


Abstract
Gene set enrichment (GSE) analysis plays an essential role in extracting biological insight from genome-scale experiments. ORA (overrepresentation analysis), FCS (functional class scoring), and PT (pathway topology) approaches are three generations of GSE methods along the timeline of development. Previous versions of KOBAS provided services based on just the ORA method. Here we presented version 3.0 of KOBAS, which is named KOBAS-i (short for KOBAS intelligent version). It introduced a novel machine learning-based method we published earlier, CGPS, which incorporates seven FCS tools and two PT tools into a single ensemble score and intelligently prioritizes the relevant biological pathways. In addition, KOBAS has expanded the downstream exploratory visualization for selecting and understanding the enriched results. The tool constructs a novel view of cirFunMap, which presents different enriched terms and their correlations in a landscape. Finally, based on the previous version's framework, KOBAS increased the number of supported species from 1327 to 5944. For an easier local run, it also provides a prebuilt Docker image that requires no installation, as a supplementary to the source code version. KOBAS can be freely accessed at http://kobas.cbi.pku.edu.cn, and a mirror site is available at http://bioinfo.org/kobas.
Graphical Abstract
Graphical Abstract.
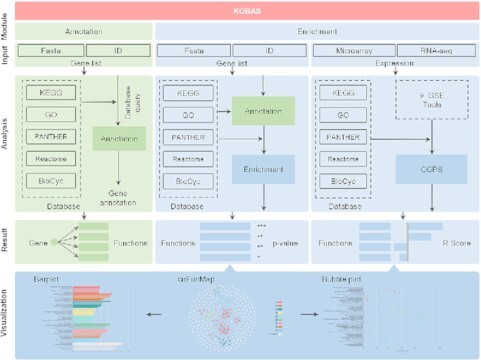
Framework of KOBAS.
INTRODUCTION
Gene set enrichment (GSE) is the optimal approach to understanding the underlying biological functions of different genes or proteins. It reduces the complexity of molecular data and improves the interpretability of biological insights. Generally, existing GES methods are divided into three types (1). Among them, overrepresentation analysis (ORA), the first-generation GSE method, is the most commonly used method. The representative tools of the ORA method are KOBAS (2), DAVID (3), clusterProfiler (4), g:Profiler (5), Enrichr (6), modEnrichr (7), agriGo (8), GeneTrail (9), GOrilla (10), ToppGene (11) and GOstat (12). These tools use various statistical analyses such as Hypergeometric test and Fisher's exact test, to evaluate whether the user-input gene list is overrepresented in a specific functional gene set. KOBAS is one of the first widely used ORA-based tools to perform GSE in its former versions 1.0 (2) and 2.0 (13).
The second-generation method is the functional class scoring (FCS) method. The massive pooling of microarray and RNA-seq data has increased the prevalence of the FCS method. This method calculates the functional score using the expression of all genes within a specific gene set rather than setting particular thresholds to select up- or downregulated differential genes, which is unavoidable in the ORA method. The representative tools of the FCS method are GSEA (14), GSVA (15), GSA (16), PADOG (17), PLAGE (18), GAGE (19), GLOBALTEST (20) and SAFE (21).
The third-generation analysis method is the pathway topology (PT)-based method. It has been developed to utilize the additional topology information in a given network of pathway, which is ignored by the ORA and FCS methods (1). The common tools for the PT-based GSE method consist of SPIA (22), GANPA (23) and CEPA (24). Although it has been demonstrated to have a better performance in some investigations (25), one obvious problem of the PT-based method is that true pathway topology is dependent on the type of cell due to cell-specific gene expression, which is rarely available and is fragmented in knowledge bases (1). Hence, this approach is still not widely used and has a long way to go before becoming mainstream.
Although the ORA method is more straightforward and faster, it has certain limitations for interpreting microarray and RNA-seq data. To better meet the enormous demands of analyzing expression data, KOBAS introduces a novel machine learning-based approach we published earlier, named Combined Gene set analysis incorporating Prioritization and Sensitivity (CGPS) (26). It is an ensemble method that integrates the results from seven widely used FCS and two prominent PT tools into a single ensemble score (R score), to optimize the prioritization of biologically relevant pathways from expression data.
