

Abstract
Single cell RNA-seq (scRNA-seq) is emerging as a powerful technology to examine transcriptomes of individual cells. We determined whether scRNA-seq could be used to detect the effect of environmental and pharmacologic perturbations on osteoblasts. We began with a commonly used in vitro system in which freshly isolated neonatal mouse calvarial cells are expanded and induced to produce a mineralized matrix. We used scRNA-seq to compare the relative cell type abundances and the transcriptomes of freshly isolated cells to those that had been cultured for 12 days in vitro. We observed that the percentage of macrophage-like cells increased from 6% in freshly isolated calvarial cells to 34% in cultured cells. We also found that Bglap transcripts were abundant in freshly isolated osteoblasts but nearly undetectable in the cultured calvarial cells. Thus, scRNA-seq revealed significant differences between heterogeneity of cells in vivo and in vitro. We next performed scRNA-seq on freshly recovered long bone endocortical cells from mice that received either vehicle or Sclerostin-neutralizing antibody for 1 week. We were unable to detect significant changes in bone anabolism-associated transcripts in immature and mature osteoblasts recovered from Sclerostin-neutralizing antibody treated mice; this might be a consequence of being underpowered to detect modest changes in gene expression, since only 7% of the sequenced endocortical cells were osteoblasts and a limited portion of their transcriptomes were sampled. We conclude that scRNA-seq can detect changes in cell abundance, identity, and gene expression in skeletally derived cells. In order to detect modest changes in osteoblast gene expression at the single cell level in the appendicular skeleton, larger numbers of osteoblasts from endocortical bone are required.
INTRODUCTION
New technologies enable investigators to characterize the transcriptomes of thousands of individual cells in a single experiment [1–3]. These technologies are collectively referred to as single cell RNA sequencing (scRNA-seq) and have been used to determine the diversity of cell types in a complex tissue [1, 4–7], identify novel cell types [8, 9], and identify cells at different stages of differentiation [10–13]. We determined the utility of using scRNA-seq to detect changes in the abundance, differentiation, and transcriptional activity of cells recovered from neonatal mouse calvarium and adult mouse long bone endosteum.
The neonatal mouse calvarium is enriched for bone-forming osteoblasts and their progenitors. A traditional approach in studying osteoblast biology has therefore been to harvest and culture calvarial cells. When supplemented with pro-osteogenic media containing ascorbic acid and β-glycerophosphate, these cells follow a well-defined differentiation cascade towards a mature osteoblast phenotype in vitro [14–16]. We performed scRNA-seq on freshly isolated calvarial cells that had been cultured and induced to mineralize in vitro to identify similarities and differences between calvarial cells in vivo and in vitro.
Sclerostin (encoded by the gene SOST) is highly expressed by osteocytes and inhibits WNT-signaling by binding to the receptors LRP5 and LRP6 [17, 18]. In humans, SOST deficiency causes two high bone mass disorders Sclerosteosis and Van Buchem disease [19, 20], which are recapitulated by Sost-null mice [21]. The administration of an antibody that neutralizes Sclerostin induces new bone formation in multiple species, including humans [22–26]. Consequently, an anti-Sclerostin monoclonal antibody has recently been approved by United States Food and Drug Administration to treat patients with osteoporosis [27]. By performing bulk RNA sequencing on mRNA recovered from mouse cortical long bone, we observed that mice with deficient WNT-signaling (i.e., Lrp5−/−) had a lower abundance of transcripts associated with bone anabolism (e.g., Col1a1, Col1a2, and Bglap) compared to controls. Conversely, we observed increased bone anabolic transcripts in bulk RNA recovered from the cortical long bones of mice that received Sclerostin-neutralizing antibody (SclAbIII) [28, 29]. Because bulk RNA sequencing cannot determine if a difference in transcript abundance is caused by a change in the number of osteoblasts or a change in the activity of individual osteoblasts, we investigated whether scRNA-seq could detect a change in the number of osteoblasts and/or a change in an osteoblasts’ activity.
METHODS
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committees at Boston Children’s Hospital, Massachusetts General Hospital and Weill Cornell Medical College. The experimental design is depicted in Figure 1. For calvarial cell isolation (Figure 1a), twelve P4 pups born to a CD-1 dam (CRL stock #2200) were euthanized by hypothermia and decapitation. For long bone endocortical cell isolation, 11-week-old male C57Bl/6J mice were purchased from The Jackson Labs (Stock # 000664) and acclimatized for 1 week under standard housing conditions. Four mice were then given 2 subcutaneous injections, 3 days apart, of PBS and 4 mice were similarly given subcutaneous injections of SclAbIII (Amgen, Thousand Oaks, CA; 25 mg/kg in PBS). Four days after the 2nd injection the animals were euthanized by CO2 inhalation. (Figure 1b).
Figure 1: Single cell RNA-seq study design.

(a) Two sets of biologic replicates, each composed of cells that had been isolated CD-1 neonatal mouse calvarium by collagenase-digestion and EDTA treatment (6 calvaria/sample), were collected. An aliquot from each sample was immediately subjected to single cell RNA-seq; the remaining cells were plated, allowed to reach confluence, and cultured under osteogenic conditions for 12 days prior to undergoing single cell RNA-seq. (b) 12-week-old C57Bl/6J male mice received 2 SclAbIII (n=4) or 2 PBS (n=4) injections, 3 days apart. Four days later, tibial and femoral diaphyses were collected from each mouse and the endocortical cells were recovered by collagenase digestion.
Calvarial Cell Isolation
Calvaria were recovered from P4 pups immediately after decapitation. Two biological replicates, each consisting of calvaria from 6 pups were digested in 10 ml collagenase solution (1 mg/ml Collagenase type I and type II (1:3 ratio, Worthington Biochemical Corp., Lakewood, NJ), 1 mM CaCl2, 0.1% BSA, 25mM HEPES in αMEM) with gentle agitation at 37°C inside a 5% CO2 incubator. The collagenase was replaced every 15 minutes for 5 cycles, followed by 5 mM EDTA (with 0.1% BSA in PBS) treatment for 1 cycle, collagenase for 1 cycle and 5 mM EDTA for the final cycle. Cells released into the medium during the final collagenase cycle and EDTA cycle were combined, mixed with an equal volume of cell culture medium (αMEM + 10% FBS + 1% anti-mycotic) and centrifuged at 500g for 10 min at 4°C. Pelleted cells were re-suspended in cell culture medium and an aliquot from each sample was used to create an scRNA-seq library. The remaining cells in each sample were seeded in 6-well plates at 50,000 cells/well and allowed to reach confluence, while changing the media every 1–2 days. Cells reached confluence at day 5, and were then washed with PBS, separated from the plate via trypsin treatment, transferred to collagen-coated plates (Corning, Corning, NY ), and supplemented with 50 μg/ml ascorbic acid and 1 mM β-glycerophosphate for another 7 days in order to initiate osteogenic differentiation. After a total of 12 days in culture, cells were recovered by collagenase digestion for 30 min at 37°C, pelleted and resuspended in cell culture medium, and used to create scRNA-seq libraries. Biological replicates comprised originally of calvaria from 6 pups each were used to create n=2 freshly isolated calvarial cell and n=2 cultured calvarial cell scRNA-seq libraries.
