


Abstract
Prime editing technology is capable of generating targeted insertions, deletions, and base conversions. However, the process of designing prime editing guide RNAs (pegRNAs), which contain a primer binding site and a reverse-transcription template at the 3′ end, is more complex than that for the single guide RNAs used with CRISPR nucleases or base editors. Furthermore, the assessment of high-throughput sequencing data after prime editors (PEs) have been employed should consider the unique feature of PEs; thus, pre-existing assessment tools cannot directly be adopted for PEs. Here, we present two user-friendly web-based tools for PEs, named PE-Designer and PE-Analyzer. PE-Designer, a dedicated tool for pegRNA selection, provides all possible target sequences, pegRNA extension sequences, and nicking guide RNA sequences together with useful information, and displays the results in an interactive image. PE-Analyzer, a dedicated tool for PE outcome analysis, accepts high-throughput sequencing data, summarizes mutation-related information in a table, and provides interactive graphs. PE-Analyzer was mainly written using JavaScript so that it can analyze several data sets without requiring that huge sequencing data (>100MB) be uploaded to the server, reducing analysis time and increasing personal security. PE-Designer and PE-Analyzer are freely available at http://www.rgenome.net/pe-designer/ and http://www.rgenome.net/pe-analyzer/ without a login process.
Graphical Abstract
Graphical Abstract.
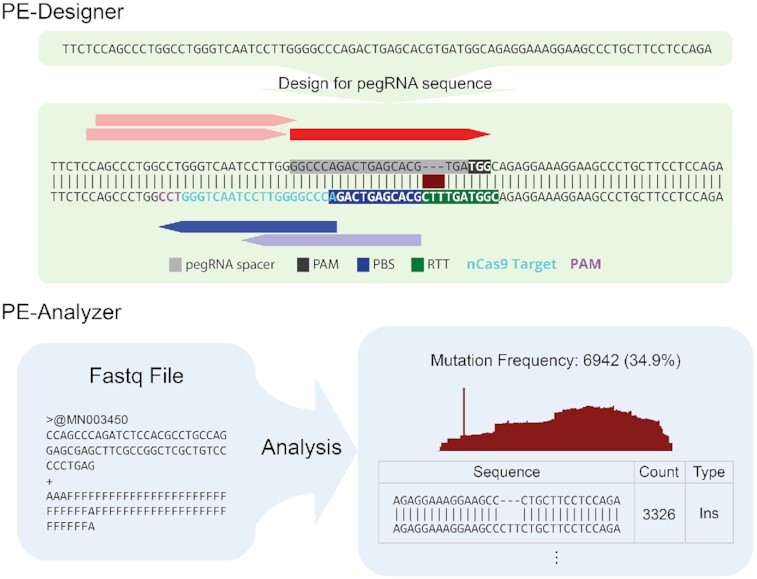
Overviews for input and output of PE-Designer and PE-Analyzer.
INTRODUCTION
Since CRISPR–Cas9 nucleases were first harnessed for targeted gene editing in cells, they have been applied in numerous areas in biology, biotechnology, and medicine (1–3). CRISPR–Cas9 nucleases fundamentally induce DNA double strand breaks (DSBs) at a target site, frequently resulting in insertions and deletions (indels) at the cleaved region (4–8). Although DNA substitutions can be realized using CRISPR–Cas9 nucleases with the addition of a donor DNA, the editing efficiency is usually very low (9). To overcome this deficiency, base editors (BEs), including cytosine base editors and adenine base editors, were developed (10–12). BEs can introduce substitutions at the target site at a high rate (13), but have several limitations; they cannot generate indels or transversion mutations and the desired mutation at the target nucleotide is frequently accompanied by unwanted bystander mutations (14–16).
As an alternative, the Liu group developed prime editors (PEs), which consist of Cas9 nickase linked with a reverse transcriptase (Figure 1A) and are capable of generating insertions, deletions, and substitutions in the targeted sequence without a need for any donor DNA (17). Because of their versatile editing abilities, PEs are attracting a great deal of attention in the genome editing field (18–21), but are more complex in design than other related tools such as CRISPR nucleases and BEs; furthermore, the analysis of PE-generated results requires special considerations. PEs use prime editing guide RNAs (pegRNAs) that are programmed with information about the desired mutation and are more complex than the guide RNAs used with other CRISPR-derived tools. pegRNAs contain a protospacer sequence for recognizing the target sequence, a reverse transcriptase template (RTT) that contains the desired edit, and a primer binding site (PBS) for the activation of reverse transcriptase. Several types of PEs have been developed. In contrast with PE2, which requires only a pegRNA, PE3 also requires a nicking guide RNA (ngRNA) to increase the prime editing efficiency. In their analysis of PE3-mediated outcomes, researchers should consider not only the desired mutations at the target site, but also additional possible mutations derived from the presence of the ngRNA.
