

Abstract
The presence of polyfunctional CD4+ T cells is often associated with favorable antitumor immunity. We report here that persistent activation of signal transducer and activator of transcription 5 (STAT5) in tumor-specific CD4+ T cells drives the development of polyfunctional T cells. We showed that ectopic expression of a constitutively active form of murine STAT5A (CASTAT5) enabled tumor-specific CD4+ T cells to undergo robust expansion, infiltrate tumors vigorously and elicit antitumor CD8+ T cell responses in a CD4+ T-cell adoptive transfer model system. Integrated epigenomic and transcriptomic analysis revealed that CASTAT5 induced genome-wide chromatin remodeling in CD4+ T cells and established a distinct epigenetic and transcriptional landscape. Single-cell RNA sequencing analysis further identified a subset of CASTAT5-transduced CD4+ T cells with a molecular signature indicative of progenitor polyfunctional T cells. The therapeutic significance of CASTAT5 came from our finding that adoptive transfer of T cells engineered to co-express CD19-targeting chimeric antigen receptor (CAR) and CASTAT5 gave rise to polyfunctional CD4+ CAR T cells in a mouse B-cell lymphoma model. Notably, the optimal therapeutic outcome was obtained when both CD4+ and CD8+ CAR T cells were transduced with CASTAT5, indicating that CASTAT5 facilitates productive CD4 help to CD8+ T cells. Furthermore, we provide evidence that CASTAT5 is functional in primary human CD4+ T cells, underscoring its potential clinical relevance. Our results implicate STAT5 as a valid candidate for T cell engineering to generate polyfunctional, exhaustion-resistant, and tumor-tropic antitumor CD4+ T cells to potentiate adoptive T-cell therapy for cancer.
One sentence summary:
Persistent STAT5 activation endows antitumor CD4+ T cells with desirable functional features that optimize adoptive T cell therapy.
Introduction
Recent advances in adoptive T-cell therapy (ACT), especially CD19-targeting chimeric antigen receptor (CAR) T-cell therapy (CD19CART), have highlighted the potential of immunotherapy to achieve durable and curative patient outcomes(1, 2. However, even for the well-developed CD19CART, many patients failed to respond to the treatment, or succumbed to late relapse after initial response(3). Moreover, by far ACT in general has not been effective in treating most solid tumors. The major barriers to effective ACT include deficiencies in donor T cell expansion, persistence and tumor-infiltration, as well as loss of effector functions in the immunosuppressive tumor microenvironment (TME)(4–6). There is increasing demand for further strategies that can overcome these barriers so as to improve the efficacy of ACT.
The infusion products of ACT usually contain both CD8+ and CD4+ T lymphocytes, and CD8+ T cells are considered as the major effector population that mediates the tumor-killing effects. Recent studies demonstrate that tumor-specific CD8+ T cells are susceptible to functional exhaustion in the TME, a process characterized by progressive loss of effector functions, which may contribute to failed tumor growth control(7, 8). CD4+ T cells are regarded as helper cells that act to enhance CD8+ T cell responses(9–12). With relevance to CD19CAR T cell therapy, the co-presence of CD4+ and CD8+ T cells in the infusion products was found to correlate with favorable patient outcomes(13), although the mechanism of CD4+-CD8+ T cell cooperation awaits to be elucidated. A likely possibility is that CD4+ helper T cells can prevent CD8+ T cell exhaustion in the TME, as it has been demonstrated in the setting of chronic viral infection(14). Apart from their role as helpers, CD4+ T cells can act as the driver of antitumor immune responses in ACT. Case report studies showed that adoptive transfer of ex vivo expanded tumor-specific CD4+ T cell clones can lead to tumor regression in patients with metastatic tumors(15, 16). A recent clinical study demonstrated the safety and efficacy of administering CD4+ T cells genetically engineered to express tumor-specific T cell receptors (TCRs)(17). CD4+ T cell-based CAR T cell therapy also showed robust antitumor effects in some preclinical models(18, 19). Whether CD4+ T cells are used as helpers or “stand-alone” effectors in ACT, their functional status appears to be a key determinant of the therapy outcome. The presence of CD4+ T cells with a polyfunctional phenotype, characterized by concomitant production of multiple inflammatory cytokines, has been associated with favorable immunological events, including dendritic cell activation, antigen epitope spreading, immunostimulatory cytokine milieu, CD8+ T cell activation, and ultimately, improved antitumor effects in tumor-bearing mice or cancer patients(15, 16, 20–25). However, CD4+ T cells are not immune to tumor-induced dysfunction or exhaustion(26), and exhausted CD4+ T cells may exacerbate CD8+ T cell dysfunction(27). Thus, finding ways to generate exhaustion-resistant, polyfunctional CD4+ T cells may have important implications for ACT.
We previously reported that CD4+ T cells exposed to interleukin 7 (IL-7) during antigenic stimulation in vitro can acquire polyfunctionality in a STAT5-dependent manner(28). However, inducing CD4 polyfunctionality with IL-7 may encounter some limitations in clinical settings. For instance, patient-derived T cells, which presumably contain exhausted CD4+ T cells, may not respond to IL-7 and become polyfunctional. Moreover, it is uncertain whether IL-7-induced CD4 polyfunctionality, acquired during in vitro stimulation, can sustain in the TME after T cell infusion. We hypothesized that the limitations restricting the efficiency of IL-7 induction of CD4 polyfunctionality can be overcome by genetically modifying T cells to allow cell-intrinsic, continuous engagement of the IL-7 signaling pathway through STAT5 activation. In the current study, we reveal the impact of persistent STAT5 signaling, transmitted by a constitutively active form of STAT5A (CASTAT5)(29), on CD4+ T cell phenotype and function in the context of ACT. We report that overexpression of CASTAT5 induces genome-wide transcriptional and epigenetic remodeling in tumor-specific CD4+ T cells, endowing these cells polyfunctionality, exhaustion-resistance and tumor-infiltrating capability. Importantly, CASTAT5 can markedly improve the expansion and persistence of CD19CAR T cells, resulting in high cure rate in mice with advanced lymphoma. These findings validate persistent activation of STAT5 as a viable strategy to generate polyfunctional CD4+ T cells for the purpose of cancer immunotherapy.
Results
CASTAT5-transduced CD4+ T cells undergo robust expansion and acquire a polyfunctional phenotype upon adoptive transfer
We set out to examine the impact of persistent STAT5 activation, transmitted by CASTAT5, on T cell expansion, functional status and migration in vivo in a mouse model of ACT. Mice with large established HA-expressing B-cell lymphoma A20 tumors (A20HA) received lymphodepleting chemotherapy (CTX) with cyclophosphamide followed by adoptive transfer one day later of either CASTAT5-transduced (CASTAT5 CD4) or mock-transduced (control CD4) HA-specific CD4+ T cells derived from 6.5 TCR Tg mice (Fig. 1A). To evaluate donor T cell expansion in vivo, tail blood from each mouse was collected weekly for flow cytometry analysis. CASTAT5 CD4+ T cells, which exhibited increased levels of phosphorylated STAT5 (pSTAT5) (Fig. 1B), underwent robust and sustained expansion, reaching a plateau (19±4.6%) about 3 weeks after transfer, whereas control CD4+ T cells only had transient and modest expansion (Fig. 1C). The proliferative advantage of CASTAT5 CD4+ T cells over the control CD4+ T cells was also confirmed in vitro. Upon serial repeated stimulations, CASTAT5 CD4+ T cells showed increased cell division and accumulation compared to control CD4+ T cells (fig. S1A–C), suggesting enhanced survival capability.
Fig. 1. CASTAT5-transduced CD4+ T cells undergo vigorous expansion, acquire a polyfunctional phenotype and robustly infiltrate the tumor upon adoptive transfer.
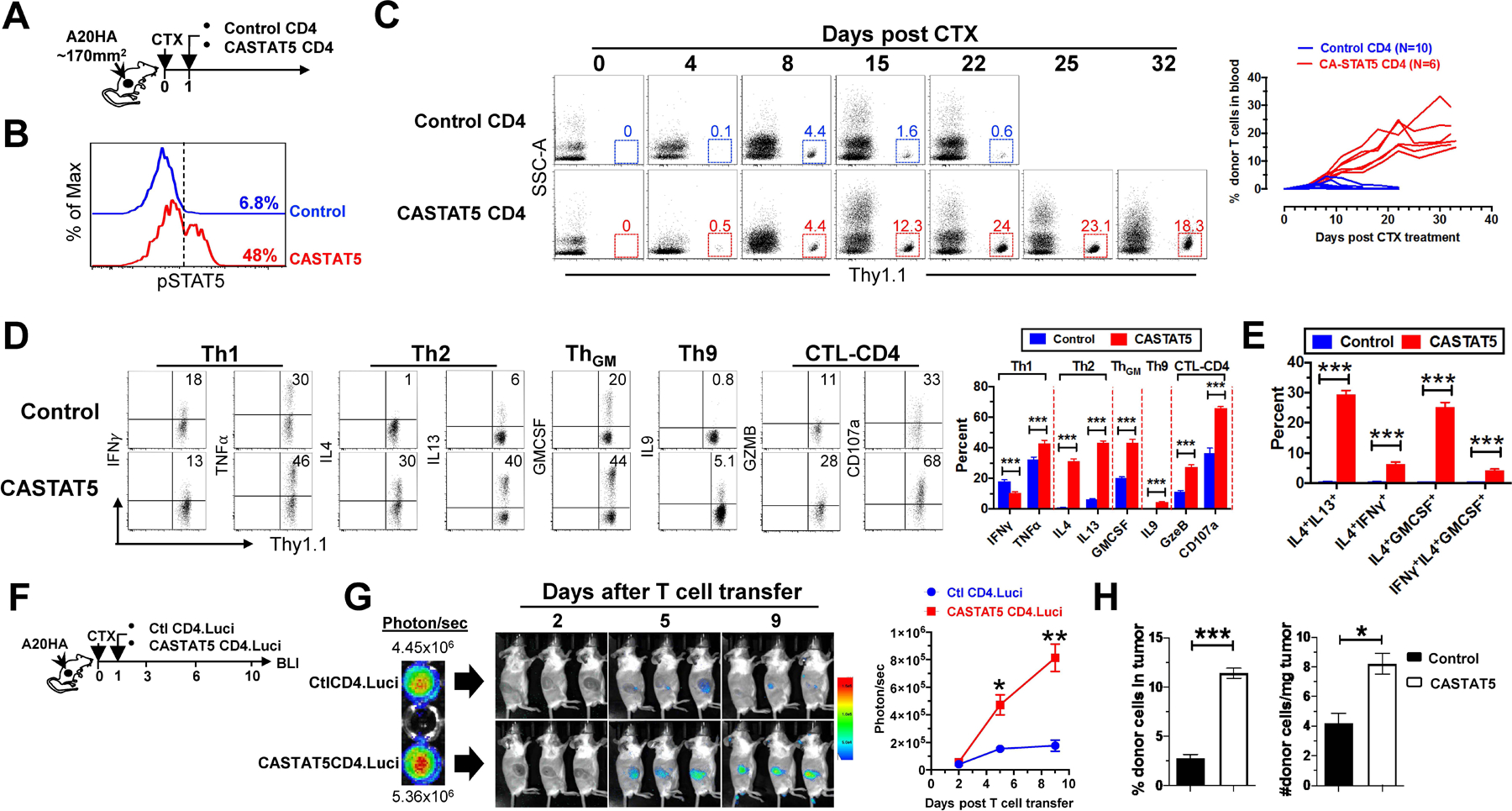
(A) The schema depicts the timeline of experimental procedures. (B) Detection of pSTAT5 in donor CD4+ T cells before adoptive transfer. The levels of phosphorylated STAT5 (pSTAT5) in mock-transduced (Control) or CASTAT5-transduced HA-specific CD4+ T cells were determined by flow cytometry. Numbers indicate the percent of pSTAT5+ cells in total CD4+ T cells. (C) Kinetics of donor CD4+ T cell expansion in peripheral blood. At the indicated time points, blood was collected from each mouse via tail vein and analyzed for the frequency of the donor T cells by flow cytometry. Representative dot plots at specific time points are shown, and the numbers represent the percentages of donor CD4+ T cells. The graph at right summarizes the frequencies of donor T cells against time. The number of mice in each group is given. (D) Cytokine profiles of donor CD4+ T cells. Ten days after T cell transfer, spleen cells were harvested and subjected to intracellular cytokine staining (ICS) following a transient stimulation with HA peptide. Representative dot plots shown depict the cytokine profiles of the donor CD4+ T cells. Numbers indicate the percentages of cytokine-producing cells in total donor CD4+ T cells. The results shown as mean ± s.d. are summarized in the bar graph at right. Data are pooled from three independent experiments. (E) Polyfunctional status of the donor CD4+ T cells. Bar graph summarizes the percent of donor CD4+ T cells co-expressing the indicated cytokines. (F-H) Migration and retention of donor CD4+ T cells in tumor. (F) The schema depicts the experimental procedures. HA-specific CD4+ T cells were transduced with luci-carrying retrovirus alone (Ctl CD4.Luci) or co-transduced with luci- and CASTAT5-carrying retrovirus (CASTAT5 CD4.Luci). Before T cell transfer, BLI was conducted for T cells in vitro to evaluate the transduction efficiency of luciferase gene (G, left panel). After T cell transfer, BLI was conducted periodically to visualize luciferase-expressing donor CD4+ T cells in vivo. Representative images of mice in each group at specific time points are shown (G, middle panel). Results of in vivo T cell luciferase signal intensity quantified as mean ± s.d. of three mice per group are summarized in graph shown at right. The enhanced infiltration and accumulation of CASTAT5 CD4+ T cells in tumor compared to control CD4+ T cells was confirmed by flow cytometry analysis and cell counting (H). Following the experimental procedures depicted in (A), tumor masses were excised and analyzed 7 days after T cell transfer. The frequencies of tumor-infiltrating donor CD4+ T cells were examined by flow cytometry and summarized in bar graph (H, left panel). The absolute donor CD4+ T cell numbers in tumor are normalized to the weight of tumor mass (H, right panel). The formula for calculating donor CD4+ T cell numbers is: total cell number x percent of donor CD4+ T cells. Data are shown as mean ± s.d. of three mice per group. *p < 0.05, **p < 0.01, ***p < 0.001.
A cohort of mice were sacrificed 10 days after T cell transfer and donor T cells in the spleens were assessed for cytokine production by intracellular cytokine staining (ICS). Fig. 1D shows that CASTAT5 CD4+ T cells had somewhat mixed changes in Th1 cytokine productions compared to the control CD4+ T cells, for example, slight reduction in IFN-γ but modest gain in TNF-α; notably, CASTAT5 CD4+ T cells had markedly elevated productions of cytokines characteristic of the Th2 (IL-4, IL-13), Th9 (IL-9) and ThGM (GM-CSF, also known as Csf2) lineages, in addition to cytolytic molecule granzyme B (GzmB) and degranulation marker CD107a (Lamp1), which are hallmarks of cytolytic CD4+ T cells (CTL-CD4). Cytokine co-staining analysis revealed at the single cell level that CASTAT5 enabled CD4+ T cells to concurrently express 2 or 3 cytokines characteristic of different Th lineages, exhibiting a unique polyfunctional phenotype (Fig. 1E).
CASTAT5 enhances CD4+ T cell tumor infiltration and retention
We next examined how CASTAT5 affects the tumor-infiltrating capacity of tumor-specific CD4+ T cells. To track and quantify tumor-infiltrating donor T cells, T cells were engineered to express luciferase (luci) so that they can be visualized by bioluminescence imaging (BLI). As shown in Fig. 1F schema, tumor-specific CD4+ T cells, transduced with the luci-carrying viral vector alone as control (Ctl CD4.luci) or co-transduced with luci and CASTAT5-carrying viral vectors (CASTAT5 CD4.luci), were transferred into CTX-conditioned tumor-bearing mice. The control and CASTAT5 CD4+ T cells showed comparable photon intensities before transfer, indicating similar transduction efficiency of the luciferase gene (Fig. 1G left panel). Notably, the bioluminescent photon signals from CASTAT5 CD4+ T cells were readily detectable in tumors on day 5 and intensified on day 9, whereas the BLI signals from intratumoral control CD4+ T cells were modest on both time points (Fig. 1G BLI imaging and summary graph). To confirm the imaging results, we conducted a separate experiment to enumerate the number of tumor-infiltrating donor T cells on day 7. As shown in Fig. 1H, CASTAT5 CD4+ T cells showed enhanced accumulation in tumors, reflected by their increased frequencies and absolute numbers, compared to the control CD4+ T cells. The results suggest that CASTAT5 enhances CD4+ T cell tumor infiltration and retention.