It should also be recognized that genes are involved in complex biological functions (27). As a result, a few differentially expressed genes may cause abnormalities in multiple pathways (28). Therefore, the enriched terms in GSE analysis may still be too numerous, usually more than 100. Even though they can be sorted accurately, these redundant terms constrain GSE analysis results to be explanatory (29). Therefore, several tools have provided downstream visualization of these enriched terms, including WebGestalt (29), Metascape (30), Enrichment map (31), WEGO (32) and GOPlot (33), in selecting and understanding the enriched results. KOBAS has constructed a novel view of cirFunMap (circular function map), which presents different enriched terms and their correlations in a landscape, expanding the downstream exploratory visualization.
Despite increasing research concerns arising to the nonmodel species from omics studies, only a few tools, such as DAVID (3) and g:Profiler(5) support nonmodel species. Taking advantage of the KEGG Orthology Based Annotation System (KOBAS) framework (34) and sequence similarity mapping, the current KOBAS could expand its supported species from 1327 to 5944. It provides curated sequences and KEGG pathway knowledge for 5944 species, and Gene Ontology annotations for 71 popular research species.
RESULTS
Framework of KOBAS
Overall, the current KOBAS consists of two parts called the ‘annotation module’ and the ‘enrichment module’ (called ‘identify module’ in the previous version) (Figure 1). The annotation module accepts the gene list as input, including IDs or sequences, and generates annotations for each gene based on multiple databases of pathways, diseases, and GO information. The enrichment module gives an answer about which pathways and GO terms are statistically significantly associated with the input gene list or expression. Two different enrichment analyses are available, named gene-list enrichment and exp-data enrichment. The former follows the ORA method of KOBAS 1.0 and 2.0, and takes the gene list as input. The latter represents the newly added machine learning-based approach CGPS, which is dedicated to grouped expression data (Figure 2). The output of the enrichment module could be visualized downstream by a novel landscape view of cirFunMap.
Figure 1.

Framework of KOBAS. KOBAS consists of two parts called the ‘annotation module’ and the ‘enrichment module’. The GSE analysis is performed on a gene-list or expression input, and the output enrichment results can be viewed by visualization.
Figure 2.

Online operation flow for running analysis of exp-data enrichment. Click ‘exp-data enrichment’ on top of the homepage, then upload the expression profile and group information to start an analysis. After obtaining the enrichment results, click the ‘visualization’ button to browse the cirFunMap.
Intelligent prioritization of biological functions from the expression data
The former ORA-based KOBAS versions 1.0 and 2.0 are not able to deal with the expression data. Current KOBAS innovatively introduced the FCS/PT-based ensemble method to support expressing data as input. The newly added approach CGPS, which our team published earlier (26), is the first GSE ensemble method built based on a priori knowledge of pathways and phenotypes using a machine learning approach. It integrates seven widely used FCS methods: GSEA (14), GSA (16), PADOG (17), PLAGE (18), GAGE (19), GLOBALTEST (20) and SAFE (21) and two prominent PT methods: GANPA (23), and CEPA (24), into a single ensemble score (R score). This score is a measure of relevance for a gene set to an experimental condition. A large positive R score value usually denotes the high relevance.
CGPS is not only a statistical ensemble model but also is a biological learning model that has the capacity to intelligently learn from the relationship between known target pathways and treated samples. Compared with ten widely used individual methods and two ensemble methods, the ensemble score (R score) in CGPS can better prioritize relevant pathways for a comprehensive evaluation of 120 simulated datasets and 45 real datasets (26). This may benefit the discovery of essential biologically relevant functions missed by other GSE methods.
