

SUMMARY
Chromatin is organized in the nucleus via CTCF loops and compartmental domains. Here, we compare different cell types to identify distinct paradigms of compartmental domain formation in human tissues. We identify and quantify compartmental forces correlated with histone modifications characteristic of transcriptional activity and previously underappreciated roles for distinct compartmental domains correlated with the presence of H3K27me3 and H3K9me3, respectively. We present a computer simulation model capable of predicting compartmental organization based on the biochemical characteristics of independent chromatin features. Using this model, we show that the underlying forces responsible for compartmental domain formation in human cells are conserved and that the diverse compartmentalization patterns seen across cell types are due to differences in chromatin features. We extend these findings to Drosophila to suggest that the same principles are at work beyond humans. These results offer mechanistic insights into the fundamental forces driving the 3D organization of the genome.
Graphical Abstract
In brief
Using high-resolution Hi-C data and computer simulations, Nichols and Corces show that compartments arise as a consequence of interactions among proteins that correlate with the presence of H3K27ac, H3K27me3, and H3K9me3, suggesting that human cells contain at least three distinct compartments. The same principles apply to other organisms.
INTRODUCTION
The highly organized nature of the eukaryotic nucleus has been evident since experiments using immunofluorescence microscopy to determine the subnuclear distribution of various proteins and histone modifications showed the existence of several types of nuclear bodies (Matera et al., 2009). These nuclear locations, where proteins with related functional properties accumulate, have been described more recently as biomolecular condensates created as a consequence of liquid-liquid phase separation due to the presence of high concentrations of multivalent proteins bound to DNA and RNA, dividing the nucleoplasm into functionally distinct compartments (Banani et al., 2017). Some of these nuclear bodies appear to be involved in RNA processing or sequestration, but others, such as the nucleolus, contain chromatin. These bodies represent distinct nuclear environments that regulate exposure of the DNA to various proteins of the nucleoplasm and are therefore essential to controlling the activity of genes. For example, active genes can be present in hubs called transcription factories, in which expressed genes aggregate together with the transcriptional machinery (Mitchell and Fraser, 2008). Features of chromatin that are associated with transcriptional silencing also cluster with one another. Polycomb bodies form via the agglomeration of polycomb repressive complex 1 (PRC1) and PRC2 protein complexes that silence genes, in part by the trimethylation of H3K27 (Pirrotta and Li, 2012). In addition, transcriptionally silenced pericentric heterochromatin colocalizes within the nucleus to form chromocenters in some cells, strongly enriched for HP1a and H3K9me3 (Wang et al., 2019). Several studies have now shown the ability of chromatin components to drive liquid-liquid phase separation in vitro and in vivo. H3K9me3 and HP1 together produce the compartmentalization of heterochromatin, and this is also the case for the intrinsically disordered regions found in PRC1, RNA polymerase II, and many transcription factors (Boija et al., 2018; Ladouceur et al., 2020; Plys et al., 2019; Wang et al., 2019).
With the advent of Hi-C it has become possible to query the organization of the entire genome simultaneously at the sequence level (Lieberman-Aiden et al., 2009). Hi-C identifies all of the interactions in the genome after fixation with formaldehyde. The resulting contact frequency maps prominently display self-associating domains formed by short-range interactions among contiguous segments of the genome and can be visualized as “triangles” present at the diagonal of Hi-C heatmaps. Classically, these contact domains are called compartments and topologically associating domains (TADs). These two types of domains originally described distinct phenomena apparent at different length scales in Hi-C maps using different computational methodologies, but which have now been shown to overlap significantly (Rowley and Corces, 2018). Compartments are defined by principal-component analysis (PCA), normally using Hi-C data binned at a 0.5- to 1.0-Mb resolution, and, as a consequence, compartments are normally considered to be >1 Mb in size. Compartments can contain sequences in an active (A) or silenced (B) transcriptional state and they interact with other compartments in the same state to give the plaid pattern observed in the Hi-C heatmap. The term “compartment” is used to refer to both the self-interacting contact domains present at the diagonal and the ensemble of all of the inter-domain interactions among all of the domains in the same transcriptional state. To avoid confusion, we use the term “compartmental domains” to refer to self-interacting contact domains present at the diagonal of Hi-C heatmaps and “compartment” to refer to all of the interactions among compartmental domains in the same transcriptional state. Different from compartmental domains, TADs are defined using algorithms that detect switches in the directionality of interactions. Analysis of Hi-C data at 1-kb resolution indicates that TADs actually correspond to two different types of domains: CCCTC-binding factor (CTCF) loops and compartmental domains (Rao et al., 2017; Rowley et al., 2017). CTCF loops are formed by the interruption of cohesin extrusion due to the presence of convergent CTCF-bound sites. CTCF loops can be visualized in Hi-C heatmaps by strong punctate signals at the summit of the domain, whereas compartmental domains lack this signal. CTCF loops disappear from Hi-C heatmaps obtained in cells depleted of CTCF or RAD21, whereas compartmental domains remain (Nora et al., 2017; Rao et al., 2017). Furthermore, compartmental domains are present in regions of the genome containing sequences in the same active or inactive transcriptional state and can be identified by PCA using 5- to 10-kb bin sizes. Neighboring regions in separate compartmental domains interact less frequently and represent a compartmental switch or border. In this way, the compartmentalization of the genome creates both local compartmental domains and distant compartmental interactions. Domains referred to as TADs in the literature are either compartmental domains, CTCF loops, or a combination of the two (Rowley and Corces, 2018).
As described above, compartmental domains can be captured by PCA of the Pearson correlation maps of each chromosome. The first principal component (PC1) or eigenvector captures the dimension with the highest variance. This vector has been classically divided into two sets of values because the corresponding sequences in the genome correlate well with transcriptionally active and inactive regions, and thus are called A and B, respectively (Lieberman-Aiden et al., 2009). This categorization performs well across mammalian cell types and therefore the compartmentalization of the genome is generally thought of as binary. However, results from microscopy and epigenetic information (i.e., covalent histone modifications and/or DNA methylation) suggest that the transcriptional state of the genome is more complex, and that the two-state classification is an oversimplification. While PCA remains the standard and most common method for calling compartmental domains, some analyses have more closely examined compartmentalization using more sophisticated techniques. The use of a hidden Markov model to cluster interchromosomal interactions resolved the active A and inactive B compartments into 2 active and 4 inactive compartment subtypes in GM12878 cells. These subcompartments differ according to various chromatin features, including post-translational histone modifications, replication timing, and measures of nucleolar and lamin association (Rao et al., 2014). However, the subcompartments within A and B do not differ substantially in which chromatin features are enriched. Other studies have also successfully used polymer simulations to reproduce the compartmental organization of the genome using chromatin-defined states (Annunziatella et al., 2018; Buckle et al., 2018; Chiang et al., 2019; Cook and Marenduzzo, 2018; Falk et al., 2019; Nuebler et al., 2018; Qi and Zhang, 2019). PCA-derived compartment calls in diverse cell lines, and tissues invariably find A/B compartmentalization patterns, but the epigenetic features enriched in those A/B patterns can differ between cell types. Notably, several studies have found the heterochromatin-associated histone modification H3K9me3 strongly enriched in B compartments (Dixon et al., 2015; Falk et al., 2019). However, this modification is only found enriched in the single B4 subcompartment in GM12878 cells, which is predominantly found only on chromosome 19 (Rao et al., 2014). Therefore, the binary A/B compartmentalization of the genome is far simpler than what would be predicted from microscopy analyses of nuclei in which a large variety of biomolecular condensates composed of different epigenetic features have been observed.
Here, we examine two cell types, GM12878 and HCT-116, with divergent compartmental definitions in an attempt to better understand the different patterns of compartmental domains seen between different human cells and investigate the potential role of the forces responsible for this aspect of genome organization, with the goal of reconciling observations derived from Hi-C analyses and microscopy-based studies. The results suggest a consistent model of genome organization and offer insights into the mechanistic underpinnings of 3D genome compartmentalization.
