

Abstract
Defects in stem cell renewal or progenitor cell expansion underlie ageing-related diseases such as osteoporosis. Yet much remains unclear about the mechanisms regulating progenitor expansion. Here we show that the tyrosine kinase c-Abl plays an important role in osteoprogenitor expansion. c-Abl interacts with and phosphorylates BMPRIA and the phosphorylation differentially influences the interaction of BMPRIA with BMPRII and the Tab1-Tak1 complex, leading to uneven activation of Smad1/5/8 and Erk1/2, the canonical and non-canonical BMP pathways that direct the expression of p16INK4a. c-Abl deficiency shunts BMP signalling from Smad1/5/8 to Erk1/2, leading to p16INK4a upregulation and osteoblast senescence. Mouse genetic studies revealed that p16INK4a controls mesenchymal stem cell maintenance and osteoblast expansion and mediates the effects of c-Abl deficiency on osteoblast expansion and bone formation. These findings identify c-Abl as a regulator of BMP signalling pathways and uncover a role for c-Abl in p16INK4a expression and osteoprogenitor expansion.
Bone homeostasis is sustained by bone marrow mesenchymal stem cells (MSCs), which have the capacity of self-renewal and differentiation into osteoblasts and other cell types1,2. In the osteogenic differentiation process, driven by growth factors such as bone morphogenetic proteins (BMPs), there exist osteoprogenitors that expand through cellular proliferation3,4. Expansion of osteoprogenitors as well as other cells is limited by their proliferation capacity as primary cells have a finite lifespan and eventually undergo replicative senescence5,6. In addition, cells also undergo senescence under oxidative stress, genotoxic stress or constitutive activation of the Ras–MAPK pathway7,8. Both replicative senescence and stress-induced senescence are genetically controlled by the p53/p21 and p16INK4a pathways9,10. Cell senescence is believed to cause ageing but suppress tumorigenesis11–13.
c-Abl is a non-receptor tyrosine kinase that is either associated with plasma membrane (type IV) or localized in the cytoplasm and nucleus (type I; refs 14,15). The oncogenic form of Abl, BCR-ABL, is constitutively activated and causes chronic myeloid leukaemia16 (CML). Biochemical studies indicate that c-Abl can be activated by genotoxic/oxidative stress, platelet-derived growth factor and other stimuli, to regulate proliferation, apoptosis and cytoskeleton dynamics17,18. c-Abl−/− mice exhibit perinatal lethality, growth retardation, thymus and spleen atrophies, lymphopenia, infertility and osteoporosis19–22. c-Abl regulates T/B-cell activation and survival through multiple signalling pathways including the T-cell receptor pathway23–26. Mice deficient for c-Abl and its paralogue Arg, a cytoplasmic protein, are embryonic lethal owing to neural tube closure defects27. c-Abl also regulates neurite outgrowth, synapse formation, and survival through CDK5, F-actin, PSD-95 and Parkin28–33. Yet, how c-Abl regulates other developmental aspects, especially bone remodelling, remains unidentified34. In this study, we investigated the molecular mechanisms behind the development of osteoporosis in c-Abl−/− mice and identified a link between c-Abl and the signalling pathways activated by BMPs, which are secreted by osteoblasts (residing in the bone) and are detectable in the bloodstream35,36. We show that c-Abl, through phosphorylating BMP receptor IA (BMPRIA), differentially regulates the Smad1/5/8 and Tak1–Mek1/2–Erk1/2 pathways, to control p16INK4a expression and thereafter osteoblast expansion and bone formation, thus establishing osteoprogenitor expansion defects as a cause of senile osteoporosis.
RESULTS
c-Abl −/− osteoblasts show reduced proliferation capacity and premature senescence
We previously showed that c-Abl−/− mice developed senile osteoporosis, accompanied by defective osteoblast differentiation21. We report here that c-Abl−/− mice had a decreased number of osteoblasts in their bones as well (Fig. 1a). BrdU labelling experiments revealed that c-Abl−/− bones (trabecular regions) had fewer cells in S phase (Fig. 1b). c-Abl−/− mice also showed a modest decrease in the number of alkaline phosphatase (ALP)-positive MSCs (ref. 21). These in vivo data support a positive role for c-Abl in regulating osteoblast proliferation/expansion. We then compared the growth potential of c-Abl−/− and wild-type (WT) calvarial osteoblasts using a modified 3T3 procedure37. Primary osteoblasts were isolated from single calvaria and plated onto a 35 mm plate (passage 1), which were expanded to a 60 mm plate (passage 2) and then to a 100 mm plate (passage 3), before being used for further experiments. These cells have the capacity to proliferate but have not yet expressed differentiation markers; yet they could differentiate into mature osteocytes in response to BMPs, and are therefore considered osteoprogenitors (referred to as osteoblasts in this report). No significant difference between c-Abl−/− and WT osteoblast cultures was observed in the number of cells isolated (data not shown), the proliferation rates (doubling time 36.5 ± 5.7 h for +/+ versus 37.2 ± 4.5 for −/−, n = 3) or the percentage of dead cells (7.1 ± 1.4% for +/+ versus 7.4 ± 1.5% for −/−, n = 3) at early passage (p1–3).
Figure 1.

c-Abl−/− osteoblasts show reduced proliferation potential in vivo and undergo premature senescence ex vivo. (a) c-Abl−/− bone sections showed a reduced number of osteoblasts. Femur bones of 2-month-old c-Abl−/− and WT mice were used for histomorphometric analysis. (b) c-Abl−/− bone sections (newborn pups) showed a decrease in the number of S-phase cells. BrdU-positive cells (brown) were counted in equal areas underneath the growth plate. (c) Growth of c-Abl−/− and WT osteoblasts. Calvarial osteoblasts were cultured as described and the numbers of cells are plotted against passage numbers. (d) The effects of c-Abl deficiency, c-Abl reconstitution, p16INK4a deficiency and ectopic expression of Id1 on the lifespan of osteoblasts. Primary calvarial osteoblasts were isolated from p16INK4a−/−, c-Abl−/− p16INK4a−/− mice and their control littermates. The doubling times (until senescence) were counted after passage 3. For retroviral expression of c-Abl, see Supplementary Fig. S2a. (e) Quantification results of cells positive for histochemical staining of senescence-associated SA-β-Gal. WT, c-Abl−/− and c-Abl reconstituted or empty vector infected c-Abl−/− osteoblasts were cultured, fixed and stained at a neutral pH for SA-β-Gal. Percentages of SA-β-Gal-positive cells at each passage of mutant and control osteoblast cultures are shown. Scale bar, 50 μm. Data are means ± s.e.m. (a, n = 5; b, n = 4; c–e, n = 3). *P < 0.05 when compared to WT counterparts. **P < 0.05 when compared to c-Abl−/− osteoblasts or mice.
Further culture of the cells revealed that both mutant and WT osteoblasts underwent senescence, yet c-Abl−/− osteoblasts ceased growing at much earlier passages and had reduced proliferating capacity, which was confirmed by BrdU labelling experiments (Fig. 1c,d). Both mutant and WT cultures also showed an increase in cells positive for senescence-associated β-galactosidase (SA-β-Gal), a widely used senescence marker38, at later passages, with c-Abl−/− cultures showing more senescent cells (Fig. 1e). Moreover, treatment with c-Abl inhibitor STI571 (Gleevec, imatinib mesylate) led to decreased proliferation capacity and increased senescence in normal osteoblasts (Supplementary Fig. S1a,b). c-Abl reconstitution using retrovirus vector rescued the reduced proliferation capacity and increased senescence of c-Abl−/− cultures (Fig. 1d,e, for c-Abl expression, see Supplementary Fig. S2a). These results, taken together, support a role for c-Abl in directly regulating osteoblast senescence and expansion.
p16INK4a upregulation underlies premature senescence of c-Abl −/− osteoblasts
Senescence is controlled by the p16INK4a and p19/p53/p21 pathways, with p16INK4a also acting as an ageing marker9. The levels of p19 or p21 did not significantly change between passage 4 and 6 (when mutant cells ceased dividing) or between c-Abl−/− and normal osteoblasts, yet the levels of p16INK4a were higher in c-Abl−/− osteoblasts, which were further elevated when cells entered senescence (Fig. 2a–c), suggesting that upregulation of p16INK4a may play a role in initiating and/or maintaining senescence in osteoblasts and that c-Abl regulates p16INK4a expression. STI571 treatment or c-Abl knockdown also led to p16INK4a upregulation in normal osteoblasts (Supplementary Fig. S1c). Moreover, the bone sections of c-Abl−/− mice exhibited increased levels of p16INK4a in the trabecular region (Fig. 2d), and the RNA extracted from c-Abl−/− mouse femurs also showed an increase in p16INK4a messenger RNA levels (Fig. 2e). The sites of p16INK4a upregulation are correlated to the localization of c-Abl expression, at the endochondral ossification region39 (Supplementary Fig. S2b). These in vivo and ex vivo results suggest that not only are the c-Abl−/− osteoblasts at a more advanced stage of senescence but also that c-Abl represses p16INK4a expression.
Figure 2.