Exploratory visualization of the enriched results
Although the results of enriched terms are sorted, the complexity of cellular biological processes may cause messy organization (35). Here, KOBAS brought a novel view form named cirFunMap, which can present different enriched terms and their correlations in a landscape. This view is generated in three steps: First, the correlation between two gene sets is calculated using the Jaccard index (36). It is a measure of similarity for the enriched terms, ranging from 0 to 1, to compare members for two sets to see how much proportion is shared. Second, all the enriched terms are connected to each other to construct a network by the user-defined correlation threshold. Finally, the network nodes are painted with different colors according to the module partition by the Infomap algorithm (37), which divides a network into clusters with possible overlaps to reduce the entire system's information entropy.
Then, we ranked all of these clusters in cirFunMap by their median P-value or R score calculated for all of the enriched terms within this specific cluster. Each cluster has one distinct color, and the user can choose the top N clusters to be colored. The terms are laid out in a circular net with mutually exclusive gravity forces. Terms in the same color (also cluster) can be hidden or displayed by switching on/off the color button on the right. The node size is determined by different significance levels or R scores in the original enriched list. It is clear that cirFunMap can explore the data from different perspectives to create a fresh view in an interactive way, which may stimulate the expert's visual thinking and should support novel insights extracting.
Quantity expansion of total species
Stepped by the data collection protocol built in KOBAS 2.0, the gene sequences and pathway/GO annotations were greatly improved. Initially, we retrieved the protein sequences in the target species from the KEGG GENES database (38), and built the BLAST database using the ‘makeblastdb’ command. Subsequently, we downloaded the raw data files from pathway databases, including KEGG (28), PANTHER (39), Reactome (40), BioCyc (41), and GO databases (42) (Table 1). For each pathway or disease database, we retrieved the gene-term mappings by parsing the raw data files. For GO curation, only the directed gene to GO term mapping was retained. To integrate across different databases, we mapped the genes in all databases to KEGG GENES. These gene-pathway and gene-GO data are then stored in our backend SQLite3 relational database. For easy download and use, each species' data were stored separately in a distinct database. In total, 5944 species were annotated with KEGG pathways, 41 species with PANTHER pathways, 14 species with Reactome pathways, 18 species with BioCyc pathways and 71 species with GO annotations (Table 1, Figure 3). The detailed statistical table is available on the download page of the website.
Table 1.
Pathway and GO databases supported by KOBAS
| Database | Data content | NO. of species | File format | URL |
|---|---|---|---|---|
| KEGG PATHWAY | Pathway | 5944 | Text | http://www.genome.jp/kegg/pathway.html |
| PANTHER | Pathway | 41 | Table | http://www.pantherdb.org/ |
| Reactome | Pathway | 14 | Table | https://reactome.org/ |
| BioCyc | Pathway | 18 | Table | http://biocyc.org/ |
| GO | Gene ontology | 71 | Text | http://amigo.geneontology.org/amigo/search/annotation |
Figure 3.
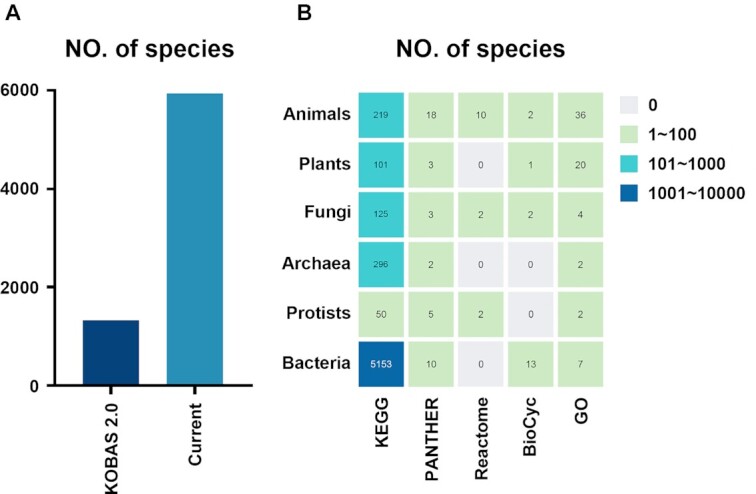
Pathway and GO databases supported by KOBAS. (A) Number of supported species in KOBAS 2.0 and current KOBAS. (B) Number of supported species with pathway and GO annotations in different species classes.