Endosteal Cell Isolation
Femora and tibiae were collected immediately after euthanasia. The distal and proximal epiphyses were cut away with a scalpel and bones were centrifuged at 13,000 rpm for 1 minute at room temperature to remove bone marrow (Supplementary Figure 1). Periosteum was aseptically removed with a scalpel and the bone was bisected lengthwise to expose the endosteum. Tibiae and femora from each mouse were combined and incubated with 4 ml of collagenase solution (3 mg/ml Collagenase Type IV in PBS, Worthington Biochemical Corp., Lakewood, NJ) inside a 5% CO2 chamber at 37°C for 15 min under continuous agitation. Cells recovered from this initial digestion were discarded since they contained a large fraction of red blood cells (Supplementary Figure 2). The long bones were digested in two additional 4 ml collagenase solutions for 30 minutes each, and the cells recovered with each digestion were mixed with an equal volume of α-MEM (w/10%FBS + 1% anti-mycotic) and combined. After pelleting the cells at 2,000 x g for 5 minutes, the pellet was resuspended in 0.7 ml of PBS (w/ 0.04% BSA) and then used to prepare scRNA-seq libraries. To verify that collagenase digestion efficiently removed cells from the endosteal bone surface, the bones were fixed in 10% formalin, decalcified with 10% EDTA, processed and embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E), and compared to similar sections from bones that had been placed in PBS (Supplementary Figure 3).
Single Cell Capture and RNA-seq Library Preparation
We used the Chromium Single Cell 3’ system (v1 for the endosteal cells and v2 for the calvarial cells) to capture and sequence single cell mRNA, following the manufacturer’s instructions (10X Genomics, Pleasanton, CA). We loaded ~10,000 cells per endosteal specimen and ~6,000 cells per calvarial specimen. Individual cells were captured inside oil droplets along with barcoding beads, such that the mRNA contents of each cell were ligated with cell-specific oligo-barcodes (Supplementary Figure 1). Following single cell capture, barcoded mRNA and reverse-transcription solutions were immediately transferred to a thermal cycler, wherein barcoded cDNA was generated. The pooled cDNAs were then chemically fragmented, ligated with sequencing adapters, amplified and purified with magnetic beads (Beckman Coulter, Brea, CA). Quality checks for libraries were performed with gel electrophoresis and TapeStation analysis (Agilent Technologies, Santa Clara, CA).
Sequencing and Data Analysis
Single cell RNA-seq libraries were pooled and run on the Illumina NextSeq platform. Data analysis was performed using the Cellranger and Seurat pipelines [30]. Briefly, raw sequence data were de-multiplexed into specimen-specific bins, and mapped to the mouse genome (mm10) with STAR aligner [31]. The mapped sequence data and the associated unique molecular identifiers (UMI) were used to determine the number of captured cells, and the transcriptome of each cell. We utilized the aggr function of the Cellranger pipeline to combine data from biological replicates prior to downstream analysis with Seurat.
We excluded cells that had higher-than-expected mitochondrial transcripts and transcriptional diversity (indicated by the number of unique transcripts per cell). We combined the fresh calvarial cell and cultured calvarial cell datasets, performed tSNE analysis [1, 30], and used the unbiased cluster-detection algorithm of Seurat [30] to identify transcriptionally distinct cell populations. We performed similar analyses with the SclAbIII- and PBS-treated endocortical cell datasets. We regressed cell-cycle associated transcriptional signals from our data, as cells going through mitotic division might be registered as distinct populations. We then identified the transcripts that set each cell population apart from the others. We quantified the number of cells in each cell population, in a sample-specific manner. We also quantified the mRNA expression levels in each cell population with respect to their sample of origin, and ultimately the treatment group of origin. We utilized the Monocle trajectory analysis algorithm (v2) to infer potential lineage-relationships between distinct clusters of cells with a semi-supervised approach [32].
Differential Gene Expression Analysis Between Long Bone Endocortical Cell Specimens
We performed differential expression analysis between the SclAbIII- and PBS-treated groups, in a cell cluster-specific manner, using edgeR [33]. We determined significance with p < 0.05 after correction for multiple hypothesis testing. As scRNA-seq can detect a limited portion of each cell’s transcriptome, we imposed an additional detectability threshold, such that > 50% of the cells in either group had to have > 0 expression of the tested gene.
Fluorescence-Activated Cell Sorting (FACS)
We quantified macrophages among the retrieved calvarial cells by FACS. Following digestion, live cells were counted, pelleted and re-suspended and blocked (1:100 dilution, BD Biosciences #553142) in FACS buffer (PBS + 2% FBS + 2mM EDTA). Cells were then incubated in the dark and on ice for 30 minutes, with an antibody cocktail containing primary antibodies for CD45 (1:100 dilution, Biolegend, #103128) and F4/80 (1:100 dilution, Biolegend, #123110), as well as DAPI. Cells were then washed with FACS buffer, re-suspended and analyzed using the BD FACSCanto system (BD Biosciences, San Jose, CA). Compensation was determined with control beads (Thermo Fisher Scientific, #01–2222-42) and gating was determined based on unstained negative controls. Live single cells were evaluated to quantify the proportion of CD45+ and F4/80+ cells. At each time-point of interest, n=3 replicates were tested with at least 100,000 events recorded per replicate.
Immunocytochemistry
Calvarial cells seeded in 48-well plates were used for the immunocytochemistry protocol 7 days after confluency. Briefly, following aspiration of medium, cells were rinsed twice with PBS at room temperature. Cells were fixed with cold 4% paraformaldehyde in PBS for 20 minutes and then washed with PBS three times. Cells were blocked and permeabilized at room temperature for 30 minutes with 10% normal donkey serum (Jackson ImmunoResearch, 017–000-121) and 0.3% Triton-X 100 in PBS. Rat anti-F4/80 antibody (1:500 dilution, Novus Biologicals, NB600–404SS) was then added to each well and plates were incubated in the dark overnight at 40C. Negative-control wells were incubated with only the block solution. Next day, each well was rinsed with 1x PBS and donkey anti-rat A488 secondary antibody (1:200 dilution, Jackson ImmunoResearch, 712–545-150) was added to each well. Following a 1-hour incubation at room temperature, each well was again washed three times with PBS and DAPI was added to the second wash as a nuclear counter-stain. Samples were immediately imaged using an inverted microscope (Zeiss Axio Observer 7) equipped with an Axiocam 506 camera at 10x and 20x magnifications using the Zen Blue software. Collected images were adjusted for brightness and contrast using the Fiji software.