Figure 1.

Schematic of a PE and the PE-Designer results panel. (A) Schematic of a PE, highlighting the pegRNA structure. (B) Interactive summary figure on the PE-Designer results page. The possible pegRNA target sequences are colored red and additional ngRNA sequences are colored blue. Each bar can be clicked to select that target sequence. When the amino acid sequence is changed, the altered residue is colored red. (C) Table of possible target sequences provided by PE-Designer. Checking the select button next to a target sequence displays a list of pegRNA extension sequences and additional ngRNA sequences. A red background means that the target is not recommended because the indicated parameter is unfavorable for prime editing.
However, reliable and dedicated tools to aid researchers in choosing pegRNAs to target DNA sequences of interest and to assess prime editing outcomes from high-throughput sequencing data are limited. To address this need, here we present two dedicated web-based prime editing toolkits: PE-Designer and PE-Analyzer. PE-Designer provides all possible protospacer sequences and 3′ extension sequences, as well as additional ngRNA sequences, with useful information including potential off-target sites for 544 organisms including GRCh38 (http://www.ensembl.org/Homo_sapiens) and GRCh37 (https://grch37.ensembl.org/Homo_sapiens). For convenience, PE-Designer directly displays the possible target sites and selected information as an interactive image. PE-Analyzer, an analysis tool for high-throughput sequencing data, can analyze four sequencing data sets at once in the user's web browser without a requirement for uploading very large data sets or installing associated programs; this is possible because PE-Analyzer is fully constructed using the JavaScript language. On the results page, PE-Analyzer displays a table and interactive graphs summarizing the mutation patterns, as well as a sequence information table showing the alignment results of query sequences with the wild-type sequence or a desired sequence.
MATERIALS AND METHODS
Construction of PE-Designer and PE-Analyzer in the CRISPR RGEN Tools server
Both PE-Analyzer and PE-Designer are incorporated into the CRISPR RGEN Tools server, which provides several computational tools and databases for CRISPR nucleases and BEs (22–30). The web server was developed using the Python web framework Django (v 3.1.0, Python 3.6.9) and the web interface was developed using web Bootstrap 3.0 and the JavaScript framework JQuery (v 1.11.0). Access is provided by Nginx (v 1.14.0) with uWSGI (v 2.0.18).
Target searching algorithm of PE-Designer
PE-Designer finds three different elements for pegRNA design: (i) protospacer sequences, with information about their potential associated off-target sites identified using Cas-OFFinder (23), (ii) sequences at the 3′ end of the pegRNA, consisting of a PBS and an RTT with the intended edit for reverse transcription and (iii) additional ngRNA sequences that are used for inducing additional nicks in the non-edited strand to increase the prime editing efficiency of PE3.
Visualizations provided by PE-Designer
In an input panel, PE-Designer uses EMBOSS needle (https://www.ebi.ac.uk/Tools/psa/emboss_needle) to align a given input reference sequence with desired sequences. The alignment results are displayed as an interactive graph to allow direct confirmation of the desired mutation. On the results page, PE-Designer displays a table of results and interactive graphs developed using D3.js library. The data can be filtered without refreshing the results page because AJAX (Asynchronous JavaScript and Extensible Markup Language) is used.
Sequence analysis algorithm used by PE-Analyzer
PE-Analyzer accepts next-generation sequencing (NGS) data that consist of either a pair of Fastq files or a single Fastq file. If the NGS data are compressed, PE-Analyzer reads the compressed Fastq file using JavaScript library ‘pako’ (http://nodeca.github.io/pako/). When PE-Analyzer accepts a pair of Fastq files, PE-Analyzer merges the paired-end reads using JavaScript port of fastq-join, a part of ea-utils (https://code.google.com/archive/p/ea-utils/).
