-DAD. As can be seen, norvoacangine formation starts after the first hour of reaction, when voacamine has not been completely consumed. No ibogaine was detected in the recorded time, and voacangine concentration remained almost constant after the first hour of reaction. It was decided to run the reaction for 1 and 2 h, and a similar voacangine isolated yield was obtained (48% and 51%, entries 10 and 9). The whole process was scaled up to convert 1 g of voacamine to render a similar molar yield of 49% of voacangine. Finally, in order to test that voacamidine could be also cleaved, the same reaction conditions were applied to a mixture of voacamidine:voacamine (4:1) to render after 1.5 h of heating, voacangine in a 47% isolated molar yield. In this manner, this optimized procedure allows for the transformation of the iboga-vobasinyl dimers mixture in order to increase the amount of voacangine available (thus giving an amount that could almost duplicate the amount of this alkaloid originally found in the root bark).
Figure 4.
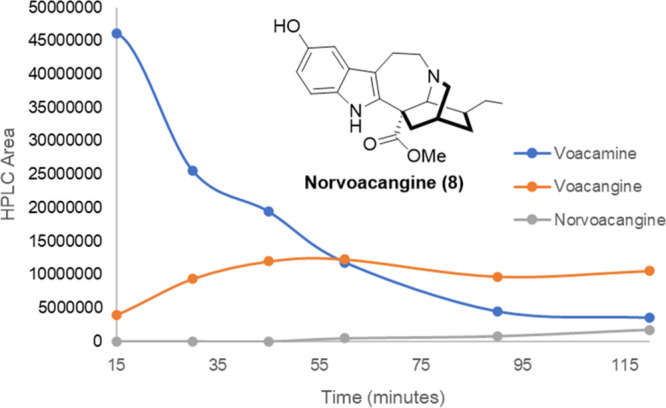
Time profile for voacamine consumption and voacangine and norvoacangine formation. Although vocamine is not totally consumed after 60 min, voacangine formation is not further increased until 120 min. The formation of other products, as norvoacangine, can explain this reaction profile.
Conclusions
A procedure for the isolation of the major alkaloids from the root bark of Voacanga africana was developed in order to optimize voacangine production and gain better access to the iboga scaffold. The direct acetone-based extraction protocol simplified the operational steps of the classical acid–base alkaloid extraction methodology, allowing to isolate (in a 0.5 kg scale): voacangine in 0.82% of root bark dried weight, voacristine in 0.45%, and 3.7% of a mixture of voacamine, voacamidine, and a minor unidentified dimer. Also, a cleavage reaction in acid media for voacamine and voacamidine was optimized (using up to 1 g of voacamine) to produce voacangine in ∼50% isolated yield. This allows us to obtain an extra amount of voacangine through the transformation of the iboga-vobasinyl dimers present as the main alkaloid fraction of the root bark. In this manner, the total amount of voacangine obtained by application of the whole procedure to the plant material (extraction and dimer cleavage) could almost duplicate (∼2.0% of voacangine) the content originally found in the root bark. Finally, the total amount of ibogaine that could potentially be obtained using this protocol is approximately 1.8 g per 100 g of Voacanga africana root bark. This represents a six-fold increase compared to the isolated yield that is currently obtained using direct extraction from Tabernanthe iboga root bark.