Comparison to existing webservers
As reviewed in a recently published GSE benchmark study (43), there are various existing popular webservers for GSE analysis. Since different tools have special characteristics and advantages, we have compared KOBAS to its alternative tools to assist users in selecting particular GSE tools. Totally 10 webservers are summarized, including 8 GSE webservers from the above benchmark study, one another webserver agriGO, and KOBAS (Table 2). It is illustrated that the ORA method is the most commonly used analysis method and is utilized by all the servers investigated, while FCS or PT methods are utilized by half of the servers, including KOBAS and another four servers. Different forms of visualizations are adopted by each of these servers. However, cirFunMap is a distinct form that only available in KOBAS.
Table 2.
Comparison to existing webservers
| Tool | Year created | Citationsa | Gene setsc | No. of speciesc | Method | Input data | Visualization | Availability |
|---|---|---|---|---|---|---|---|---|
| KOBAS | 2006 | 2566 | -GO | 5,944 | -ORA | -gene list | -cirFunMap | -webserver |
| -KEGG | -ensemble learning | -expression | -barplot | -Python program | ||||
| - +3 more | -bubble plot | |||||||
| WebGestalt | 2005 | 4146 | -GO | 12 | -ORA | -gene list | -barchart | -webserver |
| -KEGG | -FCS | -expression | -volcano plots | -R package | ||||
| - +20 more | -PT | -DAG | -API | |||||
| -GSEA plot | ||||||||
| -pathway view | ||||||||
| g:Profiler | 2007 | 2987 | -GO | 711 | -ORA | -gene list | -Manhattan plot | -webserver |
| -KEGG | -heatmap | -R package | ||||||
| - +7 more | -API | |||||||
| GeneTrail | 2007 | 413 | -GO | 12 | -ORA | -gene list | -interactive tables | -webserver |
| -KEGG | -FCS | -expression | -API | |||||
| - +64 more | -PT | |||||||
| DAVID | 2009 | 39510 | -GO | >65,000 | -ORA | -gene list | -GoCharts | -webserver |
| -KEGG | -KeggCharts | -program | ||||||
| - +38 more | -DomainCharts | -API | ||||||
| GOrilla | 2009 | 2710 | -GO | 8 | -ORA | -gene list | -DAG | -webserver |
| ToppGene | 2009 | 1846 | -GO | 2 | -ORA | -gene list | -barplot | -webserver |
| - +115 more | -API | |||||||
| agriGO | 2010 | 2934 | -GO | 45 | -ORA | -gene list | -barplot | -webserver |
| -FCS | -expression | -DAG | ||||||
| PANTHER | 2003 | 2368b | -GO | 142 | -ORA | -gene list | -pie chart | -webserver |
| - +6 more | -FCS | -expression | -bar chart | -API | ||||
| Enrichr | 2013 | 5867 | -GO | 6 | -ORA | -gene list | -barplot | -Webserver |
| -KEGG | -grid view | -API | ||||||
| -+167 more | -network | |||||||
| -clustergram | ||||||||
| -Enrichr Appyter |
aGoogle Scholar, April 2021. Citations to papers of server update or usage protocols were taken into statistics.
bCitations to usage protocols of gene function analysis using PANTHER were taken into statistics.
cThis information were summarized based on the online service of the webservers in April 2021.
Locally run with Docker image
The source code of KOBAS is available on the download page. As the source code stand-alone version is troublesome to install, especially when using the exp-data enrichment by CGPS, we have prebuilt a Docker image of local KOBAS. Users could import and enter the Docker environment which is the same as online KOBAS running without any need for preinstallation. The required BLAST and annotation SQLite3 files could be downloaded from FTP.