RESULTS
Calvarial Cell Recovery and Sequencing
We collected ~3 million cells from each sample of 6 pooled calvaria, aliquots of which were used to generate scRNA-seq libraries. We obtained an average of ~194 million reads from the fresh and cultured calvarial single cell libraries, which represented ~75,000 reads/cell, and ~2,522 cells per library following pre-processing and filtering.
Seurat’s unbiased cluster detection algorithm detected 11 distinct cell populations within the freshly collected calvarial cells and 8 distinct cell populations within the cultured calvarial cells (Figure 2a). The majority of the fresh calvarial cell population consisted of mesenchymal cells (clusters #3, #4, #7, #10 and #17, which represented >75% of all cells in each sample). Clusters #3, #4 and #7 exhibited transcriptional gradients in osteoblast markers (e.g., Col1a1, Bglap and Dmp1), suggesting they represent osteoblasts at different stages of differentiation. Consistent with this inference, 18 transcription factors had a significant change in expression (p < 0.05, absolute fold change > 2) between clusters #3, #4 or #7, including Mef2c, Satb2, Sp7 (Supplementary Figure 4). Clusters #10 and #17 exhibited transcriptional profiles compatible with chondrocytes and alpha smooth muscle actin-expressing (αSMA+) smooth muscle cells, respectively (Figure 2b). The remaining fresh calvarial cell clusters expressed transcripts found in endothelial cells (Pecam1/Cd31, #18), red-blood-cells (Hbb-bs, #12 and #15), granulocytes (#14), B-cells (#16) and myeloid cells (#13). A trajectory analysis of mesenchymal cells (#3, #4, #7, #10 and #17) suggested that the cells constituting clusters #10 (chondrocytes) and #3 (osteoblast precursors) represent 2 separate branches of primitive cells at the beginning of differentiation timeline, whereas cells constituting clusters #4 and #7 (osteoblasts) represent 2 branches of differentiated cells at the end of pseudotime (Supplementary Figure 5).
Figure 2: Cell clusters identified among cultured and fresh calvarial cells.
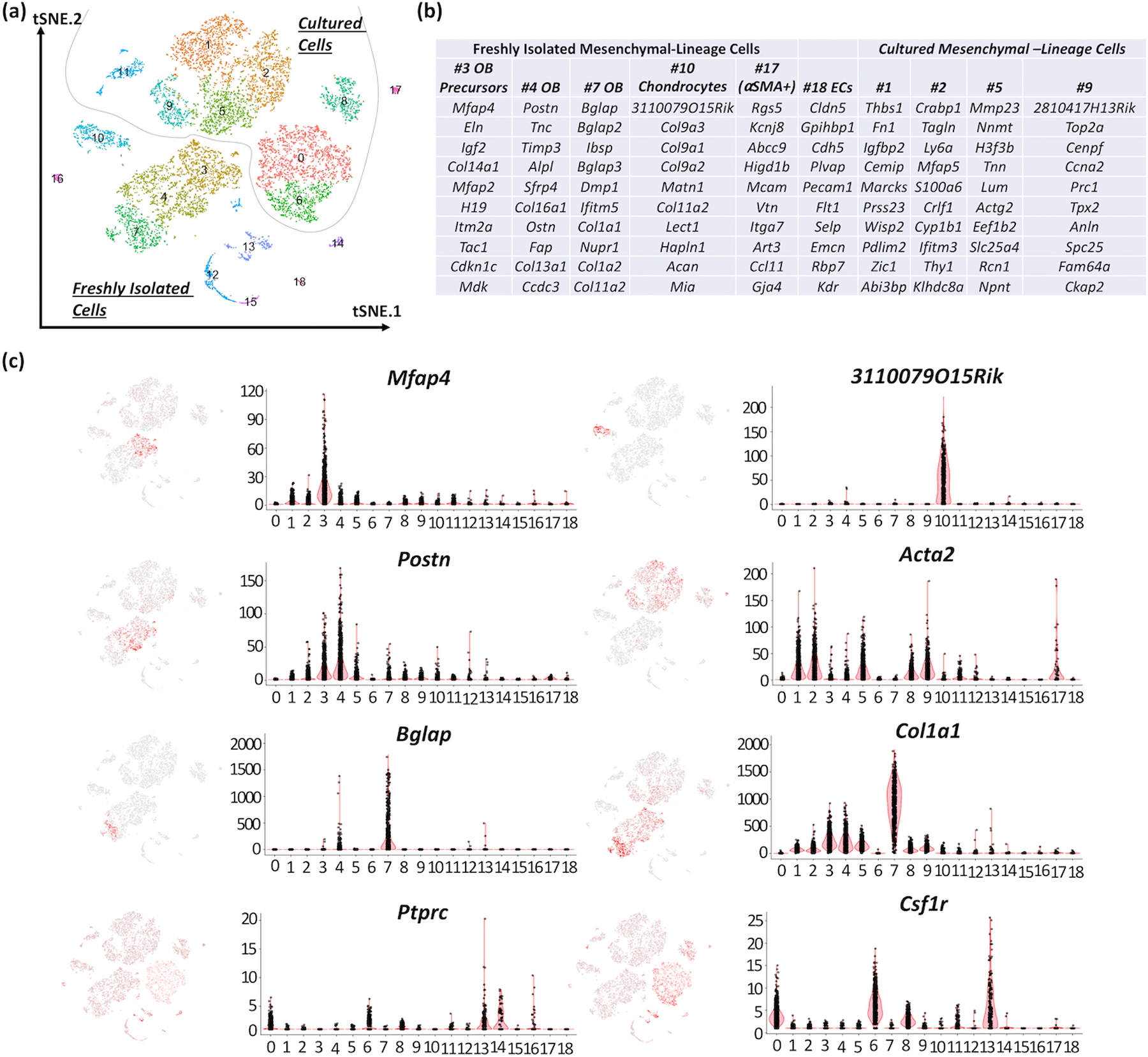
(a) tSNE plot wherein each dot represents a single cell, and cells sharing the same color code indicate discrete populations of transcriptionally similar cells. Cultured and freshly isolated cells form separate clusters (dashed line). Of note, cells in cluster #8 express transcripts associated with mesenchymal (e.g. Col1a1, Acta2) and hematopoietic (Ptprc and Csf1r) lineages, have higher than expected UMI counts, and likely represent artifact due to 2 cell types being captured in a single droplet. (b) Top 10 transcript markers corresponding to each cell cluster as determined by Seurat (OB: osteoblast). (c) Heatmaps of gene expression (left) and associated violin plots (right) indicate cluster-specific expression of representative genes. Osteoblast-lineage cells among freshly isolated calvarial cells are distinctly marked by the expression of Mfap4 (#3), Postn (#4) and Bglap (#7). Similarly, cultured osteoblast-lineage cells express Acta2 and Col1a1 (#1, 2, 5 and 9), whereas hemaotopoietic cells express Csf1r and Ptprc (#0, 6 and 8).