Experimental Section
General Experimental Methods
All solvents were distilled prior to use; chemicals and reagents were purchased from Sigma-Aldrich and used as received. Voacanga africana root bark was purchased from CapeLabs, Cape Analytical Services Laboratories (Cape Town, South Africa). Mass spectra (MS) were recorded on a HPLC MS/MS Shimadzu LCMS 8040, with LC-20AD HPLC pump, DAD SPD-M20A detector, CTO-20A oven, and SIL-20A injector; the software used to process data was LabSolutions LCMS. NMR spectra were obtained in CDCl3 on a Bruker Avance DPX-400 instrument. Proton chemical shifts (δ) are reported in ppm downfield from TMS as an internal reference, and carbon chemical shifts are reported in ppm relative to the center line of the CDCl3 triplet (77.0 ppm). Analytical TLC was performed on silica gel 60F-254 plates and visualized with UV light (254 nm) and/or p-anisaldehyde in acidic ethanolic solution. Flash column chromatography was performed using silica gel (Kieselgel 60, EM reagent, 230–400 mesh). Centrifugation was performed in a Thermo Sorvall RC6 plus Centrifuge, using the conditions specified in the protocol. The extraction of 0.5 kg was carried out in a 10 L Reactor GR-10 Greatwall equipped with a high temperature circulator Greatwall SY series. Microwave reactions were made in a CEM Discovery reactor. The voacamine/voacamidine cleavage of 1 g was carried out in a Synthware round bottom pressure vessel with PTFE bushing (150 mL, O.D. × L 60 mm × 139 mm, bushing #15). Sonication was applied using a Jeio Tech ultrasonic bath (Labcompanion).
Acid–Base Extraction
A 50 g dry Voacanga africana root bark powder was added to an Erlenmeyer flask (1000 mL) followed by 250 mL of HCl (1%). The suspension was mechanically stirred for 30 min and then filtered through cellulose-based NWF (Nonwoven fabric), and the plant residue was recovered and another 250 mL of fresh HCl (1%) were added; this procedure was repeated six times. The combined acidic extracts were treated with concentrated NH4OH under constant stirring until pH 10–11 was reached. The alkaloids precipitated forming an opaque suspension, which was centrifuged at 5000 rpm during 15 min at 10 °C. The supernatant was discarded, while the solid residue was dried at 60 °C overnight (12–14 h). The dried brown amorphous solid was dissolved in acetone and filtered through paper to afford a clear solution. Silica gel was added and the organic solvent was removed in vacuo; the resulting solid was used to load a preparative chromatography column (mobile phase gradient: (9:1) → (8:2) → (6:4) → (1:1) → (3:7) (Hex:EtOAc) + 1% NH4OH). As a result, 429 mg (0.85%) of voacangine (2), 192 mg (0.38%) of voacristine, and 1203 mg (2.41%) of a mixture voacamine:voacamidine:unknown dimer (3.5:1:0.3) were obtained (see Figure S1).
Organic Solvent Screening
A 0.3 g dry Voacanga africana root bark powder was added to a 5 mL vial followed by with 30 mg (10% w/w) of solid NaHCO3 and 4 mL of the chosen solvent. The resulting suspension was stirred 1 h at room temperature and then filtrated through Whatman filter paper. The process was repeated four times with the same plant material. The combined organic extracts were evaporated in vacuo to afford a crude extract that was dissolved in CDCl3 (0.7 mL) with 5 μL of trichloroethylene (TCE) as an internal standard. The extracts were analyzed by 1H NMR; the comparison with the 1H spectrum of the pure compounds voacangine (2) and voacamine (3) allowed for the selection of characteristic and well-resolved signals to quantify the amount of alkaloids in the sample, using TCE as an internal standard. Selected signals: (s, 3.71 ppm), (s, 3.84 ppm), (d, 6.93 ppm, J = 2.3 Hz), and (dd, 6.80 ppm, J = 8.7, 2.4 Hz). Ethyl acetate and acetone show the best results (see Figure S3 and Table S1).
Acetone-Based Extraction (100 g Scale)
A 100 g dry Voacanga africana root bark powder was added to an Erlenmeyer flask (1000 mL) followed by 10 g of NaHCO3 and 800 mL of acetone. The suspension was sonicated at room temperature for 30 min and filtrated through paper. The plant residue was recovered, and the same procedure was repeated until no alkaloids were detected in the supernatant by TLC analysis [SiO2, (8:2 Hex:EtOAc) + 1% NH4OH(cc)]. The combined organic extracts were concentrated in vacuo to afford a dark brown solid, which accounts for 9–10% of the initial mass of the plant material. The extract was loaded on silica gel to run a preparative chromatography column (mobile phase gradient: (95:5) → (9:1) → (8:2) → (6:4) → (1:1) → (3:7) (Hex: EtOAc) + 1% NH4OH). As a result, after three independent replicates of this procedure, 1.06 ± 0.17% of voacangine (2), 0.46 ± 0.02% of voacristine (5), and 2.92 ± 0.21% of a mixture of dimers voacamine (3):voacamidine (4):unidentified dimer (1.9:1:0.5) were obtained.