PE-Analyzer finds cleavage sites and defines 15-nt indicator sequences on both sides of the given input reference sequence within a user-defined comparison range (R) parameter. PE-Analyzer selects the sequences, including both indicator sequences with up to a 1 bp mismatch, from the uploaded NGS data. PE-Analyzer counts the number of identical sequences and excludes the sequences with a lower count than a minimum frequency (the default value is 1) because such sequences are considered to include sequencing errors. Then, each sequence is respectively aligned to the reference sequence and the user-defined desired sequence using EMBOSS needle. The filtered sequences are classified into wild-type, substitution, insertion, deletion, and prime edited groups, considering the length of the sequence, a wild-type marker that is sequenced around the cleavage target site, and the additional ngRNA sequence. For example, if a query sequence has the same length as the reference sequence and contains the wild-type marker, it is classified into the wild-type group. If the length of the query and reference sequences is the same but there is no wild-type marker in the query sequence, it is classified into the substitution group. If the length of the query sequence is different than that of the reference sequence, the query is classified into either the insertion or deletion group. If the query sequence contains the desired mutation and no additional mutations, it is classified into the prime edited group.
Visualizations provided by PE-Analyzer
In an input panel, PE-Analyzer accepts the reference sequence and sequences containing the desired mutation(s) from users and displays the alignment results with an interactive graph. PE-Analyzer can run four NGS data sets simultaneously in the user‘s web browser using web worker API in JavaScript. Using the web worker API, PE-Analyzer continuously uploads the progress of the analysis in the browser. In the results page, PE-Analyzer provides buttons to select the data set. PE-Analyzer shows a summary and alignment table and displays the interactive graph using Plotly.js (https://plot.ly/javascript/).
PE-Analyzer also uses EMBOSS needle to align the reference sequences with the desired, prime-edited sequences. The alignment results are displayed as an interactive graph to allow direct confirmation of the desired mutation and can be filtered without refreshing the results page using AJAX.
RESULTS
Web-based design tool for PEs: PE-Designer
Using PE-Designer, researchers can obtain a list of all possible pegRNAs and additional ngRNAs (option for PE3) for inducing prime editing in a given input reference sequence, along with useful information including potential off-target sites associated with each target sequence.
In the input panel, PE-Designer provides five protospacer adjacent motifs (PAMs) recognized by SpCas9 and SpCas9 variants: SpCas9 (5′-NGG-3′ or 5′-NRG-3′), SpCas9-VQR (5′-NGA-3′), SpCas9-VRER (5′-NGCG-3′), NG-Cas9 (5′-NG-3′), and xCas9 3.7 (5′-NGN-3′) and supports pegRNA design for 543 organisms including vertebrates, plants, insects, and bacteria (31–33). Users can input both the reference sequence before prime editing and the expected sequence containing the desired mutation after prime editing of the reference sequence (Supplementary Figure S1). PE-Designer directly aligns the sequences and displays the aligned results with the expected amino acid sequence as an image so that users can confirm the input sequence. The input sequence should consist of ‘ATGC’ to translate to amino acid codons.
After the submit button is clicked, PE-Designer instantly shows all possible protospacer sequences, sequences at the 3′ end of the pegRNA, and additional ngRNA sequences with useful information summarized in a table; when the user makes a change, the tool instantly updates the selected and possible sequences in an interactive image (Figure 1B and C). Certain properties, such as high or low GC contents in the PBS or RTT, extension sequences containing C as the first base, and a short distance between the cleavage site and additional nicking site, are unfavorable for prime editing, and PE-Designer highlights such properties in red. PE-Designer supports this filter and shows the filtered data without a need to refresh the results page. If the selected genome has an Ensembl URL, PE-Designer provides a link to the Ensembl genome browser to display detailed information about potential off-target sites.
Web-based analysis tool for PE outcomes: PE-Analyzer
After PEs are introduced into cells, the mutation efficiency and patterns of mutations can be assessed by high-throughput sequencing. It has been reported that PEs infrequently generate unintended mutations in the newly edited region; thus, it is necessary to analyze the targeted region to know whether the desired edits or unintended mutations are present. With PE-Analyzer, NGS data can be easily analyzed by researchers with just one click in their web browsers without uploading any data to a server and without local tool installation.
PE-Analyzer accepts the NGS data and basic information such as the reference sequence, the target DNA sequence, and information about the expected mutation after prime editing (Supplementary Figure S2). Information about the expected mutation can be received in two formats: (i) the mutation pattern represented by the mutation type, position, and sequence, which is similar to the format used in a previous paper (17) or (ii) the edited sequence showing the desired mutation in the reference sequence. For experiments involving PE3, PE-Analyzer will also receive the ngRNA sequence and will automatically change the analysis parameters so that there are no missing data due to the ngRNA recognizing a site far from the site of the desired mutation.