scRNA-seq Indicates that Calvarial Cell Populations and Transcriptomes Change in vitro
Cultured calvarial cells clustered separately from freshly isolated cells (Figure 2a). Within the cultured calvarial cell populations, clusters #1, #2, #5 and #9 grouped near one-another and expressed transcripts (e.g. Col1a1, Runx2, Sp7 and Alpl) that suggest they are mesenchymal cells differentiating along the osteoblast lineage. However, in contrast to the freshly isolated calvarial cells, cultured calvarial cells did not express transcripts typically associated with mature osteoblasts, such as Bglap, Dmp1 or Ifitm5, at a level detectable by single cell RNA-seq. These clustering patterns were not altered when we performed the tSNE analysis on freshly isolated and cultured cells separately (Supplementary Figure 6). Also, whereas only 10 and 12% of freshly isolated calvarial cells appeared hematopoietic in origin (clusters #12 to #16, as indicated by the expression of transcripts including Csf1r and Ptprc), these percentages increased to 45 and 48% in the cultured cells (clusters #0 and #6), respectively. We did not observe cell death or Casp3 expression in mesenchymal cells in culture. Instead, 6% of mesenchymal cells and 10% of hematopoietic cells expressed the cell proliferation-associated transcript Mki67, suggesting both cell types proliferated in culture.
In order to verify the increase in hematopoietic cells (macrophages in particular) in culture found by scRNA-seq, we performed FACS analysis of freshly retrieved and cultured calvarial cells (Figure 3). Consistent with the scRNA-seq data, we found CD45+, F4/80+ macrophages represent 2% of all cells prior to culture and ~51% of all cells 12 days later (Figure 3a). We additionally verified the increased presence of macrophages after in vitro culture by immunostaining calvarial cells with a F4/80-recognizing antibody (Figure 3b).
Figure 3: Fluorescence activated cell sorting (FACS) and immunocytochemistry analyses detect macrophages among cultured calvarial cells.

(a) Representative FACS plots indicate a significant expansion of CD45+, F4/80+ macrophages in calvarial cell culture. (b) Fluorescent imaging of cultured calvarial cells at 10x (left) and 20x (right) magnification depict a substantial number of macrophages stained with the F4/80-recognizing antibody (Scale bar: 50um).
Long Bone Endocortical Cell Recovery and Sequencing
H & E staining indicated that our collagenase/EDTA cell recovery method removed nearly all endosteal surface cells, but did not remove embedded osteoblasts or osteocytes (Supplementary Figure 3). We recovered ~1 million cells from the pooled tibiae and femora of individual mice. We generated ~ 65 million reads per scRNA-seq library, which after deconvolution, alignment, and cell-specific gene expression represented an average of ~47,000 reads/cell, and 1053 cells/mouse following filtering for outlier cells based on mitochondrial and unique transcript content.
Seurat’s unbiased cluster detection algorithm defined 22 cell populations within the long bone endocortical samples of PBS- and SclAbIII-treated mice (Figure 4a). We utilized the single cell transcriptome database (specifically bone marrow scRNA-seq data) developed by the Tabula Muris Consortium [34] to assign identities to these populations. Three cell clusters (#6, #7, and #14) from the long bone endocortical cell libraries, representing ~13% of the cells, had transcriptional profiles consistent with their being mesenchymal cells and osteoblasts (Figure 4b–c). The most abundant cell population among these, cluster #6 contains peri-arteriole stromal cells (also known as PαS cells[35]), which express Col1a1, Pdgfra and Ly6a/Sca1. Clusters #7 and #14, representing ~7% of the sequenced endocortical cells in each animal, are contiguous and express osteoblast-associated transcripts (e.g., Bglap, Ifitm5, and Dmp1). Differences in expression between these two clusters suggest cluster #14 comprises more mature osteoblasts (Figure 4b–c).
Figure 4: Endosteal cell clusters detected by scRNA-seq.

(a) tSNE plot wherein each dot represents a single cell, and cells sharing the same color code indicate discrete populations of transcriptionally similar cells. Five clusters of cells (highlighted inside the dashed line) represent mesenchymal progenitors and osteoblasts at different stages of differentiation, as indicated by their individual expression profiles depicted in (b) and (c). (b) Top 10 transcript markers corresponding to each of the mesenchymal and endothelial cell clusters as determined by Seurat. (c) Heatmaps of gene expression (left) and associated violin plots (right) indicate cluster-specific expression of representative genes. Increasing expression of Col1a1 and Bglap across clusters #7 and #14 suggest that these clusters represent osteoblasts. Cells in cluster #6 are marked by Pdgfra and Ly6a/Sca1 expression, cluster #21 is marked by Lepr and high levels of Cxcl12 expression, cluster #12 is marked by Ly6a/Sca1 expression, and cluster #19 is marked by Acta2/aSMA expression (Ob: Osteoblast).
In marked contrast to freshly recovered calvarial cells, the majority of cells (>80%) that we recovered from cortical long bone were hematopoietic in origin. Four clusters, representing ~ 40% of the cells, express transcripts associated with granulocytes and pro-granulocytes. Six clusters, representing ~30% of the cells express transcripts associated with T-cells, B-cells, neutrophils, basophils, dendritic cells and macrophages. Two clusters, representing ~12% of the cells, express transcripts associated with erythroblasts and erythrocytes.
Two other previously described cell types, Cxcl12-Abundant-Reticular (CAR) cells [36] and peri-arteriole smooth muscle (αSMA+) cells [37] comprise clusters #21 and #19, respectively (Figure 4c). Interestingly, cells in cluster #21 express transcripts seen in mature osteoblasts (e.g., Ibsp, Serpinh1/Hsp47, Gja1/Connexin43, Pcolce), but they do not express Col1a1 or Bglap at a level detectable by scRNA-seq (Supplementary Table 1).
No Discernable Effect of SclAbIII Treatment on Endocortical Cells Measured by scRNA-seq
There was no discernable difference in the relative proportions of the different cell clusters from the long bones of PBS and SclAbIII-treated mice (Figure 5a). We did not identify mesenchymal cells undergoing cell-cycling in either group. We also did not observe a significant difference in the single cell expression levels of osteoblast-associated transcripts such as Col1a1, Bglap, and Cpz in clusters #7 or #14 between PBS- and SclAbIII-treated mice (Figure 5b). Instead, we only detected significantly altered expression of 9 protein-coding, non-ribosomal, transcripts in cluster #7 (Hint1, Oaz1, Fos, H3f3b, Cst3, Tceb2, Sep15, Dynll1 and Omd) and 5 protein-coding, non-ribosomal, transcripts in cluster #14 (Cst3, Fau, Ftl1, Gpx3 and Loxl2) following SclAbIII treatment.
Figure 5: Reproducibility of endocortical scRNA-seq data across biologic replicates.