CASTAT5 CD4+ T cells eradicate advanced tumors through eliciting antitumor CD8+ T cell responses
The antitumor efficacy of ACT was evaluated by monitoring tumor growth. Fig. 2A shows that CTX conditioning alone transiently delayed tumor growth compared to untreated mice; although adoptive transfer of control CD4+ T cells after CTX resulted in further delay in tumor growth, all mice later succumbed to tumor relapse. In sharp contrast, adoptive transfer of CASTAT5 CD4 T cells after CTX led to complete regression of large well-vascularized tumors in all treated mice (Fig. 2A–B). CD4+ T cells can activate a multitude of immune cells, especially cytotoxic CD8+ T cells, to mediate tumor rejection(12). To investigate the role of endogenous CD8+ T cells in the curative effect of CASTAT5 CD4+ T cells, mice were injected with depleting antibody to remove CD8+ T cells after receiving CASTAT5 CD4+ T cell infusion. Fig. 2C shows that CD8+ T cell depletion abolished the curative effect of CASTAT5 CD4+ T cells, indicating that the host CD8+ T cells were a critical component of CASTAT5 CD4+ T cell-induced antitumor immunity. The result implied that the endogenous CD8+ T cells might be primed against tumor-associated antigens. To examine CD8+ T cell reactivity toward tumors, host CD8+ T cells were isolated from mice 21 days after receiving control or CASTAT5 CD4+ T cell infusion, at which time mice in the former group started to have relapse while mice in the latter group had complete tumor regression (Fig. 2A). Purified host CD8+ T cells were labeled with violet dye and co-cultured with irradiated tumor cells (Fig. 2D schema). 6 days after culture, CD8+ T cells were evaluated for cell division (violet dye dilution) and activation markers (CD25 and CD44). Fig. 2D shows that host CD8+ T cells from the mice receiving control CD4+ T cells barely divided (1.4±0.3%) in response to A20HA, the original tumor inoculants; in contrast, a significant fraction (40.7±3.1%) of CD8+ T cells from the mice receiving CASTAT5 CD4+ T cells underwent multiple rounds of cell division and expressed high levels of CD25 and CD44 in response to A20HA. The results imply that a sizable fraction of endogenous CD8+ T cells were primed against A20HA cells in vivo in the presence of CASTAT5 CD4+ T cells. Moreover, a smaller but noticeable fraction of host CD8+ T cells (10.5±3.2%) from the mice receiving CASTAT5 CD4+ T cells responded to A20WT cells (the parental wild-type A20 cells that don’t express HA), whereas the fraction of A20WT-reactive CD8+ T cells from the mice receiving control CD4+ T cells was marginal (4.2±2.3%). Of note, host CD8+ T cells from neither group were reactive to MOPC315 cells, a plasmacytoma cell line unrelated to A20 cells. The results suggest that the endogenous CD8+ T cells were primed against not only HA, the antigen targeted by CASTAT5 CD4+ T cells, but also other A20-derived tumor-associated antigens, indicating the occurrence of epitope spreading in the presence of CASTAT5 CD4+ T cells. It has been well-established that CD8αCD11c+CD103+ dendritic cells can effectively activate CD8+ T cells through cross-priming(30, 31). Indeed, this DC subset increased significantly in both tumor draining lymph nodes (dLNs) and spleens in mice receiving CASTAT5 CD4+ T cells (Fig. 2E), suggesting the involvement of DCs in bridging the crosstalk between the donor CASTAT5 CD4+ T cells and the endogenous CD8+ T cells.
Fig. 2. CASTAT5 CD4+ T cell-mediated tumor rejection is dependent on host CD8+ T cells and associated with antigen epitope spreading and DC activation.
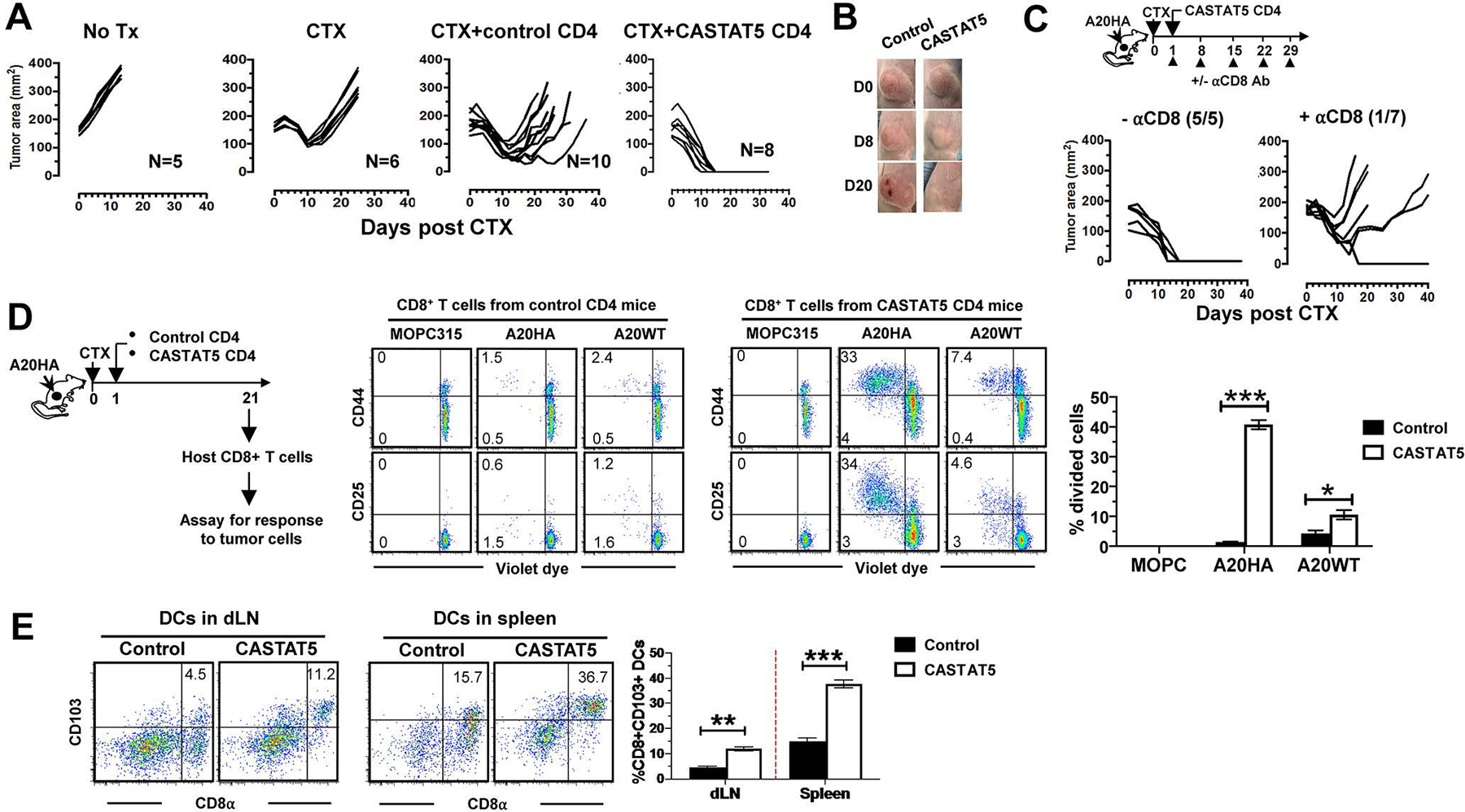
(A) CASTAT5 CD4+ T cells mediate complete rejection of advanced tumors. Following the experimental time line depicted in Fig. 1A, mice with established A20HA tumors were randomly assigned into four groups to receive the specified treatments. Tumor growth curves of mice in each group are shown. The number of mice in each group is given. (B) Representative images of tumors in mice before treatment and at different time points after treatment. (C) Host CD8+ T cells contribute to CASTAT5 CD4+ T cell-mediated tumor rejection. The schema depicts the timeline of experimental procedures. Tumor growth curves indicate the impact of host CD8+ T cells on CASTAT5 CD4+ T cell-mediated tumor rejection. The numbers denote the number of tumor-free mice out of all treated mice. (D) Host CD8+ T cells are reactive to HA and other tumor-associated antigens. Following the timeline depicted in the schema, purified, violet dye-labeled host CD8+ T cells were incubated with equal number of irradiated A20HA, A20WT or MOPC315 cells. 6 days later, CD8+ T cells were evaluated for cell division (violet dye dilution) and activation (CD25 and CD44) by flow cytometry. Representative dot plots are shown. The numbers indicate the percentage of cells in the corresponding quadrant. The frequencies of divided CD8+ T cells under each culture condition are summarized in bar graphs and shown as mean ± s.d. of triplicate cultures. Data shown are representative of two independent experiments. (E) Frequencies of CD8α+CD103+CD11c+ dendritic cells in mice after treatment. Tumor-bearing mice were treated as described in Fig. 1A. 10 days after T cell transfer, mice were sacrificed to collect tumor draining lymph nodes (dLNs) and spleens for flow cytometry analysis. Representative dot plots are shown. The results are summarized in the bar graph shown as mean ±s.d. of three sample per group. Date are representative of two independent experiments. **p < 0.01, ***p < 0.001.
Aberrant activation of STAT5 has been associated with leukemic transformation(32–37), raising the question whether CASTAT5-transduced T cells would become leukemic. To address this, we examined whether CASTAT5-transduced T cells become immortal in vitro. We found that CASTAT5 CD4+ T cells died out when cultured in the absence of antigenic stimulation or growth factors, though in a delayed pace compared to control CD4+ T cells, (fig. S1D). We further evaluated the leukemic potential of CASTAT5-transduced T cells in vivo. Splenocytes isolated from mice (CD45.2+/+) that became tumor-free after receiving CASTAT5 CD4+ T cells were adoptively transferred into naïve CD45.1+/+ recipients (fig. S1E). No gross abnormality was observed in the recipient mice 60 days after splenocyte infusion, and the percentage of CASTAT5 CD4+ T cells in the CD4+ T cell population of the input splenocytes remained unchanged (fig. S1F–G), arguing against leukemia development of CASTAT5-transduced CD4+ T cells within the experiment timeframe. Together, these data suggest that CASTAT5 transduction by itself may not be sufficient to make primary T cells immortal or leukemic.
CASTAT5 drives genome-wide transcriptional and epigenetic remodeling in CD4+ T cells
To understand at the molecular level the biological impact of CASTAT5 on CD4+ T cells, we performed bulk RNA sequencing (RNAseq) and assay for transposase-accessible chromatin using sequencing (ATACseq) on FACS-sorted donor CD4+ T cells (Fig. 3A). RNAseq analysis demonstrates a clear separation of CASTAT5 CD4+ T cells from control CD4+ T cells (fig. S2A–B). Using absolute log2 fold change (log2FC) ≥ 1 and adjusted p-value <0.05 as cutoff values, we identified 1695 up-regulated and 643 down-regulated genes in CASTAT5 CD4+ T cells as compared to control CD4+ T cells, respectively (fig. S2C and Supplementary Table 1). Heatmap indicates the markedly different gene expression patterns between control CD4+ and CASTAT5 CD4+ T cells (fig. S2D). Similarly, ATACseq analysis identified 9438 and 4913 peaks with increased or decreased chromatin accessibility in CASTAT5 CD4+ T cells as compared to control CD4+ T cells (absolute log2FC ≥ 1 and adjusted p-value <0.05), respectively (fig. S3A–B) and reveals clear epigenetic differences as shown in the heatmap (fig. S3C). A majority (about 60%) of the differential ATACseq peaks are located in the non-coding regions of the genome (fig. S3D). Gene set enrichment analysis (GSEA) plots show that genes associated with more accessible chromatin had more overlaps with up-regulated genes than down-regulated genes, and vice versa (Fig. 3B), suggesting that gene expression changes are highly correlated with chromatin changes. We compared the genome-wide alterations in chromatin accessibility landscape with the corresponding gene expression changes. As illustrated in Fig. 3C, the increases in read densities seen in CASTAT5 as compared to control CD4+ T cells are positively correlated with increases in gene expression and vice versa. Furthermore, ingenuity pathway analysis (IPA) of RNAseq data and ATACseq data results in highly consistent pathway identification (Fig. 3D).
Fig. 3. Chromatin accessibility and transcriptome profiling identifies key regulatory molecules.
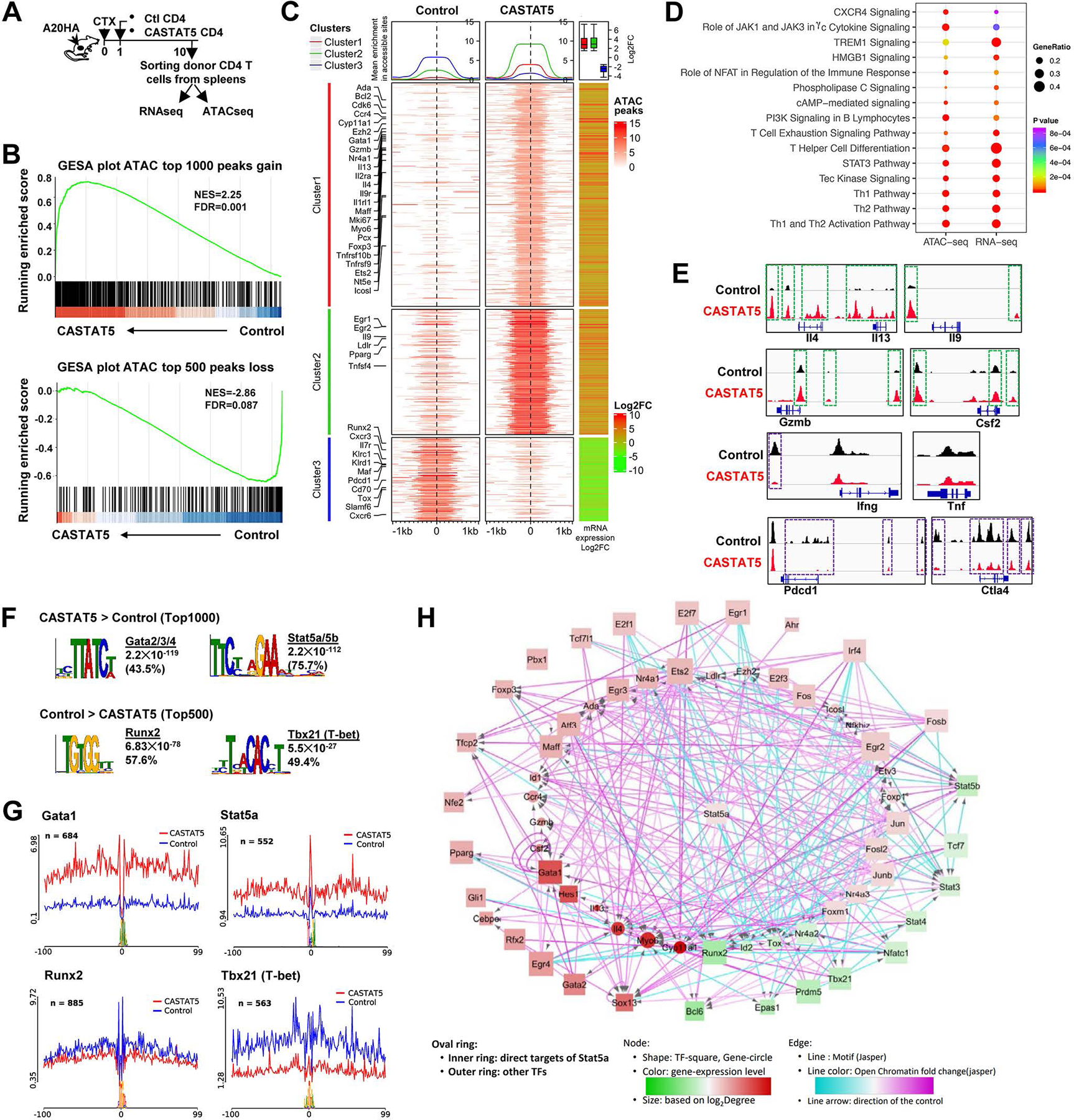
(A) The schema depicts the experimental procedures. (B) GSEA results demonstrating the correlation between chromatin states and transcription activities. Genes associated with top 1000 ATAC peaks gained and top 500 peaks lost in CASTA5 CD4+ T cells were used as separate gene sets and compared with rank sorted gene expression from matched samples. (C) The left two heatmaps illustrate differential chromatin accessibility at 2kb windows centered at the summit of the ATAC peaks (absolute log2FC ≥2and adjusted p-value <0.05). The ATACseq data from 3 biological replicates were merged and used to plot the heatmap. Genes are grouped into 3 clusters with clusters 1 and 2 showing variable degree gain of peaks while cluster 3 showing loss of peaks, indicative of increased and reduced chromatin accessibility, respectively, in CASTAT5 CD4+ T cells compared to control CD4+ T cells. The line graphs on top of the heatmap demonstrate the average accessibility profiles of each cluster. The RNAseq data (absolute log2FC ≥1.5and adjusted p-value <0.05) are used to generate the column on the right which shows matched average expression log2FC of 3 replicates, and the results are summarized in the box plot shown on top of the column. Representative genes with significant changes in both chromatin accessibility and gene expression are listed beside the heatmap. (D) IPA results from both ATACseq and matched RNAseq datasets. The genes associated with differential ATACseq peaks (absolute log2FC ≥ 1, adjusted p-value <0.05) and differentially expressed genes (absolute log2FC ≥1, adjusted p-value <0.05) are used in the IPA analysis. (E) The chromatin accessibility tracks of genes related to polyfunctionality. Tracks in red and blue correspond to CASTAT5 and control CD4+ T cells, respectively. The green rectangles mark ATAC peaks showing increased signal intensity in CASTAT5 CD4+ T cells compared to control CD4+ T cells, while the purple rectangles mark ATAC peaks that disappeared or reduced in intensity in CASTAT5 CD4+ T cells. Each ATACseq track showing was randomly selected from one of three biological replicates. (F) The results from MEME-Chip motif analysis identified transcription factor consensus binding sites significantly enriched in the top 1000 and top 500 ATAC peaks with gain or loss of chromatin accessibility in CASTAT5 CD4+ T cells, respectively. (G) Results from the transcription factor footprinting analysis using the ATACseq data. The line plot indicates the normalized chromatin accessibility signals centered at the 200bp window flanking the indicated transcription factor binding sites. (H) A transcription regulatory network built on the ATACseq and RNAseq data from this study, as well as a published Stat5a ChIP-seq data (GSE79518).