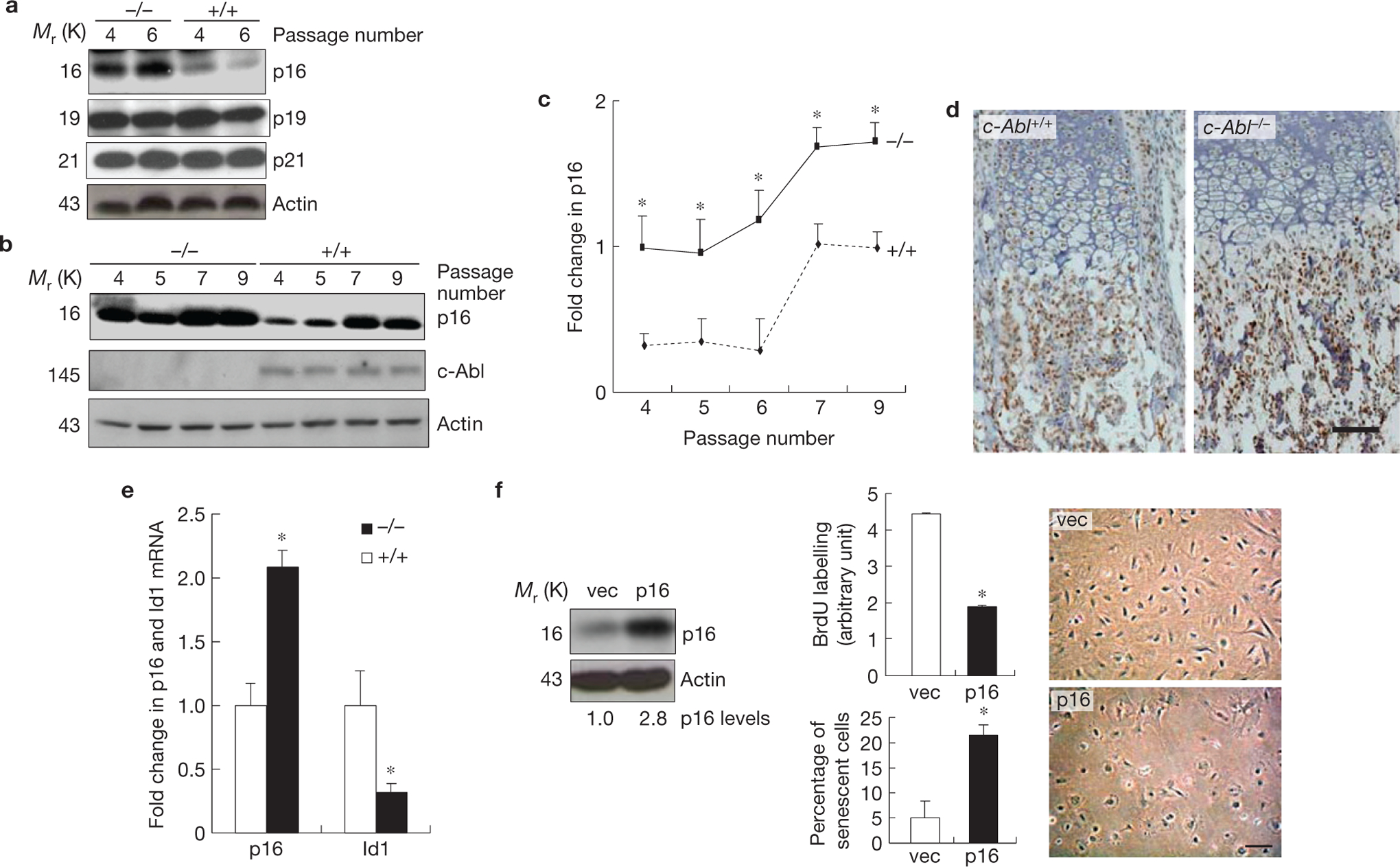
c-Abl−/− osteoblasts showed p16INK4a upregulation, which is responsible for premature senescence. (a) c-Abl−/− osteoblasts expressed increased levels of p16INK4a, but not p19 or p21, during replicative senescence. Mutant and WT osteoblasts were cultured as in Fig. 1c. At day three of the passages indicated, cells were collected and the same amounts of total protein were analysed by western blot. (b) Expression of p16INK4a in c-Abl−/− and control osteoblasts during the passaging of the cells. (c) Quantification results of a and b combined. (d) Immunohistological staining of p16INK4a on bone sections of newborn pups. Note that the difference mainly exists in the area occupied by osteoblasts, but not areas occupied by chondrocytes. For c-Abl expression, see Supplementary Fig. S2b. (e) Real-time PCR results show that c-Abl−/− femurs exhibited an increase in the mRNA levels of p16INK4a and a decrease in Id1. Total RNA was isolated from femurs of three c-Abl−/− and WT mice. The smaller increase in p16INK4a than cell cultures might be caused by inclusion of non-osteoblast cells, which may not behave like osteoblasts in Id1 and p16INK4a expression. (f) Ectopic expression of p16INK4a led to senescence-like phenotypes. WT osteoblasts were infected with empty retroviruses or viruses expressing p16INK4a, selected against puromycin, and cultured. Left panel: western blot showing the levels of p16INK4a; middle panels: BrdU incorporation (upper) and percentages of SA-β-Gal-positive cells (bottom); right panel: cell morphology change in p16INK4a-expressing cells. Scale bars, 50μm. Data are means ± s.e.m. (n = 3). *P < 0.05 when compared to WT counterparts. Uncropped images of blots are shown in Supplementary Fig. S8.
Functionally, ectopic expression of p16INK4a resulted in senescence-like phenotypes in primary WT osteoblasts: flattened morphology, reduced BrdU incorporation and an increase in SA-β-Gal-positive cells (Fig. 2f), suggesting that elevated expression of p16INK4a was able to induce senescence in osteoblasts. Moreover, p16INK4a deficiency could rescue the premature senescence phenotype of c-Abl−/− osteoblasts as c-Abl−/− p16INK4a−/− osteoblasts, like p16INK4a−/− osteoblasts, showed a modestly extended lifespan when compared with WT osteoblasts (Fig. 1d). These results suggest that p16INK4a upregulation is responsible for premature senescence of c-Abl−/− osteoblasts.
p16INK4a upregulation is attributable to enhanced Erk1/2 activation in c-Abl −/− osteoblasts
p16INK4a expression can be induced by the constitutively active Ras–Raf–MAPK pathway, repressed by the helix–loop–helix protein Id1 and regulated by chromatin remodelling40–42. We indeed found that c-Abl−/− osteoblasts exhibited constantly higher levels of activated Erk1/2 (Fig. 3a). Note that overnight cultures showed much higher Erk1/2 activation than 3-day cultures (compare passage ‘o/n’ and p4), probably owing to growth factors in the fresh serum. Also note that we are looking at the steady-state levels of activated Erk1/2 rather than transient activation, with the former usually inducing senescence and the latter promoting cell proliferation43,44. Inhibition of c-Abl with STI571 also enhanced Erk1/2 activation and reconstitution of c-Abl in c-Abl−/− osteoblasts resulted in reduced Erk1/2 activation (Supplementary Figs S1d and S2c). Activation of Mek1/2, immediate upstream kinases for Erk1/2, was also enhanced in c-Abl−/− osteoblasts, and inhibition of Mek1/2 with U0126 repressed p16INK4a expression in WT and c-Abl−/− osteoblasts (Fig. 3b,c), suggesting that enhanced Erk1/2 activation contributes to p16INK4a upregulation in c-Abl−/− osteoblasts. The incomplete suppression by U0126 suggests there exist additional factors that regulate p16INK4a expression. We also found that long-term U0126 treatment suppressed osteoblast proliferation (Supplementary Fig. S2d), confirming that activation of Erks is required for cell growth as well43,44.
Figure 3.

c-Abl−/− osteoblasts show enhanced Erk1/2 activation in response to BMP2, which contributes to p16INK4a upregulation. (a) Mutant and WT cells were cultured as in Fig. 1c and one plate was collected at day three of each passage. Passage ‘o/n’ stands for cells that have been grown overnight. Erk activation was analysed by western blot. As p-Erk1/2 levels are similar in different passages of WT or c-Abl−/− osteoblasts, an average is shown in the right panel. (b) Inhibition of Mek1/2 activation decreased the levels of p16INK4a. Osteoblasts (passage 2) were treated for 24 h with different concentrations of U0126 and western blot analysis was used to determine the protein levels of p16INK4a. Right panel: quantification of p16INK4a. (c) c-Abl deficiency altered BMP2-induced Erk1/2 activation. c-Abl−/− and control osteoblasts were stimulated with 100 ng ml−1 BMP2 for different periods of time and western blot analysis was used to determined the activation of these kinases. Right panel: quantification of active Erk1/2. (d) Blockade of BMP2 action with noggin/chordin led to Erk1/2 activation. c-Abl−/− and WT osteoblasts were treated with BMP2 (B; 100 ng ml−1) or noggin/chordin (N/C; 0.5 μg ml−1 and 1.0 μg ml−1, respectively) for 2 days and western blot was used to determine the activation of Erk1/2 and Tak1. Note that p-Tak1 is difficult to detect in WT cells. Right panel: quantification. Data are means ± s.e.m. (n = 3) *P < 0.05, compared to untreated cells or WT cells. Uncropped images of blots are shown in Supplementary Fig. S8.