Acetone-Based Extraction (0.5 kg Scale)
A 0.5 kg dry Voacanga africana root bark powder was added to a 10 L reactor followed by 50 g of NaHCO3 and 4 L of acetone (see Figure S5). The resulting suspension was stirred and heated at 200 rpm and 40 °C. After 45 min, the suspension was filtrated through paper. The plant residue was recovered, and the same procedure was repeated five times. The combined organic extracts were concentrated in vacuo to afford a dark brown solid, which accounts for 9–10% of the initial mass of the plant material. The extract was loaded on silica gel to run a preparative chromatography column (mobile phase gradient: (95:5) → (9:1) → (8:2) → (6:4) → (1:1) → (3:7) → (0:1) (Hex:EtOAc) + 1% NH4OH). After several chromatographic separations (see Figure S4), it was obtained 0.82% of voacangine (2), 0.45% of voacristine (5), and 3.70% of a mixture voacamine (3):voacamidine (4):unidentified dimer (1.7:1:0.4) were obtained (see Figure S4).
Cleavage of Voacamine Using Microwave-Assisted Heating (30–100 mg) (Optimization of Reaction Conditions): General Technique
Voacamine (35 mg, 0.049 mmol) followed by 1 mL of HCl 3 M (or the appropriate solvent) were added to a CEM Discovery reactor’s tube (6 mL). The heating program was set on the equipment [100 °C, 100 W, 9 min] (or the appropriate conditions), and the tube was sealed and kept with constant stirring. After the irradiation was completed, the system was allowed to reach room temperature, and the solution was neutralized with the addition of solid NaHCO3, extracted with EtOAc (×3), and the combined organic phases dried over Na2SO4. The solvent was removed in vacuo, and the resulting crude was purified by column chromatography (mobile phase gradient: (8:2) → (6:4) → (1:1) → (3:7) (Hex:EtOAc) + 1% NH4OH).
Cleavage of Voacamine Using Conventional Heating (0.5–1.0 g): General Technique
Voacamine (0.5 g, 0.709 mmol) was added followed by triisopropylsilane (0.43 mL, 2.12 mmol), and a HCl aqueous solution (3 M, 50 mL) to a 150 mL round-bottom pressure flask. The suspension was heated at 110 °C for the indicated time under constant stirring. Then, the mixture was cooled to room temperature and basified by adding solid NaHCO3. The resulting suspension was extracted exhaustively with ethyl acetate. Combined organic layers were dried using Na2SO4, and the solvent was distilled in vacuo to obtain a crude reaction mixture, which was purified using column chromatography (SiO2, Hex:EtOAc, 9:1/1% NH4OH) to obtain voacangine as a pure solid with 51% molar yield. The same protocol was used to convert 1.0 g of voacamine (voacangine molar yield: 49%) and for 34 mg of a voacamidine/voacamine (4:1) mixture (voacangine molar yield: 47%).
Ibogaine (1): Hydrolysis and Decarboxylation of Voacangine (2)
In a two-neck round-bottom flask, voacangine (0.547 g, 1.486 mmol) followed by (EtOH:H2O) (3:2) (37 mL, 0.04 M) and KOH (0.832 g, 14.860 mmol, 10 eq) were added. The solution was bubbled with argon for 15 min at room temperature and then heated at reflux temperature over 12–15 h. Consumption of the starting material was verified by TLC (8:2) (Hex:EtOAc) + 1% NH4OH). The solution was allowed to cool down to room temperature, and ethanol was removed in vacuo. Then, HCl 5 N (11.3 mL, 56.47 mmol, 37 eq) was added, and the solution was refluxed for 15 min. The solution was allowed to cool down to room temperature and was neutralized by adding solid NaHCO3. The aqueous phase was extracted with EtOAc (×4), and the combined organic layers were dried over Na2SO4, the solvent was removed in vacuo and purified by column chromatography (8:2) (Hex:EtOAc) + 1% NH4OH. Ibogaine free base was obtained as a white solid (414 mg, 90% yield).