We examined the chromatin accessibility of genes related to T cell polyfunctionality. Notably, cytokines that showed significantly increased productions in CASTAT5 CD4+ T cells, including Il4, Il13, Il9, Csf2 and GzmB, all had markedly increased chromatin accessibility in the corresponding gene loci (Fig. 3E). The Ifng gene locus showed reduced chromatin accessibility in CASTAT5 CD4+ T cells, in line with the slight reduction in Ifng expression in these cells detected by ICS. No obvious chromatin structure changes were observed in the Tnf gene locus, although modest gain in TNF-α expression was detected by ICS in CASTAT5 CD4+ T cells. In contrast, genes associated with T cell dysfunction, such as Pdcd1 and Ctla4, showed reduced chromatin accessibility in CASTAT5 CD4+ T cells, indicating a widespread yet selective remodeling of the epigenetic landscape by CASTAT5.
We further analyzed our RNAseq and ATACseq datasets to establish the concordance between gene transcription profile and chromatin accessibility status. A selection of genes (n=90) distributed across multiple important functional categories demonstrate highly correlative RNAseq and ATACseq data (fig. S4A). We performed additional flow cytometry analysis for CASTAT5 and control CD4+ T cells to confirm the differential expressions, as predicted by ATACseq and RNAseq data, of a panel of molecules at the protein level (fig. S4B–C). Altogether, our data indicate that CASTAT5 CD4+ T cells have acquired a distinct epigenetic landscape that is in concordance with gene transcription and protein expression profiles.
It is known that STAT5 can bind directly to gene regulatory regions to mediate gene expression(38, 39). By comparing with a published STAT5A ChIPseq dataset in murine pro-B cell line Ba/F3 stimulated with or without STAT5 inducer IL3(40), we observed that about 9% of the differential ATACseq peaks in our study directly overlap with STAT5A ChIPseq peaks and most of them (1164 out of 1328) are associated with gain of chromatin accessibility in CASTAT5 CD4+ T cells (fig. S5A upper panel). More importantly, by aligning our RNAseq dataset with the same Stat5a ChIP-seq dataset, we found that 763 out of 1695 (45%) up-regulated genes and also 259 out of 643 (40%) down-regulated genes in CASTAT5 CD4+ T cells, are potential direct targets of Stat5a (fig. S5A bottom panel). Similar results were obtained when we compared our ATACseq results with another pan-STAT5 ChIPseq dataset generated in murine CD4+ T cells(41) (fig. S5B). These comparative analyses underscored the impact of CASTAT5 on transcription and chromatin accessibility of some known STAT5 target genes, such as Bcl2, Junb and Id2, in the donor CD4+ T cells (fig. S5C). To test if other transcription factors (TFs) are also involved in epigenetic reprogramming, we performed TF motif analysis using two most-used MEME-Chip and HOMER software packages. Analysis of the top 1000 ATACseq peaks (Supplementary Table 2) with increased chromatin accessibility in CASTAT5 CD4+ T cells using MEME-Chip revealed enrichment for GATA family TFs and STAT5A/B consensus binding motifs, while analysis of the top 500 less accessible ATACseq peaks (Supplementary Table 2) revealed enrichment for Runx2 and Tbx21 (T-bet) binding motifs (Fig. 3F). Similar results were obtained using HOMER (fig. S6). One of the advantages of ATACseq is that it allows us to perform TF footprinting analysis. As shown in Fig. 3G, differential TF footprinting analysis using Rgt-HINT software package demonstrates significant higher Tn5 cutting site signals at the TF binding site of STAT5A/B and Gata1, and reduced signals at Runx2 and Tbx21 consensus sites. Additional TFs with increased or decreased activities in CASTAT5 CD4+ T cells compared to control CD4+ T cells were identified (fig. S7). By integrating the predicted TF binding map and expression changes of these TFs, we constructed an integrated regulatory network to explore the transcriptional circuitry in CASTAT5 CD4+ T cells (Fig. 3H). This network identifies augmented activities of Stat5, Gata1, Ap1 and decreased activities of Nfat, Runx and Tbx21. STAT5A appears to directly activate Gata1, Jun, Junb, Fos, Fosl2 and Ezh2, while repressing Runx2, Id2, Nr4a2 and Tox. In sum, our study provides comprehensive information on how persistent STAT5 activation reprograms CD4+ T cell epigenetic and transcriptomic landscapes, leading to the formation of polyfunctional CD4+ T cells.
CASTAT5 CD4+ T cells exhibit similar epigenetic features in spleen and tumor
For the analyses described above, donor T cells from the spleens of mice were used to ensure collection of sufficient number of donor T cells for downstream experimental procedures. It is known that T cells residing in different organs can have different properties. We asked whether the epigenetic changes seen in spleen-derived donor T cells can be found in tumor-infiltrating donor T cells. To address this, ATACseq analyses were performed for CASTAT5 and control CD4+ T cells which were either collected right before adoptive transfer (in vitro), or were FACS-sorted from the spleens or tumors of mice 10 days after T cell transfer. A pair-wise comparison between CASTAT5 and control CD4+ T cells revealed that CASTAT5 induced overt changes in chromatin accessibility in both spleen and tumor samples, although the changes in tumor-infiltrating T cells were less profound (Fig. 4A), suggesting that the epigenetic landscape of T cells is subjected to regulation by the microenvironment where the T cells reside. To identify the epigenetic changes unique to each cell population, we generated a data matrix containing ATACseq peaks identified from all samples and selected the top 5% peaks with largest variation among them. Then we selected the genes of interest identified by bulk RNAseq as shown in Fig. 3C to re-plot the heatmap (Fig. 4B). Notably, the majority of the epigenetic differences between CASTAT5 versus control CD4+ T cells recovered from the spleen were also found in donor T cells recovered from the tumor, although to a lesser extent in tumor-infiltrating T cells. In contrast, the in vitro cultured CASTAT5 and control CD4+ T cells did not differ in epigenome. Fig. 4C illustrates the typical pattern of epigenetic changes in donor T cells recovered from the spleen versus tumor for 3 representative genes.
Fig. 4. The differential chromatin accessibility profiles observed in spleen-derived CASTAT5 and control CD4+ T cells are detected in tumor-derived samples.
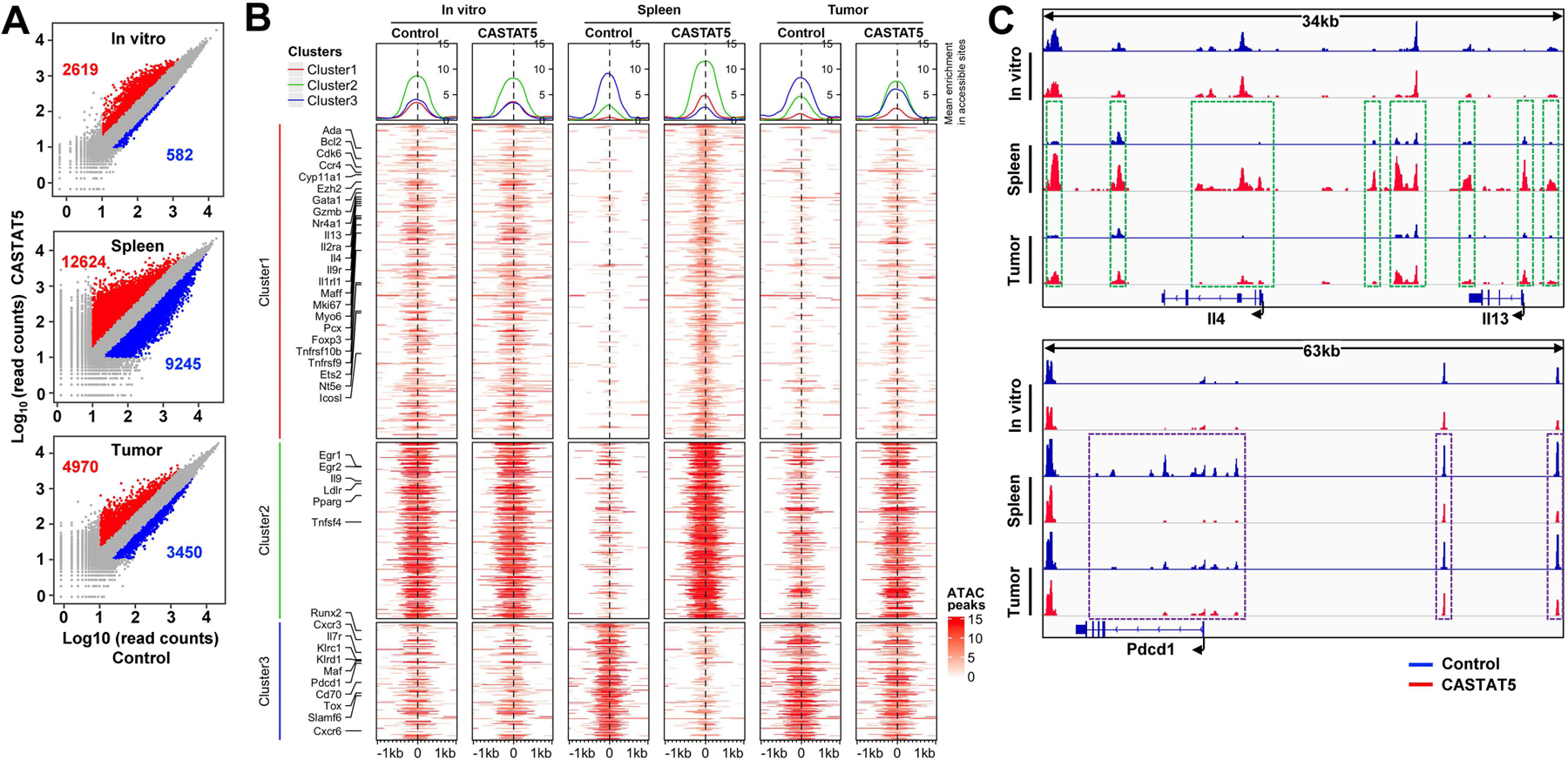
Following the experimental procedures depicted in Fig. 3A, spleen and tumor samples were collected separately 10 days after T cell transfer. For each group, donor CD4+ T cells were flow cytometry-sorted separately from 3 different mice. Equal numbers of sorted cells from the 3 biological replicates were pooled together and processed for ATACseq. Control and CASTAT5 CD4+ T cells collected in vitro before adoptive transfer were also processed for ATACseq. (A) Scatter plots comparing the ATACseq peak signals between paired CASTAT5 and control samples isolated in vitro and from spleen and tumor samples. The numbers in each panel indicate the number of peaks with gain (red) or loss (blue) of chromatin accessibility, respectively. (B) Heatmaps that illustrate differential chromatin accessibility at 2kb windows centered at the summit of the ATACseq peaks identified between different samples. The heatmaps were generated using the same ATACseq peak regions from Fig. 3C and plotted in the same orders. Line graphs on top of the heatmaps demonstrate the average accessibility profiles of clusters 1–3, respectively. (C) Chromatin accessibility tracks of representative genes. Tracks in red and blue correspond to CASTAT5 and control CD4+ T cells, respectively. The green rectangles on spleen and tumor-derived samples mark ATACseq peaks showing increased signal intensity in CASTAT5 CD4+ T cells compared to control CD4+ T cells. The purple rectangles on spleen and tumor-derived samples mark ATACseq peaks that disappeared or reduced in intensity in CASTAT5 CD4+ T cells. These differential ATACseq peak profiles between CASTAT5 and control CD4+ T cells are not observed in in vitro samples.
Single cell RNAseq reveals the heterogeneity of CASTAT5-induced polyfunctional CD4+ T cells
To further understand the heterogeneity of the T cell population and the transcriptome associated with polyfunctionality, we conducted single-cell RNA sequencing analysis (scRNAseq) for CASTAT5 and control CD4+ T cells using the 10x Genomics platform. As shown in Fig. 5A, 1,531 CASTAT5 and 1,632 control CD4+ T cells from pooled samples can be clearly separated apart on the t-distributed stochastic neighbor embedding (t-SNE) plot. Using unsupervised cluster analysis, the 3163 single cells can be further partitioned into 6 clusters based on their transcriptomes. Cells in clusters 1–3 belonged to control CD4+ T cells, whereas cells in clusters 4–6 were mostly CASTAT5 CD4+ T cells. Consistent with the bulk RNAseq data and ICS results, Il4, Il13, Il9, Csf2, Gzmb and Lamp1 (CD107a) transcripts were predominantly detected in CASTAT5 CD4+ T cells (clusters 4–6) while Ifng and Tnf transcripts were present in all clusters (Fig. 5B). Based on scRNAseq data, we computed the frequency of cells expressing one or more of the effector molecules, including the aforementioned 6 cytokines plus Gzmb and Lamp1 (CD107a) as displayed in Fig. 5B. A significant increase in the number of cells expressing 2 or more effector molecules was observed in CASTAT5 CD4+ T cells compared to control CD4+ T cells. Particularly, nearly 75% of cells in cluster 6 expressed at least 2 and about 48% of them expressed 3 or more effector molecules simultaneously (Fig. 5C). Thus cluster 6 represented a unique population highly enriched for polyfunctional CD4+ T cells. The scRNAseq analysis identified the gene signatures associated with each cluster (Fig. 5D and Supplementary table 3). Notably, the signature genes defining cluster 6 include genes regulating cell survival (Birc5), proliferation (Mki67, Pclaf), and chromatin modification (Uhrf1, Ezh2) (Fig. 5E). Following the same categorization schema used for bulk RNAseq and ATACseq, we examined the expression patterns of additional genes at the single-cell level (fig. S8). In general, the scRNAseq results are in congruence with the bulk RNAseq data and ATACseq data. To further identify modules of co-expressed genes in each cluster, the scRNAseq data were subjected to co-expression network analysis and a total of 18 modules were identified (fig. S9A–B). The gene co-expression network associated with cluster 6 features multiple genes involved in regulating cell cycle progression, chromosome segregation and spindle checkpoint activity (Fig. 5F).