A role for c-Abl in BMP-induced Erk1/2 activation
BMPs activate Erk1/2 by recruiting the Tab1–Tak1 complex to BMPRIA through adaptor proteins including Traf6 and Xiap (refs 45,46), in addition to the Smad1 pathway35,47,48. In normal osteoblasts, BMP2 stimulation led to rapid Erk1/2 activation that lasted about 30 min, followed by repression of Erk1/2 activation. However, c-Abl−/− osteoblasts showed elevated basal levels and enhanced transient activation of Erk1/2, and impeded Erk1/2 repression in response to BMP2 (Fig. 3b,c), which were restored by c-Abl reconstitution (Supplementary Fig. S2e). Blockade of BMP action with noggin/chordin led to Erk1/2 activation in WT cells, but much less in c-Abl−/− osteoblasts, whereas long-term BMP2 presence led to Erk1/2 inactivation in WT osteoblasts but not in c-Abl−/− osteoblasts (Fig. 3d). Moreover, activation of Tak1, upstream kinase of Mek1/2, was also enhanced in the absence of c-Abl (Fig. 3d). These results, taken together, suggest that Erk1/2 activation in c-Abl−/− osteoblasts is modulated by BMPs.
p16INK4a upregulation is also attributable to reduced expression of BMP-target gene Id1
A number of studies have demonstrated that Id1 inhibits cell senescence in a p16INK4a-dependent manner49,50. We found that Id1 was downregulated in c-Abl−/− osteoblasts and femur bones (Figs 4a, 3b and 2e). Id1 is a direct target gene of BMPs. On BMP2 treatment, both c-Abl−/− and WT osteoblasts showed a pronounced Id1 induction, with the former showing significantly less (Fig. 4b). c-Abl−/− osteoblasts also showed a decrease in Id1 mRNA levels, which was restored by c-Abl reconstitution (Fig. 4c). The role for c-Abl in BMPs-induced Id1 transcription was supported by the observations that overexpression of c-Abl enhanced Id1 induction by BMP2 in the osteoblast cell line MC3T3–E1 (Supplementary Fig. S3a); and reporter assays showed that v-Abl and c-Abl activated the Id1 promoter and the BMP-responsive SBE–Luc reporter, which were suppressed by STI571 or c-Abl deficiency51 (Supplementary Fig. S3b–e). Besides Id1, c-Abl−/− osteoblasts also showed compromised expression of BMP target genes including Id3, Smad6, Smad7 and Jun B at the mRNA levels, which could be restored by c-Abl reconstitution (Supplementary Fig. S3f–i).
Figure 4.
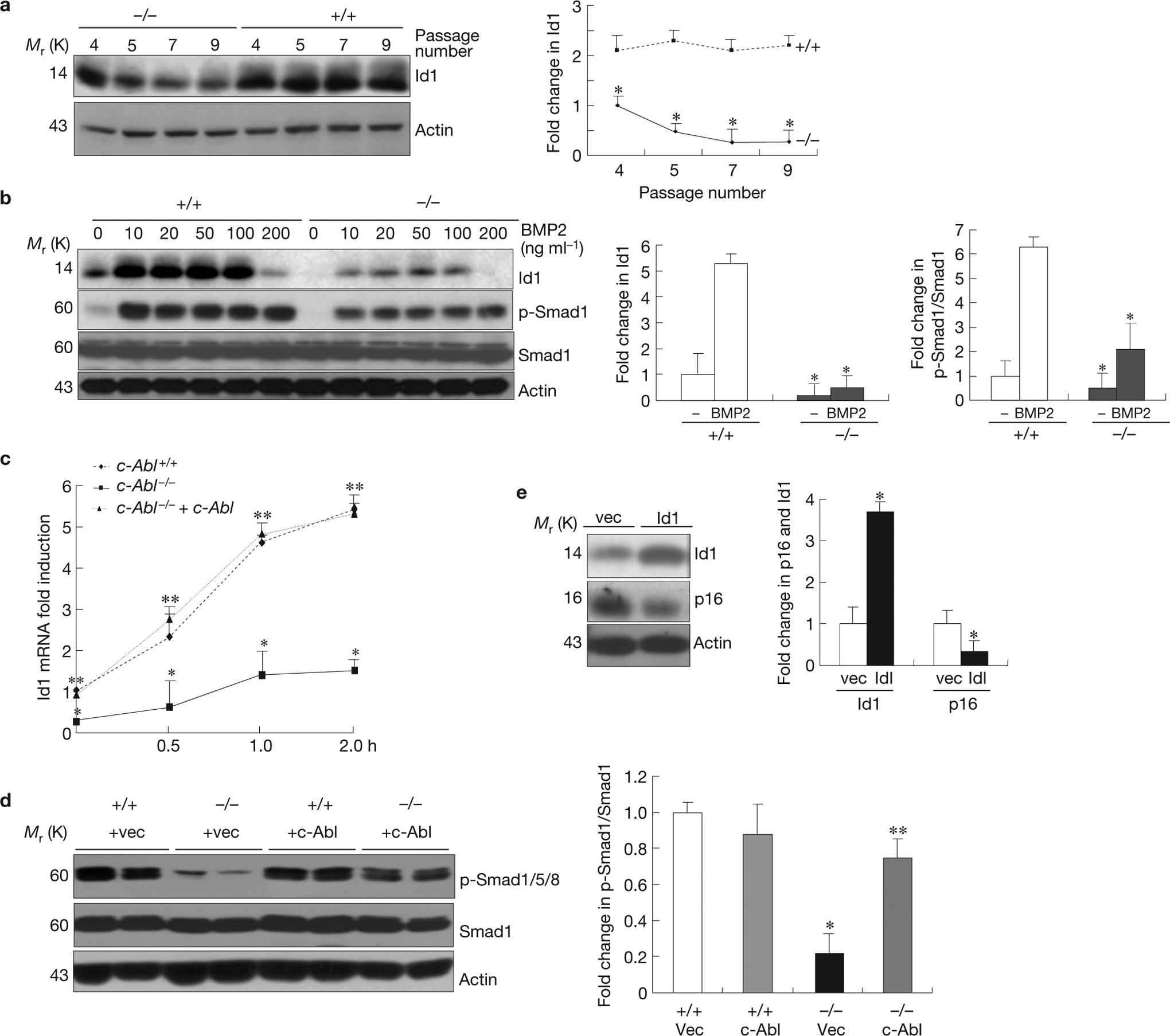
c-Abl positively regulates the expression of Id1 through BMP Smad1/5/8 signalling, which contributes to p16INK4a upregulation. (a) Downregulation of Id1 in c-Abl−/− osteoblasts. The experiment was carried out as described in Fig. 2b and the protein levels of Id1 were determined by western blot analysis. Right panel: quantification. (b) Reduced Id1 induction and Smad1 activation by BMP2 in c-Abl−/− osteoblasts. c-Abl−/− and control osteoblasts were serum-starved, stimulated with different concentrations of BMPs for 4 h and the levels of Id1 were analysed by western blot analysis. BMP2 at 200 ng ml−1 failed to induce Id1, probably owing to a feedback regulation in cell response to high concentrations of BMP2. Right panel: quantification. As p-Smad1 levels and Id levels are similar in response to different doses of BMP2, an average is shown in the right panel. (c) Real-time PCR assays revealed reduced induction of Id1 by BMP2 at the mRNA level in c-Abl−/− osteoblasts. WT, c-Abl−/− and c-Abl reconstituted osteoblasts were stimulated with 5 ng ml−1 BMP2 for different periods of time and the levels of Id1 mRNA were assessed by real-time PCR. (d) Reduced basal levels of p-Smad1/5/8 in c-Abl−/− osteoblasts (two lines from different embryos and control littermates) were restored by c-Abl reconstitution (see Supplementary Fig. S2a for c-Abl expression). Right panel: quantification. (e) Ectopic expression of Id1 with retrovirus led to downregulation of p16INK4a in c-Abl−/− osteoblasts. Right panel: quantification data. Data are means ± s.e.m. (n=3). *P < 0.05, compared to WT cells, **P < 0.05, compared with c-Abl−/− cells. Uncropped images of blots are shown in Supplementary Fig. S8.
All of these results suggest that c-Abl is involved in BMP–Smad1 signalling. Indeed, we found that the basal levels of phosphorylated Smad1/5/8, but not TGFβ-responsive Smad2/3, were significantly reduced in c-Abl−/− cells, which was rescued by c-Abl reconstitution (Fig. 4d and data not shown). Moreover, c-Abl−/− osteoblasts also showed reduced Smad1/5/8 activation in response to BMP2 (Fig. 4b), and STI571 also suppressed Smad1 activation (Supplementary Fig. S1d). These results suggest that c-Abl is involved in BMP–Smad1/5/8 activation.