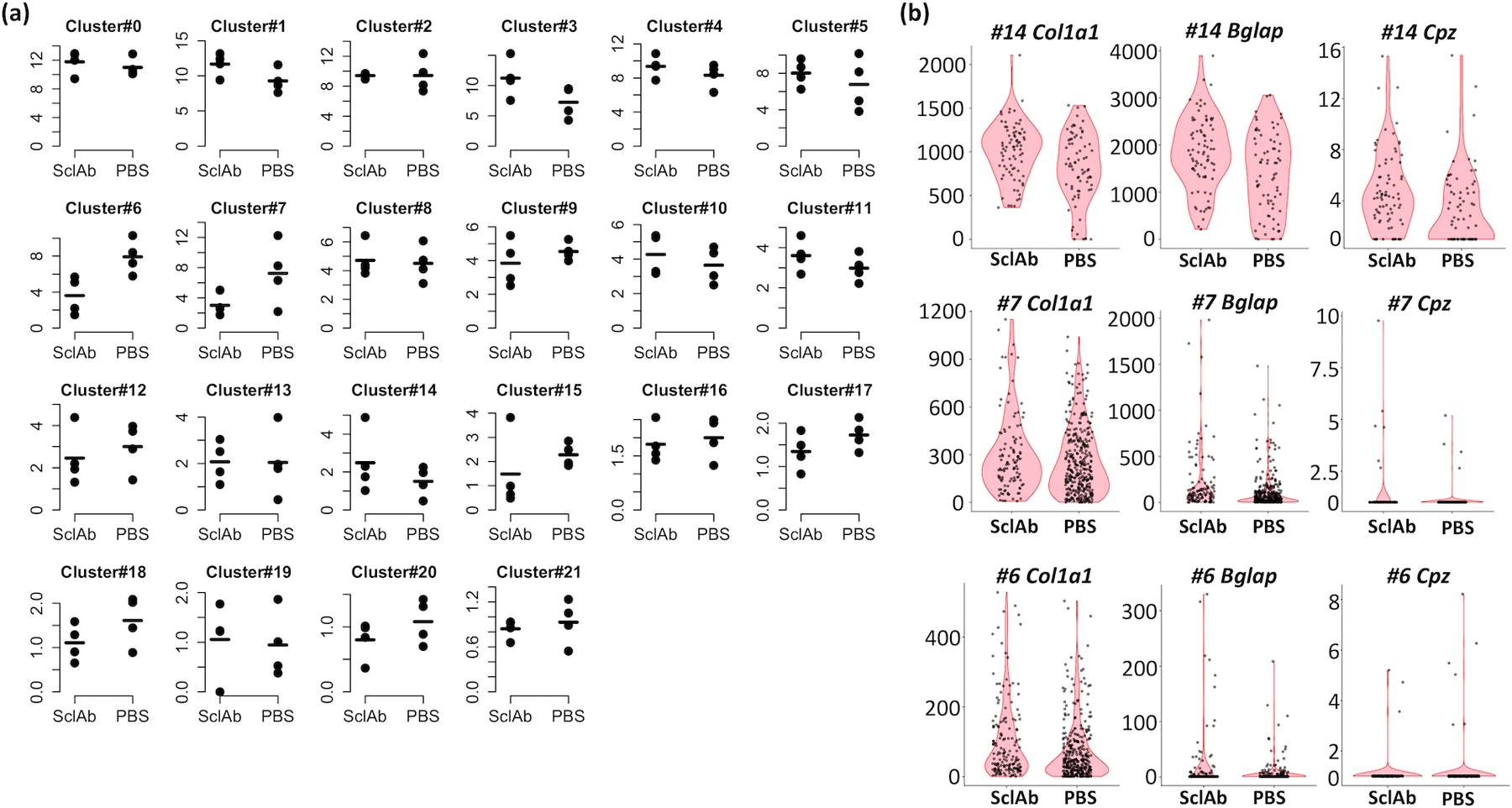
(a) Dot plots depict the percentage of cells in each cluster across endocortical cells collected from 8 mice (n=4 SclAbIII-treated and n=4 PBS-treated), suggesting high reproducibility in the quantification of transcriptionally distinct cells. (b) Violin plots show no significant difference in the expression of 3 genes associated with osteoblast anabolism (Col1a1, Bglap, and Cpz) due to SclAbIII treatment in the mesenchymal or osteoblast cell clusters.
Osteoblasts Freshly Isolated from Calvaria and Cortical Long Bone Have Similar Transcriptomes
In contrast to the scRNA-seq data that indicated freshly isolated calvarial osteoblasts had different transcriptomes from those of cultured calvarial osteoblasts (Figure 2a), we found strong concordance when we compared the transcriptomes of freshly isolated osteoblasts from calvaria and long bone (Figure 6).
Figure 6: High concordance between the transcriptomes of freshly isolated calvarial and long bone endocortical osteoblasts.

(a) Heatmap depicts the mean expression levels of the top 40 gene markers for calvarial osteoblast cluster #7, across the osteoblast-lineage cells in endosteal and calvarial biological replicates. (b) Violin plots depict the changes in the expression of top osteoblast markers Bglap, Ibsp, Dmp1 and Ifitm5 across the long bone endosteal (left) and calvarial (right) cell clusters.
Robustness of Bone-derived Single Cell RNA-seq Data
Obtaining endocortical cells from 8 individual mice allowed us to assess the consistency with which we recovered different cell types and to determine whether gene expression within a cell type correlated between animals. We observed that the relative proportions of cell types were similar across all animals and there was a high Pearson correlation coefficient between the transcriptomes of each cluster across animals (Supplementary Figure 7). The only exceptions to these observations are for clusters #19 and #21 where lower correlations across replicates were observed (R2 range: 0.37–0.79), likely due to the low number of cells (<90 cells) that were contained in these clusters. Although we only had 2 biologic replicates to compare for the freshly obtained calvarial cells, these specimens also were closely matched, and we calculated high Pearson correlation coefficients when transcriptomes of each cluster were compared between samples. (Supplementary Figure 8). Importantly, we did not control for sex when collecting and culturing calvarial cells. However, we did not observe a clustering bias due to sex, as measured by Xist-expression which appears uniform across the calvarial cells.
Using whole tissue RNA-seq, we had previously identified a set of transcripts, whose abundances were dramatically elevated when long bone tissue was exposed to collagenase [28]. These transcripts included Fos, Fosb, Jun, Il6, possibly indicating a stress response. When we interrogated our scRNA-seq data, we found that these stress response transcripts were at low or undetectable levels (Supplementary Figures 9 and 10, Supplementary Table 1). We infer that the cells expressing these stress-response transcripts were not sampled by scRNA-seq, most likely because cells that remained embedded express these transcripts.
DISCUSSION
Single cell RNA-seq can identify discrete cell types in cultured cells and in complex tissues, based on each individual cell’s transcriptome. The ability to interrogate the transcriptomes of large numbers of individual cells has led to the discovery of previously unrecognized cell types [8, 9] and has detected changes in cellular diversity in response to genetic or environmental perturbation [38–40]. Recovering individual cells from skeletal tissue is more challenging than from cultured cells or from other tissues whose cells can be easily separated by brief digestion and/or physical disruption. For this reason, in vitro studies of osteoblast differentiation and mineralization typically expand cells in culture and then chemically induce matrix mineralization [15, 16]. In order to study freshly isolated osteoblasts or their precursors, skeletal tissues need to be subjected to sequential enzymatic digestions for an hour or longer [14, 15, 41].