RESULTS
Dynamic compartmentalization across human cells
To better understand the mechanisms underlying the formation of compartmental domains and their compartmental interactions, we compared high-resolution Hi-C datasets containing 2.5–5 billion contacts from two cell types: the lymphoblastoid GM12878 cell line and colorectal carcinoma HCT-116 cells. We took the Pearson correlation of the distance-normalized interaction maps to display the correlation of interactions of each bin with each other bin. This method can be used to visualize compartmental domains and their interactions because genomic regions in the same compartment will have highly correlated interaction frequencies and will have a high score in the correlation map. Figures 1A and 1B show the Pearson correlation maps for chromosome 4 of GM12878 and HCT-116, respectively. This chromosome shows very different organizations between the two cell lines. Both possess clear compartmental domains along the diagonal of the map and compartmental interactions as seen by the plaid pattern away from the diagonal, but both differ considerably between the two cell lines. We sought to explore whether differences in the epigenetic profiles of the chromosomes of these two cell types could explain their distinct compartmentalization patterns. We compared the distribution of H3K27ac, which is correlated with transcriptional activity, H3K27me3, which is correlated with transcriptional silencing, and H3K9me3, which is also correlated with transcriptionally inactive sequences, to the Pearson correlation maps in these cells. These three histone modifications were chosen due to their largely non-overlapping distributions, substantial presence in the genome, and observed correlations with compartmental features. We also tested H3K4me1, H3K4me2, H3K4me3, H3K36me3, H3K9ac, H3K79me2, H4K20me1, and global run-on sequencing (GRO-seq), but these modifications do not correlate as well with compartmental features (Figures S1A–S1C). We also performed PCA at 100-kb resolution and show PC1 in both cell lines (Figures 1A and 1B). Remarkably, GM12878 and HCT-116 show very different distributions of H3K9me3. In HCT-116 cells, large H3K9me3-rich domains correlate with prominent compartmental domains that are strongly correlated with one another in their Hi-C interaction frequencies. GM12878, however, lacks these large H3K9me3-rich domains and correspondingly lacks the prominent compartmental domains associated with them.
Figure 1. GM12878 and HCT-116 cells show different compartmental patterns.
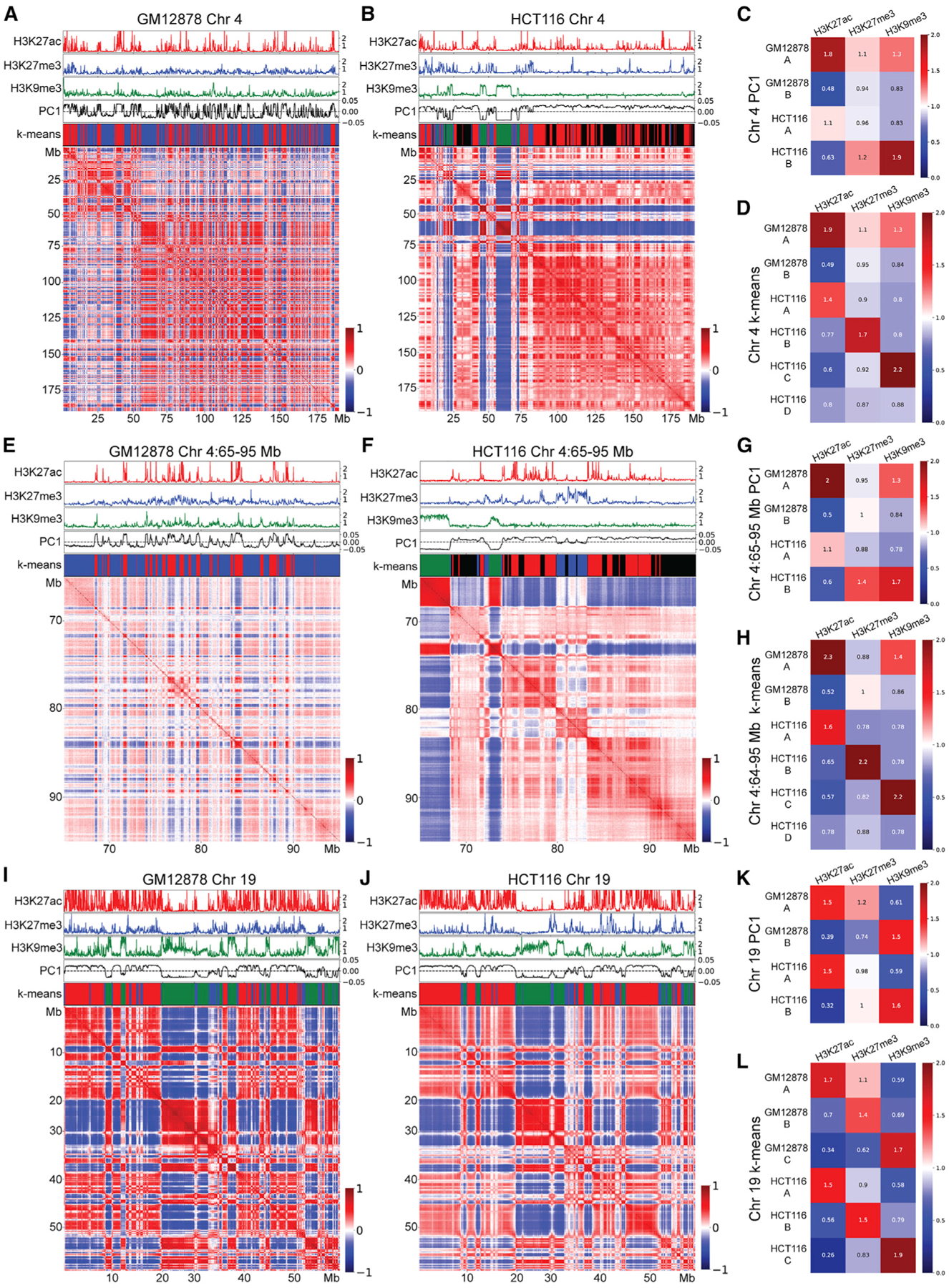
Pearson correlations of distance-normalized Hi-C interaction frequency maps corresponding to various chromosome regions in GM12878 and HCT-116 cells. On top of each Hi-C map from top to bottom: fold change over control shown for H3K27ac (red), H3K27me3 (blue), and H3K9me3 (green); PC1 component from PCA (black), and k-means cluster classifications corresponding to compartments A (red), B (blue), C (green), and D (black).
(A) Chromosome 4 from GM12878 cells.
(B) Chromosome 4 from HCT-116 cells.
(C) Fold enrichment of each histone modification within each compartment defined by PCA in chromosome 4 from GM12878 and HCT-116 cells.
(D) Fold-enrichment of each histone modification within each compartment defined by k-means clustering on chromosome 4 from GM12878 and HCT-116 cells.
(E) Region spanning 65–95 Mb of chromosome 4 from GM12878 cells.
(F) Region spanning 65–95 Mb of chromosome 4 from HCT-116 cells.
(G) Fold enrichment of each histone modification within each compartment defined by PCA on the region spanning 65–95 Mb of chromosome 4 from GM12878 and HCT-116 cells.
(H) Fold enrichment of each histone modification within each compartment defined by k-means clustering on the region spanning 65–95 Mb of chromosome 4 from GM12878 and HCT-116 cells.
(I) Chromosome 19 from GM12878 cells.
(J) Chromosome 19 from HCT-116 cells.
(K) Fold enrichment of each histone modification within each compartment defined by PCA on chromosome 19 from GM12878 and HCT-116 cells.
(L) Fold enrichment of each histone modification within each compartment defined by k-means clustering on chromosome 19 from GM12878 and HCT-116 cells.
To quantify these observations, we used PC1 to call A and B compartmental domains in GM12878 and HCT-116 cells and measured the relative enrichments of chromatin features on their chromosomes (Figure 1C). In GM12878 cells, A/B compartmentalization strongly follows transcriptional activity/inactivity, with histone modifications associated with active transcription such as H3K27ac, H3K4me3, and H3K36me3 all enriched in A compartmental domains and depleted in the B compartment of chromosome 4, while modifications associated with silenced chromatin such as H3K27me3 show an inverse pattern (Figures 1C and S1A). In HCT-116 cells, however, A compartmental domains in chromosome 4 are not strongly enriched for histone modifications associated with transcriptional activity, including GRO-seq, which is a direct measure of transcription. Instead, binary compartmental delineation using PC1 divides the chromosome into a transcriptionally inactive H3K9me3-rich portion and the remainder, which consists of both transcriptionally active and inactive regions enriched in H3K27me3. In contrast, H3K9me3 in chromosome 4 of GM12878 is enriched in A compartmental domains and presents very differently on the chromosome as sporadic peaks rather than contiguously enriched domains (Figures 1C and S1A). Given that B compartmental domains in chromosome 4 of HCT-116 cells are depleted of transcriptionally active sequences, we asked why the corresponding A compartmental domains are not strongly enriched for transcribed sequences. A simple explanation for this phenomenon is that transcriptionally inactive regions of HCT-116 not containing H3K9me3 correlate more closely in their interaction frequencies with transcriptionally active regions. Thus, the A compartment in these cells as defined by PCA is composed of a conglomeration of all H3K9me3-poor chromatin, both transcriptionally active and inactive, leading to only a mild enrichment for active histone modifications. These observations differ from canonical definitions of A compartmental domains, which are enriched for transcriptionally active features, indicating that binary PCA compartmental classifications in HCT-116 differ fundamentally from those of GM12878.