On the other hand, BCR-ABL-positive K562 myeloid cells showed enhanced Smad1/5/8 activation and elevated Id1 expression when compared with the BCR-ABL-negative myeloid cell line HL-60, which were diminished by STI571 or by noggin and chordin (Supplementary Fig. S3j–m). The expression of p16INK4a is silenced in K562 cells (data not shown), as well as in leukaemic cells of many CML patients, indicating the importance of downregulating p16INK4a in CML development16. These findings suggest that activated Abl could enhance Id1 expression through BMP–Smad1 signalling.
Functionally, knockdown of Smad1 with short interfering RNA led to a decrease in Id1 and an increase in p16INK4a expression in primary osteoblasts, validating a role for Smad1 in regulating p16INK4a expression (Supplementary Fig. S4a). Moreover, overexpression of Id1 in c-Abl−/− osteoblasts using a retroviral vector was able to downregulate p16INK4a and extended the lifespan of c-Abl−/− cells by three doublings (Fig. 4e,1d).
We then investigated the function of BMPs in p16INK4a expression and found that blockade of BMP action with noggin and chordin decreased Id1 expression and resulted in an increase in p16INK4a (Supplementary Fig. S4b), and BMP2 downregulated p16INK4a in osteoblast cultures, but to a lesser extent, which was affected by Id1 knockdown or Erk1/2 inhibition (Supplementary Fig. S4c). These results suggest that BMPs regulate p16INK4a expression through Erks and Id1.
c-Abl phosphorylates BMPRIA to regulate the lifespan of osteoblasts
We then wanted to study how c-Abl simultaneously regulates BMP–Erk1/2 and BMP–Smad1/5/8 activation. c-Abl deficiency did not alter the expression of BMPRIA, BMPRIB or BMPRII (data not shown). Yet c-Abl could phosphorylate co-expressed BMPRIA and BMPRIB, but not BMPRII (Fig. 5a and data not shown). In WT cells, endogenous BMPRIA was tyrosine phosphorylated, but the level of this phosphorylation was significantly reduced in c-Abl−/− osteoblasts and c-Abl−/− mouse embryonic fibroblasts (MEFs), and almost abolished in c-Abl−/− Arg−/− MEFs (Fig. 5b and Supplementary Fig. S5a,b). An in vitro kinase assay showed that immunoprecipitated c-Abl, but not the kinase-dead c-Abl, could phosphorylate purified BMPRIA, which was not significantly affected by BMP2 treatment (Fig. 5c). Furthermore, we found that endogenous or ectopically expressed c-Abl and BMPRIA formed a complex, which was not altered by BMP2 treatment (Fig. 5d and Supplementary Fig. S5c,d). These results suggest that c-Abl associates with and phosphorylates BMPRIA at the basal levels.
Figure 5.

c-Abl phosphorylates BMPRIA, which plays a role in osteoblast expansion. (a) Co-expression with c-Abl led to BMPRIA tyrosine phosphorylation. BMPRIA (HA tagged) was expressed alone or co-expressed with c-Abl in Cos7 cells. BMPRIA was immunoprecipitated (IP) using anti-HA antibodies and its tyrosine phosphorylation was determined by immunoblotting (IB). KD, kinase dead. (b) Reduced tyrosine phosphorylation of BMPRIA in c-Abl−/− osteoblasts. Endogenous BMPRIA was immunoprecipitated from the cell lysate of the mutant and control cells, and its tyrosine phosphorylation was determined by immunoblotting. Right panel: quantification. (c) The in vitro kinase assay shows that immunoprecipitated c-Abl, but not kinase-dead c-Abl, was able to phosphorylate purified BMPRIA on tyrosine residues. Right panel: quantification. (d) Interaction between endogenous c-Abl and BMPRIA in osteoblasts. Osteoblasts were treated with 100 ng ml−1 BMP2 for 1 h or untreated; cell lysates were divided into three parts for analysis of BMPRIA and c-Abl expression by western blot (WB; left panel), immunoprecipitation of BMPRIA (upper right) or immunoprecipitation of c-Abl (lower right), with IgG as control antibodies. (e) A diagram showing the alignment of the C-terminal regions of BMPRIA of different species. (f) Mutagenesis revealed that all four tyrosine residues could be phosphorylated with Tyr 453/467 being more preferably phosphorylated by c-Abl. BMPRIA with different combinations of tyrosine to phenylalanine mutations was co-expressed with c-Abl and their phosphorylation status was determined by western blot analysis. m1, Y453/457/458F; m2, Y457/458/467F; m3, Y453/467F; m4, 453/457/458/467F (see Supplementary Fig. S5e lower panel for positioning of these mutations). (g) Bmpr1a−/− osteoblasts expressing YF BMPRIA showed a decrease in lifespan when compared with cells expressing WT BMPRIA. For the levels of WT and mutant BMPRIA, see Supplementary Fig. S6a. Data are means ± s.e.m. (n = 3). *P < 0.05, compared with WT BMPRIA. Uncropped images of blots are shown in Supplementary Fig. S8.
Serial deletion experiments revealed that tyrosine phosphorylation occurred at the extreme carboxy terminus of BMPRIA (Supplementary Fig. S5e). Sequence alignment of this region revealed four highly conserved tyrosine residues in a cluster (Fig. 5e). Mutagenesis experiments of single or multiple tyrosine residues to phenylalanine indicated that all 4 tyrosine residues could be phosphorylated with Tyr 453/467 being preferential sites (Fig. 5f). The phosphorylation sites were confirmed by mass spectrometric analysis, as these tyrosine residues on two peptides (amino acids 448–462 and 456–471) could be phosphorylated by purified c-Abl (Supplementary Fig. S5f–h).
To investigate the function of these phosphorylation sites, we infected Bmpr1a f/f (homozygous for the floxed allele of Bmpr1a) osteoblasts with retrovirus expressing Cre (puromycin selection marker) and retroviruses expressing WT BMPRIA or the mutant BMPRIA (all four tyrosine residues mutated to phenylalanine, designated the YF mutant; hygromycin selection marker). The cells selected against both antibiotics were Bmpr1a−/− osteoblasts expressing the foreign WT or YF BMPRIA (Supplementary Fig. S6a). We found that cells expressing YF BMPRIA had a reduced lifespan when compared with cells expressing WT BMPRIA (Fig. 5g), revealing an important role for BMPRIA phosphorylation in cell expansion.
It seems that BMPRIA YF mutation has different effects from BMPRIA deletion. Osteoblast-specific BMPRIA knockout mice initially have a normal number of osteoblasts52–54. This was confirmed by our finding that 6-week-old osteoblast-specific BMPRIA knockout mice (Bmpr1a f/f, Osx1–GFP::Cre; ref. 55) had a normal number of osteoblasts, and that deletion of BMPRIA with Cre-expressing retrovirus in Bmpr1a f/f osteoblasts did not alter their lifespan (Supplementary Fig. S6a–c). This can be attributable to the fact that BMPRIA deletion has a minor or no effect on p16INK4a expression and Erk activation (Supplementary Fig. S6d). The observation that osteoblast-specific BMPRIA deletion leads to increased bone mass in older mice owing to decreased osteoclastogenesis and bone resorption, whereas c-Abl−/− mice showed osteoporosis, could be explained by the findings that Bmpr1a−/− osteoblasts showed a decrease in RANKL but an increase in OPG production52,54, whereas c-Abl−/− mice did not show a significant change in osteoclastogenesis/bone resorption21, or the expression of RANKL/OPG in osteoblasts (Supplementary Fig. S6e). Recent studies showed that Smad1 and Erk both play positive roles in RANKL expression56. Thus, the effects of Erks and Smad1 on RANKL expression might be negated in c-Abl−/− osteoblasts.
BMPRIA phosphorylation differentially regulates Erk1/2 and Smad1 pathways
We then investigated the roles of BMPRIA phosphorylation in regulating canonical and non-canonical BMP pathways and found that Bmpr1a−/− osteoblasts expressing YF BMPRIA showed a decrease in Smad1 activation and Id1 expression, but an increase in Erk activation and p16INK4a expression when compared with the cells expressing WT BMPRIA, whereas cells expressing YD BMPRIA (phosphomimetic mutation of all 4 tyrosines to aspartic acid) showed the opposite (Fig. 6a,b). These results validate a critical role for tyrosine phosphorylation of BMPRIA in regulating BMP downstream pathways. Moreover, ectopic expression of YF BMPRIA also enhanced Erk1/2 activation in osteoblasts or C2C12 cells when compared with WT BMPRIA (Supplementary Fig. S2f,g).
Figure 6.