Fig. 5. Heterogeneity of CASTAT5 and control CD4+ T cells revealed by scRNAseq analysis.
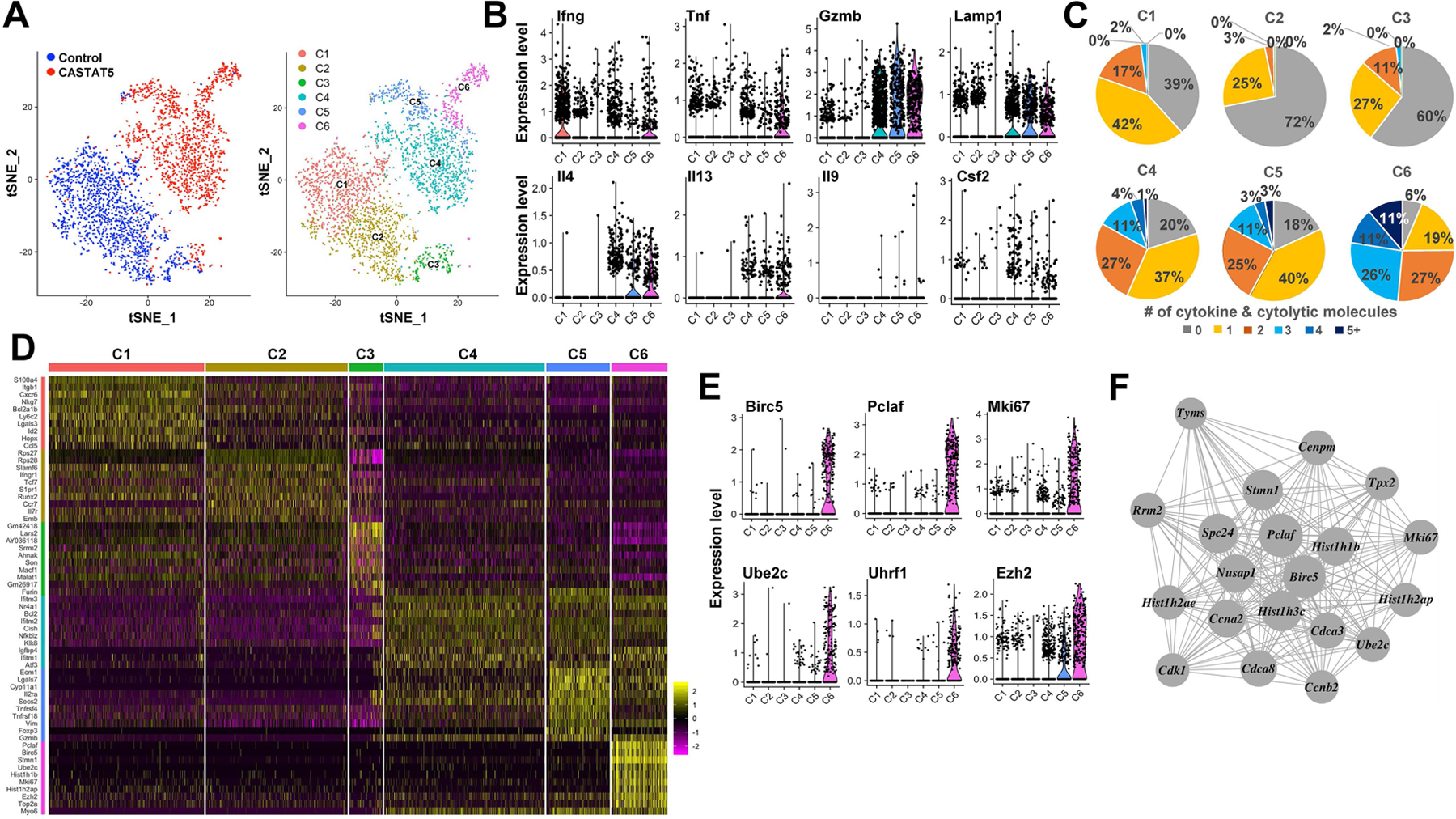
The t-SNE plots generated using scRNAseq data show clear separation of CASTAT5 and control CD4+ T cells (A, left panel), and each of them can be further projected into three sub-clusters based on gene expression profiles (A, right panel). (B) The violin plots show differential expression of 8 polyfunctionality-related genes among the six clusters of CD4+ T cells. (C) The pie charts show the percent of CD4+ T cells expressing varied number (0 to ≥5) of cytokines and cytolytic molecules in each cluster. The number of cells with positive expression of each effector molecule was counted based on the presence or absence of raw sequencing reads mapped to the mRNA sequences of each relevant gene. Cells with at least 1 read mapped to the relevant gene were called as positive for the corresponding gene. (D) Heatmap of top 10 genes expressed in each cluster. The columns represent cells and the rows present genes. Cells are grouped by clusters and top 10 most significant markers are shown. (E) Violin plots of six unique marker genes representing cluster 6. Each dot represents one cell and the violin shows the distribution of probability density at each value. (F) A gene co-expression network specific to cells of cluster 6 is identified using WGCNA.
We calculated cell cycle phase scores based on the expression patterns of 97 genes involved in regulating the cell cycle. CASTAT5 CD4+ T cells displayed a unique cell cycle status with most cluster 6 cells in G2/M and S phases, while most cells of cluster 4 and cluster 5 were in G1 phase (Fig. 6A). These distinct cell cycle phase distribution patterns prompted us to investigate the transcriptional dynamics of CASTAT5 CD4+ T cells. We performed RNA velocity analysis, in which the transcription kinetics is estimated by distinguishing the spliced and unspliced mRNA and used to predict the future state of individual cells on the time scale of hours(42, 43). As shown in Fig. 6B, the cluster 6 cells at the border with clusters 4 and cluster 5 exhibited long arrows (large RNA velocities) pointing towards cluster 4 and cluster 5 cells, suggesting that cluster 6 cells have self-renewal capacity and can differentiate into cluster 4 and cluster 5 cells. On the contrary, most of the cells in cluster 1 and cluster 2, which mostly consisted of control CD4+ T cells, showed short or no arrows (small RNA velocity), suggesting their status as terminally differentiated cells. In line with the results of RNA velocity analysis, pseudotime trajectory analysis showed that CASTAT5A CD4+ T cells were ordered along a trajectory with cluster 6 cells having the shortest pseudo-differentiation time (Fig. 6C). Cluster 6 cells were positioned as the progenitors of cells in other clusters based on the pseudo-time trajectory projection (Fig. 6D). We performed a branch point analysis to identify the critical molecular changes that may be important to the transition between cell states. As a result, 133 genes were identified as branch-dependent differentially expressed genes (Fig. 6E). For example, several transcription factors including Egr1, Jun, and Runx2 may play an important role at the first branchpoint (Fig. 6F). These results support the notion that cluster 6 cells represent a resource population, or progenitor-like cells, which may give rise to more polarized but less polyfunctional effector CD4+ T cells.
Fig. 6. Cell cycle, RNA velocity and pseudotime trajectory analyses reveal dynamic transition of cell cycle and transcription programming in CASTAT5, but not in control CD4+ T cells.
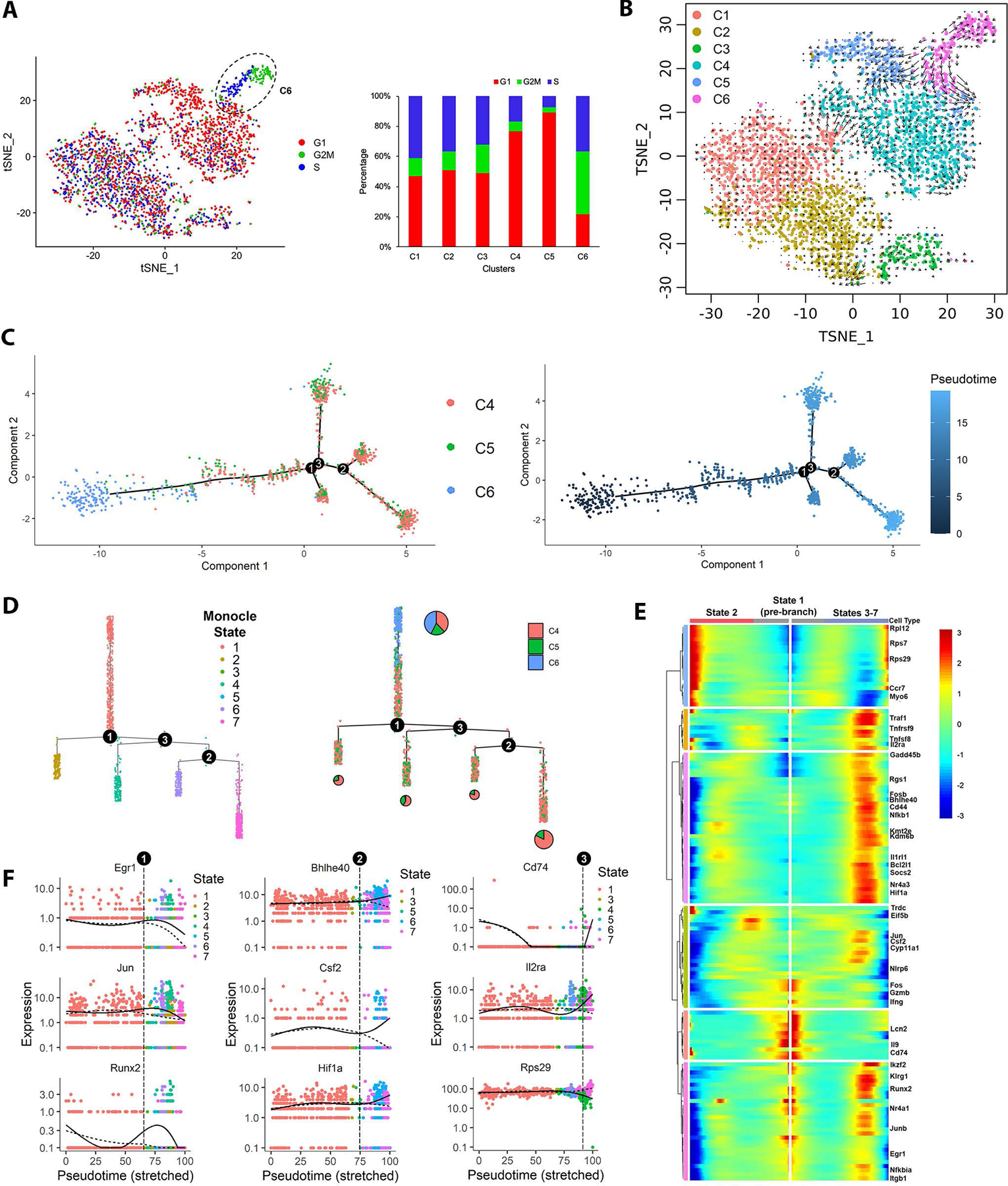
(A) The t-SNE plots of CASTAT5 and control CD4+ T cells are constructed with cell cycle scores calculated using Seurat based on a group of 97 cell cycle genes. The color key indicates the cell cycle phase. The bar graph at right summarizes the percent of cells in G1, G2/M and S phase according the cell cycle scores in each cluster as defined in Fig. 5A. (B) The RNA velocity plot describes the dynamic mRNA transcription states in CASTAT5 and control CD4+ T cells. The RNA velocity field is projected on the tSNE space in Fig. 5A. The direction of cell state transition, and RNA velocity are indicated by the vectors (arrows) and their lengths, respectively. (C) The plot on the left shows the order of CASTAT5 CD4+ T cells from 3 different clusters along the pseudotime trajectory determined by Monocle. Each dot represents one cell. The colors indicate the clusters to which the cells belong, as defined in Fig. 5A. The plot on the right shows the trajectory of CASTAT5 CD4+ T cells plotted by pseudo differentiation time. The trajectory branchpoints are marked by numbered circles. (D) Monocle trajectory ordered as a cluster tree. Cells in the left panel are labeled by monocle cell states, while cells in the right panel are labeled by Seurat clusters as defined in Fig. 5A. The pie charts within the right panel describe the proportion of cells in the Seurat clusters distributed among branches of the cluster tree. (E) Heatmap of the branchpoint-dependent differentially expressed genes (adjusted p-value <0.001) identified by Monocle showing a trend of gradual changes of gene expression along the pseudotime trajectory at the first branchpoint. The columns show gene expression levels at each point in pseudotime, the rows indicate the corresponding genes. The starting point of pseudotime is placed in the center of the heatmap. The left-half of the heatmap, from center to left, shows the gene expression trend in pseudotime changing from cell state 1 towards cell state 2. The right-half of the heatmap, from center to right, shows the gene expression trend in pseudotime changing from cell state 1 towards cell states 3–7. (F) The expression levels of branchpoint-dependent genes ordered along the pseudotime trajectory. The gene shown in the left, middle and right panels are representative genes associated with cell fate determination at the first, second and third branchpoints, respectively.
It should be noted that the donor CD4+ T cells sorted for NGS may contain both transduced (CASTAT5 positive) and untransduced (CASTAT5 negative) T cells, raising the question to what extent our data reflected the effects of CASTAT5 on CD4+ T cells. We found that the two specific STAT5A mutations (H299R and S711F), which were introduced during mutagenesis to create CASTAT5(29), were prevalent (mutant allele frequency ≥ 89%) in both the bulk and single-cell sequencing reads generated from CASTAT5 CD4+ samples, whereas these mutations were not detected in control CD4+ samples (fig. S10). The results indicate that at the time of analysis, the sorted CASTAT5 CD4+ samples contained predominantly transduced T cells, likely due to better expansion and survival of CASTAT5-transduced T cells.
CASTAT5 potentiates the efficacy of CD19CAR T cell therapy
CD4+ T cell polyfunctionality was found to correlate with the clinical outcomes of CD19CART therapy(25). We postulated that CASTAT5 can be used to enhance the efficacy of CD19CAR T cells. We tested this using a mouse model of CD19CART therapy for B-cell lymphoma(44, 45). As shown in Fig. 7A, mice with established subcutaneous A20 tumors were conditioned with CTX, followed by infusion of T cells transduced to express CD19CAR alone, or co-transduced to express both CD19CAR and CASTAT5. Fig. 7B shows that adoptive transfer of CD19CAR T cells resulted in only transient tumor regression, simulating an ineffective treatment scenario frequently observed in the clinic; In contrast, adoptive transfer of CASTAT5 CD19CAR T cells was curative to nearly all mice. In this model, tumor relapse was not due to the outgrowth of antigen-loss variants because the relapsed tumors from CD19CAR T cell-treated mice expressed the same levels of CD19 as the tumors from untreated mice (Fig. 7C). Fig. 7D shows that CASTAT5 CD19CAR T cells caused long-term B cell aplasia, whereas B cells in mice receiving control CD19CAR T cells began to increase when these mice started to have tumor relapse (>day 20). To examine the persistence of CD19CAR T cells, bone marrow aspirates were collected from mice 30 days after T cell infusion and analyzed by a sensitive PCR-based approach. CD19CAR vector DNA was detected in the bone marrows recovered from mice that had received CASTAT5 CD19CAR T cells, but not in the bone marrows from untreated naïve mice or mice that had received the control CD19CAR T cells (Fig. 7E). Furthermore, mice cured by CASTAT5 CD19CAR T cells were resistant to tumor re-challenge (Fig. 7F), indicating the formation of immune memory. We further tested the usefulness of CASTAT5-modified CAR in a typical solid tumor model in which CD19-expressing CT26 colon cancer cells were used for tumor inoculation (fig. S11A). Enhanced donor T cell expansion and persistence were observed for CASTAT5-transduced CD19CAR T cells compared to control CD19CAR T cells (fig. S11B). CASTAT5-transduced CD19CAR T cells also led to significantly improved antitumor effects (fig. S11C), although curative outcome was not achieved due to the outgrowth of CT26 tumors that lost CD19 expression (fig. S11D).