A demo case
To illustrate the new features of KOBAS, especially exp-data enrichment and exploration visualization, we have analyzed one public microarray dataset of Alzheimer's disease (GSE1297) (44), which was also tested in KOBAS’s previous versions and the CGPS algorithm. This dataset included nine severe Alzheimer's disease patients and seven healthy people as controls. The demo input files are available on the download page, and the running parameter settings can be found in Table 3.
Table 3.
Basic information for current demo case
| Gene-list enrichment demo | Exp-data enrichment demo | |
|---|---|---|
| Input | 600 gene symbols | 1) Expression profile |
| 2) Group Information | ||
| Database | KEGG | KEGG |
| GSE Analysis | Gene-list enrichment | Exp-data enrichment |
| GSE Analysis parameters | Default | Sequencing Technology = ‘ Microarray data’ and Expression Data Type = ‘ Normalized data’; Others = Default |
| Running time | >5 s | >1000 s |
| Analysis task id | 4bb0dfeba38d4ecc9ce1d3f0e82a25f6 | ea311ada58b94f42ba4368b2274a7364 |
| Visualization parameter | correlation >0.35 and top N = 7 | correlation >0.25 and top N = 7 |
| Visualization URL | $HOMEa/retrieve/visualization/?app = gene_list&taskid = ac84235ea9124228971198fbe4a2effe | $HOMEa/retrieve/visualization/?app = exp_data&taskid = ed73ed528aa547c4ab7de03cf3d71806 |
a$Home could be http://kobas.cbi.pku.edu.cn (primary site) or http://bioinfo.org/kobas (mirror site).
We conducted the gene-list enrichment and exp-data enrichment separately. For the gene-list enrichment, we screened up- and downregulated significant (P < 0.05) by t-test. A total of 600 differentially expressed genes were identified and submitted to the gene-list enrichment using the KEGG pathway database. The output 286 terms with calculated enriched P-values (no filter) were then visualized using cirFunMap (correlation > 0.35 and top N = 7). Interestingly, the KEGG pathway hsa05010 (Alzheimer disease) ranked 36 in the total terms whereas it was the top 3 cluster viewed by cirFunMap (Figure 4A). This suggests that cirFunMap may pull biological functions ranked low back to the user's attention. Additionally, cluster 4 containing the most terms was related to the immune functions, which was consistent with the finding of strong neuroinflammation in Alzheimer's disease (45).
Figure 4.

cirFunMap visualization of two demo enrichment results. (A) cirFunMap visualization of gene-list enrichment results. Left is the circular network view; the node color represents different clusters; the node size represents six levels of P-value, node size from small to large: [0.05,1], [0.01,0.05), [0.001,0.01), [0.0001,0.001), [1e-10,0.0001), [0,1e-10); the edge represents correlations larger than the user-defined threshold (0.35 in the demo). Right is the barplot of the P-value for terms in different clusters. (B) cirFunMap visualization of exp-data enrichment results. Left is the circular network view; the node color represents different clusters; the node size is linearly positively correlated with the R score; the edge represents correlations greater than the user-defined threshold (0.25 in the demo). Right is the barplot of the R score for terms in different clusters, while R score is a measure of relevance for a gene set to an experimental condition.
For exp-data enrichment, the expression matrix and group information are passed into exp-data enrichment as two inputs using the KEGG pathway as a concerned database (Sequencing Technology = ‘ Microarray data’ and Expression Data Type = ‘ Normalized data’). The output 286 terms with calculated enriched P-value (no filter) were then visualized using cirFunMap (correlation > 0.25 and top N = 7) (Figure 4B). The exp-data enrichment result was somewhat different from the previous results, showing that hsa05010 (Alzheimer disease) ranked first in the enriched terms. In addition, it was in the top cluster 1 viewed by cirFunMap, together with hsa05010 (Alzheimer disease), hsa05012 (Parkinson disease), hsa05016 (Huntington disease), hsa04932 (Non-alcoholic fatty liver disease), and hsa00190 (Oxidative phosphorylation). Another top cluster 6 containing the most terms was related to the synapse, which was consistent with synapse damage or loss in Alzheimer's disease (46).