To examine more closely the mechanisms underlying the formation of compartmental domains, we focused on a 65- to 95-Mb region on chromosome 4 and used a resolution of 25 kb to call compartments using PCA (Figures 1E and 1F). In HCT-116 cells, this region contains instances of all 4 clusters found in chromosome 4. Compartmental domains present in the A compartment defined by PCA in GM12878 cells are highly enriched in H3K27ac with respect to those in the B compartment, whereas H3K27me3 and H3K9me3 are similarly enriched in both A and B compartments (Figures 1E, 1G, and S1B). However, in HCT-116 cells there is a clear enrichment of both H3K9me3 and H3K27me3 in the B compartment corresponding to compartmental domains and interactions absent in GM12878 cells (Figures 1F, 1G, and S1B). These findings, showing differential enrichment of active and repressive histone modifications in the A and B compartments in different cell lines, are surprising, since it is generally assumed that the A compartment contains transcriptionally active genes and the B compartment is enriched in silenced sequences independent of the cell type. This suggests that the canonical binary classification of A and B compartments is insufficient to represent the properties and mechanisms by which compartmental domains form in these cells.
Conserved principles underlie dynamic compartmentalization
To further explore the complex compartmentalization logic observed in GM12878 and HTC-116 cells, which appears to follow different rules in the two cell lines and cannot be explained by a simple binary division of PC1, we used the unsupervised k-means clustering algorithm to identify compartmental clusters in both cell types (Pedregosa et al., 2011). The primarily binary A/B organization of chromosome 4 in GM12878 cells obtained by PCA can be reproduced by two clusters obtained via unsupervised k-means clustering (Figures 1A and 1E). However, four clusters are required to produce a meaningful classification of chromosome 4 that correlates with the Hi-C heatmap in HCT-116 cells (Figures 1B and 1F). As expected, one of these four clusters corresponds directly to H3K9me3-rich regions, whereas a second one correlates strongly with H3K27ac. Surprisingly, the two other clusters are both transcriptionally inactive with distinct chromatin features, one highly enriched for H3K27me3 and the last lacking all three histone modifications (Figures 1D and 1H). Only H3K9me2 is enriched in this compartment (Figure S1A). Therefore, chromosome 4 in HCT-116 appears to have four distinct compartmental domains. Interactions among each type give rise to the complex plaid pattern in the Hi-C heatmap, forming compartment A (transcriptionally active), B (H3K27me3-rich), C (H3K9me3-rich), and D (enriched in H3K9me2 but depleted of standard active and silencing histone modifications). This 4-compartment nomenclature corresponding to enrichment in each of these different histone modifications is used in the rest of the article. We note that the enrichments of epigenetic features in chromosome 4 A and D clusters of HCT-116 cells correspond well to the A and B compartments of chromosome 4 in GM12878 cells, with the exception of H3K9me3 enrichment in the A compartment of GM12878 (Figures 1C and 1D).
We then sought to understand why H3K9me3 is enriched in different compartmental domains in HCT-116 versus GM12878 cells. The distribution of H3K9me3 in these two cell types is very different, with GM12878 chromosomes typically having narrow peaks of signal, whereas HCT-116 chromatin tends to have large consistently enriched plateaus. At least two possible hypotheses could explain these different distribution patterns. One possibility is that H3K9me3 regions compartmentalize differently in the two cell types due to different nuclear environments determined by cell identity and physiology. A second explanation is that H3K9me3 regions compartmentalize differently due to distinct distributions of this histone modification as a consequence of transcriptional differences between the two cell types. Analysis of chromosome 19 offers an opportunity to distinguish between these two possibilities, since this chromosome contains large domains of H3K9me3 in both GM12878 and HCT-116 cells. Chromosome 19 of GM12878 cells shows a strong correlation between the presence of H3K9me3 and the formation of strong compartmental domains (Figure 1I), as was seen on chromosome 4 of HCT-116 cells, and these H3K9me3 compartmental domains are similar in chromosome 19 of both cell types (Figure 1J). The similarity between H3K9me3 domains in chromosome 19 of GM12878 and HCT-116 cells suggests that the nuclear environment is not responsible for the differences observed in other chromosomes. Strikingly, the histone modification profiles and the A/B compartments defined by PC1 also closely match between the two cell lines (Figure 1K). k-means clustering was then performed in both cell types with three clusters, since chromosome 19 lacks large regions devoid of any signals that would fall into the D cluster. The resulting cluster calls closely match each other (Figure 1L). The high correspondence in k-means cluster definitions is mirrored by the relative signal enrichments in each cluster. In contrast to chromosome 4, the compartments called by PCA and by k-means showed similar enrichments for various histone modifications in chromosome 19 (Figures 1L and S1C). The alignment of chromosome 19 in histone modification profiles and compartmental organization in HCT-116 and GM12878 fits a model in which these histone modifications, or a chromatin feature correlated to them, drive chromosomal compartmentalization, and suggests that the underlying forces driving compartmentalization are consistent between these cell types. We suggest that the diverging compartments seen in chromosome 4 are the result of distinct chromatin profiles and that the resulting conflicting histone modification enrichments for the A and B compartments of chromosome 4 between these cell lines do not reflect differences in the underlying chemistry driving compartmentalization. The enrichment of H3K9me3 in the active compartment of GM12878 chromosome 4 can be explained as the inability of small narrow peaks of H3K9me3 to drive compartmentalization against entropic mixing. Their proximity to other active compartmentalizing features may further inhibit their self-segregation. Alternatively, since these short regions containing H3K9me3 are much smaller than the 25-kb bins used to perform the clustering analyses, it is possible that compartmental calls performed at a higher resolution may resolve H3K9me3-associated compartmentalization in these small regions. This possibility highlights the potential biases introduced by the resolution of the data used in the analyses when interpreting Hi-C information and the need for improved conformation data.
We then considered whether either of these cell lines perhaps exemplify an outlier unrepresentative of normal human tissues, and thus we investigated H3K9me3 distribution in numerous immortalized and primary human cells. We found that neither the H3K9me3 pattern observed in GM12878 and HCT-116 cells is unrepresented in other cell types or in primary tissues but rather exemplifies two ends of a continuum of H3K9me3 present across human cells (Figures S1D and S1E). These findings reveal that H3K9me3 domains are highly dynamic and give rise to significant changes in compartmentalization patterns across tissues.
Compartmental domains correlate directly with chromatin features
With the understanding that binary models are insufficient to represent human compartmentalization and that the forces responsible for the formation of compartmental domains appear to correlate closely with chromatin features, particularly transcriptional activity, H3K27me3, and H3K9me3, we sought to more precisely visualize and quantify the forces driving compartmentalization. To this end, we used chromosome 14 from HCT-116 cells as an example and sorted the Pearson correlation map by various features (Figure 2A). A similar analysis for chromosome 14 from GM12878 is shown in Figure S2A. These assemblies are produced by reordering the rows and columns of the correlation matrix so that instead of being placed in their natural order, bins are arranged by increasing signal of the chosen feature. This allows us to compare the ability of a particular sequence to form a specific type of compartmental domain with its epigenetic features. This approach also provides a means of visualizing the frequency of interactions within and among compartmental domains or “compartmentalization strength.” Sorting by PC1 values, we observe a clear segregation of chromosome 14 into three types of compartmental domains A, B, and C in HTC-116 cells (Figure 2B) and, to a lesser extent, in GM12878 cells (Figure S2B). One type of compartmental domain, called compartment C above, corresponds to negative values of PC1 and contains sequences enriched in H3K9me3 but lacking H3K27me3 and H3K27ac in HTC-116, but not in GM12878 cells due to the different distribution of H3K9me3 in these cells with respect to HTC-116. A second type of compartmental domain, compartment B, corresponds to intermediate positive PC1 values and contains sequences enriched in H3K27me3 but lacking H3K9me3 and H3K27ac. Finally, a third A compartment contains sequences with high positive PC1 values, and it is enriched in sequences with high levels of H3K27ac but lacking the other two silencing histone modifications. We note that A and C compartmental domains are highly self-correlated but strongly anticorrelated with each other in interaction frequency, while B compartmental domains self-correlate and have intermediate levels of correlation with both A and C domains (Figure 2B). B compartmental domains have a higher correlation with A than with C domains, indicating that they interact more similarly to A and would be grouped with A by a binary classification system (Figure 2B). The existence of these three compartments is supported by results from k-means clustering (see green/blue/red bar in Figures 2B, S2B, and other panels). Surprisingly, this indicates that PC1, while typically used to delineate binary compartments, could be used to call more compartmental domain types using different thresholds. Chromosome 14, like chromosome 19, largely lacks regions that would fall into the D compartment. Sorting the chromosome by H3K9me3 instead of PC1 values reproduces the compartmentalization of the H3K9me3-rich C compartment as well as PC1 itself; however, it was unable to distinguish between the A and B compartments (Figure 2C). This is not observed in GM12878 cells due to the different distribution of H3K9me3 in these cells (Figure S2C). Sorting by H3K27ac as a marker of transcriptional activity also results in compartmental segregation, although this is less precise (Figures 2D and S2D). This is likely a function of H3K27ac signal’s lacking the consistency and continuity of H3K9me3 domains within transcriptionally active regions. Sorting by H3K27me3 organized only the regions most enriched for H3K27me3 and was unable to organize the rest of the chromosome (Figures 2E and S2E). These results show that covalent histone modifications strongly correlate with and are predictive of compartmental organization.