We employed scRNA-seq to assess similarities and differences between osteoblasts that were freshly obtained from calvaria versus long bone endocortices, between fresh calvarial cells and those that had been expanded in culture, and between long bone endocortical cells from PBS- and SclAbIII-treated animals. We observed greater enrichment for osteoblasts that had been freshly isolated from calvaria compared to those that had been freshly isolated from long bone endocortices. More than 40% of calvarial cells had transcriptomes expected of osteoblasts, but only 7% of endocortical cells exhibited these profiles (Figures 2a and 4a). Reassuringly, however, the transcriptomes of freshly isolated calvarial and endocortical osteoblasts were highly similar, suggesting that osteoblast precursors converge to a shared transcriptional phenotype during differentiation, regardless of their origin (Figure 6). Although we cannot exclude the possibility that many of these similarities are solely due to collagenase induced transcription, we think this is unlikely since most of the transcripts used to define these cells as osteoblasts are present in cortical bone bulk RNA-seq data and are seen in osteoblasts in situ [28]. Consistent with our findings, other recent studies on bone marrow stromal cells have identified cell populations that are highly similar to those we observed, including multiple osteoblast populations (Bglap-high, Ifitm5+, Dmp1+ and Bglap-low, Postn+, Col3a1+ populations) , smooth muscle cells (Acta2+, Tagln+), CAR cells (Lepr+, Cxcl12+, Kitl+, Gdpd2+) and PαS cells (Pdgfra+, Ly6a+) [4, 6]. Finally, we observed high Pearson correlation coefficients within individual cell populations from 2 freshly collected or cultured calvarial specimens and 8 endocortical specimens, indicating that we are reproducibly recovering cells (Supplementary Figures 7–8 and 11–12). Calvarial cells collected after 120 minutes (i.e. 8 cycles) of enzymatic digestion from P4 mice are enriched for chondrocytes, osteoblasts and their precursors. Although the primary mechanism of mineralization in calvaria is intramembraneous ossification, cartilage has previously been observed in parietal bones [42, 43], which is likely the source of chondrocytes in our dataset. In our hands, ~6% of the freshly isolated cells are macrophages (cluster #13, Figure 2a), based on the expression of genes such as Ptprc/Cd45, Adgre1 (also known as F4/80), Cd68 and Csf1r. However, after we expanded the calvarial cells in culture and began to induce differentiation, we found that 34% of the cells were macrophages (clusters #0 and #6). Our results are consistent with those of Chang et al., who describe a population of cells they termed “osteomacs,” which accounted for ~16% of freshly collected calvarial cells and proliferated in culture [44]. Colony stimulating factor 1 (CSF1) had previously been proposed as a mediator of signaling between osteoblasts and osteomacs [45, 46]; consistent with this model, Csf1 and Csf1r expression distinctly mark cells we transcriptionally identified as osteoblasts and macrophages, respectively. Moreover, our scRNA-seq data identifies other cell type-specific transcripts (such as Cd68, Cd14, Ccl9) that may also be involved in paracrine signaling. We did not detect Bglap-expressing cells among the calvarial osteoblasts that had been expanded in culture and induced to differentiate. This contrasts with freshly isolated calvarial cells that exhibited abundant Bglap expression. These data are consistent with previous studies that indicate freshly isolated calvarial osteoblasts initially de-differentiate in vitro [14, 47, 48].
Although we previously used bulk RNA-seq of cortical bone to observe significant increases in transcripts associated with bone anabolism in mice given a short course of SclAbIII [29], we did not observe an increase using scRNA-seq in our long bone endocortical samples. Several factors may account for this discordance. First, we administered 4 doses of neutralizing antibody for our bulk RNA-seq studies but only 2 doses for the scRNA-seq study. Second, our scRNA-seq data were derived from osteoblasts, whereas our earlier bulk RNA-seq data were enriched for osteocytes. However, other investigators have reported changes following short term treatment with SclAbIII and the effects of SclAbIII on osteoblasts[49]. Therefore, the most likely explanation for our findings is that our endosteal cell recovery protocol was underpowered to detect modest increases in gene expression. Bulk RNA-seq sampled millions of cells per mouse and detected the expression of ~10,000 genes. In contrast, our scRNA-seq data indicated we recovered an average of only 75 osteoblasts/mouse (6% of all recovered cells) with each cell expressing ~1,500 genes (Supplementary Figure 13). Therefore, with only 75 osteoblasts/mouse and 4 biologic replicates/treatment group, modest changes in gene expression (e.g. ~1.2-fold change in Bglap, Bglap2, Col1a1 and Col1a2 expression) did not reach statistical significance. However, if the scRNA-seq data remained consistent and we had sampled 250 osteoblasts/mouse, then these data would have become statistically significant, even after controlling for multiple hypothesis testing. Therefore, more efficient osteoblast enrichment protocols will be needed to make scRNA-seq sensitive for detecting modest gene expression changes in response to genetic, pharmacologic, or environmental perturbations. One possible approach to this could be to utilize existing reporter mouse strains (such as Bglap.eGFP[50] or 2.3ColGFP[51]) and enrich for fluorescently-labeled osteoblasts with flow cytometry following enzymatic digestion of bone tissue.
Increasing sample size also needs to be considered when employing scRNA-seq to identify changes in the distribution of cell types following genetic, pharmacologic, or environmental perturbations. For example, reactivation of bone-lining cells to become active osteoblasts has previously been reported following SclAbIII administration in mice [49]. Assuming these cells are represented by cluster #14 in our endocortical bone scRNA-seq datasets, one-way ANOVA analyses of our existing data indicate we would need 18 mice/treatment group to observe a significant difference after controlling for multiple hypothesis testing.
In conclusion, we describe the use of scRNA-seq to show that freshly recovered osteoblasts from newborn calvaria and adult endocortical long bone have highly correlated transcriptomes, and that freshly isolated calvarial cells undergo changes in their relative abundances and transcriptomes when expanded and differentiated in vitro. We found that scRNA-seq is not yet sensitive for detecting changes in osteoblast gene expression as bulk RNA-seq. However, as better methods for recovering and enriching for osteoblasts from endocortical samples are developed, and greater depths of coverage for transcripts from individual cells are obtained, we anticipate scRNA-seq will become a useful tool for monitoring the effects of genetic, environmental, and pharmacologic perturbations on endocortical bone cells.
Supplementary Material
Supplementary Figure 4: We identified 18 transcription factors whose expression changed significantly between calvarial cell clusters #3, #4 or #7.