DISCUSSION
As one of the common GSE tools, KOBAS has served the scientific community for 15 years and its core ORA-based algorithm has been online since the first version. Generally, current version of KOBAS has been greatly improved in enrichment algorithm, visualization function, species numbers and platform stability.
Driven by the development of sequencing techniques and the explosion of omics data, it is very urgent to construct more intelligent GSE algorithms. Here, KOBAS introduced a machine learning-based method CGPS for microarray and RNA-seq data, which we published earlier (26), and it better prioritizes the relevant biological pathways among the different datasets. CGPS is a preliminary exploration of intelligent enrichment analysis. It has also been proven that introducing artificial intelligence methods such as machine learning into GSE is quite valuable and feasible. As more a priori pathway knowledge accumulates, PT-based algorithms employing machine learning or deep learning technology would be a future direction for KOBAS. Thus, we will continue to develop cutting-edge GES algorithms further and integrate them into our server.
Additionally, exploratory visualization was constructed by cirFunMap to present different enriched terms and their correlations from different perspectives in a landscape view in KOBAS. This improvement is an efficient downstream supplement to former GSE algorithms. In addition to the current partition and coloring scheme from the information entropy algorithm Infomap, other schemes designed for module finding such as SPICi (47), affinity propagation (48), and weighted set cover (49) could also be utilized. Additionally, a more accurate ensemble score could be calculated for cluster ranking rather than using the median P-value or R score. In addition, we will make cirFunMap more public by opening its API in the future. Thus, users will be allowed to import their GSE results locally and then explore insights and export publication-level figures using cirFunMap.
Moreover, the gene sequences and KEGG pathway annotation were curated for 5944 model or nonmodel species in KOBAS. Among the 5944 species, only 71 species were curated with GO ontology annotation. As GO is the world's largest information source for functional genes and is widely used in GSE analysis, we are planning to integrate more GO contents in the next version of KOBAS, such as its hierarchical structure and different types of relationships. Beyond KEGG-based visualization, GO-based visualization will be further extended.
In addition, the entire code related to the web service has been rewritten and changed from PHP to the Python Flask framework under the REST API schema to provide a more stable online service. Considering the platform's stability and queuing speed, we split the previous task queue into distinguishing the gene-list enrichment task and the exp-data enrichment task. As the BLAST tasks would cause a long time CPU occupancy and affect other users' task queuing, datasets greater than 10M are limited for online service. Furthermore, route monitoring was added to completely record all users' API evoking and to report abnormal information quickly. To overcome the problem of potential access latency to KOBAS in different network environments, we provide a mirror website (http://bioinfo.org/kobas) as an alternative to the current primary site. Moreover, our team will continue to maintain and upgrade KOBAS to provide more accurate and stable services for its scientific users.
ACKNOWLEDGEMENTS
We thank Dr Yajie Xiao from The Chinese University of Hong Kong for language polishing of the manuscript.
Contributor Information
Dechao Bu, Pervasive Computing Research Center, Institute of Computing Technology, Chinese Academy ofSciences, Beijing, 100190, China.
Haitao Luo, Translational Medicine Collaborative Innovation Center, The Second Clinical Medical College (Shenzhen People's Hospital), Jinan University, Shenzhen 518020, China.
Peipei Huo, Chinese Academy of Sciences, LuoYang Branch of Institute of Computing Technology, Luoyang, 471000, China.
Zhihao Wang, Chinese Academy of Sciences, LuoYang Branch of Institute of Computing Technology, Luoyang, 471000, China.
Shan Zhang, Chinese Academy of Sciences, LuoYang Branch of Institute of Computing Technology, Luoyang, 471000, China.
Zihao He, School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, ChaoYang District, Beijing 100029, China.
Yang Wu, Pervasive Computing Research Center, Institute of Computing Technology, Chinese Academy ofSciences, Beijing, 100190, China.
Lianhe Zhao, Pervasive Computing Research Center, Institute of Computing Technology, Chinese Academy ofSciences, Beijing, 100190, China.