Figure 2. Results from sorting sequences from chromosome 14 of HCT-116 cells based on the magnitude of PC1 and the levels of various histone modifications.

Pearson correlations of distance-normalized Hi-C interaction frequency map of chromosome 14 from HCT-116 cells. Shown above each Hi-C map from top to bottom are the following: fold change over control for H3K27ac (red), H3K27me3 (blue), and H3K9me3 (green); PC1 (black), and k-means cluster classifications A (red), B (blue), and C (green).
(A) Interactions among sequences from chromosome 14 arranged in natural order.
(B) Interactions among sequences from chromosome 14 sorted according to PC1 values from lowest to highest.
(C) Interactions among sequences from chromosome 14 sorted according to H3K9me3 levels from lowest to highest.
(D) Interactions among sequences from chromosome 14 sorted according to H3K27ac levels from lowest to highest.
(E) Interactions among sequences from chromosome 14 sorted according to H3K27me3 levels from lowest to highest.
An unusual feature of chromosome 14 is the existence of a trimodal distribution of H3K9me3 in HCT-116. While most chromosomes in GM12878 do not show 100-kb regions strongly enriched for H3K9me3 (Figures S3A and S3C), most chromosomes in HCT-116 cells possess a bimodal distribution of H3K9me3 signal leading to H3K9me3-rich and -poor regions (Figure S3B), while chromosome 19 shows this bimodal distribution in both cell types (Figures S3E and S3F). In HCT-116, chromosomes 13 and 14 have distinct strong and weak H3K9me3 domains (Figures 2A and S3D). Comparing their correlations in the sorting of chromosome 14 by H3K9me3 shows that weak-H3K9me3 and strong-H3K9me3 regions correlate better with regions of similar strength (arrowheads in Figure 2C). This phenomenon, along with the varying correlation strength seen in the heatmaps obtained by sorting various features, suggests that compartmentalization is more accurately thought of as a quantitative rather than a categorical feature of the genome and that the classification of chromatin into categories is a simplification that neglects to consider the effects of varying epigenetic signal strengths in the formation of compartmental domains. Sorting by a single signal does not result in perfect compartmentalization because multiple, independent forces drive the process of compartmental domain formation and long-range interactions among domains of the same type. The most significant of these forces may be those involved in interactions between sequences containing H3K9me3 and proteins and histone modifications associated with active transcription. These observations suggest a model of compartmentalization in which the genome is organized by multiple independent forces directly correlated with chromatin features that attract and repel one another.
Independent contributions of chromatin features can reproduce compartmental organization
We next sought to test the hypothesis that formation of compartmental domains and establishment of long-range interactions among domains of the same type is driven by the independent contributions of chromatin features. To approach this question, we created a regression-based machine learning model to reproduce Hi-C interaction maps using epigenetic features. This new simulation represents both a methodological and theoretical advance over our previous work (Rowley et al., 2017), which was optimized by manual adjustment, whereas the parameters of the current simulation are learned algorithmically. The results described above suggest that the presence of H3K27ac as an indicator of transcriptionally active regions, H3K27me3 and H3K9me3 as indicators of different types of silenced sequences, or the absence of these three modifications can account for all of the compartmental domains we observe in chromosomes of human cells. Other histone modifications or chromatin signals correlated with these compartmental domains can be used in place of these three particular chromatin features; however, these features correlated best with their respective compartments in our analysis (Figures S1A–S1C). To enable comparison across cell types and experiments, we binned these three epigenetic signals into quantiles at 100-kb resolution. For each normalized signal, an algorithm then learned an attraction-repulsion relationship for each pair of quantiles using a maximum likelihood estimation approach. This attraction-repulsion mapping effectively represents the average enrichment or depletion between all bins with the corresponding level of each chromatin feature. The estimated contact frequency in the simulated map is then derived by the simple addition of the estimated effect of each signal and multiplied by a distance-dependent constant representing the average interaction frequency at each genomic distance (Figure 3A). This relatively straightforward model, which is only capable of representing the independent attractive and repulsive forces of each chromatin feature, tests to what extent this framework is capable of recapitulating the 3D organization of the genome represented by the compartmental domains and their interactions.
Figure 3. Histone modifications can predict compartmentalization using learned attraction-repulsion relationships.
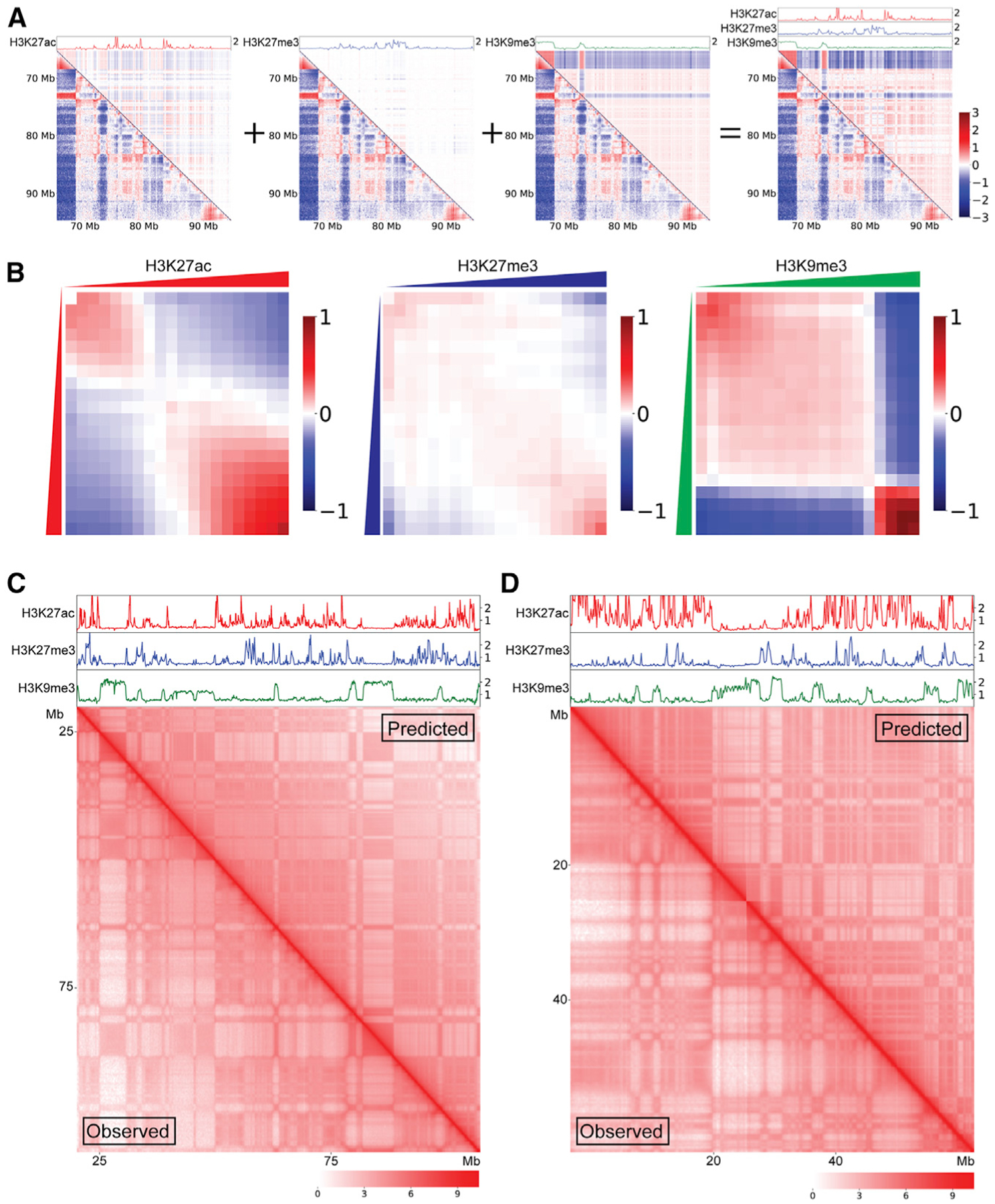
(A) Log(observed/expected) of Hi-C interaction maps showing the 65- to 95-Mb region of chromosome 4 from HCT-116 cells. Bottom left triangles are observed Hi-C interaction maps, while upper right triangles are simulations using only the components shown above as tracks. From left to right H3K27ac (red), H3K27me3 (blue), H3K9me3 (green), and all 3 combined.