Fig. 7. CASTAT5 potentiates the efficacy of CD19CAR T cell therapy in a murine B-cell lymphoma model.
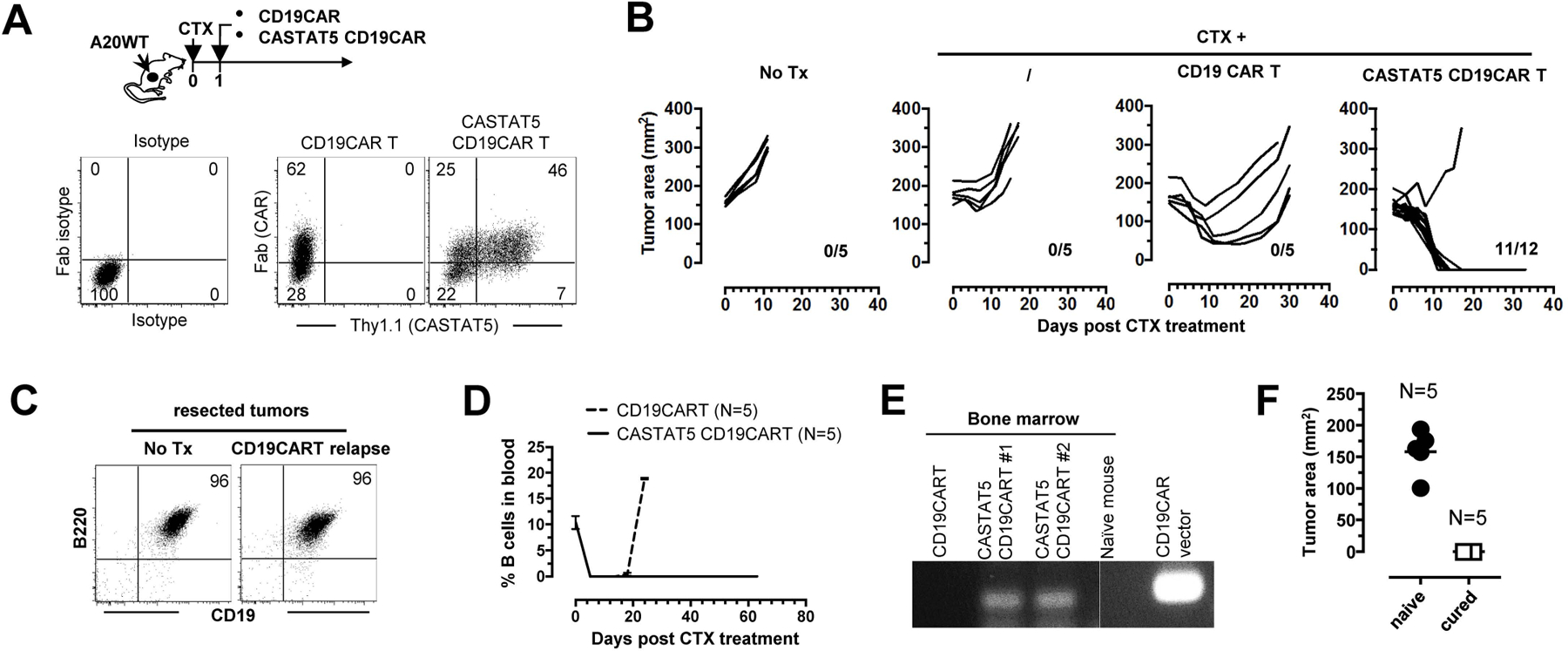
The schema depicts the timeline of the experimental procedures. A20WT tumor cells were subcutaneously inoculated to mice. When tumor sizes reached 170 mm2, mice were randomly grouped and received either no treatment (No Tx), CTX only, CTX + control CD19CAR T cells, or CTX + CASTAT5 CD19CAR T cells. (A) Expression of CAR and CASTAT5 in transduced T cells. Total T cells purified from CD45.1 mice were transduced to express CD19CAR alone or co-express CD19CAR and CASTAT5. Representative dot plots show the levels of CD19CAR and CASTAT5 in transduced T cells. CD19CAR was measured by anti-Fab stain and CASTAT5 was evaluated by Thy1.1 expression. (B) Tumor growth curves of each mouse under each condition are shown. The numbers indicate the number of tumor-free mice among treated mice at the end point. (C) Relapsed tumors did not lose CD19 expression. Mice treated with CD19CAR T cells had initial tumor regression followed by relapse. Relapsed tumors were resected and processed into single cell suspension for evaluation of CD19 expression by flow-cytometry. Tumors from untreated mice were used for comparison. Representative dot plots shown represent the co-stain of CD19 and B220. (D) Frequencies of host B cells in blood. At the indicated time points, tail blood was collected and the presence of host B cells in blood was evaluated by CD19 and B220 co-stain. (E) Detection of CD19CAR viral vector in bone marrow by PCR. 30 days after T cell transfer, a cohort of mice were killed and bone marrow aspirates were collected. The presence of CD19CAR T cells was evaluated by PCR detection of the CD19CAR viral vector. (F) Mice cured by CTX+CASTAT5 CD19CAR T cells were resistant to A20WT re-challenge. Live A20WT tumor cells were subcutaneously injected to cured mice and a group of naive mice used as controls. Tumor sizes in mice 3 weeks after tumor inoculation are shown.
Concurrent expression of CASTAT5 in CD4+ and CD8+ CD19CAR T cells leads to optimal therapeutic outcomes
Since our CD19CAR T cell infusion products contained both CD4+ and CD8+ T cells, we sought to determine how expression of CASTAT5 in each subset affected CD19CAR T cell expansion, persistence and efficacy. To this end, splenic CD4+ and CD8+ T cells, separately isolated from normal mice, were transduced with CD19CAR only, or co-transduced with CD19CAR and CASTAT5 (Fig. 8A). As depicted in Fig. 8B, mice with established A20 tumors were grouped and conditioned with CTX, followed by infusion of CD8+ CD19CAR T cells, either CASTAT5-null (Control CD8) or CASTAT5-positive (CASTAT5 CD8), in the absence or presence of equal numbers of CD4+ CD19CAR T cells that were either CASTAT5-null (Control CD4) or CASTAT5-positive (CASTAT5 CD4). Longitudinal tail blood flow cytometry staining analysis revealed that CASTAT5-null CD8+ CD19CAR T cells transferred alone had minimal expansion in recipient mice (Fig. 8C group I), and that this poor expansion cannot be rescued by co-transfer of CASTAT5-null or CASTAT5-positive CD4+ CD19CAR T cells (Fig. 8C groups II&III). When transferred alone, CASTAT5-positive CD8+ CD19CAR T cells expanded initially, reaching the peak (~10% in blood) around day 8, but underwent a rapid contraction and returned to the baseline by day 14 (Fig. 8C groups IV). Co-transfer of CASTAT5-null CD4+ CD19CAR T cells did not enhance the expansion but slightly slowed down the contraction of CASTAT5 CD8+ CD19CAR T cells (Fig. 8C groups V). Notably, in the presence of CASTAT5-positive CD4+ CD19CAR T cells, CASTAT5 CD8+ CD19CAR T cells underwent more robust expansion (up to 20% in blood), and remained readily detectable (~5% in blood) 40 days after infusion (Fig. 8C groups VI). B cell aplasia, an on-target off-tumor effect of CD19CAR T cells, occurred rapidly (~5 days after T cell infusion) in all groups, but persisted only in group VI in which mice received a mixture of CASTAT5 CD4+ and CASTAT5 CD8+ CD19CAR T cells (Fig. 8D). Corresponding to the robust expansion and persistence of donor T cells, mice in group VI had the best therapeutic outcome, measured by percentage of tumor-free mice on day 40 (Fig. 8E). A cohort of mice were sacrificed 10 days after T cell transfer and splenocytes were subject to phenotypic analysis by flow cytometry. Cytokine profile analysis revealed that CASTAT5 CD19CAR CD4+ T cells acquired polyfunctionality as evidenced by simultaneous production of IFN-γ, IL-4, IL-13 and GM-CSF (Fig. 8F). CD8+ CD19CAR T cells transferred alone (group I) showed high levels of co-inhibitory molecules including PD-1, Tim3, Tigit and Lag3 (Fig. 8G). These molecules were significantly downregulated in CASTAT5-transduced CD8+ CD19CAR T cells, transferred either alone (group IV) or together with CASTAT5 CD4+ CD19CAR T cells (group VI). CASTAT5 CD8+ CD19CAR T cells appeared to express lower levels of exhaustion-associated markers in the presence of CASTAT5 CD4+ CD19CAR T cells, though the differences did not reach statistical significance. In contrast, CASTAT5 CD8+ CD19CAR T cells co-transferred with CASTAT5 CD4+ CD19CAR T cells had significantly higher level of granzyme B than those transferred alone, while the control CD8+ CD19CAR T cells showed the lowest granzyme B expression. Altogether, the results indicate that CASTAT5 expression is required in both CD4+ and CD8+ CD19CAR T cells to achieve optimal synergistic therapeutic effects.
Fig. 8. Concurrent expression of CASTAT5 in CD4+ and CD8+ CD19CAR T cells leads to optimal therapeutic outcomes.
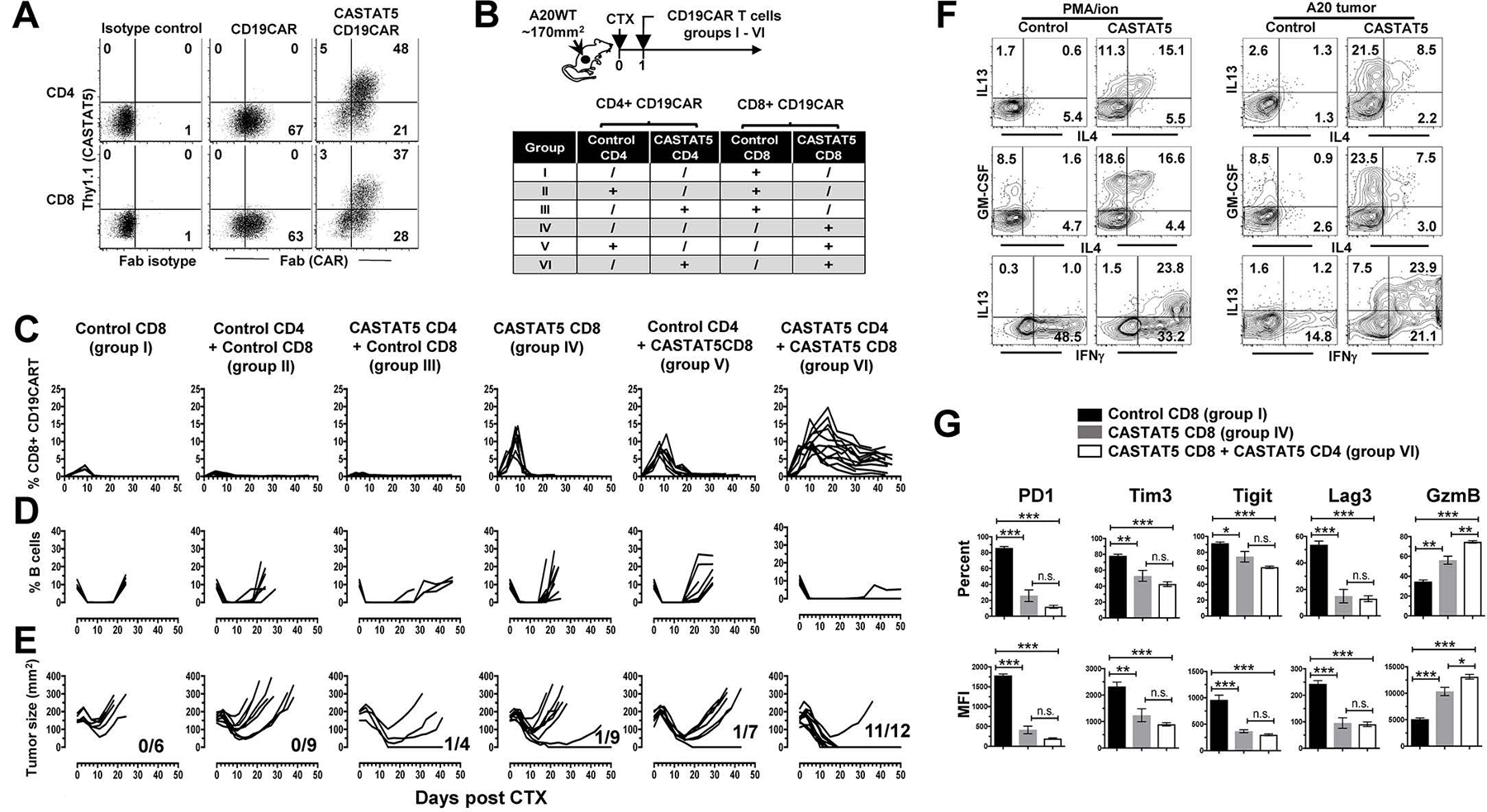
(A) Viral transduction efficiency in CD4+ and CD8+ T cells. CD4+ and CD8+ T cells were separately purified from CD45.1 mice. These cells were transduced to express CD19CAR alone or co-express CD19CAR and CASTAT5. Representative dot plots show the levels of CD19CAR and CASTAT5 in transduced T cells. CD19CAR was measured by anti-Fab stain and CASTAT5 was evaluated by Thy1.1 expression. The numbers in plots indicate the percentage of cells in the corresponding quadrant. (B) Experimental setup and treatment conditions. The schema depicts the experimental procedures. The different combinations of engineered CD4+ and CD8+ CD19CAR T cells used for adoptive transfer are outlined in the chart. (C) The frequencies of donor CD8+ T cells in recipient mice during the course of CD19CAR T cell therapy. At the indicated time points, tail blood was collected from mice and subjected to flow cytometry to detect the donor CD8+ T cells (CD45.1+CD8+). The frequencies of donor CD8+ T cells were plotted against time. The frequencies of host B cells (CD19+B220+) in blood were also evaluated by flow cytometry and summarized in (D). (E) Tumor growth curves of mice in each group. The numbers indicate the number of tumor-free mice among treated mice at the end point of experiment. (F and G) Phenotypic characterization of donor CD4+ and CD8+ CD19CAR T cells in selected groups. Ten days after T cell transfer, splenocytes isolated from a cohort of mice from the specified groups (3 mice/group) were subject to flow cytometry analysis. Splenocytes from groups V and VI were stimulated with PMA/ionomycin or A20 tumor cells for ICS to examine the cytokine profiles of control CD4+ and CASTAT5 CD4+ CD19CAR T cells (F). Numbers in representative dot plots indicate percentage of cells in the corresponding quadrant. Donor CD8+ T cells from groups I, IV and VI, which expressed CD19CAR alone, or co-expressed CASTAT5 and CD19CAR in the absence or presence of CASTAT5 CD4+ CD19CAR T cells, respectively, were examined for expression of the specified markers. The frequencies of cells positive for each of the markers and the mean fluorescence intensity (MFI) of these markers are summarized in bar graphs shown as mean ± s.d. of five samples per group. (G). n.s., not significant; *p < 0.05, **p < 0.01, ***p < 0.001.