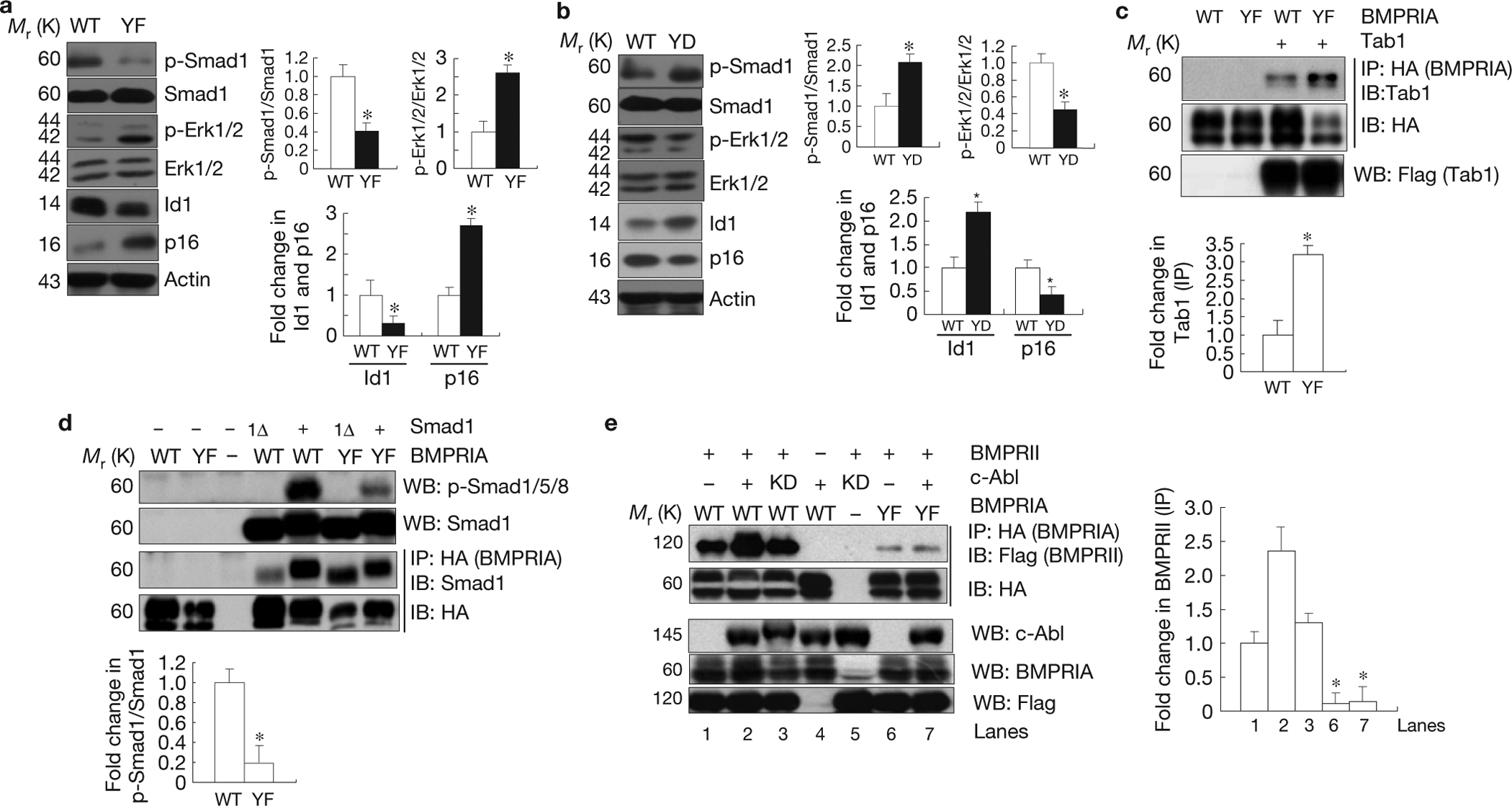
c-Abl-mediated BMPRIA phosphorylation negatively regulates Erk1/2 activation but positively regulates Smad1/5/8 activation. (a) Bmpr1a−/− osteoblasts expressing the mutant YF BMPRIA showed an increase in Erk1/2 activation and 16INK4a expression, but a decrease in Smad1 activation and Id1 expression. Right panel: quantification (fold change in p-Smad1/Smad1, p-Erk/Erk, Id1, and p16). (b) Bmpr1a−/− osteoblasts expressing YD mutant BMPRIA showed an increase in Smad1 activation and Id1 expression, but a decrease in Erk1/2 activation and p16INK4a expression. Right panel: quantification (fold change in p-Smad1/Smad1, p-Erk/Erk, Id1, and p16). (c) Enhanced interaction between mutant BMPRIA and Tab1. Mutant or WT BMPRIA was co-expressed with Tab1 in Cos7 cells cultured in normal medium. The mutant and normal BMPRIA were immunoprecipitated with anti-HA antibodies and the co-purified Tab1 was detected by western blot analysis. Lower panel: quantification. (d) The YF mutant BMPRIA had compromised activity in activating Smad1. The WT and YF BMPRIA were co-expressed with Smad1 or Smad1 carrying a deletion of the last 11 amino acids (Smad1Δ) in Cos7 cells cultured in normal medium. Western blot analysis was used to determine the activation of Smad1. The WT and YF BMPRIA were immunoprecipitated with anti-HA antibodies and the associated Smad1 was detected by anti-Smad1 antibodies. Lower panel: quantification. (e) c-Abl facilitated the interaction between BMPRII (Flag tagged) and BMPRIA. The WT and YF BMPRIA were co- expressed with BMPRII and c-Abl (or c-Abl KD) in Cos7 cells cultured in normal medium. BMPRIA was immunoprecipitated with anti-HA antibodies and the associated BMPRII was detected with an anti-Flag antibody. Right panel: quantification. Data are means ± s.e.m. (n = 3). *P < 0.05, compared with WT BMPRIA. Uncropped images of blots are shown in Supplementary Fig. S8.
MAPK activation by BMPs is mediated by recruitment of the Tab1–Tak1 complex to BMPRIA (ref. 45). Co-immunoprecipitation experiments showed that YF BMPRIA had a higher affinity for Tab1 than WT BMPRIA (Fig. 6c), suggesting that c-Abl deficiency might facilitate recruitment of the Tab1–Tak1 complex to BMPRIA, hence promoting Erk1/2 activation. On the other hand, in co-expression experiments, YF BMPRIA showed diminished Smad1 activation, without affecting the interaction between BMPRIA and Smad1 or the mutant Smad1Δ (with 11 amino acids deleted from the C terminus and therefore cannot be phosphorylated by BMPRIA; Fig. 6d). Instead, we found that c-Abl facilitated the interaction between BMPRIA and BMPRII, and YF BMPRIA showed reduced interaction (Fig. 6e), suggesting that c-Abl-mediated BMPRIA phosphorylation facilitates BMP receptor complex formation to augment Smad1/5/8 activation.
A role for p16INK4a in MSC expansion and in rescuing bone phenotypes of c-Abl −/− mice
We then investigated a potential role for p16INK4a in osteoblast expansion. Two-month-old p16INK4a−/− mice did not show any significant change in the bone volume, bone formation rate, and the number and differentiation of osteoblasts (Fig. 7a and Supplementary Fig. S7). However, 12-month-old p16INK4a−/− mice showed increased numbers of osteoblasts (3.3±0.7 for +/+ versus 6.7±1.2 for −/−, n=6;P < 0.05), accompanied by a doubled number of osteoclasts (data not shown). This might explain why p16INK4a−/− mice did not show a significant change in bone volume (data not shown). Moreover, p16INK4a−/− mice showed a significant increase in the number of MSCs at 12 months but not at 2 months of age, compared with WT mice (Fig. 7b and data not shown). Consistently, p16INK4a mRNA and protein are upregulated at 12 months in bone marrow cells exclusive of cells of haematopoietic origin (Fig. 7c). Thus, p16INK4a upregulation might be an important mechanism regulating MSC ageing.
Figure 7.