(B) Average of attraction-repulsion relationship maps learned by maximum likelihood estimation from every chromosome of HCT-116 cells.
(C) Comparison of HCT-116 chromosome 8 logged Hi-C interaction maps. The bottom left below the diagonal corresponds to observed interactions and the upper right above the diagonal represents simulated contacts.
(D) Comparison of HCT-116 chromosome 19 logged Hi-C interaction maps. The bottom left below the diagonal corresponds to observed interactions and the upper right above the diagonal represents simulated contacts.
The averaged attraction-repulsion relationships learned from every chromosome of HCT-116 cells are shown in Figure 3B. The learned relationship between the levels of a specific histone modification and the interaction frequency of the corresponding sequence is similar to that observed experimentally described above (Figures 2C–2E). Genomic regions containing high levels of a given histone modification show increased interactions with other regions high in the same modification. The model learns and predicts that pairs of regions in which one is high in such a signal and the other is low will not be attracted and have reduced interaction frequency. The minimal model using three histone modifications, H3K27ac, H3K27me3, and H3K9me3, is able to recapitulate most aspects of 3D genome organization while remaining easy to interpret. All three signals show a degree of attraction between the highest quantile bins, as seen by the enrichment in the bottom right of the attraction-repulsion maps, as well as repulsion between the highest and lowest quantiles as seen by the depletion in the upper right and bottom left corners (Figure 3B). Importantly, the strength and nature of these maps differ significantly, indicating that the forces driving compartmentalization vary for each feature. H3K9me3 maps show a strong attraction among the most enriched quarter of the genome, which strongly repels the rest of the genome equally. This shows the existence of a single critical threshold of H3K9me3 density and quantity and reflects the generally bimodal distribution of this modification in HCT-116 cells. H3K27ac, however, shows the greatest attraction among the highest quantiles with a more gradual reduction in attraction with reduced signal. H3K27me3 primarily shows only attraction between the highest quantiles of signal and otherwise contributes little to the organization of the genome. When interpreting this information, it is important to consider that the distributions of these three histone modifications are interrelated, and that regions of the genome lacking one of the modifications may contain one of the other two.
Simulations were generated at 100-kb resolution using the average of the attraction-repulsion maps learned from every chromosome except the one being simulated. A comparison of observed and simulated maps reveals close agreement on the majority of large compartmental features (Figures 3C, 3D, S4A, and S4B). We quantified the accuracy of the model using the Pearson correlations between the observed and simulated maps after dividing by the average distance. Due to the power-law decay of interaction frequency with respect to distance in Hi-C maps, any simulation that accurately reproduces this decay will have a high correlation. As this would not represent the ability of the model to reproduce compartmental organization, we normalized for distance to eliminate the natural correlation driven by the accurate representation of the distance decay. Simulations of chromosomes using the average maps derived from all other chromosomes varied in correlation by chromosome, but generally performed well with correlations in the range of 0.5–0.7 (Figure S4C). Using this same methodology to compare biological replicates of a Hi-C experiment resulted in similar ranges of correlation scores across the chromosomes (Figure S4D). The fact that Hi-C maps can be predicted by only modeling the attraction and repulsion of chromatin features against themselves suggests a direct role of these features, or a chromatin component correlated with these features, in compartmentalizing the nucleus.
Conserved forces give rise to diverse genomic organizations
Given the accuracy of our model in reproducing the 3D organization of HCT-116 cells, we then applied the same model to GM12878 cells. These cells have different distribution of H3K9me3, and their 3D genome organization as seen in PCA enrichment analysis is also different (Figure 1C). We found that with the exception of H3K9me3, the average of the attraction-repulsion maps learned across all of the chromosomes in GM12878 were strikingly similar to those learned in HCT-116 (Figure 4A). The incongruence of H3K9me3 attraction-repulsion maps between these cell types was expected as the distribution of H3K9me3 is very different between most of their chromosomes. We simulated the Hi-C interaction maps of GM12878 chromosomes using the average of the attraction-repulsion maps learned from all of the other chromosomes (Figures 4B, S5A, and S5B). However, simulating chromosome 19 in GM12878, which is unique compared to other chromosomes in these cells due to its large H3K9me3 domains, was less successful when using the attraction-repulsion maps learned from the rest of the chromosomes, with the simulation failing to capture the role of H3K9me3 in compartmentalization (Figure 4C). Pearson correlations between the real and simulated maps were significantly lower in GM12878 cells than in HCT-116, indicating that the simulation was less successful at reproducing the Hi-C maps in GM12878, most likely due to the absence of the strong organizing feature of H3K9me3 (Figure S5C). Nevertheless, the similarity between the attraction-repulsion maps of these two cell types suggests that the fundamental forces underlying compartmentalization are largely conserved between these cells. If this is true, then the attraction-repulsion maps learned in one cell type should be able to accurately model the organization of another cell type, showing that dynamic compartmentalization is largely a consequence of diverging distributions of chromatin features.
Figure 4. Attraction-repulsion relationships are consistent across cell types.

(A) Average of attraction-repulsion relationship maps learned by maximum likelihood estimation from every chromosome of GM12878 cells.
(B) Comparison of GM12878 chromosome 8-logged Hi-C interaction maps. The bottom left below the diagonal corresponds to observed interactions and the upper right above the diagonal represents simulated contacts using attraction-repulsion maps learned from GM12878.
(C) Comparison of GM12878 chromosome 19-logged Hi-C interaction maps. The bottom left below the diagonal corresponds to observed interactions and the upper right above the diagonal represents simulated contacts using attraction-repulsion maps learned from GM12878.
(D) Comparison of GM12878 chromosome 8-logged Hi-C interaction maps. The bottom left below the diagonal corresponds to observed interactions and the upper right above the diagonal represents simulated contacts using attraction-repulsion maps learned from HCT-116.
(E) Comparison of GM12878 chromosome 19-logged Hi-C interaction maps. The bottom left below the diagonal corresponds to observed interactions and the upper right above the diagonal represents simulated contacts using attraction-repulsion maps learned from HCT-116.
To test this idea, we used the attraction-repulsion maps learned from HCT-116 chromosomes to simulate the 3D organization of GM12878 chromatin and found a high correspondence between the observed and simulated maps (Figures 4D and S5D–S5F). This is remarkable, given that the Hi-C interaction maps of most chromosomes between these cell types are very different. This ability of compartmental forces learned from one cell type to successfully predict compartmentalization in another indicates that the same underlying forces directly correlated with chromatin features are largely conserved between these two cell types. This is further support that the radical differences between the respective Hi-C maps of these cell lines are a consequence of different chromatin features, primarily the presence and absence of large H3K9me3-rich domains.
As the simulation of chromosome 19 of GM12878 cells using attraction-repulsion maps learned with the rest of the chromosomes is poor, we asked whether chromosome 19 would be better simulated by the attraction-repulsion relationships learned from HCT-116 than from GM12878. The histone modification profiles of chromosome 19 in GM12878 cells, particularly the presence of large H3K9me3 domains, more closely resemble the patterns seen in most chromosomes of HCT-116 cells (Figure 1G). The simulation of chromosome 19 is significantly more accurate using HCT-116 attraction-repulsion maps (Figure 4E; compare with Figure 4C). In agreement, the Pearson correlations between the observed and simulated maps of chromosome 19 are higher when simulated with HCT-116 (Figure S5F). These results again support the idea that the 3D organization of chromosomes is a consequence of the distribution patterns of 1D epigenetic information.