CASTAT5 is functional in transduced primary human CD4+ T cells
The constitutively active STAT5A mutant used in our study was created by random mutagenesis in murine STAT5A(29). Although naturally occurring equivalent mutations in human STAT5A have not been reported, it has been shown that the murine origin CASTAT5 was functional in CASTAT5-transduced human hematopoietic stem cells and erythroid progenitors(46–48). To examine the activity of CASTAT5 in primary human T cells, total T cells isolated from the peripheral blood mononuclear cells (PBMC) of a healthy donor were mock-transduced (control) or CASTAT5-transduced (CASTAT5), and adoptively transferred to immunodeficient NOD-Scid IL-2Rγ-null mice (NSG). Two weeks later, the mutant murine STAT5A, reflected by Thy1.1, was still detectable in a fraction of CASTAT5-transduced human CD4+ T cells recovered from the spleens of mice (fig. S12A). Interestingly, the level of human BCL2, a known downstream target of STAT5A, was significantly higher in the fraction of human CD4+ T cells expressing murine CASTAT5 (Thy1.1+) compared to the CASTAT5-negative fraction (Thy1.1-) or mock-transduced T cells (fig. S12B). In addition, CASTAT5-transduced human CD4+ T cells exhibited a polyfunctional cytokine profile compared to mock-transduced T cells (fig. S12C). The results suggest the potential of using CASTAT5 to engineer polyfunctional human CD4+ T cells.
Discussion
The molecular mechanisms governing CD8+ T cell exhaustion have been extensively studied in recently years(49, 50). These studies provide a plethora of information on the transcriptional and epigenetic programs controlling the phenotype and function of exhausted murine and human CD8+ T cells induced in the setting of chronic viral infections or cancer. A number of transcription factors, including NFAT, TOX and NR4A1, are found to play critical roles in inducing and maintaining CD8+ T cell exhaustion(51–58). The overarching consensus is that the exhaustion initiation transcription factor NFAT1, in the absence of it partner AP-1 (Fos/Jun), induces and interacts with NR4A1 and TOX to drive a transcriptional program leading to exhaustion(51, 58, 59). Exhausted CD8+ T cells are a heterogeneous population containing subsets of progenitor exhausted and terminally exhausted cells that can be distinguished by varied expression levels of a panel of genes including Tcf7, Tbx21, Eomes and Slamf6(60–63). As the knowledge on exhausted CD8+ T cells rapidly accumulates, our understanding of exhaustion in CD4+ T cells is relatively limited. Transcriptomic profiling of virus-specific CD4+ T cells during chronic viral infections revealed that exhausted CD4+ T cells exhibit a molecular profile distinct from exhausted CD8+ T cells, though some common features of CD4+ and CD8+ T cell exhaustion exist(26). Our study sheds light on the molecular mechanisms dictating whether tumor-specific CD4+ T cells become polyfunctional or exhausted. In the absence of CASTAT5, tumor-specific CD4+ T cells had limited cytokine production, reduced proliferative capacity, high levels of Pdcd1 and Ctla4, and failed to eradicate tumors in mice. In addition, these CD4+ T cells expressed both Nfatc1 and Nfatc2, but were in paucity of Fos and Jun. These CD4+ T cells also expressed Tox and Slamf6, molecules associated with CD8+ T cell exhaustion. These features implicate a population of CD4+ T cells destined to exhaustion. Our data indicate that CASTAT5-induced transcriptional reprogramming can divert the fate of CD4+ T cells from exhaustion to polyfunctionality. Notably, CASTAT5 upregulated the levels of Fos and Jun but repressed the expressions of Tox, Pdcd1, Ctla4, Haver2 (Tim3), Lag3, Tigit and Slamf6, suggesting that CASTAT5 enables T cells to acquire resistance to exhaustion. Interestingly, we did not find prominent level of Nr4a1, a key regulator of exhaustion in CD8+ T cells(51, 52, 57), in exhausted tumor-specific CD4+ T cells; instead, CASTAT5 markedly upregulated Nr4a1 but reduced Nr4a2 in polyfunctional CD4+ T cells (fig. S8H). Moreover, some other TFs used to distinguish exhausted virus-specific CD4+ T cells from their memory counterparts, including Batf, Eomes, Bcl6 and Prdm1 (Blimp)(26), did not show the expected patterns between exhausted tumor-specific CD4+ T cells and polyfunctional CD4+ T cells as revealed by scRNAseq. The cause and biological implication of these discrepancies warrant further investigation.
Numerous studies have shown that CD4 help to CD8+ T cells is essential to the success of cancer immunotherapy(9–12). Emerging evidence indicates that the efficacy of CD19CART therapy benefits from the co-presence of CD4+ and CD8+ T cells in the infusion products(13, 64). A recent study reported that in patients with non-Hodgkin lymphoma (NHL) treated with CD19CAR infusion products, the clinical outcomes were associated with CD19CAR T-cell polyfunctionality, and that the correlation was greater with the polyfunctionality of CD4+ CAR T cells compared to that of CD8+ CAR T cells(25, 65). Although in the current study we focused on the impact of CASTAT5 on CD4+ T cells, CASTAT5-transduced CD8+ CD19CAR T cells alone appeared to expand better and express reduced levels of multiple co-inhibitory molecules than control CD8+ CD19CAR T cells, suggesting that CASTAT5 can benefit CD8+ T cells as well. Importantly, our study provides clear evidence that CASTAT5 not only enables CD4+ CD19CAR T cells to be superior helpers, but also renders CD8+ CD19CAR T cells more responsive to CD4 help. Some recent studies showed CD4 help via IL-21 production or relayed by activated CD70-expressing DCs(66, 67). We did not observe increased IL-21 transcription in CASTAT CD4+ T cells, instead our data suggest that CD4 help may involve CD8α+CD103+ DCs. The exact molecular mechanisms underlying the productive communications between DCs, CD4+ and CD8+ CAR T cells await further investigation.
It has been reported that CAR T cells made to secrete IL-7, or respond to exogenous IL-7, exhibited improved expansion, persistence and antitumor activities in preclinical models(68–71). Shum et al recently reported that CAR T cells engineered to express a constitutively active IL-7Rα, which subsequently activated STAT5, exhibited enhanced antitumor efficacy in preclinical models. Our study further underscores the importance of the STAT5 signaling pathway in generating and maintaining T cells with the desirable functional features and provides a potential strategy to manipulate this pathway in T cells for therapeutic benefits. Importantly, our data not only demonstrate the critical contribution of polyfunctional CD4+ T cells to effective antitumor immunity, but also reveal the gene regulatory network induced by CASTAT5 to drive CD4 polyfunctionality. The results may lead to better utilization of tumor-reactive CD4+ T cells in cancer immunotherapy. One safety concern associated with persistent STAT5 activation is the leukemic potential of CASTAT5-transduced T cells(32–37). We did not observe development of leukemia in mice cured long after (3 months) receiving CASTAT5-transduced CD19CAR T cells. It should be noted that leukemogenesis is often driven by mutations in signaling pathways acting upstream of STAT5, such as Janus kinases, FLT3 and BCR/ABL, which result in abnormal STAT5 activation in hematopoietic precursor cells(72–74). Moreover, mutations in STAT5B, but not STAT5A, have been frequently found in human leukemia(75–77), likely due to more prevalent expression of the former isoform(41), although transgenic mice expressing a constitutively active form of STAT5B did not report development of T cell leukemia(78). Our data suggest that persistent STAT5 activation (CASTAT5) per se may not be sufficient to drive T cells into leukemic cells. For adoptive T cell therapy using the CASTAT5 strategy, the safety concern can be further addressed by incorporating additional safety measures such as inducible CAR T cell suicide system(79).
In summary, our study sheds light on the heterogeneity of polyfunctional CD4+ T cells and reveals the transcriptomic and epigenomic programs that dictate CD4+ T cell polyfunctionality. Our results imply that persistent activation of STAT5 in CD4+ T cells represents a promising strategy to potentiate the efficacy of ACT including CAR T cell therapy.
Materials and Methods
Study Design
The objective of this study was to investigate the functional fate and therapeutic effectiveness of tumor-specific CD4+ T cells expressing a constitutively active form of Stat5a (CASTAT5) via retroviral transduction. To achieve this goal, we designed a CD4+ T-cell adoptive transfer model system using a murine B-cell lymphoma model and performed ATACseq and bulk RNAseq to interrogate how CASTAT5 reshaped CD4+ T cell phenotypic and functional features using FACS-sorted tumor-specific CD4+ T cells recovered from the spleens and tumors of mice. Our datasets revealed that CASTAT5 induced genome-wide transcriptional and epigenetic remodeling in tumor-specific CD4+ T cells. Subsequent single-cell RNAseq analysis revealed the heterogeneity of CASTAT5-transduced CD4+ T cells and identified a subset of cells bearing a gene signature indicative of progenitor polyfunctional cells. We validated the beneficial effects of CASTAT5-transduced CD4+ T cells in CD19CAR T cell therapy mouse tumor models. We demonstrated that CASTAT5 was also functional in transduced primary human T cells. Mice with established tumors were randomly grouped before receiving the indicated treatment. Sample sizes for the in vivo experiments were determined based on previous studies performed in our lab, and the suitability for statistical analysis and independent repeats. Information regarding the sample size and number of replicates for each experiment is provided in the relevant figure legend.
Mice
BALB/c mice (Thy1.2) of 4 to 6 weeks of age were purchased from Charles River. 6.5 TCR-Tg mice on a BALB/c (Thy1.1) background expressing an αβTCR specific for amino acids 110–120 from influenza HA presented by MHC class II molecule I-Ed were described previously(22). The CD45.1 mice on a BALB/c background and immunocompromised NOD-Scid IL-2Rγ-null mice (NSG) mice were purchased from Jackson Laboratory. All mice were housed under specific pathogen-free (SPF) conditions by Laboratory Animal Services of the Augusta University. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Augusta University.
Antibodies and reagents
The following anti-mouse antibodies, including CD4 (GK1.5), CD3 (17A2), CD90.1 (OX-7), PD-1 (RMP1–14), CD127 (A7R34), CD45R/B220 (RA3–6B2), CD19 (6D5), CD44 (IM7), CD62L (MEL-14), IL-33Rα (ST2), CD39 (Duha59), ICOS (15F9), CCR4 (2G12), CXCR3 (CXCR3–173), OX40 (OX-86), CD103 (2E7), I-A/I-E (M5/114.15.2), CD11C (N418), CD45.1 (A20), anti-IL-2 (JES6–5H4), anti- TNF-α (MP6-XT22), anti-IFN-γ (XMG1.2), anti-IL-4 (11B11), anti-IL-9 (RM9A4), anti-GM-CSF (MP1–22E9), anti-Granzyme B (GB11), anti-GATA3 (16E10A23) and anti-STAT5 Phospho (Tyr694) (A17016B), and anti-human antibodies, including CD4 (OKT4), CD8α (HIT8a) and Bcl2 (100), were purchased from Biolegend. Anti-mouse CD25 (PC61), EZH2 (11/EZH2) and Ki-67 antibodies were purchased from BD Biosciences. Anti-mouse antibodies IL-13 (eBio13A), FOXP3 (FJK-16s), T-bet (eBio4B10), CD107a (eBio1D4B) and Bcl-2 (Bcl2/100) were purchased from eBioscience. Anti-PPARγ (81B8) antibody was purchased from Cell Signaling Technology. Alexa Fluor 488-conjugated goat anti-rabbit IgG antibody was purchased from Thermo Fisher Scientific. FITC-conjugated polyclonal mouse anti-rat F(ab’)2 antibodies and FITC-conjugated polyclonal mouse IgG antibodies were purchased from Jackson ImmunoResearch. CD8 depletion antibody (2.43,) was purchased from Bio X Cell. Intracellular fixation and permeabilization solution kit was purchased from BD. Dynabeads mouse T-activator, Dynabeads human T-activator, transcription factor staining buffer set and violet cell proliferation kit were purchased from Thermo Fisher Scientific. Cyclophosphamide monohydrate used for CTX was purchased from Tokyo Chemical Industry. Retronectin was from Takara. D-luciferin monosodium salt was purchased from Thermo Scientific Pierce. Lipofectamine 2000 was purchased from Invitrogen.
Cell preparation and flow cytometry analysis
Peripheral blood was collected via tail vein bleeding. Spleen and tumor samples were digested and gently dissociated into single-cell suspensions. Red blood cells were lysed by ACK lysing buffer. For surface molecule detection, cells were stained with fluorochrome-conjugated antibodies for 10 minutes at room temperature in the dark. To detect intranuclear molecules (Foxp3, EZH2, Tbet, GATA3, Ki67 and PPARγ), transcription factor staining buffer set (eBioscience) was used following manufacturer’s instruction. For intracellular cytokine staining, donor T cells in splenocytes were stimulated with 10 μg/ml cognate HA peptide or PMA/ionomycin in the presence of GolgiStop for 4 hours at 37°C. When tumor cells were used as antigen-presenting cells, donor T cells were cultured with irradiated tumor cells at 1:1 ratio for 24 hours with GolgiStop being added 3 hours before harvest. Cells were harvested and surface stained, followed by cytokine staining with intracellular fixation and permeabilization kit (BD Biosciences). For detection of CD19CAR expression in transduced T cells, polyclonal mouse anti–rat-F(ab’)2 antibody was used to detect 1D3 scFv with normal polyclonal mouse IgG antibody as isotype control. To detect phosphorylated STAT5 (pSTAT5) in T cells, cells were fixed in 2% formaldehyde followed by permeabilization with ice-cold methanol. After wash with PBS, cells were stained with pSTAT5 antibody at room temperature for 20 minutes before wash and flow cytometry analysis. All FACS data were acquired on a LSRII instrument (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Cell lines
B-cell lymphoma cell line A20 and murine colorectal cancer cell line CT26 were obtained from American Type Culture Collection (ATCC). HA-expression A20 tumor cell line (A20HA) was generated as described previously(22). CD26-CD19 cells were generated by transfecting cells with a vector encoding murine CD19 (pCMV3-mCD19, SinoBiological Inc.) using Lipofectamine 2000. CD19+ tumor cells were FACS-sorted, expanded and cryopreserved. Plasmacytoma cell line MOPC315.GFP was described previously(80). Tumor cells were cultured in RPMI 1640 (HyClone Laboratories) supplemented with 10% fetal bovine serum albumin (FBS), 1% penicillin/streptomycin (HyClone Laboratories), 1% non-essential amino acids and 1% glutamine (Corning) at 37 °C in a 5% CO2 incubator.
Mouse and human T cell retroviral transduction
The murine stem cell virus (MSCV) retroviral vector containing CASTAT5 and Thy1.1 marker interspaced by IRES was a gift from Dr. Susan Kaech. The retroviral vector MSGV-1D3–28Z.1–3 (anti-CD19CAR), in which the 1st and 3rd ITAMs of the CD3-ζ molecule are inactivated to reduce apoptosis, was provided by Dr. James N. Kochenderfer(81). Retroviral vector MSCV-luciferase-IRES-Thy1.1 was generated as described previously(28). To produce retroviral supernatants, 293FT cells were infected with retroviral vector and pcLEco packaging plasmid using Lipofectamine 2000. Mouse T cell retroviral transduction was done as described previously(28). Briefly, T cells were purified from mouse splenocytes and stimulated with mouse CD3/CD28 activation Dynabeads in the presence of 30 U/ml hIL-2 overnight. Cells were harvested and transduced twice in two consecutive days with retroviral supernatant using retronectin. 24 hours after the second transduction, anti-CD3/CD28 magnetic beads were removed, and cells were washed with PBS before infusion to mice via tail vein. To detect the presence of CD19CAR vectors in mouse tissues by PCR, genomic DNA used as template was isolated from bone marrow aspirates using Qiagen Blood and Tissue Kit. PCR was carried out on an Eppendorf cycler with the primers specific to the CD19CAR vector (forward primer: 5’-ATCCATGGAGATCTATGAAGTGGCC-3’, and reverse primer: 5’-ATGTCGACGTTAACTCATCTGGGG-3’).