On the results page, the analysis results are summarized in a table displaying eight categories of information (Figure 2A): (i) ‘Total Sequences’ is the number of reads in the received NGS file, (ii) ‘With both indicator sequences’ is the number of reads having both indicator sequences, (iii) ‘More than minimum frequency’ is the sum of the number of reads with a count higher than the minimum frequency when a given sequence is counted, (iv–vi) ‘Insertions’, ‘Deletions’, and ‘Substitutions’ are the number of reads for each type of mutation, (vii) ‘Mutation frequency’ is the combined number of reads of insertions, deletions, and substitutions, (viii) ‘Prime editing’ is the number of reads containing the desired sequence without unintended mutations in the region near the desired sequence or the site targeted by an additional ngRNA. The distributions of each type of mutation pattern are displayed as interactive graphs, in which the x-axis is the reference sequence or expected sequence (Figure 2B). The alignment results are categorized by mutation pattern and displayed as a table at the bottom of the page. The alignment table can be filtered by clicking the button for the appropriate category or by inputing a specific sequence (Figure 2C). PE-Analyzer also offers the ability to undertake multiple analyses for researchers who have several NGS data sets for the same target. Researchers can control the number of data sets by selecting ‘slot count’ on the main page and can see the results of each analysis by selecting the button at the top of the results page. Users can also test PE-Analyzer using an example data set available at http://www.rgenome.net/pe-analyzer/example.
Figure 2.

PE-Analyzer results panel. (A) Summary table indicating the number of sequence reads for each type of mutation. ‘Prime editing’ indicates the count of the desired sequence. (B) Bar graphs showing the distribution of insertions, deletions, and substitutions. The graphs can be changed by clicking the buttons above the graphs to switch the alignment of query sequences against either reference or expected sequences. (C) Table of alignment results. The results can be changed by clicking the buttons to switch the alignment of query sequences against either reference or expected sequences, or to filter the results by the mutation type; specific sequences can be found by entering that information.
DISCUSSION
In this paper, we introduce two easy-to-use web tools for studies involving PEs: PE-Designer and PE-Analyzer. PE-Designer provides all possible pegRNA sequences and additional ngRNA sequences for efficiently generating desired mutations, along with useful information such as the GC content and potential off-target sites. Recently, a few web-based tools, including PrimeDesign (34), pegFinder (35) and Prime Editing Design Tool (https://primeedit.nygenome.org/), were developed for pegRNA design. PrimeDesign provides useful information such as a pegRNA secondary structure and scores based on the predicted specificity. pegFinder also provides ranking system based on predicted on-target/off-target scores and Prime Editing Design Tool is a dedicated platform for conducting disease modelling or therapeutic editing via PEs. In addition, DeepPE (36) can predict PE2 efficiency of each pegRNA, which is calculated by deep learning-based computational models. Although PE-Designer does not support a scoring system, it can strictly find off-target sites for pegRNAs and ngRNAs using Cas-OFFinder. Furthermore, all information and selected options are summarized in an interactive image, so that appropriate PE targets can be easily selected. In PE-Designer, the length of the input sequence is presently limited to ≤1000 nucleotides, because the server becomes overloaded when several users input longer sequences, with many possible target sequences, at the same time. This limitation will be solved by a server upgrade in the future. Alternatively, for more than 1000 nucleotides, users can use a command-line version of PE-Designer (https://github.com/Gue-ho/PE-Designer).