Transcriptional activity and H3K9me3 also compartmentalize the Drosophila genome
Given the ability of our model to reproduce the 3D organization of multiple human cell types from a single universal set of attraction-repulsion maps, we sought to determine the applicability of this model outside of humans. Our previous foundational work showed that transcriptional activity was predictive of compartmental organization in multiple representative eukaryotic genomes (Rowley et al., 2017). We hypothesized that, as in humans, the same forces responsible for compartmental domain formation and the establishment of distinct nuclear compartments via interactions among compartmental domains with the same epigenetic features may be at work in other organisms. Thus, we applied our model to explore whether the distribution of H3K27ac, H3K27me3, and H3K9me3 could reproduce the 3D organization of the Drosophila genome.
Hi-C maps from Drosophila Kc cells using datasets with ~1 billion reads were simulated using the attraction-repulsion model. Due to the higher read count and smaller genome size, we were able to simulate the genome at 10-kb resolution. A small modification was made to the model to limit the simulation to <2 Mb, as Drosophila compartmental domains are smaller than those of mammals and long-range interactions among domains decay rapidly beyond this distance. While distinct, the learned attraction-repulsion maps resemble those learned in human cells (Figure 5A). The model learns attraction between features with similar histone modifications and repulsion between genomic regions with dissimilar ones.
Figure 5. Attraction-repulsion relationships explain compartmentalization in Drosophila.

(A) Average of attraction-repulsion relationship maps learned by maximum likelihood estimation from chromosomes 2 and 3 of Kc167 cells.
(B) Comparison of Kc167 chromosome 2-logged Hi-C interaction maps. The bottom left below the diagonal corresponds to observed interactions and the upper right above the diagonal represents simulated contacts using attraction-repulsion maps learned from chromosome 3 of Kc167 cells.
(C) Comparison of Kc167 chromosome 3-logged Hi-C interaction maps. The bottom left below the diagonal corresponds to observed interactions and the upper right above the diagonal represents simulated contacts using attraction-repulsion maps learned from chromosome 2 of Kc167 cells.
As Drosophila only has two autosomes of significant size, we simulated each chromosome with the attraction-repulsion maps of the other. The resulting simulations largely reproduce the compartmental patterning of these chromosomes (Figures 5B, 5C, and S6A). Moreover, high-resolution maps indicate that the simulation reproduces short-range interactions within compartmental domains. This indicates that, as in humans, these chromatin features or others strongly correlated with them drive the formation of compartmental domains and their interactions. These observations suggest a universal model of compartmentalization in which the fundamental underlying forces driving this process are conserved between organisms.
DISCUSSION
Interphase chromosomes of vertebrates are generally thought to be organized into large domains whose sequences can be in one of two states, active or inactive (Lieberman-Aiden et al., 2009). Within these large domains are smaller TADs (Dixon et al., 2012), some of which are flanked by CTCF sites in convergent orientation, and therefore correspond to CTCF loops formed by cohesin extrusion, whereas others lack CTCF at their boundaries and are thus formed by different, unknown mechanisms (Rao et al., 2014). Smaller domains called sub-TADs can also be observed within TADs (Phillips-Cremins et al., 2013). Results reported here address the question of what the unit of eukaryotic chromosome organization is and how we can explain its formation using known biochemical and biophysical forces operating in the nucleus. Answers to this question provided by chromosome conformation studies should account for results obtained using microscopy or biochemical approaches. However, results from Hi-C studies seem to contradict well-established concepts in nuclear biology. Non-transcribed regions of the genome do not simply interact with one another, as one would conclude from the checkerboard pattern observed in Hi-C heatmaps representing long-range interactions among sequences located in the B compartment. Rather, non-transcribed regions in the genome contain either H3K27me3/PCR1/PCR2 or H3K9me3/HP1, or lack either of these histone modifications. Immunofluorescence localization experiments show that Pc-containing regions interact with one another to form Pc bodies, both in Drosophila and mammals (Bantignies et al., 2011; Lanzuolo et al., 2007; Li et al., 2011; Tolhuis et al., 2011). The same is true for regions containing H3K9me3 and HP1 that form chromocenters (Huang et al., 2010; Spierer et al., 2005) and actively transcribed regions to form transcription factories (Jackson et al., 1993; Mitchell and Fraser, 2008). Furthermore, recent results indicate that multivalent proteins present in these three types of genomic regions are able to form biomolecular condensates by liquid-liquid phase separation (Boehning et al., 2018; Larson et al., 2017; Strom et al., 2017; Tatavosian et al., 2019). Therefore, compartmental domains and their interactions detected by Hi-C must be more complex than is generally assumed and this complexity must reflect existing observations from microscopy and biochemistry.
The results presented here reveal an underappreciated diversity of compartmental domains, the conserved forces underlying their establishment, and the long-range interactions responsible for the formation of nuclear compartments. Multiple independent forces organize the genome into compartments. Transcriptional activity and H3K9me3 correlate with the strongest of these forces. H3K27me3, while weaker, also correlates directly with compartmental patterns. Finally, sequences lacking any of these histone modifications and enriched in H3K9me2 represent a fourth class of sequences that form their own independent compartment. The powerful nature of these chromatin features across cell types allows for dynamic compartmentalization during cell differentiation. While both transcriptional activity and H3K9me3 have been previously reported as highly correlated with the formation of nuclear compartments, here we show that each of the biochemical forces correlated with the presence of these histone modifications drives compartmentalization independently and can do so in the absence of the other (Falk et al., 2019; Lieberman-Aiden et al., 2009). Previous in-depth Hi-C analyses of GM12878 cells suggested a unique H3K9me3-correlated compartmentalization of chromosome 19 and categorized it as a subcompartment of inactive B chromatin (Rao et al., 2014). Here we show that this compartment, while unique in GM12878, is widespread in other cell lines and is the strongest compartmentalizing force wherever large domains of H3K9me3 are found. On chromosomes where large H3K9me3-rich domains exist, the segregation between these domains and the rest of the H3K9me3-poor chromosome represent the strongest feature of the Hi-C maps. As such, using PCA to delineate binary compartments on the Pearson correlation maps divides the genome into H3K9me3 rich and poor, rather than along the expected lines of transcriptional activity. The inclusion of H3K9me3-poor transcriptionally inactive regions into the A compartment defined by a binary segregation of PC1 in these cells leads to incompatible and confusing definitions of genomic compartments between cell lines and tissues.
While PCA is a powerful tool to investigate compartments, the PC1-based canonical binary classification of compartments is inadequate to represent the true compartmentalization of the genome. We suggest shifting away from naive unsupervised classification techniques in single cell lines to a categorization informed by the breadth of organizational diversity seen across human samples, which will improve the accuracy and generalizability of future studies. Our findings fit a model of compartmentalization where the attraction of similar chromatin states drives interactions among them to the exclusion of other chromatin types. The dependence of H3K9me3 compartmentalization in GM12878 cells on the levels of this modification, in which small, discrete peaks fail to strongly compartmentalize through most of the genome while large domains seen on chromosome 19 do, suggests the existence of a compartmentalization threshold. The forces driving compartmentalization must overcome the entropy of mixing in the nucleus and, therefore, some minimum quantity of compartmentalizing signal must exist below which the attractive forces at work are insufficient to overcome entropy. We propose that the distinctive patterns of H3K9me3 in GM12878 cells reflect this threshold where the discrete chromatin immunoprecipitation sequencing (ChIP-seq) peaks seen in most of the genome fail to strongly compartmentalize, while in the same nuclear environment, the larger domains present in chromosome 19 do. As further evidence of the quantity-dependent nature of compartmentalization, several chromosomes in HCT-116 cells possess both weak and strong H3K9me3 domains, which correspondingly compartmentalize weakly and strongly. These findings suggest that compartmentalization is more accurately represented as a continuum, in which each sequence is driven to interact according to the strength, corresponding to the quantity, of chromatin forces driving it, rather than discrete chromatin types (Imakaev et al., 2012).