For retroviral transduction of human T cells, Phoenix Amphotropic (Phoenix-AMPHO) cells were transfected with CASTAT5 retroviral vector using Lipofectamine 2000 to produce viral supernatant. Human T cells were isolated from the buffy coat of healthy donors using EasySep human T cells isolation kit (Stemcell Technology). Purified T cells were activated by human CD3/CD28 activation Dynabeads in the presence of 30 U/ml hIL-2 for 48 h. Activated T cells were spin-infected twice in two consecutive days with CASTAT5 retroviral supernatant using retronectin. Anti-CD3/CD28 Dynabeads beads were removed 24 h after the 2nd transduction. T cells were further expanded for 7 days in the presence of 100 U/ml hIL-2 before adoptive transfer.
Mice tumor inoculation and treatment
Tumor cells were subcutaneously injected to the right flank of mice (5 x 106 tumor cells/mouse in 100 μl PBS). The tumor size was monitored by caliper every other day, and expressed as the product of two perpendicular diameters in square millimeters. When tumor sizes reached the desired sizes, mice were randomly assigned into groups to receive the specified treatments. CTX was dissolved in PBS and i.p. injected to mice at a dose of 150 mg/kg one day before T cell transfer. For adoptive T-cell transfer, in vitro cultured T cells were harvested, wash twice with PBS and injected into each recipient via tail vein. To deplete host CD8+ T cells, CD8α depleting antibodies were injected via i.p. (200 μg per injection) once a week for 5 weeks with the first injection right after T cell transfer.
Cell preparations for next-generation sequencing
10 days after T cell transfer, spleen and tumor samples from mice were collected and processed into single cell suspensions. Cells were stained with anti-CD4 and anti-Thy1.1 Abs and subjected to cell sorting on a FACSAria sorter. The purity of sorted donor CD4+ T cells was normally greater than 98%. For bulk RNAseq and ATACseq analyses using spleen samples, donor CD4+ T cells derived from three individual mice under each treatment condition were used as biological replicates. For single-cell RNAseq, 1.7 x105 CD4+ T cells sorted from three individual mice were admixed as pooled sample for each treatment condition. Pooled samples were also used for ATACseq analysis designed to compare the chromatin accessibility status in donor CD4+ T cells collected before adoptive transfer versus those recovered from the spleen and tumor 10 days post transfer.
RNAseq library preparation, sequencing and analysis
Total RNA was isolated from FACS-sorted donor CD4+ T cells using TRIzol reagent (Thermo Fisher). 100 ng of total RNA was used for RNAseq library preparation by following the Illumina TruSeq stranded mRNA sample preparation guide. The RNAseq libraries were subjected to quantification process, pooled for cBot amplification, and subsequently sequenced on a HiSeq3000 (Illumina) sequencer with 50bp single-end cycling. Demultiplexing with Bcl2fastq2 was employed to generate the fastq file for each sample. 23–32 million single-end reads were obtained for each sample, respectively. The RNAseq analysis was performed on Galaxy main server (https://usegalaxy.org). The raw reads were mapped to mouse genome mm10 using HISTAT2 (Galaxy Version 2.1.0) with default settings. Reads mapped to annotated features were counted using featureCounts (Galaxy v1.6.3). The DEseq2 (Galaxy Version 2.11.40.6) was used to normalize the raw accounts and identify differentially expressed genes. The PCA plot was generated using plotPCA function within DESeq2 package. Volcano plots and heatmaps were generated using EnhancedVolcano v1.2.0 and ComplexHeatmap v2.0.0 in R3.6.0, respectively.
ATACseq profiling
The ATACseq libraries were prepared based on the Omni-ATACseq protocols with some minor modifications(82). Briefly, 60,000 FACS-sorted CD4+ T cells were used to prepare nuclei. The transposition reaction was performed by incubating nuclei with Nextera Tn5 transposase (Illumina) at 37 °C for 1hr. The transposition mixture was purified using a Zymo DNA Clean and Concentrator kit. The ATAC libraries were amplified for 11–13 cycles using NEBNext 2X MasterMix and Nextera Index primers. The amplified libraries were size-selected using AMPure beads to remove the large fragment of DNA and then purified using Zymo DNA Clean and Concentrator kit. Six ATAC libraries were pooled and sequenced on a NextSeq500 instrument in a pair-end 75 cycle run. 60–100 million read pairs were obtained for each sample.
ATACseq and ChIPseq analysis
The ATACseq analysis was performed on the Galaxy server. Adaptor and quality trimming was performed using Trim Glore! Modules (Galaxy v0.4.3.1). Trimmed reads were mapped to mouse genome mm10 using Bowtie2 (Galaxy v2.3.4.3) using default settings. Reads mapped to the mitochondria genome were removed using Filter SAM or BAM function under the samtools module (Galaxy v1.8), and PCR duplicates were removed using MarkDuplicates from Picard tool v2.20.2. The mapped reads overlapping with ENCODE blacklist regions (version 2) were filtered before calling peaks. ATACseq peak calling was performed using MACS2 v2.1.2 with the following command “-p 0.01 --nomodel --shift --100 --extsize 200 -B --SPMR --keep-dup all --call -summits”. BigWig files were generated by bedGraphtoBigwig and visualized in the Integrated genome viewer (IGV) v2.5.2. The Bed file contains common peaks among three biological replicates that were generated using bedTools Multiple Intersect function for CASTAT5 and control samples, respectively. The two bed files were combined using bedtools MergeBED function (Galaxy v1.2.0) to generate a single bed file that contains all peaks identified in both CASTA5 and control samples. Finally, the number of paired fragments that overlap with the intervals identified featureCounts (Galaxy v1.6.3) for each of six ATACseq samples, respectively. DEseq2 (Galaxy Version 2.11.40.6) was used to call differential peaks(83), and ChIPseeker v1.20.0. was used to annotate the differential peaks(84). DiffBind (Galaxy v. 2.6.6.4)(85) was used to identify differential peaks with a fixed window length of 400bp (summit ± 200bp), which were used for motif analysis by MEME-ChIP v5.05 and HOMER v4.9. The Stat5a and pan-stat5 ChIPseq data sets were downloaded from the NCBI short read archive (SRA) according to the corresponding GEO accession number (GSE79518 and GSE77656, respectively). The raw sequencing reads in the fastq format were mapped to the mouse reference genome (mm10) using the same pipeline as described above. MACS2 was used for peak calling, and ChIPseeker v1.20.0. was used to annotate the Stat5a and Stat5-bound peaks.
Integration of RNAseq, ATACseq, and ChIPseq data
The integration of RNAseq with ATACseq or Stat5 ChIPseq data was carried out by matching gene names. The overlapping regions between ATACseq and Stat5 ChIPseq peaks were identified using the findOverlaps function in GenomicRanges 1.38.0 package. Venn diagram was used to show the number of overlapped genes or peaks. For generating integrated heatmaps between ATACseq and RNAseq data, we first matched the differential ATACseq peaks with their nearest differentially expressed genes by matching gene names. Then, we selected those differential peaks positively correlated with the differential expression status of their nearest genes. For genes associated with multiple differential ATACseq peaks, only the peaks with the highest log2FC values compared to the control group were used for plotting heatmaps. For plotting the extensive heatmaps in Fig. 3C and 4B, we first identified the matched ATACseq peak data and RNAseq expression value as described above. Then we identified the center of each peak (summit) and created a 2000bp (summit±1000bp) window. The ATACseq signals within each 2000bp window were collected from the narrowpeak file generated by MACS2 and plotted side-by-side with matched RNAseq expression data. For samples with biological replicates, the merged ATACseq data was used to plot the heatmap. For plotting the heatmap of a panel of candidate genes in Fig.S4A, we sorted the ATACseq peaks associated with each candidate gene by log2FC values from largest to smallest. Then we only selected the ATACseq peak with the largest log2FC value for each gene so that each row corresponds to a unique peak and associated with only one unique gene in the heatmap.
Single cell RNA sequencing (scRNAseq)
FACS-sorted donor CD4+ T cells were processed for scRNAseq libraries using the Chromium Controller (10X Genomics). scRNAseq libraries were generated for approximately 2,000 cells per sample using 10X 3’ single cell mRNAseq V3 reagents. The scRNAseq libraries were sequenced using Illumina NextSeq500 sequencer to collect approximately 80k reads per cell. Each cell was tagged with a 16bp barcode sequence, which represents the identity of each single cell throughput of the analysis pipeline. The raw reads in the fastq format were processed using the 10X genomics cellranger analysis package. CASTAT5 and control samples were pooled using the cellranger aggr pipeline. The cellranger pipeline outputs containing gene-by-cell expression data from the aggregated libraries were imported into R package Seurat 3.1.4 to create a Seurat object(86). Quality control measures were implemented in Seurat to filter out cells expressing > 6000 genes. Cells with a higher percentage of mitochondrial genes (percent of mt >0.3) were also excluded from the subsequent analysis. Normalization and scaling were performed within Seurat before subsequent analysis such as PCA, umap, tSNE, and clustering analyses. Visualization of the scRNA data was performed using Seurat tSNEplot, Vinplot, Featureplot, Dotplot, and DoHeatmap functions. Cell cycle analysis was performed using the Seurat cell cycle scoring function. A cell cycle gene signature containing 97 cell cycle-related genes(87) were used to derive the G1/S and G2/M scores.
RNA velocity analysis
RNA velocity analysis was performed using R package nlvelo, which employs a modified dynamical model to relate the dynamics of unspliced pre-mRNA and spliced mature RNA(42, 43). The bam files generated by the cellranger pipeline were pre-processed using the velocyto command-line tool (CLI) with default parameters specific for 10X genomics data. CASTAT5 and control cells from the Seurat objects were selected for the analysis. A modified dynamic model based on Michaelis-Menten kinetics was used to relate the abundance of pre-mRNA to the abundance of mature mRNA(42). RNA velocity was estimated using a gene-relative model with k-nearest neighbor cell pooling (k = 30). Velocity was visualized on tSNE embedding from the Seurat object using velocity vector fields. The size of the neighborhood cells used for projecting the velocity was set to 500.
Pseudotime and trajectory analysis
The pseudotemporal and trajectory analysis was performed using Monocle 2 with default settings. The Seurat object with cluster-IDs was exported as CellDataSets (CDS) and directly imported into Monocle 2.14.0 (88). CASTAT5 CD4+ T in clusters C4-C6 were analyzed for pseudotime trajectory inference. The marker genes identified by Seurat FindAllMarkers function were used in ordering cells along the pseudotime trajectory. DDRTree method was utilized for dimension reduction and trajectory inference. Branch analysis was performed using the Monocle2 BEAM function. 133 genes (adjusted p-value < 0.001) with expression patterns that are branch dependent was used to plot branchpoint heatmaps. 6 gene blocks were chosen according to the distinct patterns of gene expression change toward the 2 different cell states.
Weighted gene expression network analysis (WGCNA)
We performed WGCNA using R package WGCNA v1.67(89) to identify highly correlated gene modules and establish the regulatory network in the scRNAseq data. The power parameter was determined by the pickSoftThreshold function to allow scale-free topology. The adjacency matrix was calculated to measure the single cell correlation network connectivity. Hierarchical clustering was used to group genes with similar expression profiles into modules based on the dissimilarity of topological overlap matrix (TOM) with a minimum number of 30 genes included in each module. Gene correlation network was visualized using R package igraph v 1.2.4.1.
Transcription factor footprinting analysis
ATACseq footprinting analysis was performed using the Rgt-HINT package(90). The BAM files from the six ATACseq experiments (three biological replicates each from the CASTAT5 and control CD4+ T cells) were merged. The merged two BAM files were used to call peaks with MACS2, as described above(91). The narrowpeak and BAM files were then analyzed using rgt-footprinting, followed by rgt-hint differential modules. The transcription factor footprinting line plots were generated by the analysis. The Mus musculus transcription factor motifs downloaded from HOmo sapiens COmprehensive MOdel COllection (HOCOMOCO) v11 were used in the analysis.
Gene regulatory network construction
The transcription factor binding sites overlapping with predicted footprints were identified by rgt-footprinting as described above. We intersected this map with our RNAseq data and ATACseq data to identify transcription factor binding sites located in the promoter regions (-3000 to +3000bp) around the transcription start sites of genes with positively correlated chromatin accessibility and gene expression changes. Further intersection with ChIP-seq data (GSE79518) revealed a set of regulatory interactions from 45 transcription factors to 888 genes. Then we used this data to construct our regulatory network models in Cytoscape 3.7.
Pathway analysis
Ingenuity pathway analysis (IPA) was performed using the Ingenuity pathway analysis suite spring 2019 release (Qiagen). Go-term analysis, Wikipathway and KEGG pathway enrichment and gene-set enrichment analysis (GSEA) were performed using the R package clusterProfiler v3.12.0(92).
Statistical analysis
Data were analyzed using Prism 7.0 (GraphPad Software, Inc.). For comparison between two groups, the statistical significance of the results was determined using unpaired two-tailed Student’s t test. One-way ANOVA was used to determine statistical differences among three or more groups. For multiple testing correction in sequencing analyses, the false discovery rate (FDR) method was used. The adjusted p-value described in this manuscript refers to FDR.
Supplementary Material
Table S1. List of differentially expressed genes between CASTAT5 and control CD4+ T cells.
Table S3. List of marker genes for the six clusters identified by scRNAseq of CASTAT5 and control CD4+ T cells.
Table S2. List of differential ATACseq peaks between CASTAT5 and control CD4+ T cells.
Table S4. Raw data file (Excel spreadsheet).
Fig. S1. Characterization of CASTAT5-transduced CD4+ T cell survival in vitro and in vivo.
Fig. S2. The transcriptomic sequencing analysis of CASTAT5 and control CD4+ T cells.
Fig. S3. The ATACseq analysis of CASTAT5 and control CD4+ T cells.
Fig. S4. CASTAT5 CD4+ T cells exhibit an epigenetic landscape in concordance with gene and protein expression profiles.
Fig. S5. RNAseq and ATACseq data are consistent with published STAT5A Chip-seq data.
Fig. S6. Motif analysis results from HOMER analysis.
Fig. S7. Transcription factor footprinting analysis identifies key transcription factors.
Fig. S8. scRNAseq feature plots of individual genes.
Fig. S9. Gene co-expression network analysis using WGCNA package.
Fig. S10. Identification of STAT5A H299R and S711F mutations in CASTAT5 CD4+ T cells in both bulk and single-cell RNAseq datasets.
Fig. S11. CASTAT5-transduced CAR T cells exert enhanced antitumor effects in a solid tumor model.
Fig. S12. Transduction of primary human T cells with CASTAT5.
Acknowledgments:
We thank Dr. Toshio Kitamura for providing the CASTAT5 sequence information. We thank Jeanene Pihkala for cell-sorting, Drs. Roni Bollag and Nahid Mivechi for providing reagents for human T cell experiments. We appreciate the assistance from Drs. Wenduo Gu, Qiangbo Xu, Suoqin Jin on scRNAseq analysis.
Funding: This work was supported by National Institutes of Health grants 1R01CA215523 and 1R01CA238514 to G.Z., 1R03CA212829 to H.S., R01 HL56067 and R37AI34495 to B.R.B.
Footnotes
Competing interests: B.R.B. receives remuneration as an advisor to Kamon Pharmaceuticals, Inc, Five Prime Therapeutics Inc, Regeneron Pharmaceuticals, Magenta Therapeutics and BlueRock Therapeutics; research support from Fate Therapeutics, RXi Pharmaceuticals, Alpine Immune Sciences, Inc, Abbvie Inc., BlueRock Therapeutics, Leukemia and Lymphoma Society, Children’s Cancer Research Fund, KidsFirst Fund and is a co-founder of Tmunity. G.Z. is the inventor on a patent application (63/061,339) held/submitted by Augusta University Research Institute, Inc that covers “Chimeric Antigen Receptor T Cells And Methods Of Use Thereof.” All other authors declare no competing interests.