Jingjia Liu, Cancer Center, Ningbo Institute of Life and Health Industry, University of Chinese Academy of Sciences, Zhejiang 315000, China.
Jincheng Guo, School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, ChaoYang District, Beijing 100029, China.
Shuangsang Fang, School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, ChaoYang District, Beijing 100029, China.
Wanchen Cao, School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, ChaoYang District, Beijing 100029, China.
Lan Yi, Pervasive Computing Research Center, Institute of Computing Technology, Chinese Academy ofSciences, Beijing, 100190, China.
Yi Zhao, Pervasive Computing Research Center, Institute of Computing Technology, Chinese Academy ofSciences, Beijing, 100190, China.
Lei Kong, Center for Bioinformatics, State Key Laboratory of Protein and Plant Gene Research, School of Life Sciences, Peking University, Beijing 100871, China.
FUNDING
National Key R&D Program of China [2016YFB0201700]; National Natural Science Foundation of Zhejiang Province [LY20C060001, LY21C060003]; The National Natural Science Foundation of China [32070670]; National Key R&D Program of China [2017YFC0908404]; Zhejiang Provincial Research Center for Cancer Intelligent Diagnosis and Molecular Technology [JBZX-202003]; National Natural Science Foundation for Young Scholars of China [31701149, 31701141]. Funding for open access charge: National Key R&D Program of China [2016YFB0201700].
Conflict of interest statement. None declared.
REFERENCES
- 1. Khatri P., Sirota M., Butte A.J.. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol. 2012; 8:e1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu J., Mao X., Cai T., Luo J., Wei L.. KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006; 34:720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang da W., Sherman B.T., Lempicki R.A.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu G., Wang L.G., Han Y., He Q.Y.. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J.. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019; 47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A.et al.. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuleshov M.V., Diaz J.E.L., Flamholz Z.N., Keenan A.B., Lachmann A., Wojciechowicz M.L., Cagan R.L., Ma’ayan A. modEnrichr: a suite of gene set enrichment analysis tools for model organisms. Nucleic Acids Res. 2019; 47:W183–W190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tian T., Liu Y., Yan H., You Q., Yi X., Du Z., Xu W., Su Z.. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017; 45:122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gerstner N., Kehl T., Lenhof K., Müller A., Mayer C.. GeneTrail 3: advanced high-throughput enrichment analysis. Nucleic Acids Res. 2020; 48:W515–W520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z.. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009; 10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen J., Bardes E.E., Aronow B.J., Jegga A.G.. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009; 37:W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beissbarth T., Speed T.P.. GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004; 20:1464–1465. [DOI] [PubMed] [Google Scholar]
- 13. Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C.Y., Wei L.. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011; 39:316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hänzelmann S., Castelo R., Guinney J.. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Efron B., Tibshirani R.. On testing the significance of sets of genes. Ann. Appl. Stat. 2007; 1:107–129. [Google Scholar]
- 17. Tarca A.L., Draghici S., Bhatti G., Romero R.. Down-weighting overlapping genes improves gene set analysis. BMC Bioinformatics. 2012; 13:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tomfohr J., Lu J., Kepler T.B.. Pathway level analysis of gene expression using singular value decomposition. BMC Bioinformatics. 2005; 6:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luo W., Friedman M.S., Shedden K., Hankenson K.D., Woolf P.J.. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009; 10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goeman J.J., van de Geer S.A., de Kort F., van Houwelingen H.C.. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004; 20:93–99. [DOI] [PubMed] [Google Scholar]
- 21. Barry W.T., Nobel A.B., Wright F.A.. Significance analysis of functional categories in gene expression studies: a structured permutation approach. Bioinformatics. 2005; 21:1943–1949. [DOI] [PubMed] [Google Scholar]