Our model provides insight into the fundamental mechanisms of genome 3D organization. That the compartmentalization of the genome can be predicted using only the distribution of a few histone modifications strongly implies that these chromatin features are either directly or indirectly responsible for the spatial separation of chromatin types in the genome. In our previous prediction algorithm, we used GRO-seq signal alone to perform simulations and predict 3D chromatin organization from 1D epigenetic information in Drosophila and human GM12878 cells. However, the existence of transcriptionally inactive regions with extremely divergent compartmental interaction patterns in HCT-116 cells could not be captured by GRO-seq alone, while H3K9me3 and H3K27me3, respectively, could explain this phenomenon. A GRO-seq-based approach performs reasonably well in reproducing the Hi-C maps of GM12878, but mischaracterizes inactive regions in HCT-116. Our finding that the patterns of attraction and repulsion are largely consistent between cell types with divergent compartments shows that these underlying forces behave consistently and that the fundamental forces shaping chromatin organization are steady between cells. These attraction-repulsion mappings do not provide direct insight into the physical nature of these forces as either attractive or repulsive because in our model any attraction between genomic regions is also a repulsion against all other regions and vice versa. The ability of the same model to reproduce the organization of Drosophila chromosomes suggests that the attraction and repulsion of chromatin by the independent contributions of compartmentalizing forces may be a universal driver of compartmentalization across Animalia. The widespread use of canonical A/B compartmentalization is largely a product of the pioneering work done in GM12878, but it does not reflect the epigenetic diversity of human cell types. Informing future analyses of compartments with this understanding and approaching compartmental organization from the perspective of chromatin state-driven attraction and repulsion will allow for reproducible and comparable definitions of compartments across human tissues and beyond to other organisms.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Victor G Corces (vgcorces@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The code generated during this study is hosted on GitHub: https://github.com/5centmike/CIFSIM/.
This study did not generate new unique datasets.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
No model organisms or subjects were used for the study. All data was obtained from other sources as described in the Key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| GM12878 Gro-seq | Core et al., 2014 | GSE60454 |
| GM12878 H3K27ac ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF180LKW |
| GM12878 H3K27me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF594HSG |
| GM12878 H3K36me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF662QFK |
| GM12878 H3K4me1 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF682WPF |
| GM12878 H3K4me2 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF828CQV |
| GM12878 H3K4me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF674QZB |
| GM12878 H3K4me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF818GNV |
| GM12878 H3K79me2 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF396JIR |
| GM12878 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF776OVW |
| GM12878 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF846MPX |
| GM12878 H4K20me1 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF831WYD |
| HCT-116 GRO-seq | Andrysik et al., 2017 | GSE86165 |
| IMR90 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF272ZUB |
| Kc167 H3K27ac ChIP-seq | Yang et al., 2013 | GSE36374 |
| Kc167 H3K27me3 ChIP-seq | Rowley et al., 2017 | GSE89244 |
| HCT-116 H3K27ac ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF225QAB |
| HCT-116 H3K27me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF030SYQ |
| HCT-116 H3K36me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF062CBC |
| HCT-116 H3K4me2 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF563OKQ |
| HCT-116 H3K4me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF057GFU |
| HCT-116 H3K4me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF859AQD |
| HCT-116 H3K79me2 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF127XQD |
| HCT-116 H3K9me2 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF854QMM |
| HCT-116 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF402WZH |
| HCT-116 H4K20me1 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF355TAA |
| HCT-116 H3K4me1 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF285DIL |
| IMR90 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF272ZUB |
| K562 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF834YLI |
| Kc167 H3K9me3 ChIP-seq | Rowley et al., 2017 | GSE89244 |
| Primary B cell H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF740QXN |
| H9 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF108MOZ |
| HCT-116 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF402WZH |
| Hepatocyte H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF354EPR |
| HepG2 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF881LOL |
| IMR90 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF272ZUB |
| K562 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF834YLI |
| Keratinocyte H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF242SAO |
| Liver H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF552IQT |
| Neutrophil H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF202TBY |
| Osteoblast H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF598YQV |
| PBMC1 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF319EBK |
| Stomach5 H3K9me3 ChIP-seq | ENCODE Project Consortium, 2012 | ENCFF995YEN |
| GM12878 Hi-C | Rao et al., 2014 | GSE63525 |
| HCT-116 Hi-C | Rao et al., 2017 | GSE104333 |
| Kc167 Hi-C | Cubeñas-Potts et al., 2017 | GSE80702 |
| Software and algorithms | ||
| seaborn | Seaborn, 2020 | https://seaborn.pydata.org/ |
| scikit-learn | Pedregosa et al., 2011 | https://scikit-learn.org/ |
| numpy | Oliphant, 2006 | https://numpy.org |
| scipy | Virtanen et al., 2020 | https://www.scipy.org/ |
| pandas | McKinney, 2010 | https://pandas.pydata.org |
| matplotlib | Hunter, 2007 | https://matplotlib.org |
| Juicebox | Durand et al., 2016 | https://www.aidenlab.org/juicebox/ |
| CIFSIM | This paper | https://github.com/5centmike/CIFSIM |
METHOD DETAILS
ChIP-seq quantile normalization
Two replicates of each ChIP-seq data described in the Key resources table were combined in bigwig format and the fold-change over control was determined. 100 kb bins with no reads mapped in any ChIP-seq were excluded from analysis. The remaining bins were normalized into twenty discrete quantiles according to their signal compared genome-wide.
Hi-C quality control
Hi-C maps were generated from data obtained from Rao et al. (2014, 2017) using reads with quality score > Q30. Genomic bins were removed from the maps and all subsequent analyses according to several criteria calculated for each chromosome as follows. Bins removed from ChIP-seq quantiles were also removed from the Hi-C. Bins with a total read sum greater than 3 standard deviations above or less than 3 standard deviations below the average bin read sum were dropped. Bins with non-zero interactions with bins with non-zero interactions greater than 3 standard deviations above or less than 3 standard deviations below the average bin were dropped.
Hi-C normalization and Pearson correlation
Hi-C maps were balanced using Knight-Ruiz normalization. For some analyses such as Pearson correlation, the Hi-C maps were distance-normalized by dividing each interaction by the average of all interactions at that distance. This produces an observed/expected value for all interaction bins. Pearson correlations of Hi-C maps were then generated from these distance normalized matrices.
Principal component analysis and compartment calls of Hi-C data
Principal Component Analysis was performed on the chromosome Pearson correlation maps. The first principal component (PC1) is defined as the eigenvector with the largest eigenvalue. All bins with positive values in PC1 were assigned to one compartment while all negative values were assigned to the other. The compartment with the largest enrichment for Gro-seq signal was defined as the A compartment and the other one as B.
k-means clustering of Hi-C data
To dissect the complex compartmentalization of the genome based on Hi-C information, we employed the unsupervised k-means clustering algorithm to identify clusters in GM12878 and HCT-116 cells. We used MiniBatchKMeans from scikit-learn’s machine learning library with varying numbers for k depending on the features of the given chromosome. Clustering was performed on the Pearson correlation maps of each chromosome separately and clusters were identified as A, B, C, or D by their enrichments for H3K27ac, H3K27me3, H3K9me3, and H3K9me2, respectively.
Compartmentalization by independent forces to simulate interaction maps
To enable comparison across cell types and experiments, we first binned all epigenetic signals into quantiles at 100 kb resolution. For each normalized signal, the model learned, using a Maximum Likelihood Estimation approach, an attraction-repulsion relationship for each pair of quantiles. This attraction-repulsion mapping effectively represents the average enrichment or depletion between all bins with the corresponding level of signal. The model then predicts the number of reads at each bin by summing the attraction-repulsion scores for each signal and multiplying by a constant distance factor to account for the power law decay of genomic interactions.
We model the compartmentalization of the genome as the independent contributions of individual 1D epigenetic signals and use a machine learning method, Maximum Likelihood Estimation, to learn the relationship between the signals and interaction frequencies in the Hi-C map. In this way we hope to quantify the nuclear forces driving compartmentalization. Our model treats each epigenetic signal as an independent but additive effect on Hi-C interaction frequency according to the equation:
where the expected interaction frequency E between any two genomic loci i and j is the sum of the weight effect of each signal. Using Maximum Likelihood Estimation we find the optimal values of the weights of each signal such that the expected result E is as close to the observed value as possible. This produces a trained model that can reproduce the 3D organization of the genome from 1D epigenetic signals by quantifying the expected contributions of each correlated compartmentalizing force.
We use the Maximum Likelihood Estimation approach to optimize the values of a vector β where each entry in β corresponds to an entry in the attraction-repulsion maps such that for all possible pairs of quantiles for each of the three chromatin signals H3K27ac, H3K27me3, and H3K9me3, there is a corresponding weight in β. We then construct a sparse matrix X where each row corresponds to an interaction bin in the flattened Hi-C matrix and each column a weight in β. Each row in X is zeros except in the 3 columns corresponding to the entries in β that describe the pair of signal quantiles of genomic bins. If we take an observed Hi-C map which has been normalized for distance by dividing by the expected value at each distance, which we term y, the approximation of the model of the normalized observed map is then:
Treating the normalized interaction frequency as a normal distribution we derive a likelihood function the log of which we will maximize to optimize the weights of β in a Maximum Likelihood Estimation approach:
where N is the total number of unique interacting bins in the linearized Hi-C matrix excluding interactions of each genomic bin with itself. To optimize the parameters of β we iteratively solve starting with initial values of 0 for all weights in β. We use the Newton-Raphson method to update the weights by gradient descent. With each iteration we account for one final feature of the Hi-C matrix. Due to the colocalization of compartmentalizing features along the chromosome, the frequency of intra-compartmental interactions is enriched at short ranges. Two genomic bins within one Mb are more likely to be enriched for compartmental interactions than two bins many Mb apart. This bias leads to aberrant distance normalization and, thus the distance normalization is updated after each iteration to account for the average compartmental enrichment the model predicts at each distance. The distance normalization is divided by the average enrichment so that it more accurately reflects the true effect of distance on interaction frequency.