PE-Analyzer is a web tool for the rapid analysis of NGS data obtained to assess PE outcomes; multiple data sets can be analyzed at one time in a user's web browser without a need to upload data to a server or install an additional program. Previously, we and other groups developed web-based analysis platforms, such as CRISPResso v1 (37), v2 (38), and Cas-Analyzer (22) for original CRISPR nucleases and BE-Analyzer (24) for BEs. Those pre-existing tools might be used for PE2 outcome analysis but they are confined to evaluate NGS data of PE3 system. PE-Analyzer is a dedicated assessment tool for PEs, including both PE2 and PE3. PE-Analyzer can automatically find a pegRNA and a ngRNA in input sequences optional for PE3, and the NGS data are aligned to the reference sequence as well as the expected sequence. Furthermore, using PE-Analyzer, multiple data sets can be analyzed at one time in a user's web browser without a need to upload data to a server or install an additional program, which substantially reduces the analysis time as a web-based tool. As an example, PE-Analyzer can analyze four data sets (1.03GB each, 4.12GB in total) at once in 233 seconds in a Chrome browser with our computing materials (AMD Ryzen 7 3800X and 32GB RAM). PE-Analyzer displays a table summarizing the results, interactive graphs, and alignment results categorized by each type of mutation pattern on the results page. We are continuously communicating with users to make updates to include additional organisms and PAMs. We believe that our web-based prime editing toolkits will be quite useful for many researchers in the genome editing field.
DATA AVAILABILITY
PE-Designer and PE-Analyzer are freely available at http://www.rgenome.net/pe-designer and http://www.rgenome.net/pe-analyzer without a login process. PE-Designer for standalone version is available at https://github.com/Gue-ho/PE-Designer.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Heather McDonald for English language editing.
Contributor Information
Gue-Ho Hwang, Department of Chemistry and Research Institute for Convergence of Basic Sciences, Hanyang University, Seoul, South Korea.
You Kyeong Jeong, Department of Chemistry and Research Institute for Convergence of Basic Sciences, Hanyang University, Seoul, South Korea.
Omer Habib, Department of Chemistry and Research Institute for Convergence of Basic Sciences, Hanyang University, Seoul, South Korea.
Sung-Ah Hong, Department of Chemistry and Research Institute for Convergence of Basic Sciences, Hanyang University, Seoul, South Korea.
Kayeong Lim, Center for Genome Engineering, Institute for Basic Science, Daejeon, South Korea.
Jin-Soo Kim, Center for Genome Engineering, Institute for Basic Science, Daejeon, South Korea; Department of Chemistry, Seoul National University, Seoul, South Korea.
Sangsu Bae, Department of Chemistry and Research Institute for Convergence of Basic Sciences, Hanyang University, Seoul, South Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institute for Basic Science [IBS-R021-D1 to J.-S.K.]; National Research Foundation of Korea [2021M3A9H3015389]; Korea Institute for Advancement of Technology project [P0015362]; New Breeding Technologies Development Program [PJ01487401202001 to S.B.]. Funding for open access charge: National Research Foundation of Korea.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wu Y., Liang D., Wang Y., Bai M., Tang W., Bao S., Yan Z., Li D., Li J.. Correction of a genetic disease in mouse via use of CRISPR–Cas9. Cell Stem Cell. 2013; 13:659–662. [DOI] [PubMed] [Google Scholar]
- 2. Sun Y., Zhang X., Wu C., He Y., Ma Y., Hou H., Guo X., Du W., Zhao Y., Xia L.. Engineering Herbicide-Resistant Rice Plants through CRISPR/Cas9-Mediated homologous recombination of acetolactate synthase. Mol Plant. 2016; 9:628–631. [DOI] [PubMed] [Google Scholar]
- 3. Hsu P.D., Lander E.S., Zhang F.. Development and applications of CRISPR–Cas9 for genome engineering. Cell. 2014; 157:1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doudna J.A., Charpentier E.. Genome editing. The new frontier of genome engineering with CRISPR–Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 5. Sander J.D., Joung J.K.. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014; 32:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O.. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014; 156:935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim H., Kim J.S.. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014; 15:321–334. [DOI] [PubMed] [Google Scholar]
- 9. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R.. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016; 533:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R.. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017; 551:464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y.et al.. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016; 353:aaf8729. [DOI] [PubMed] [Google Scholar]
- 13. Kim H.S., Jeong Y.K., Hur J.K., Kim J.S., Bae S.. Adenine base editors catalyze cytosine conversions in human cells. Nat. Biotechnol. 2019; 37:1145–1148. [DOI] [PubMed] [Google Scholar]