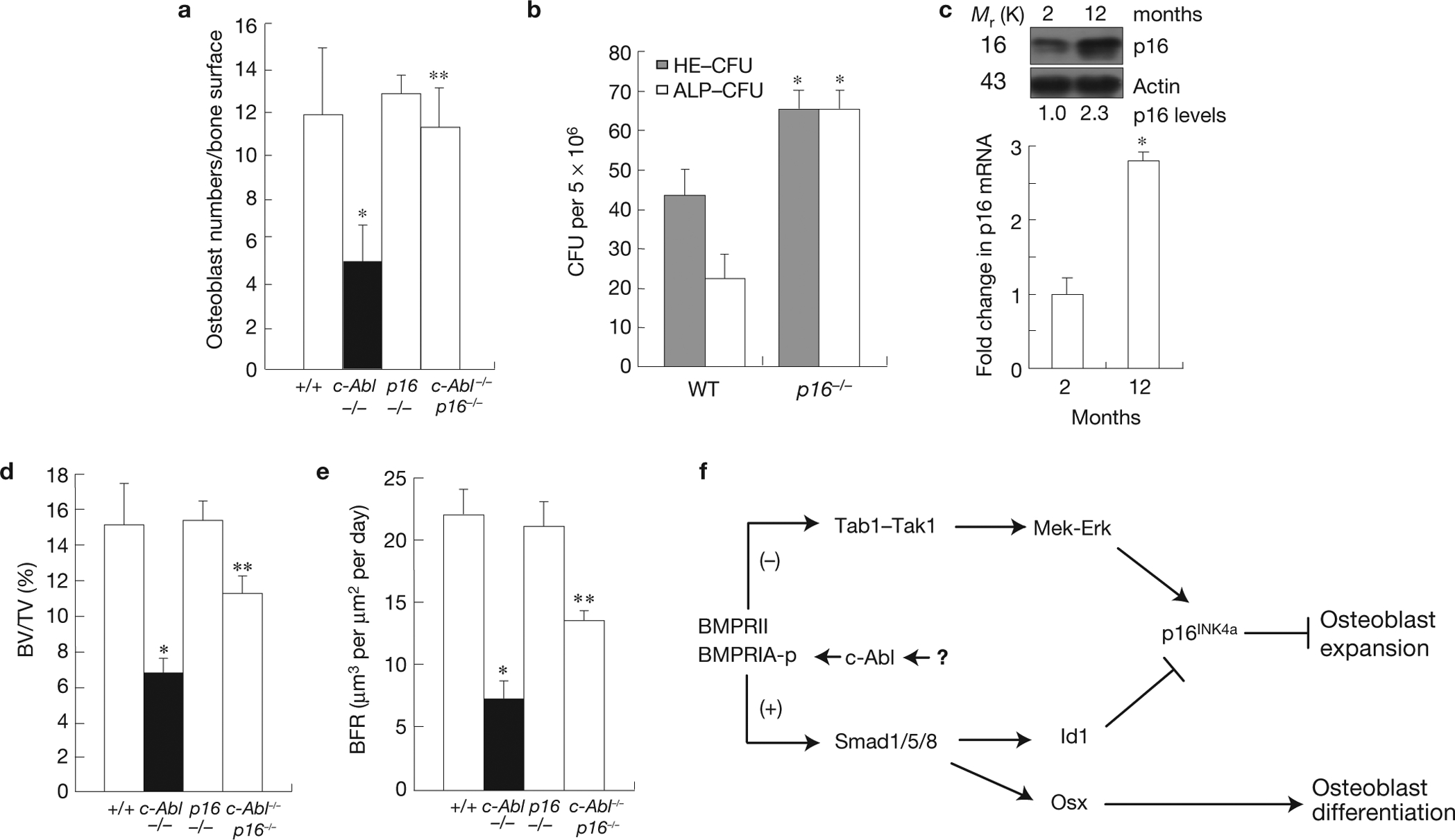
A role for p16INK4a in MSC maintenance and osteoblastogenesis in normal and c-Abl−/− mice. (a) The decrease in the number of osteoblasts in c-Abl−/− mice was rescued by p16INK4a deficiency. Two-month-old mice were used in this analysis. (b) An increase in ALP CFU (ALP-positive CFU, osteogenic) and HE-CFU (haematoxylin/eosin-positive CFU, total) in 12-month-old p16INK4a−/− mice. (c) An increase in p16INK4a in 12-month-old normal mice. Bone marrow of 2- or 12-month-old mice was directly used to analyse the protein and mRNA levels of p16INK4a by western blot (upper panel) and real-time PCR (bottom panel) respectively. (d) The decrease in bone volume (BV/TV) in c-Abl−/− mice (2-month-old) was partially rescued by p16INK4a deficiency. (e) The decrease in bone formation rates (BFR) in c-Abl−/− mice (2-month-old) was partially rescued by p16INK4a deficiency. (f) A diagram showing how c-Abl might be involved in regulating osteoblast expansion and differentiation in response to BMPs. Data are means ± s.e.m. (a,d,e, n = 6; b,c, n = 4). *P < 0.05, compared with WT mice, **P < 0.05, compared with c-Abl−/− mice. Uncropped images of blots are shown in Supplementary Fig. S8.
We then crossed p16INK4a−/− mice and c-Abl−/− mice to generate double knockout mice. Whereas none of c-Abl−/− mice survived to 6 weeks, about 30% of the c-Abl−/− p16INK4a−/− mice did (15 out of 48 c-Abl−/− p16INK4a−/− versus 0 out of 76 c-Abl−/− mice). In contrast, none of the c-Abl−/−p53−/− mice was viable, as previously reported57, suggesting that p16INK4a upregulation could be part of the reason for neonatal lethality of c-Abl−/− mice. More importantly, p16INK4a deficiency rescued the reduction in osteoblast population and partially rescued the reduction in bone formation rates and bone volumes in 2-month-old c-Abl−/− mice (Fig. 7a,d,e), suggesting that p16INK4a upregulation mediated the defects in osteoblast expansion and bone formation of c-Abl−/− mice. The incomplete rescue is probably due to the fact that p16INK4a deficiency does not rescue the differentiation defect of c-Abl−/− osteoblasts (Supplementary Fig. S7 and data not shown).
DISCUSSION
The bone undergoes constant remodelling. In human adults, it is estimated that 25% of the trabecular bone and 3% of the cortical bone is replaced with newly formed bone each year58. We show that c-Abl deficiency causes defective osteoprogenitor expansion and an inadequate supply of functional osteoblasts, which lead to decreased bone formation and development of osteoporosis. Thus, c-Abl−/− mice represent a model that bridges defective osteoblast expansion and senile osteoporosis.
This study also highlights the importance of p16INK4a in osteoblast ageing and expansion. p16INK4a is increased in various aged cells and tissues and p16INK4a−/− cells are resistant to ageing42,59–63. We show that p16INK4a not only plays a critical role in MSC maintenance and osteoblast expansion, but also mediates the effects of c-Abl in regulating osteoblast expansion. As a result, p16INK4a deficiency partially rescues the defects in bone formation and bone mass, and perinatal lethality of c-Abl−/− mice. The partial rescue is consistent with our observation that c-Abl deficiency impedes osteoblast differentiation through the BMP–Smad1/5/8 pathway in a p16INK4a-independent manner. The differentiation defect is probably caused by reduced expression of Osx, a target gene of the BMPs–Smad1 pathway (Fig. 7f; ref. 1).
BMPs are secreted by osteoblasts and are important regulators of osteogenic differentiation and bone remodelling35,64. Here we show that BMP-activated Erk1/2 and Smad1/5/8–Id1 pathways are involved in regulating p16INK4a expression, in opposite ways (Fig. 7f). A balanced activation of the canonical and non-canonical pathways, or the lack of it in the absence of BMPRIA, may control osteogenic differentiation by regulating Runx2/Osterix expression, and osteoblast-mediated osteoclastogenesis by regulating RANKL expression. However, imbalanced activation (in the absence of c-Abl) may have a greater effect on p16INK4a expression and osteoblast expansion, but a lesser effect on RANKL/OPG expression and osteoblast-mediated osteoclastogenesis (Fig. 7f). This may explain why c-Abl−/− mice (faulty BMPRIA phosphorylation) and osteoblast-specific BMPRIA knockout mice showed distinct bone phenotypes. Our findings place c-Abl at the branch point of BMP–Smad1 and BMP–MAPK pathways, where it differentially regulates these two pathways by phosphorylating BMPRIA. Further studies are needed to identify factors that activate c-Abl in the context of cell senescence. In addition, recent studies show that BMPs can activate the NF-κB pathway through Tak1, which inhibits osteoblast differentiation and bone homeostasis65, and promotes cell senescence66. Although these findings are supportive of our conclusion that the BMPR–Tak1 pathway promotes cell senescence, whether NF-κB participates in p16INK4a expression warrants further investigation.
This study uncovers a role for the non-receptor tyrosine kinase c-Abl in regulating cell senescence, which might be an underlying reason for development of senile osteoporosis and other ageing-related phenotypes in c-Abl−/− mice. This function of c-Abl seems to be mediated by p16INK4a. Mechanistically, c-Abl modifies BMPRIA and differentially regulates canonical and non-canonical BMP pathways to control p16INK4a expression.
METHODS
Mouse and cell isolation and culture.
p16INK4a−/− mice (from R. DePinho, University of Texas, MD Anderson Cancer Center, USA; ref. 67) were on the FVB N2 background and c-Abl−/− (abl1) and p53−/− deficient mice were on the C57BL/6 background. p16INK4a−/− mice were crossed to abl1 mice to generate double knockout mice. Osx1–GFP:Cre mice and Bmpr1a f/f mice were used to generate osteoblast-specific Bmpr1a knockout mice. To prepare primary osteoblasts, calvaria from 19- to 20-day-old fetuses were isolated, washed in PBS and digested in MEM alpha medium containing 0.1% collagenase type IV and 0.05% trypsin–EDTA for 15 min at 37 °C four times. The supernatant from the first digestion was discarded and supernatants from the last three digestions were pooled in MEM alpha medium supplemented with 15% FBS. The cells were washed and plated onto 6-well plates and grown until confluent. The osteoblast cultures were amplified to passage three before use in further experiments. To isolate bone marrow cells for mRNA and protein analysis, erythrocytes were lysed with 0.8% NH4Cl.
Cell proliferation assays.
To examine the proliferation potential of primary osteoblasts, cells at passage three were used as a start point. Briefly, 4×105 cells were plated onto 6 cm plates, cultured for three days and counted. [notdef]The same number of cells was re-plated, cultured for three days and counted again. The procedure was repeated until the cells stopped dividing. To assess cell proliferation by 5-bromodeoxyuridine (BrdU) labelling (Roche), cells at different passages were seeded in four replicas onto 96-well plates at a cell density of 1×104 cells. After 24 h, BrdU was added to the wells for 16 h at a final concentration of 10 mmol l−1, and proliferation was analysed colorimetrically following the manufacturer’s instructions.
Immunoprecipitation, western blot analysis and kinase assay.