Simulation of Drosophila Hi-C data
Drosophila Hi-C data was obtained from Cubeñas-Potts et al. (2017). The simulation of Drosophila Hi-C data works identically to the simulation described above for human data except for a limit on the maximum size of bins that are considered for the analysis. We excluded all interaction bins further than 2 Mb apart as the compartmental signal beyond this distance is substantially weaker.
QUANTIFICATION AND STATISTICAL ANALYSIS
Simulations were generated at 100 kb resolution using the average of the attraction-repulsion maps learned from every chromosome except the one being simulated. We quantified the accuracy of the model using the Pearson correlations between the observed and simulated maps. Due to the power law decay of interaction frequency with respect to distance in Hi-C maps, any simulation that accurately reproduces this decay will have an extremely high correlation. As this would not represent the accuracy with which the model reproduces the compartmental organization, we normalized for distance by performing a Z-norm of each diagonal in the matrix to eliminate the natural correlation driven by the accurate representation of the distance decay. We report both the Pearson correlation coefficient and the Z-normed Pearson correlation coefficient between the simulations and the observed maps in the supplemental figures associated with each simulation.
Supplementary Material
Highlights.
Human cells have at least three distinct compartments: one active and two inactive
Compartments correlate with the presence of H3K27ac, H3K27me3, or H3K9me3
Compartmentalization principles differ between chromosomes of the same cell
Compartments are predictable from one-dimensional epigenetic information
ACKNOWLEDGMENTS
We would like to thank Anthony Christodoulou and Andrea Trevino for their insights into machine learning. This work was supported by US Public Health Service Award R35 GM139408 from the National Institutes of Health. M.H.N. was supported by NIH T32 GM008490. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109330.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Andrysik Z, Galbraith MD, Guarnieri AL, Zaccara S, Sullivan KD, Pandey A, MacBeth M, Inga A, and Espinosa JM (2017). Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 27, 1645–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziatella C, Chiariello AM, Esposito A, Bianco S, Fiorillo L, and Nicodemi M (2018). Molecular dynamics simulations of the Strings and Binders Switch model of chromatin. Methods 142, 81–88. [DOI] [PubMed] [Google Scholar]
- Banani SF, Lee HO, Hyman AA, and Rosen MK (2017). Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, Tixier V, Mas A, and Cavalli G (2011). Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell 144, 214–226. [DOI] [PubMed] [Google Scholar]
- Boehning M, Dugast-Darzacq C, Rankovic M, Hansen AS, Yu T, Marie-Nelly H, McSwiggen DT, Kokic G, Dailey GM, Cramer P, et al. (2018). RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol 25, 833–840. [DOI] [PubMed] [Google Scholar]
- Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckle A, Brackley CA, Boyle S, Marenduzzo D, and Gilbert N (2018). Polymer Simulations of Heteromorphic Chromatin Predict the 3D Folding of Complex Genomic Loci. Mol. Cell 72, 786–797.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang M, Michieletto D, Brackley CA, Rattanavirotkul N, Mohammed H, Marenduzzo D, and Chandra T (2019). Polymer Modeling Predicts Chromosome Reorganization in Senescence. Cell Rep. 28, 3212–3223.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PR, and Marenduzzo D (2018). Transcription-driven genome organization: a model for chromosome structure and the regulation of gene expression tested through simulations. Nucleic Acids Res. 46, 9895–9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, and Lis JT (2014). Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet 46, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubeñas-Potts C, Rowley MJ, Lyu X, Li G, Lei EP, and Corces VG (2017). Different enhancer classes in Drosophila bind distinct architectural proteins and mediate unique chromatin interactions and 3D architecture. Nucleic Acids Res. 45, 1714–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016). Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M, Feodorova Y, Naumova N, Imakaev M, Lajoie BR, Leonhardt H, Joffe B, Dekker J, Fudenberg G, Solovei I, and Mirny LA (2019). Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Yu Z, Zhang S, Liang X, Chen J, Li C, Ma J, and Jiao R (2010). Drosophila CAF-1 regulates HP1-mediated epigenetic silencing and pericentric heterochromatin stability. J. Cell Sci 123, 2853–2861. [DOI] [PubMed] [Google Scholar]
- Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng 9, 90–95. [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, and Mirny LA (2012). Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Hassan AB, Errington RJ, and Cook PR (1993). Visualization of focal sites of transcription within human nuclei. EMBO J. 12, 1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladouceur A-M, Parmar BS, Biedzinski S, Wall J, Tope SG, Cohn D, Kim A, Soubry N, Reyes-Lamothe R, and Weber SC (2020). Clusters of bacterial RNA polymerase are biomolecular condensates that assemble through liquid-liquid phase separation. Proc. Natl. Acad. Sci. USA 117, 18540–18549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzuolo C, Roure V, Dekker J, Bantignies F, and Orlando V (2007). Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat. Cell Biol 9, 1167–1174. [DOI] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1a suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H-B, Müller M, Bahechar IA, Kyrchanova O, Ohno K, Georgiev P, and Pirrotta V (2011). Insulators, not Polycomb response elements, are required for long-range interactions between Polycomb targets in Drosophila melanogaster. Mol. Cell. Biol 31, 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Izaguire-Sierra M, Praveen K, and Rajendra TK (2009). Nuclear bodies: random aggregates of sticky proteins or crucibles of macromolecular assembly? Dev. Cell 17, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney W (2010). Data Structures for Statistical Computing in Python. https://conference.scipy.org/proceedings/scipy2010/mckinney.html.
- Mitchell JA, and Fraser P (2008). Transcription factories are nuclear subcompartments that remain in the absence of transcription. Genes Dev. 22, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton A-L, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, and Bruneau BG (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuebler J, Fudenberg G, Imakaev M, Abdennur N, and Mirny LA (2018). Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 115, E6697–E6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant TE (2006). A Guide to NumPy (Trelgol Publishing; ). [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong C-T, Hookway TA, Guo C, Sun Y, et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta V, and Li H-B (2012). A view of nuclear Polycomb bodies. Curr. Opin. Genet. Dev 22, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plys AJ, Davis CP, Kim J, Rizki G, Keenen MM, Marr SK, and Kingston RE (2019). Phase separation of Polycomb-repressive complex 1 is governed by a charged disordered region of CBX2. Genes Dev. 33, 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, and Zhang B (2019). Predicting three-dimensional genome organization with chromatin states. PLoS Comput. Biol 15, e1007024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat. Rev. Genet 19, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, and Corces VG (2017). Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol. Cell 67, 837–852.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaborn (2020). Seaborn: statistical data visualization. https://seaborn.pydata.org/.
- Spierer A, Seum C, Delattre M, and Spierer P (2005). Loss of the modifiers of variegation Su(var)3–7 or HP1 impacts male X polytene chromosome morphology and dosage compensation. J. Cell Sci 118, 5047–5057. [DOI] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavosian R, Kent S, Brown K, Yao T, Duc HN, Huynh TN, Zhen CY, Ma B, Wang H, and Ren X (2019). Nuclear condensates of the Polycomb protein chromobox 2 (CBX2) assemble through phase separation. J. Biol. Chem 294, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis B, Blom M, Kerkhoven RM, Pagie L, Teunissen H, Nieuwland M, Simonis M, de Laat W, van Lohuizen M, and van Steensel B (2011). Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet. 7, e1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. ; SciPy 1.0 Contributors (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gao Y, Zheng X, Liu C, Dong S, Li R, Zhang G, Wei Y, Qu H, Li Y, et al. (2019). Histone Modifications Regulate Chromatin Compartmentalization by Contributing to a Phase Separation Mechanism. Mol. Cell 76, 646–659.e6. [DOI] [PubMed] [Google Scholar]
- Yang J, Sung E, Donlin-Asp PG, and Corces VG (2013). A subset of Drosophila Myc sites remain associated with mitotic chromosomes colocalized with insulator proteins. Nat. Commun 4, 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code generated during this study is hosted on GitHub: https://github.com/5centmike/CIFSIM/.
This study did not generate new unique datasets.