(a) Cells represented by clusters #3, #4, #7, #10 and #17 in the tSNE plot were evaluated. Cells originating from clusters #3 and #10 were positioned as 2 separate branches at the beginning of pseudotime. Cells originating from clusters #4 and #7 were similarly positioned as 2 separate branches at the end of pseudotime. Cells originating from cluster #17 were distributed across the trajectory without a visible pattern. (b) Heatmap-style scatterplots (left) and one-dimensional pseudotime charts (right) for top markers genes associated with chondrocytes (3110079O15Rik), osteoblast precursors (Mfap4) and osteoblasts (Postn and Bglap) mark 4 distinct branches of the presumed differentiation trajectory. Notably, Monocle predicts certain positions for chondrocytes and smooth muscle cells along the pseudotime trajectory, but it is unclear if these cells actually do share a lineage-relationship with osteoblasts in the calvaria.
(a and c) tSNE plots representing freshly isolated (a) and cultured (c) calvarial cells that were subjected to single cell RNA-seq. (b and d) Cluster-specific markers identified in Figure 2 also mark distinct populations in separately analyzed calvarial cells. For example, Bglap expression labels 2 distinct clusters (#2 and #4) among freshly retrieved cells, whereas its expression is not detectable among cultured cells.
Supplementary Figure 11: Reproducibility of scRNA-seq on calvarial cells indicated by tSNE distribution.
Supplementary Figure 12: Reproducibility of scRNA-seq on endocortical cells indicated by tSNE distribution.
Supplementary Table 1: Mean gene expression levels per cell cluster in calvarial cells.
Supplementary Table 2: Mean gene expression levels per cell cluster in endocortical cells.
Supplementary Figure 1: Single cell RNA-seq workflow for endocortical and calvarial bone specimens.
Supplementary Figure 2: Two-fold reduction in bulk Hbb expression between endosteal cell collections #1 and #2, measured by qRT-PCR.
We found that there were very few cells left on the endosteal surface of bone samples treated with collagenase, whereas there were numerous clusters of cells on PBS treated surfaces (white arrows).
We calculated the mean gene expression profile in a cell cluster- and mouse-specific manner, and calculated R2 values by performing intra-group comparisons (top: SclABIII, bottom: PBS) between each experimental mouse. Our cell cluster-specific measurements were highly reproducible among the majority of the clusters, except for #19 and #21. The intra-group variability of gene expression in these clusters is likely due to low number of cells (<90 cells total in each cluster).
Supplementary Figure 8: Reproducibility of cluster-specific gene expression measurements in biologic replicates of fresh (highlighted with blue) and cultured (highlighted with red) cells.
Note that these genes are expressed at either undetectable (as in the case of Il6 and Ftl2) or low to moderate levels in all cells (as reference, mean Bglap and Col1a1 expression levels were found to be 876 and 490 in Cluster#14, respectively).
Note that these genes are expressed at either undetectable (as in the case of Ftl2) or low to moderate levels in all cells.
We found that the the magnitude of transcriptional diversity was highly variable across the 22 cell populations we identified. Hbb+ red blood cells (cluster#8) were the least diverse, whereas mature osteoblasts (cluster#14), neutrophils (cluster#10) and macrophages (cluster#11) expressed up to ~3,000 distinct genes per cell.
ACKNOWLEDGMENTS
Grant Support:
This study was funded by grants P30 AR066261, R01 AR053237, R01 AR064231, R01 AR071342 and R21AR067388 from NIAMS.
REFERENCES
- 1.Macosko EZ, et al. , Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell, 2015. 161(5): p. 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng GX, et al. , Massively parallel digital transcriptional profiling of single cells. Nat Commun, 2017. 8: p. 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zilionis R, et al. , Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc, 2017. 12(1): p. 44–73. [DOI] [PubMed] [Google Scholar]
- 4.Baryawno N, et al. , A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell, 2019. 177(7): p. 1915–1932 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shekhar K, et al. , Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell, 2016. 166(5): p. 1308–1323 e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tikhonova AN, et al. , The bone marrow microenvironment at single-cell resolution. Nature, 2019. 569(7755): p. 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolock SL, et al. , Mapping Distinct Bone Marrow Niche Populations and Their Differentiation Paths. Cell Rep, 2019. 28(2): p. 302–311 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Debnath S, et al. , Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature, 2018. 562(7725): p. 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plasschaert LW, et al. , A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature, 2018. 560(7718): p. 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan MM, et al. , Molecular recording of mammalian embryogenesis. Nature, 2019. 570(7759): p. 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanton S, et al. , Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature, 2019. 574(7778): p. 418–422. [DOI] [PubMed] [Google Scholar]
- 12.Pijuan-Sala B, et al. , A single-cell molecular map of mouse gastrulation and early organogenesis. Nature, 2019. 566(7745): p. 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popescu DM, et al. , Decoding human fetal liver haematopoiesis. Nature, 2019. [DOI] [PMC free article] [PubMed]
- 14.Zhong ZA, Ethen NJ, and Williams BO, Use of Primary Calvarial Osteoblasts to Evaluate the Function of Wnt Signaling in Osteogenesis. Methods Mol Biol, 2016. 1481: p. 119–25. [DOI] [PubMed] [Google Scholar]
- 15.Lian JB and Stein GS, Development of the osteoblast phenotype: molecular mechanisms mediating osteoblast growth and differentiation. Iowa Orthop J, 1995. 15: p. 118–40. [PMC free article] [PubMed] [Google Scholar]
- 16.Lynch MP, et al. , The influence of type I collagen on the development and maintenance of the osteoblast phenotype in primary and passaged rat calvarial osteoblasts: modification of expression of genes supporting cell growth, adhesion, and extracellular matrix mineralization. Exp Cell Res, 1995. 216(1): p. 35–45. [DOI] [PubMed] [Google Scholar]
- 17.Li X, et al. , Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem, 2005. 280(20): p. 19883–7. [DOI] [PubMed] [Google Scholar]
- 18.Semenov M, Tamai K, and He X, SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem, 2005. 280(29): p. 26770–5. [DOI] [PubMed] [Google Scholar]
- 19.Balemans W, et al. , Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet, 2001. 10(5): p. 537–43. [DOI] [PubMed] [Google Scholar]
- 20.Brunkow ME, et al. , Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet, 2001. 68(3): p. 577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li X, et al. , Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res, 2008. 23(6): p. 860–9. [DOI] [PubMed] [Google Scholar]
- 22.Li X, et al. , Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res, 2009. 24(4): p. 578–88. [DOI] [PubMed] [Google Scholar]
- 23.Li X, et al. , Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res, 2010. 25(12): p. 2647–56. [DOI] [PubMed] [Google Scholar]
- 24.Marenzana M, et al. , Sclerostin antibody treatment enhances bone strength but does not prevent growth retardation in young mice treated with dexamethasone. Arthritis Rheum, 2011. 63(8): p. 2385–95. [DOI] [PubMed] [Google Scholar]
- 25.Ominsky MS, et al. , Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res, 2010. 25(5): p. 948–59. [DOI] [PubMed] [Google Scholar]