Cells were washed in cold PBS and lysed in TNEN buffer containing 0.1% NP-40, 0.5% Triton X-100, 1 mM sodium orthovanadate, 1 mM NaF and protease inhibitors. Fifty micrograms of total protein was loaded onto an 8–15% polyacrylamide gel. Antibodies against p-Erk1/2 (9106, 1:1,000), Erk1/2 (4695, 1:1,000), p-Mek1/2 (9154, 1:1,000), Mek1/2 (8727, 1:1,000), p-Smad1/5/8 (9511, 1:1,000), Smad1 (9743, 1:1,000), Tab1 (3225, 1:1,000), p-Tak1 (9339, 1:1,000) and Tak1(5206, 1:1,000) were purchased from Cell Signing. Antibodies against p16INK4a (sc-81157, 1:1,000), actin (sc-130656, 1:1,000), HA (sc-161069, 1:1,000), c-Abl (sc-887, 1:500), p-Tyr (sc-51688, 1:1,000) and Id1 (sc-374287, 1:500) were purchased from Santa Cruz Biotechnology. Antibodies against p19 (07–543, 1:1,000) and p21 (OP76, 1:1,000) were purchased from Upstate Biotechnologies. Anti-BMPRIA (ab-38560, 1:1,000) was from Abcam and anti-Flag (F2555, 1:2,000) was from Sigma. BMP2, noggin and chordin were from R & D systems. The kinase assay for c-Abl was carried out as previously described32, using BMPRIA (Sino Biological) as a substrate, which was detected by anti-p-Tyr antibodies on western blots.
SA-β-Gal assay.
Cells were washed in TBS buffer, fixed in formaldehyde/glutaradehyde, washed with water and then stained with X-Gal at neutral pH (ref. 38).
Reverse transcription and real-time PCR and siRNA.
Total RNA was isolated from cultured cells or mouse femurs using Trizol reagent (Invitrogen), quantified and then used for reverse transcription using a kit from Roche. Oligonucleotides for real-time PCR were designed following the instructions from Roche. Id1: forward 5′-GCGAGATCAGTGCCTTGG-3′, reverse 5′-CTCCTGAAGGGCTGGAGTC-3′; Id3: forward 5′-GAGGAGCTTTTGCCACTGAC-3′, reverse 5′-GCTCATCCATGCCCTCAG-3′; Smad6: forward 5′-GTTGCAACCCCTACCACTTC-3′, reverse 5′-GGAGGAGACAGCCGAGAATA-3′; Smad7: forward 5′-ACCCCCATCACCTTAGTCG-3′, reverse 5′-GAAAATCCATTGGGTATCTGGA-3′; JunB: forward 5′-ACCCCCATCACCTTAGTCG-3′, reverse 5′-GAAAATCCATTGGGTATCTGGA-3′; p16INK4a: forward 5′-GGGTTTTCTTGGTGAAGTTCG-3′, reverse 5′-TTGCCCATCATCATCACCT-3′; RANKL: forward 5′-GCACACCTCACCATCAATGCT-3′, reverse 5′-GGTACCAAGAGGACAGAGTGACTTTA-3′; OPG: forward 5′-TGAGTGTGAGGAAGGGCGTTA-3′, reverse 5′-CCATCTGGACATTTTTTGCAAA-3′ and β-actin: forward 5′-CTAAGGCCAACCGTGAAAAG-3′, reverse 5′-ACCAGAGGCATACAGGGACA-3′. ON-TARGETplus SMARTpool siRNA targeting c-Abl (5′-TCAACAAGCTGGAGAGCAA-3′, 5′-CCATGGAGGTGGAGGAGTT-3′, 5′-TGGAGTACTTGGAGAAGAA-3′, 5′-CAGATGAGGTGGAGAAGGA-3′) and Smad1 (5′-TGAAGAAACTGAAGAAGAA-3′, 5′-GGGCGATGAAGAAGAGAAA-3′, 5′-AGAGAAATGGGCAGAGAAA-3′, 5′-GTGAAGAAACTGAAGAAGA-3′) was purchased from Thermo Scientific and used following the standard protocol from the manufacturer.
Retrovirus infection.
Retroviral constructs expressing p16INK4a, Id1, c-Abl, Cre or BMPRIA (WT or mutant) were used to transfect Plat E, a retroviral packaging cell line, using FUGENE (Roche). After two days, media containing the packaged viruses were collected and directly used for infection of primary osteoblasts at early passages. Osteoblasts were then selected against hygromycin or puromycin for 2–7 days. Expression of the virus-carrying proteins was confirmed by western blot analysis.
Tissuepreparation and immunostaining.
The femurs of day 1–3 pups were fixed in 4% paraformaldehyde in PBS overnight and equilibrated in 30% sucrose. The tissues were then frozen and sectioned (10 μm). Sections were dried on PLL-coated slides and processed immediately for immunostaining. Slides were blocked in 5% BSA in PBS for 30 min. Primary antibodies against BrdU, p16 or c-Abl were diluted in the same blocking solution and applied overnight at 4 °C; slides were then washed three times in PBS for 10 min and incubated with secondary antibodies at 1:100 in PBS for 1 h. After washing three times in PBS for 10 min, the slides were incubated with 3,3-diaminobenzidine (DAB) at room temperature without light for 10 min. Slides were then dehydrated and mounted with neutral gums. For BrdU labelling experiment, BrdU was injected (intraperitoneally) into the mice following the protocol from Roche.
Quantification and statistical analysis.
Western blot results were scanned with a Molecular Dynamics scanning densitometer. The relative levels of protein of interest were then determined by measuring the intensity of the corresponding bands. All values were averages of cell cultures isolated from at least three mutant mice and their control littermates and were normalized to the housekeeping gene actin. In all of the quantification data, the values of untreated WT cells were set as 1.0. Statistical analysis was performed using Student’s unpaired t-test (STATISTICA). Error bars show the mean ± s.e.m. *, P < 0.05 when compared with the WT counterparts or untreated cells; **, P < 0.05 when compared with the mutant counterparts.
Luciferase assay.
The Id1 promoter and the BMP-responsive SBE-OC–Luc were used in luciferase assays. v-Abl, c-Abl or c-Abl kinase-dead expression constructs, the promoter plasmid (pGL2–Id1–Luc) and Renilla plasmid were co- transfected into C2C12 cells, WT or c-Abl−/− osteoblasts. To determine the effect of BMP2, the transfected cells were serum-starved for 8 h, stimulated with BMP2 overnight, collected, washed with PBS and then lysed with passive lysis buffer (Invitrogen). The luciferase activities were measured following the manufacturer’s procedures and were normalized against the Renilla activity.
Histomorphometric analysis.
Histomorphometric parameters of cancellous and cortical bones in the femurs were measured with a digitizing morphometry system, which consists of an epifluorescent microscope (model BH-2; Olympus), a colour video camera and a digitizing pad (Numonics 2206) coupled to a computer and a morphometry program OsteoMetrics (OsteoMetrics). Measured parameters of cancellous bone included total tissue area, trabecular bone area and perimeter, single- and double-labelled perimeters and interlabelled widths. They were then used to calculate the percentage of cancellous bone volume (trabecular bone area/total tissue area × 100%) and the cancellous bone formation rate ([double-labelled perimeters + single-labelled perimeters/2] × interlabelled widths/interval time/trabecular perimeters). The region of bone measured in all groups is 1–4 mm from the growth plate in the proximal tibia. All measurements and calculations were referenced to the standard nomenclature.
Phosphorylation site identification68,69.
The peptides that contain the four tyrosine residues (amino acids 448–462 and 456–471) were synthesized by ChinaPeptides, which were used as substrates for in vitro kinase assays using purified c-Abl kinase, for 2 h at 30 °C. The products of the Abl enzymatic reaction were centrifuged to remove insoluble materials, and the 10 μl supernatant was loaded onto a Sep-Pak tc18 cartridge 50 mg (Waters) for peptide desalting. The eluted peptides were lyophilized using a SpeedVac (Thermo), and resuspended in 20 μl 0.1% formic acid/2% acetonitrile. All mass spectrometric experiments were performed on a LTQ orbitrap ‘XL’ mass spectrometer (Thermo) connected to a Paradigm MDLC nanoflow LC system through an ADVANCE Spray Source LC–MS interface (Michrom BioResources). The peptide mixture was loaded onto a 15 cm, 0.1 mm inner diameter column packed with Magic C18AQ 3 μm Reversed Phase resins (Michrom BioResources), and separated within a 60 min linear gradient from 100% solvent A (0.1% formic acid/5% acetonitrile/95% water) to 35% solvent B (0.1% formic acid/100% acetonitrile) at a flow rate of 500 nl min−1. The spray voltage was set to 1.5 kV and the temperature of the ion transfer capillary was 160 °C. The mass spectrometer was operated in positive ion mode and employed in the data-dependent mode to automatically switch between MS and MS/MS using the Tune and Xcalibur 2.5.5 software package. One full MS scan from 350 to 1,800 m/z−1 was acquired at high resolution R = 100,000 (defined at m/z = 400), followed by three data dependent MS/MS spectra in the linear ion trap from the three most abundant ions. All MS/MS ion spectra were analysed using Sequest1 (Thermo, version v.27, rev. 11) and were incorporated into the Sorcerer engine version 4.0.4 build (Sage-N Research). Sequest parameters were set up to search for the specialized peptides associated with phosphorylation (STY, +79.966 Da). Finally, pLabel 2.42 was used to mark the fragment ions assigned to the appropriate ‘y-ions’ or ‘b-ions’.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank A. Nur-E-Kamal, Y. Wan and X-H. Sun for helpful discussions, I. H. In, D. Cai, G. C. Hong, L. Soh, J. Lin and T. Cheng for technical assistance, and Novartis, J. Campisi (Buck Institute for Research on Aging, USA), J. Wang (UCSD, USA), A. Koleske (Yale University, USA), X-H. Sun (Oklahoma Medical Research Foundation, USA), C. Cepko (Harvard Medical School, USA), D. Bulavin (IMCB, Singapore) and R. DePinho (University of Texas, MD Anderson Cancer Center, USA) for reagents and mice. S.P.G. is an investigator of Howard Hughes Medical Institute. The work was supported by grants from the Ministry of Science and Technology of China (The National Key Scientific Program (2012CB966901, to B.L.)), the National Natural Science Foundation of China (81130039, 31071229 and 81121001), Shanghai Pujiang Program (10PJ1405000), Changjiang Scholars Program of the Ministry of Education and the Agency for Science, Technology and Research of Singapore.