Acknowledgments
We thank Luisina Rodríguez and Dr. Danilo Davyt for the LC-MS analysis and Horacio Pezaroglo and Dr. Gonzalo Hernandez for the NMR spectra. We also thank Prof. Graciela Mahler for discussions regarding the voacamine cleavage using microwave irradiation and Lic. Paola Rodríguez for her contributions to the acetone-based extraction optimization. Finally, we thank Juan Pablo Rodríguez Carrera for the original drawings of Tabernanthe iboga and Voacanga africana in Figure 1 and in the Graphical Abstract.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00745.
Author Contributions
∥ B.G and C.F. contributed equally to this work. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was supported by Agencia Nacional de Investigacion e Innovación (ANII-Uruguay, Fondo María Viñas FMV_1_2014_1_103488), Comisión Sectorial de Investigacion Científica (Universidad de la Republica, CSIC-Grupos I+D 1063), Programa de Desarrollo de las Ciencias Basicas (PEDEClBA - Uruguay), and Espacio Interdisciplinario, Universidad de la República. B.G. and C.F. wish to thank Comisión Académica de Posgrado – UdelaR for Ph.D. and postdoctoral scholarships, respectively.
The authors declare the following competing financial interest(s): Diver Sellanes is the CEO of SIQUIMIA S.R.L. The rest of the authors declare no competing financial interest.
After this paper was published ASAP June 24, 2021, a correction was made to the structure of compound 7 in Figure 2. The corrected version was reposted July 6, 2021.
Supplementary Material
References
- Lavaud C.; Massiot G. The Iboga Alkaloids. Prog. Chem. Org. Nat. Prod. 2017, 105, 89–136. 10.1007/978-3-319-49712-9_2. [DOI] [PubMed] [Google Scholar]
- Iyer R. N.; Favela D.; Zhang G.; Olson D. E. The Iboga Enigma: The Chemistry and Neuropharmacology of Iboga Alkaloids and Related Analogs. Nat. Prod. Rep. 2021, 38, 307. 10.1039/d0np00033g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper K. R. Ibogaine: A Review. Alkaloids Chem. Biol. 2001, 56, 1–38. 10.1016/s0099-9598(01)56005-8. [DOI] [PubMed] [Google Scholar]
- Alper K. R.; Lotsof H. S.; Frenken G. M.; Luciano D. J.; Bastiaans J. Treatment of Acute Opioid Withdrawal with Ibogaine. Am. J. Addict. 1999, 8, 234–242. 10.1080/105504999305848. [DOI] [PubMed] [Google Scholar]
- Schenberg E. E.; de Castro Comis M. A.; Chaves B. R.; da Silveira D. X. Treating Drug Dependence with the Aid of Ibogaine: A Retrospective Study. J. Psychopharmacol. 2014, 28, 993–1000. 10.1177/0269881114552713. [DOI] [PubMed] [Google Scholar]
- Brown T. K.; Alper K. Treatment of Opioid Use Disorder with Ibogaine: Detoxification and Drug Use Outcomes. Am. J. Drug Alcohol Abuse 2018, 44, 24–36. 10.1080/00952990.2017.1320802. [DOI] [PubMed] [Google Scholar]
- Mash D. C.; Duque L.; Page B.; Allen-Ferdinand K. Ibogaine Detoxification Transitions Opioid and Cocaine Abusers Between Dependence and Abstinence: Clinical Observations and Treatment Outcomes. Front. Pharmacol. 2018, 9, 529. 10.3389/fphar.2018.00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgers M.; Leenaars M.; Homberg J. R.; Ritskes-Hoitinga M.; Schellekens A. F.; Hooijmans C. R. Ibogaine and Addiction in the Animal Model, a Systematic Review and Meta-Analysis. Transl. Psychiatry 2016, 6, e826 10.1038/tp.