- 26.Padhi D, et al. , Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res, 2011. 26(1): p. 19–26. [DOI] [PubMed] [Google Scholar]
- 27.Markham A, Romosozumab: First Global Approval. Drugs, 2019. 79(4): p. 471–476. [DOI] [PubMed] [Google Scholar]
- 28.Ayturk UM, et al. , An RNA-seq protocol to identify mRNA expression changes in mouse diaphyseal bone: applications in mice with bone property altering Lrp5 mutations. J Bone Miner Res, 2013. 28(10): p. 2081–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kedlaya R, et al. , Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med, 2013. 5(211): p. 211ra158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler A, et al. , Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol, 2018. 36(5): p. 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobin A, et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 2013. 29(1): p. 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu X, et al. , Reversed graph embedding resolves complex single-cell trajectories. Nat Methods, 2017. 14(10): p. 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MD, McCarthy DJ, and Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 2010. 26(1): p. 139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tabula Muris C, et al. , Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature, 2018. 562(7727): p. 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morikawa S, et al. , Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med, 2009. 206(11): p. 2483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou BO, et al. , Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell, 2014. 15(2): p. 154–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthews BG, et al. , Osteogenic potential of alpha smooth muscle actin expressing muscle resident progenitor cells. Bone, 2016. 84: p. 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klaus J, et al. , Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat Med, 2019. 25(4): p. 561–568. [DOI] [PubMed] [Google Scholar]
- 39.Sharir A, et al. , A large pool of actively cycling progenitors orchestrates self-renewal and injury repair of an ectodermal appendage. Nat Cell Biol, 2019. 21(9): p. 1102–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szczerba BM, et al. , Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature, 2019. 566(7745): p. 553–557. [DOI] [PubMed] [Google Scholar]
- 41.Greenblatt MB, et al. , The Unmixing Problem: A Guide to Applying Single-Cell RNA Sequencing to Bone. J Bone Miner Res, 2019. 34(7): p. 1207–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holmbeck K, et al. , MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell, 1999. 99(1): p. 81–92. [DOI] [PubMed] [Google Scholar]
- 43.Zhou H, et al. , Glucocorticoid-dependent Wnt signaling by mature osteoblasts is a key regulator of cranial skeletal development in mice. Development, 2009. 136(3): p. 427–36. [DOI] [PubMed] [Google Scholar]
- 44.Chang MK, et al. , Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J Immunol, 2008. 181(2): p. 1232–44. [DOI] [PubMed] [Google Scholar]
- 45.Alexander KA, et al. , Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Miner Res, 2011. 26(7): p. 1517–32. [DOI] [PubMed] [Google Scholar]
- 46.Winkler IG, et al. , Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood, 2010. 116(23): p. 4815–28. [DOI] [PubMed] [Google Scholar]
- 47.Jonason JH and O’Keefe RJ, Isolation and culture of neonatal mouse calvarial osteoblasts. Methods Mol Biol, 2014. 1130: p. 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aubin JE, et al. , Osteoblast and chondroblast differentiation. Bone, 1995. 17(2 Suppl): p. 77S–83S. [DOI] [PubMed] [Google Scholar]
- 49.Kim SW, et al. , Sclerostin Antibody Administration Converts Bone Lining Cells Into Active Osteoblasts. J Bone Miner Res, 2017. 32(5): p. 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bilic-Curcic I, et al. , Visualizing levels of osteoblast differentiation by a two-color promoter-GFP strategy: Type I collagen-GFPcyan and osteocalcin-GFPtpz. Genesis, 2005. 43(2): p. 87–98. [DOI] [PubMed] [Google Scholar]
- 51.Kalajzic I, et al. , Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res, 2002. 17(1): p. 15–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 4: We identified 18 transcription factors whose expression changed significantly between calvarial cell clusters #3, #4 or #7.
(a) Cells represented by clusters #3, #4, #7, #10 and #17 in the tSNE plot were evaluated. Cells originating from clusters #3 and #10 were positioned as 2 separate branches at the beginning of pseudotime. Cells originating from clusters #4 and #7 were similarly positioned as 2 separate branches at the end of pseudotime. Cells originating from cluster #17 were distributed across the trajectory without a visible pattern. (b) Heatmap-style scatterplots (left) and one-dimensional pseudotime charts (right) for top markers genes associated with chondrocytes (3110079O15Rik), osteoblast precursors (Mfap4) and osteoblasts (Postn and Bglap) mark 4 distinct branches of the presumed differentiation trajectory. Notably, Monocle predicts certain positions for chondrocytes and smooth muscle cells along the pseudotime trajectory, but it is unclear if these cells actually do share a lineage-relationship with osteoblasts in the calvaria.
(a and c) tSNE plots representing freshly isolated (a) and cultured (c) calvarial cells that were subjected to single cell RNA-seq. (b and d) Cluster-specific markers identified in Figure 2 also mark distinct populations in separately analyzed calvarial cells. For example, Bglap expression labels 2 distinct clusters (#2 and #4) among freshly retrieved cells, whereas its expression is not detectable among cultured cells.
Supplementary Figure 11: Reproducibility of scRNA-seq on calvarial cells indicated by tSNE distribution.
Supplementary Figure 12: Reproducibility of scRNA-seq on endocortical cells indicated by tSNE distribution.
Supplementary Table 1: Mean gene expression levels per cell cluster in calvarial cells.
Supplementary Table 2: Mean gene expression levels per cell cluster in endocortical cells.
Supplementary Figure 1: Single cell RNA-seq workflow for endocortical and calvarial bone specimens.
Supplementary Figure 2: Two-fold reduction in bulk Hbb expression between endosteal cell collections #1 and #2, measured by qRT-PCR.
We found that there were very few cells left on the endosteal surface of bone samples treated with collagenase, whereas there were numerous clusters of cells on PBS treated surfaces (white arrows).
We calculated the mean gene expression profile in a cell cluster- and mouse-specific manner, and calculated R2 values by performing intra-group comparisons (top: SclABIII, bottom: PBS) between each experimental mouse. Our cell cluster-specific measurements were highly reproducible among the majority of the clusters, except for #19 and #21. The intra-group variability of gene expression in these clusters is likely due to low number of cells (<90 cells total in each cluster).
Supplementary Figure 8: Reproducibility of cluster-specific gene expression measurements in biologic replicates of fresh (highlighted with blue) and cultured (highlighted with red) cells.
Note that these genes are expressed at either undetectable (as in the case of Il6 and Ftl2) or low to moderate levels in all cells (as reference, mean Bglap and Col1a1 expression levels were found to be 876 and 490 in Cluster#14, respectively).
Note that these genes are expressed at either undetectable (as in the case of Ftl2) or low to moderate levels in all cells.
We found that the the magnitude of transcriptional diversity was highly variable across the 22 cell populations we identified. Hbb+ red blood cells (cluster#8) were the least diverse, whereas mature osteoblasts (cluster#14), neutrophils (cluster#10) and macrophages (cluster#11) expressed up to ~3,000 distinct genes per cell.