Data and materials availability:
The retroviral vectors used in this study, MSGV-1D3–28Z.1–3 and CASTAT5, were obtained via material transfer agreements with the National Cancer Institute and Yale University, respectively. The data for this study have been deposited in the GEO database with accession #GSE152242. All the open source software packages used in this study are freely available through Bioconductor collection or Github.com. The custom codes used to make figures in this study will be made available upon request. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Sadelain M, CAR therapy: the CD19 paradigm. J Clin Invest 125, 3392–3400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.June CH, Sadelain M, Chimeric Antigen Receptor Therapy. N Engl J Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fesnak AD, June CH, Levine BL, Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer 16, 566–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilham DE, Debets R, Pule M, Hawkins RE, Abken H, CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med 18, 377–384 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Newick K, O’Brien S, Moon E, Albelda SM, CAR T Cell Therapy for Solid Tumors. Annu Rev Med 68, 139–152 (2017). [DOI] [PubMed] [Google Scholar]
- 6.D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M, CAR-T cells: the long and winding road to solid tumors. Cell Death Dis 9, 282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schietinger A, Greenberg PD, Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol 35, 51–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pauken KE, Wherry EJ, Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36, 265–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardoll DM, Topalian SL, The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol 10, 588–594 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Bevan MJ, Helping the CD8(+) T-cell response. Nat Rev Immunol 4, 595–602 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Ding ZC, Zhou G, Cytotoxic chemotherapy and CD4+ effector T cells: an emerging alliance for durable antitumor effects. Clin Dev Immunol 2012, 890178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W, CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 18, 635–647 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG, CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest, (2016). [DOI] [PMC free article] [PubMed]
- 14.Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, Barber DL, Ye L, Sharpe AH, Freeman GJ, Ahmed R, Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci U S A 108, 21182–21187 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C, Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 358, 2698–2703 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA, Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, Yao X, Li YF, Robbins PF, Feldman SA, van der Bruggen P, Klebanoff CA, Goff SL, Sherry RM, Kammula US, Yang JC, Rosenberg SA, Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol 35, 3322–3329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gacerez AT, Sentman CL, T-bet promotes potent antitumor activity of CD4(+) CAR T cells. Cancer Gene Ther 25, 117–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, Ostberg JR, Forman SJ, Brown CE, Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, Restifo NP, Allison JP, Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 207, 637–650 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, Antony PA, Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med 207, 651–667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding ZC, Huang L, Blazar BR, Yagita H, Mellor AL, Munn DH, Zhou G, Polyfunctional CD4(+) T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood 120, 2229–2239 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong H, Lesokhin AM, Avogadri-Connors F, Yuan J, Li Y, Houghton AN, Merghoub T, Wolchok JD, Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med 209, 2113–2126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujiwara H, Ochi T, Ochi F, Miyazaki Y, Asai H, Narita M, Okamoto S, Mineno J, Kuzushima K, Shiku H, Yasukawa M, Antileukemia multifunctionality of CD4(+) T cells genetically engineered by HLA class I-restricted and WT1-specific T-cell receptor gene transfer. Leukemia 29, 2393–2401 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Rossi J, Paczkowski P, Shen YW, Morse K, Flynn B, Kaiser A, Ng C, Gallatin K, Cain T, Fan R, Mackay S, Heath JR, Rosenberg SA, Kochenderfer JN, Zhou J, Bot A, Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 132, 804–814 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, Wherry EJ, Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 40, 289–302 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang S, Cobb DA, Bhadra R, Youngblood B, Khan IA, Blimp-1-mediated CD4 T cell exhaustion causes CD8 T cell dysfunction during chronic toxoplasmosis. J Exp Med 213, 1799–1818 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding ZC, Liu C, Cao Y, Habtetsion T, Kuczma M, Pi W, Kong H, Cacan E, Greer SF, Cui Y, Blazar BR, Munn DH, Zhou G, IL-7 signaling imparts polyfunctionality and stemness potential to CD4(+) T cells. Oncoimmunology 5, e1171445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, Miyajima A, Kitamura T, Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol 18, 3871–3879 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM, Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spranger S, Dai D, Horton B, Gajewski TF, Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723 e714 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chai SK, Nichols GL, Rothman P, Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol 159, 4720–4728 (1997). [PubMed] [Google Scholar]
- 33.Spiekermann K, Bagrintseva K, Schwab R, Schmieja K, Hiddemann W, Overexpression and constitutive activation of FLT3 induces STAT5 activation in primary acute myeloid leukemia blast cells. Clin Cancer Res 9, 2140–2150 (2003). [PubMed] [Google Scholar]
- 34.Birkenkamp KU, Geugien M, Lemmink HH, Kruijer W, Vellenga E, Regulation of constitutive STAT5 phosphorylation in acute myeloid leukemia blasts. Leukemia 15, 1923–1931 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL, Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene 13, 247–254 (1996). [PubMed] [Google Scholar]
- 36.Ribeiro D, Melao A, van Boxtel R, Santos CI, Silva A, Silva MC, Cardoso BA, Coffer PJ, Barata JT, STAT5 is essential for IL-7-mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv 2, 2199–2213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pham HTT, Maurer B, Prchal-Murphy M, Grausenburger R, Grundschober E, Javaheri T, Nivarthi H, Boersma A, Kolbe T, Elabd M, Halbritter F, Pencik J, Kazemi Z, Grebien F, Hengstschlager M, Kenner L, Kubicek S, Farlik M, Bock C, Valent P, Muller M, Rulicke T, Sexl V, Moriggl R, STAT5BN642H is a driver mutation for T cell neoplasia. J Clin Invest 128, 387–401 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owen DL, Farrar MA, STAT5 and CD4 (+) T Cell Immunity. F1000Res 6, 32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wingelhofer B, Neubauer HA, Valent P, Han X, Constantinescu SN, Gunning PT, Muller M, Moriggl R, Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia 32, 1713–1726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nanou A, Toumpeki C, Lavigne MD, Lazou V, Demmers J, Paparountas T, Thanos D, Katsantoni E, The dual role of LSD1 and HDAC3 in STAT5-dependent transcription is determined by protein interactions, binding affinities, motifs and genomic positions. Nucleic Acids Res 45, 142–154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villarino A, Laurence A, Robinson GW, Bonelli M, Dema B, Afzali B, Shih HY, Sun HW, Brooks SR, Hennighausen L, Kanno Y, O’Shea JJ, Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haensel D, Jin S, Sun P, Cinco R, Dragan M, Nguyen Q, Cang Z, Gong Y, Vu R, MacLean AL, Kessenbrock K, Gratton E, Nie Q, Dai X, Defining Epidermal Basal Cell States during Skin Homeostasis and Wound Healing Using Single-Cell Transcriptomics. Cell Rep 30, 3932–3947 e3936 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, Fan J, Borm LE, Liu Z, van Bruggen D, Guo J, He X, Barker R, Sundstrom E, Castelo-Branco G, Cramer P, Adameyko I, Linnarsson S, Kharchenko PV, RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Habtetsion T, Ding ZC, Pi W, Li T, Lu C, Chen T, Xi C, Spartz H, Liu K, Hao Z, Mivechi N, Huo Y, Blazar BR, Munn DH, Zhou G, Alteration of Tumor Metabolism by CD4+ T Cells Leads to TNF-alpha-Dependent Intensification of Oxidative Stress and Tumor Cell Death. Cell Metab 28, 228–242 e226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuczma MP, Ding ZC, Li T, Habtetsion T, Chen T, Hao Z, Bryan L, Singh N, Kochenderfer JN, Zhou G, The impact of antibiotic usage on the efficacy of chemoimmunotherapy is contingent on the source of tumor-reactive T cells. Oncotarget 8, 111931–111942 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcon L, Rivat C, James C, Lacout C, Camara-Clayette V, Ugo V, Lecluse Y, Bennaceur-Griscelli A, Vainchenker W, Constitutive activation of STAT5 and Bcl-xL overexpression can induce endogenous erythroid colony formation in human primary cells. Blood 108, 1551–1554 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Schuringa JJ, Chung KY, Morrone G, Moore MA, Constitutive activation of STAT5A promotes human hematopoietic stem cell self-renewal and erythroid differentiation. J Exp Med 200, 623–635 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuringa JJ, Wu K, Morrone G, Moore MA, Enforced activation of STAT5A facilitates the generation of embryonic stem-derived hematopoietic stem cells that contribute to hematopoiesis in vivo. Stem Cells 22, 1191–1204 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Wherry EJ, Kurachi M, Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pereira RM, Hogan PG, Rao A, Martinez GJ, Transcriptional and epigenetic regulation of T cell hyporesponsiveness. J Leukoc Biol 102, 601–615 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, Lopez-Moyado IF, Georges RO, Zhang W, Onodera A, Wu CJ, Lu LF, Hogan PG, Bhandoola A, Rao A, TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A 116, 12410–12415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, Rao A, NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, Heuston EF, Kirby M, Anderson S, Cheng J, Khan O, Handon R, Reilley J, Fioravanti J, Hu J, Gossa S, Wherry EJ, Gattinoni L, McGavern DB, O’Shea JJ, Schwartzberg PL, Wu T, Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol 20, 890–901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, Wherry EJ, TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, Zamarin D, Walther T, Snyder A, Femia MR, Comen EA, Wen HY, Hellmann MD, Anandasabapathy N, Liu Y, Altorki NK, Lauer P, Levy O, Glickman MS, Kaye J, Betel D, Philip M, Schietinger A, TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, Eisenberg V, Wohlleber D, Steiger K, Merkler D, Delorenzi M, Knolle PA, Cohen CJ, Thimme R, Youngblood B, Zehn D, TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, Hao J, Alekseev A, Khong H, Chen T, Huang R, Wu J, Zhao Q, Wu Q, Xu S, Wang X, Jin W, Yu S, Wang Y, Wei L, Wang A, Zhong B, Ni L, Liu X, Nurieva R, Ye L, Tian Q, Bian XW, Dong C, Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, Lamperti ED, Ansel KM, Mempel TR, Lahdesmaki H, Hogan PG, Rao A, The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity 42, 265–278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mann TH, Kaech SM, Tick-TOX, it’s time for T cell exhaustion. Nat Immunol 20, 1092–1094 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, Wherry EJ, Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu W, Lang PA, Gattinoni L, McGavern DB, Schwartzberg PL, The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, Haining WN, Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin JX, Leonard WJ, Greenleaf WJ, Ahmed R, Goronzy JJ, Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc Natl Acad Sci U S A 116, 14113–14118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR, Chimeric antigen receptor-modified T cells derived from defined CD8 and CD4 subsets confer superior antitumor reactivity in vivo. Leukemia, (2015). [DOI] [PMC free article] [PubMed]
- 65.Davila ML, CAR T cells find strength in polyfunction. Blood 132, 769–770 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, Cui W, CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 51, 1028–1042 e1024 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, Jacobs H, Borst J, CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 47, 848–861 e845 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, Dotti G, Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res 20, 131–139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K, IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol 36, 346–351 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, Yi Z, Sauer T, Liu D, Parihar R, Castillo P, Liu H, Brenner MK, Metelitsa LS, Gottschalk S, Rooney CM, Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov 7, 1238–1247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bajgain P, Tawinwung S, D’Elia L, Sukumaran S, Watanabe N, Hoyos V, Lulla P, Brenner MK, Leen AM, Vera JF, CAR T cell therapy for breast cancer: harnessing the tumor milieu to drive T cell activation. J Immunother Cancer 6, 34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sillaber C, Gesbert F, Frank DA, Sattler M, Griffin JD, STAT5 activation contributes to growth and viability in Bcr/Abl-transformed cells. Blood 95, 2118–2125 (2000). [PubMed] [Google Scholar]
- 73.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC, A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352, 1779–1790 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sargin B, Ueker A, Bohmer FD, Berdel WE, Muller-Tidow C, Serve H, Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood 110, 370–374 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Kontro M, Kuusanmaki H, Eldfors S, Burmeister T, Andersson EI, Bruserud O, Brummendorf TH, Edgren H, Gjertsen BT, Itala-Remes M, Lagstrom S, Lohi O, Lundan T, Marti JM, Majumder MM, Parsons A, Pemovska T, Rajala H, Vettenranta K, Kallioniemi O, Mustjoki S, Porkka K, Heckman CA, Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia 28, 1738–1742 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Bandapalli OR, Schuessele S, Kunz JB, Rausch T, Stutz AM, Tal N, Geron I, Gershman N, Izraeli S, Eilers J, Vaezipour N, Kirschner-Schwabe R, Hof J, von Stackelberg A, Schrappe M, Stanulla M, Zimmermann M, Koehler R, Avigad S, Handgretinger R, Frismantas V, Bourquin JP, Bornhauser B, Korbel JO, Muckenthaler MU, Kulozik AE, The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica 99, e188–192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kollmann S, Grundschober E, Maurer B, Warsch W, Grausenburger R, Edlinger L, Huuhtanen J, Lagger S, Hennighausen L, Valent P, Decker T, Strobl B, Mueller M, Mustjoki S, Hoelbl-Kovacic A, Sexl V, Twins with different personalities: STAT5B-but not STAT5A-has a key role in BCR/ABL-induced leukemia. Leukemia 33, 1583–1597 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Burchill MA, Goetz CA, Prlic M, O’Neil JJ, Harmon IR, Bensinger SJ, Turka LA, Brennan P, Jameson SC, Farrar MA, Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: development of CD4+CD25+ regulatory T cells versus CD8+ memory T cells. J Immunol 171, 5853–5864 (2003). [DOI] [PubMed] [Google Scholar]
- 79.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G, Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 24, 1160–1170 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Riedel SS, Mottok A, Brede C, Bauerlein CA, Jordan Garrote AL, Ritz M, Mattenheimer K, Rosenwald A, Einsele H, Bogen B, Beilhack A, Non-invasive imaging provides spatiotemporal information on disease progression and response to therapy in a murine model of multiple myeloma. PLoS One 7, e52398 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA, Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 116, 3875–3886 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, Kathiria A, Cho SW, Mumbach MR, Carter AC, Kasowski M, Orloff LA, Risca VI, Kundaje A, Khavari PA, Montine TJ, Greenleaf WJ, Chang HY, An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu G, Wang LG, He QY, ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 85.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C, Carroll JS, Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin JR, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CG, Kazer SW, Gaillard A, Kolb KE, Villani AC, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jane-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA, Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL, The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Langfelder P, Horvath S, WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li Z, Schulz MH, Look T, Begemann M, Zenke M, Costa IG, Identification of transcription factor binding sites using ATAC-seq. Genome Biol 20, 45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu G, Wang LG, Han Y, He QY, clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of differentially expressed genes between CASTAT5 and control CD4+ T cells.
Table S3. List of marker genes for the six clusters identified by scRNAseq of CASTAT5 and control CD4+ T cells.
Table S2. List of differential ATACseq peaks between CASTAT5 and control CD4+ T cells.
Table S4. Raw data file (Excel spreadsheet).
Fig. S1. Characterization of CASTAT5-transduced CD4+ T cell survival in vitro and in vivo.
Fig. S2. The transcriptomic sequencing analysis of CASTAT5 and control CD4+ T cells.
Fig. S3. The ATACseq analysis of CASTAT5 and control CD4+ T cells.
Fig. S4. CASTAT5 CD4+ T cells exhibit an epigenetic landscape in concordance with gene and protein expression profiles.
Fig. S5. RNAseq and ATACseq data are consistent with published STAT5A Chip-seq data.
Fig. S6. Motif analysis results from HOMER analysis.
Fig. S7. Transcription factor footprinting analysis identifies key transcription factors.
Fig. S8. scRNAseq feature plots of individual genes.
Fig. S9. Gene co-expression network analysis using WGCNA package.
Fig. S10. Identification of STAT5A H299R and S711F mutations in CASTAT5 CD4+ T cells in both bulk and single-cell RNAseq datasets.
Fig. S11. CASTAT5-transduced CAR T cells exert enhanced antitumor effects in a solid tumor model.
Fig. S12. Transduction of primary human T cells with CASTAT5.
Data Availability Statement
The retroviral vectors used in this study, MSGV-1D3–28Z.1–3 and CASTAT5, were obtained via material transfer agreements with the National Cancer Institute and Yale University, respectively. The data for this study have been deposited in the GEO database with accession #GSE152242. All the open source software packages used in this study are freely available through Bioconductor collection or Github.com. The custom codes used to make figures in this study will be made available upon request. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.