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton S.; González B.; Rodríguez-Bottero S.; Miquel E.; Martínez-Palma L.; Pazos M.; Pedro Prieto J.; Rodríguez P.; Sames D.; Seoane G.; Scorza C. Ibogaine Administration Modifies GDNF and BDNF Expression in Brain Regions Involved in Mesocorticolimbic and Nigral Dopaminergic Circuits. Front. Pharmacol. 2019, 10, 193. 10.3389/fphar.2019.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez P.; Urbanavicius J.; Prieto J. P.; Fabius S.; Reyes A. L.; Havel V.; Sames D.; Scorza C.; Carrera I. A Single Administration of the Atypical Psychedelic Ibogaine or Its Metabolite Noribogaine Induces an Antidepressant-Like Effect in Rats. ACS Chem. Neurosci. 2020, 11, 1661–1672. 10.1021/acschemneuro.0c00152. [DOI] [PubMed] [Google Scholar]
- Gonzalez J.; Prieto J. P.; Rodriguez P.; Cavelli M.; Benedetto L.; Mondino A.; Pazos M.; Seoane G.; Carrera I.; Scorza C.; Torterolo P. Ibogaine Acute Administration in Rats Promotes Wakefulness, Long-Lasting REM Sleep Suppression, and a Distinctive Motor Profile. Front. Pharmacol. 2018, 9, 374. 10.3389/fphar.2018.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez J.; Cavelli M.; Castro-Zaballa S.; Mondino A.; Tort A. B.; Rubido N.; Carrera I.; Torterolo P. EEG Gamma Band Alterations and REM-like Traits Underpin the Acute Effect of the Atypical Psychedelic Ibogaine in the Rat. ACS Pharmacol. Transl. Sci. 2021, 517. 10.1021/acsptsci.0c00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janot M.-M.; R G.. Derivatives of the Ibogaine Alkaloids. United States Patent Office 2,813,879, 1956.
- Jenks C. W. Extraction Studies of Tabernanthe Iboga and Voacanga Africana. Nat. Prod. Lett. 2002, 16, 71–76. 10.1080/1057563029001/4881. [DOI] [PubMed] [Google Scholar]
- Alper K. R.; Lotsof H. S.; Kaplan C. D. The Ibogaine Medical Subculture. J. Ethnopharmacol. 2008, 115, 9–24. 10.1016/j.jep.2007.08.034. [DOI] [PubMed] [Google Scholar]
- Dickinson J. Iboga Root: Dynamics of Iboga′s African Origins and Modern Medical Use. HerbalGram 2016, 109, 48–57. [Google Scholar]
- Krengel F.; Dickinson J.; Jenks C.; Reyes-Chilpa R. Quantitative Evaluation of a Mexican and a Ghanaian Tabernaemontana Species as Alternatives to Voacanga Africana for the Production of Antiaddictive Ibogan Type Alkaloids. Chem. Biodiversity 2020, 17, e2000002 10.1002/cbdv.202000002. [DOI] [PubMed] [Google Scholar]
- Koroch A. R.; Juliani H. R.; Kulakowski D.; Arthur H.; Asante-Dartey J.; Simon J. E. Voacanga Africana: Chemistry, Quality and Pharmacological Activity. ACS Symp. Ser. 2009, 1021, 363–380. 10.1021/bk-2009-1021.ch020. [DOI] [Google Scholar]
- Hussain H.; Hussain J.; Al-Harrasi A.; Green I. R. Chemistry and Biology of the Genus Voacanga. Pharm Biol 2012, 50, 1183–1193. 10.3109/13880209.2012.658478. [DOI] [PubMed] [Google Scholar]
- Harada M.; Asaba K. N.; Iwai M.; Kogure N.; Kitajima M.; Takayama H. Asymmetric Total Synthesis of an Iboga-Type Indole Alkaloid, Voacangalactone, Newly Isolated from Voacanga Africana. Org. Lett. 2012, 14, 5800–5803. 10.1021/ol3027945. [DOI] [PubMed] [Google Scholar]
- Chen H. M.; Yang Y. T.; Li H. X.; Cao Z. X.; Dan X. M.; Mei L.; Guo D. L.; Song C. X.; Dai Y.; Hu J.; Deng Y. Cytotoxic Monoterpenoid Indole Alkaloids Isolated from the Barks of Voacanga Africana Staph. Nat. Prod. Res. 2016, 30, 1144–1149. 10.1080/14786419.2015.1046132. [DOI] [PubMed] [Google Scholar]
- Jenks C.Voacanga Extraction Manual. 2015.