- 14. Zuo E., Sun Y., Wei W., Yuan T., Ying W., Sun H., Yuan L., Steinmetz L.M., Li Y., Yang H.. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019; 364:289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeong Y.K., Song B., Bae S.. Current status and challenges of DNA base editing tools. Mol. Ther. 2020; 28:1938–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grunewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K.. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019; 569:433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A.et al.. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019; 576:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin Q., Zong Y., Xue C., Wang S., Jin S., Zhu Z., Wang Y., Anzalone A.V., Raguram A., Doman J.L.et al.. Prime genome editing in rice and wheat. Nat. Biotechnol. 2020; 38:582–585. [DOI] [PubMed] [Google Scholar]
- 19. Kim Y., Hong S.-A., Yu J., Eom J., Jang K., Lee S.-N., Woo J.-S., Jeong J., Bae S., Choi D.. Adenine base editing and prime editing of chemically derived hepatic progenitors rescue genetic liver disease. cell stem cell. 2020; doi:10.1016/j.stem.2021.04.010. [DOI] [PubMed] [Google Scholar]
- 20. Liu Y., Li X., He S., Huang S., Li C., Chen Y., Liu Z., Huang X., Wang X.. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020; 6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Surun D., Schneider A., Mircetic J., Neumann K., Lansing F., Paszkowski-Rogacz M., Hanchen V., Lee-Kirsch M.A., Buchholz F.. Efficient generation and correction of mutations in human iPS cells utilizing mRNAs of CRISPR base editors and prime editors. Genes (Basel). 2020; 11:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park J., Lim K., Kim J.S., Bae S.. Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics. 2017; 33:286–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bae S., Park J., Kim J.S.. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014; 30:1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hwang G.H., Park J., Lim K., Kim S., Yu J., Yu E., Kim S.T., Eils R., Kim J.S., Bae S.. Web-based design and analysis tools for CRISPR base editing. BMC Bioinformatics. 2018; 19:542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park J., Childs L., Kim D., Hwang G.H., Kim S., Kim S.T., Kim J.S., Bae S.. Digenome-seq web tool for profiling CRISPR specificity. Nat. Methods. 2017; 14:548–549. [DOI] [PubMed] [Google Scholar]
- 26. Hwang G.H., Yu J., Yang S., Son W.J., Lim K., Kim H.S., Kim J.S., Bae S.. CRISPR-sub: analysis of DNA substitution mutations caused by CRISPR–Cas9 in human cells. Comput Struct Biotechnol J. 2020; 18:1686–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Park J., Bae S.. Cpf1-Database: web-based genome-wide guide RNA library design for gene knockout screens using CRISPR-Cpf1. Bioinformatics. 2018; 34:1077–1079. [DOI] [PubMed] [Google Scholar]
- 28. Park J., Kim J.S., Bae S.. Cas-Database: web-based genome-wide guide RNA library design for gene knockout screens using CRISPR–Cas9. Bioinformatics. 2016; 32:2017–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park J., Bae S., Kim J.S.. Cas-Designer: a web-based tool for choice of CRISPR–Cas9 target sites. Bioinformatics. 2015; 31:4014–4016. [DOI] [PubMed] [Google Scholar]
- 30. Bae S., Kweon J., Kim H.S., Kim J.S.. Microhomology-based choice of Cas9 nuclease target sites. Nat. Methods. 2014; 11:705–706. [DOI] [PubMed] [Google Scholar]
- 31. Kleinstiver B.P., Prew M.S., Tsai S.Q., Topkar V.V., Nguyen N.T., Zheng Z., Gonzales A.P., Li Z., Peterson R.T., Yeh J.R.et al.. Engineered CRISPR–Cas9 nucleases with altered PAM specificities. Nature. 2015; 523:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z.et al.. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018; 556:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H.et al.. Engineered CRISPR–Cas9 nuclease with expanded targeting space. Science. 2018; 361:1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hsu J.Y., Grunewald J., Szalay R., Shih J., Anzalone A.V., Lam K.C., Shen M.W., Petri K., Liu D.R., Joung J.K.et al.. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 2021; 12:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chow R.D., Chen J.S., Shen J., Chen S.. A web tool for the design of prime-editing guide RNAs. Nat Biomed Eng. 2021; 5:190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim H.K., Yu G., Park J., Min S., Lee S., Yoon S., Kim H.H.. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 2021; 39:198–206. [DOI] [PubMed] [Google Scholar]
- 37. Pinello L., Canver M.C., Hoban M.D., Orkin S.H., Kohn D.B., Bauer D.E., Yuan G.C.. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 2016; 34:695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E.et al.. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019; 37:224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PE-Designer and PE-Analyzer are freely available at http://www.rgenome.net/pe-designer and http://www.rgenome.net/pe-analyzer without a login process. PE-Designer for standalone version is available at https://github.com/Gue-ho/PE-Designer.