- 22. Tarca A.L., Draghici S., Khatri P., Hassan S.S., Mittal P., Kim J.S., Kim C.J., Kusanovic J.P., Romero R.. A novel signaling pathway impact analysis. Bioinformatics. 2009; 25:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fang Z., Tian W., Ji H.. A network-based gene-weighting approach for pathway analysis. Cell Res. 2012; 22:565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gu Z., Wang J.. CePa: an R package for finding significant pathways weighted by multiple network centralities. Bioinformatics. 2013; 29:658–660. [DOI] [PubMed] [Google Scholar]
- 25. Fang R., Xiao T., Fang Z., Sun Y., Li F., Gao Y., Feng Y., Li L., Wang Y., Liu X.et al.. MicroRNA-143 (miR-143) regulates cancer glycolysis via targeting hexokinase 2 gene. J. Biol. Chem. 2012; 287:23227–23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ai C., Kong L.. CGPS: A machine learning-based approach integrating multiple gene set analysis tools for better prioritization of biologically relevant pathways. J. Genet. Genomics. 2018; 45:489–504. [DOI] [PubMed] [Google Scholar]
- 27. Han J.D. Understanding biological functions through molecular networks. Cell Res. 2008; 18:224–237. [DOI] [PubMed] [Google Scholar]
- 28. Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K.. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017; 45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liao Y., Wang J., Jaehnig E.J., Shi Z., Zhang B.. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019; 47:W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K.. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019; 10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Merico D., Isserlin R., Stueker O., Emili A., Bader G.D.. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010; 5:e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ye J., Zhang Y., Cui H., Liu J., Wu Y., Cheng Y., Xu H., Huang X., Li S., Zhou A.et al.. WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018; 46:71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walter W., Sánchez-Cabo F., Ricote M.. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015; 31:2912–2914. [DOI] [PubMed] [Google Scholar]
- 34. Mao X., Cai T., Olyarchuk J.G., Wei L.. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005; 21:3787–3793. [DOI] [PubMed] [Google Scholar]
- 35. Bauer S., Gagneur J., Robinson P.N.. GOing Bayesian: model-based gene set analysis of genome-scale data. Nucleic Acids Res. 2010; 38:3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jost L. Entropy and Diversity. 2010; Oikos. [Google Scholar]
- 37. Rosvall M., Bergstrom C.T.. Maps of random walks on complex networks reveal community structure. PNAS. 2008; 105:1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M.. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016; 44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mi H., Ebert D., Muruganujan A., Mills C., Albou L.P., Mushayamaha T., Thomas P.D.. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021; 49:D394–D403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R.et al.. The reactome pathway knowledgebase. Nucleic Acids Res. 2020; 48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karp P.D., Billington R., Caspi R., Fulcher C.A., Latendresse M., Kothari A., Keseler I.M., Krummenacker M., Midford P.E., Ong Q.et al.. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2019; 20:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021; 49:D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Geistlinger L., Csaba G., Santarelli M., Ramos M., Schiffer L., Turaga N., Law C., Davis S., Carey V., Morgan M.et al.. Toward a gold standard for benchmarking gene set enrichment analysis. Brief. Bioinform. 2021; 22:545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Colangelo V., Schurr J., Ball M.J., Pelaez R.P., Bazan N.G., Lukiw W.J.. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci. Res. 2002; 70:462–473. [DOI] [PubMed] [Google Scholar]
- 45. Heneka M.T., Carson M.J., El Khoury J.. Neuroinflammation in Alzheimer's disease. Lancet. Neurol. 2015; 14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Colom-Cadena M., Spires-Jones T., Zetterberg H., Blennow K., Caggiano A., DeKosky S.T., Fillit H., Harrison J.E., Schneider L.S., Scheltens P.et al.. The clinical promise of biomarkers of synapse damage or loss in Alzheimer's disease. Alzheimer's Res. Ther. 2020; 12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jiang P., Singh M.. SPICi: a fast clustering algorithm for large biological networks. Bioinformatics. 2010; 26:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Frey B.J., Dueck D.. Clustering by passing messages between data points. Science (New York, N.Y.). 2007; 315:972–976. [DOI] [PubMed] [Google Scholar]
- 49. Golab L., Korn F., Li F., Saha B., Srivastava D.. Size-constrained weighted set cover. IEEE. 2015; 31:879–890. [Google Scholar]