- Krengel F.; Mijangos M. V.; Reyes-Lezama M.; Reyes-Chilpa R. Extraction and Conversion Studies of the Antiaddictive Alkaloids Coronaridine, Ibogamine, Voacangine, and Ibogaine from Two Mexican Tabernaemontana Species (Apocynaceae). Chem. Biodiversity 2019, 16, e1900175 10.1002/cbdv.201900175. [DOI] [PubMed] [Google Scholar]
- Krengel F.; Chevalier Q.; Dickinson J.; Herrera Santoyo J.; Reyes Chilpa R. Metabolite Profiling of Anti-Addictive Alkaloids from Four Mexican Tabernaemontana Species and the Entheogenic African Shrub Tabernanthe Iboga (Apocynaceae). Chem. Biodiversity 2019, 16, e1800506 10.1002/cbdv.201800506. [DOI] [PubMed] [Google Scholar]
- Manu A.; Sofoklis R.; Ahijo M.; Song R. Isolation of Voacangine and Voacamine from Voacanga Africana and Their Activities against the Onchocerca Ochengi Worm. Glob. J. Med. Plants Res. 2016, 2, 169–176. [Google Scholar]
- Buchi G.; Manning R. E.; Monti S. A. Voacamine and Voacorine. J. Am. Chem. Soc. 1964, 86, 4631–4641. 10.1021/ja01075a023. [DOI] [Google Scholar]
- Babiaka S. B.; Simoben C. V.; Abuga K. O.; Mbah J. A.; Karpoormath R.; Ongarora D.; Mugo H.; Monya E.; Cho-Ngwa F.; Sippl W.; Loveridge J.; Ntie-Kang F. Alkaloids with Anti-Onchocercal Activity from Voacanga Africana Stapf (Apocynaceae): Identification and Molecular Modeling. Molecules 2021, 26, 70. 10.3390/molecules26010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. X.; Liu R. Q.; Zhang H. M.; Cao Z. X.; Zhu L. X.; Li Y. Y.; Ding W. J.; Chen Y. H.; Deng Y. In Vitro Anti-HIV Effect of Voacamine from Voacanga Africana Stapf, Based on the SPRi Experiment. Russ. J. Bioorg. Chem. 2020, 46, 306–311. 10.1134/S1068162020030115. [DOI] [Google Scholar]
- Seong S.; Lim H.; Han S. Biosynthetically Inspired Transformation of Iboga to Monomeric Post-Iboga Alkaloids. Chem 2019, 5, 353–363. 10.1016/j.chempr.2018.10.009. [DOI] [Google Scholar]
- Chemat F.; Rombaut N.; Sicaire A. G.; Meullemiestre A.; Fabiano-Tixier A. S.; Abert-Vian M. Ultrasound Assisted Extraction of Food and Natural Products. Mechanisms, Techniques, Combinations, Protocols and Applications. A Review. Ultrason. Sonochem. 2017, 540–560. 10.1016/j.ultsonch.2016.06.035. [DOI] [PubMed] [Google Scholar]
- Bao M. F.; Yan J. M.; Cheng G. G.; Li X. Y.; Liu Y. P.; Li Y.; Cai X. H.; Luo X. D. Cytotoxic Indole Alkaloids from Tabernaemontana Divaricata. J. Nat. Prod. 2013, 76, 1406–1412. 10.1021/np400130y. [DOI] [PubMed] [Google Scholar]
- Okuyama E.; Gao L. H.; Yamazaki M. Analgesic Components from Bornean Medicinal Plants, Tabernaemontana Pauciflora Blume and Tabernaemontana Pandacaqui Poir. Chem. Pharm. Bull. 1992, 40, 2075–2079. 10.1248/cpb.40.2075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.