Footnotes
METHODS
Methods and any associated references are available in the online version of the paper at www.nature.com/naturecellbiology
Note: Supplementary Information is available on the Nature Cell Biology website
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Harada S & Rodan GA Control of osteoblast function and regulation of bone mass. Nature 423, 349–355 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Olsen BR, Reginato AM & Wang W Bone development. Annu. Rev. Cell Dev. Biol 16, 191–220 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Ducy P, Schinke T & Karsenty G The osteoblast: a sophisticated fibroblast under central surveillance. Science 289, 1501–1504 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Canalis E, Economides AN & Gazzerro E Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr. Rev 24, 218–235 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Sharpless NE & DePinho RA Telomeres, stem cells, senescence, and cancer. J Clin. Invest 113, 160–168 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherr CJ & DePinho RA Cellular senescence: mitotic clock or culture shock? Cell 102, 407–410 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Serrano M & Blasco MA Putting the stress on senescence. Curr. Opin. Cell Biol 13, 748–753 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Ivanov A & Adams PD A damage limitation exercise. Nat. Cell Biol 13, 193–195 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Lundberg AS, Hahn WC, Gupta P & Weinberg RA Genes involved in senescence and immortalization. Curr. Opin. Cell Biol 12, 705–709 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Takahashi A et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol 8, 1291–1297 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Schmitt CA Senescence, apoptosis and therapy—cutting the lifelines of cancer. Nat. Rev. Cancer 3, 286–295 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Beausejour CM & Campisi J Ageing: balancing regeneration and cancer. Nature 443, 404–405 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Van Riggelen J & Felsher DW Myc and a Cdk2 senescence switch. Nat. Cell Biol 12, 7–9 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Van Etten RA Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends Cell Biol 9, 179–186 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Hantschel O & Superti-Furga G Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol 5, 33–44 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Wong S & Witte ON The BCR-ABL story: bench to bedside and back. Annu. Rev. Immunol 22, 247–306 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Pendergast AM The Abl family kinases: mechanisms of regulation and signaling. Adv. Cancer Res 85, 51–100 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Wang X et al. A positive role for c-Abl in Atm and Atr activation in DNA damage response. Cell Death Diff 18, 5–15 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT & Mulligan RC Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65, 1153–1163 (1991). [DOI] [PubMed] [Google Scholar]
- 20.Schwartzberg PL et al. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell 65, 1165–1175 (1991). [DOI] [PubMed] [Google Scholar]
- 21.Li B et al. Mice deficient in Abl are osteoporotic and have defects in osteoblast maturation. Nat Genet 24, 304–308 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Li B et al. Distinct roles of c-Abl and Atm in oxidative stress response are mediated by protein kinase C delta. Genes Dev 18, 1824–1837 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silberman I et al. T cell survival and function requires the c-Abl tyrosine kinase. Cell Cycle 7, 3847–3857 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y et al. The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse. Blood 112, 111–119 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzeng SJ, Bolland S, Inabe K, Kurosaki T & Pierce SK The B cell inhibitory Fc receptor triggers apoptosis by a novel c-Abl family kinase-dependent pathway. J. Biol. Chem 280, 35247–35254 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Zipfel PA, Zhang W, Quiroz M & Pendergast AM Requirement for Abl kinases in T cell receptor signaling. Curr. Biol 14, 1222–1231 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Koleske AJ et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron 21, 1259–1272 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Schlatterer SD, Acker CM & Davies P c-Abl in neurodegenerative disease. J. Mol. Neurosci 45, 445–452 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko HS et al. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc. Natl Acad. Sci. USA 107, 16691–16696 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Arce KP et al. Synaptic clustering of PSD-95 is regulated by c-Abl through tyrosine phosphorylation. J. Neurosci 30, 3728–3738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michael M, Vehlow A, Navarro C & Krause M c-Abl, Lamellipodin, and Ena/VASP proteins cooperate in dorsal rufįng of fibroblasts and axonal morphogenesis. Curr. Biol 20, 783–791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodring PJ et al. Modulation of the F-actin cytoskeleton by c-Abl tyrosine kinase in cell spreading and neurite extension. J Cell Biol 156, 879–892 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zukerberg LR et al. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 26, 633–646 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Wang JY Controlling Abl: auto-inhibition and co-inhibition? Nat. Cell Biol 6, 3–7 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Chen D, Zhao M & Mundy GR Bone morphogenetic proteins. Growth Factors 22, 233–241 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Varga AC & Wrana JL The disparate role of BMP in stem cell biology. Oncogene 24, 5713–5721 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Randle DH, Zindy F, Sherr CJ & Roussel MF Differential effects of p19(Arf) and p16(Ink4a) loss on senescence of murine bone marrow -derived preB cells and macrophages. Proc. Natl Acad. Sci. USA 98, 9654–9659 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dimri GP et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA 92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Neill AJ, Cotter TG, Russell JM & Gaffney EF Abl expression in human fetal and adult tissues, tumours, and tumour microvessels. J. Pathol 183, 325–329 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Kotake Y et al. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4α tumor suppressor gene. Genes Dev 21, 49–54 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bracken AP et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 21, 525–530 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin AW et al. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 12, 3008–3019 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cagnol S & Chambard JC ERK and cell death: mechanisms of ERK—induced cell death—apoptosis, autophagy and senescence. FEBS J 277, 2–21 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Von Kriegsheim A et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat. Cell Biol 11, 1458–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamaguchi K et al. XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1-TAK1 in the BMP signaling pathway. EMBO J 18, 179–187 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sorrentino A et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol 10, 1199–1207 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Miyazono K & Miyazawa K Id: a target of BMP signaling. Sci. STKE 2002, pe40 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y & Derynck R Regulation of Smad signalling by protein associations and signalling crosstalk. Trends Cell Biol 9, 274–279 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Zebedee Z & Hara E Id proteins in cell cycle control and cellular senescence. Oncogene 20, 8317–8325 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Ruzinova MB & Benezra R Id proteins in development, cell cycle and cancer. Trends Cell Biol 13, 410–418 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Zhao M et al. Bone morphogenetic protein receptor signaling is necessary for normal murine postnatal bone formation. J. Cell Biol 157, 1049–1060 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mishina Y et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J. Biol. Chem 279, 27560–27566 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Kamiya N et al. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 135, 3801–3811 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamiya N et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J. Bone Miner. Res 23, 2007–2017 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodda SJ & McMahon AP Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133, 3231–3244 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Usui M et al. Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J. Bone Miner. Res 23, 314–325 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Whang YE et al. c-Abl is required for development and optimal cell proliferation in the context of p53 deficiency. Proc. Natl Acad. Sci. USA 97, 5486–5491 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manolagas SC & Jilka RL Bone marrow, cytokines, and bone remodeling. Emerging insights into the pathophysiology of osteoporosis. N. Engl. J. Med 332, 305–311 (1995). [DOI] [PubMed] [Google Scholar]
- 59.Huot TJ et al. Biallelic mutations in p16(INK4a) confer resistance to Ras- and Ets-induced senescence in human diploid fibroblasts. Mol. Cell Biol 22, 8135–8143 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alani RM, Young AZ & Shifflett CB Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc. Natl Acad. Sci. USA 98, 7812–7816 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Molofsky AV et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443, 448–452 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krishnamurthy J et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443, 453–457 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Janzen V et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443, 421–426 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Tsuji K et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet 38, 1424–1429 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Chang J et al. Inhibition of osteoblastic bone formation by nuclear factor-κB. Nat. Med 15, 682–689 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawahara TL et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span. Cell 136, 62–74 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serrano M et al. Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27–37 (1996). [DOI] [PubMed] [Google Scholar]
- 68.Yates JR 3rd, Eng JK, McCormack AL & Schieltz D Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem 67, 1426–1436 (1995). [DOI] [PubMed] [Google Scholar]
- 69.Wang LH et al. pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry. Rapid Commun. Mass Spec 21, 2985–2991 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.