

Abstract
Aims
Adaptor protein p66Shc, encoded by Shc1 gene, contributes to the pathogenesis of oxidative stress-related diseases. p66Shc ability to promote oxidative stress-related diseases requires phosphorylation of serine 36 residue (Ser36) and depends on translocation of p66Shc to the mitochondria. We tested the hypothesis that abnormal p66Shc-mediated reactive oxygen species (ROS) production could be critically involved in nephrons development during nephrogenesis.
Main methods
We have generated unique mutant rats (termed p66Shc-Del), which express endogenous p66Shc with a 9-amino acid deletion, and lack regulatory Ser36. H2O2 renal production was measured by enzymatic microelectrode biosensors. Nephron numbers in 3–5 weeks old p66Shc-Del rats were quantified using the acid maceration method.
Key Findings
p66Shc-Del rats, as wild type salt sensitive rats, display increased mean arterial blood pressure following chronic exposure to a high salt diet. In contrast to wild type rats, p66Shc-Del rats display increased H2O2 renal production and are characterized by a reduction in renal function. The number of glomeruli is significantly reduced in adult p66Shc-Del rats.
Significance
Since low nephron number is an established risk factor for kidney disease and hypertension in humans and rodents, our data suggest that H2O2 renal production, caused by irregular signaling of p66Shc, could be critical in regulating nephrogenesis and that abnormal p66Shc signaling negatively impacts kidney development and renal function by increasing susceptibility to diabetic nephropathy and hypertension-induced nephropathy.
Keywords: oxidative stress, kidney, nephrology, cell signaling, development, Shc proteins, glomeruli
Introduction
Adaptor protein p66Shc is one of the three proteins, encoded by the Shc1 gene, which are named p66Shc, p52Shc, and p46Shc to match their mobility in SDS-PAGE analysis. Acronym Shc stands for Src Homology Collagen, and all Shc isoforms include identical protein-protein interaction domains with p66Shc isoform containing additional NH2-terminal collagen-homologous domain. Three Shc isoforms play remarkably different roles in cells, with p66Shc being important for progression of oxidative stress-related diseases by sensitising cells and tissues to oxidative stress (1–3). Isoforms p52Shc and p46Shc are ubiquitously expressed, while p66Shc expression is tightly regulated and is detected in glomerular cells, smooth muscle cells of renal vessels and tubular cells in diseased kidney of hypertensive rats. It is generally accepted that although multiple renal tissues have a potential to express p66Shc, p66Shc expression is significantly elevated only when some renal injury is progressing. p66Shc is however ubiquitously expressed in fetal lungs of mice, baboons and humans where it regulates morphogenesis and epithelial maturation (4,5). Not much is known about the role of p66Shc in kidney development, but it was shown that p66Shc is mechanistically linked to the expression dynamics of blastocyst lineage associated transcription factors during mouse preimplantation development (6). We tested the hypothesis that reactive oxygen species (ROS), produced as a result of irregular signaling by p66Shc, impair nephrogenesis and cause the reduction of number of glomeruli. p66Shc increases production of mitochondria ROS, following its translocation to mitochondira as a result of phosphorylation of serine 36 residue (Ser36), which causes p66Shc conformation change (7). p66Shc-mediated translocation is triggered by Ser36 phosphorylation in response to extracellular stimulus. p66Shc oxidizes cytochrome c in mitocondria (8), preventing cytochrome c mediated reduction of oxygen to water. It diverts a fraction of mitochondrial electron flow to the production of H2O2. Kidney is considered to be especially susceptible to increases in oxidative stress which plays a pathogenetic role in developmental programming of kidney diseases (9). We have previously shown that the deletion of p66Shc resulted in a restoration of renal microvascular reactivity in rats with hypertension-induced nephropathy (10) and diabetic nephropathy (11). p66Shc is also involved in early embryonic development, the stage when fine regulation of ROS production is of great importance (12). In order to test the contribution of p66Shc in nephrogenesis, we have generated a unique strain of genetically modified rats (p66Shc-Del) on the genetic background of Dahl salt-sensitive (SS) rats. Since p66Shc-Del rats express endogenous p66Shc with a 9-amino acid deletion which removes the Ser36, making its regulatory phosphorylation impossible, these rats allow to investigate the consequences of p66Shc signaling in the absence of normal regulation. Since ureteric bud branching and nephrogenesis are particularly sensitive to ROS production (9,13), we tested whether p66Shc-Del rats are susceptible to renal pathologies due to altered nephrogenesis. The question whether ROS production is beneficial or detrimental to ureteric bud branching is a complex one. Evidently, enhanced ROS production is causing lower nephron numbers in hyperglycemic environment in utero (14). However, limited ROS production is beneficial for ureteric bud branching as was shown in study when reduced NADPH oxidase-dependent ROS production was associated with reduced number of ureteric branches and tips (13). Nephron numbers were quantitated in fully developed homozygote p66Shc-Del, heterozygote p66Shc-Del and parental SS rats using the acid maceration method. While low nephron number is an established risk factor for kidney disease and hypertension in human (15,16), the mechanisms of nephron number control are not sufficiently known. We demonstrate that H2O2 renal production caused by irregular p66Shc signaling are critically involved in nephrogenesis and, moreover, constitutive signaling by p66Shc impairs kidney development and negatively impacts renal function.
Methods
Animals
All animal protocols were conducted per the National Institutes of Health Guide’s guidelines for the Care and Use of Laboratory Animals and reviewed and approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin. Transgenic rats were developed on the background of the Dahl salt-sensitive strain SS/JrHsdMcwi obtained from the colony at the Medical College of Wisconsin. Three transgenic animals were established with targeted mutagenesis using zinc-finger nucleases (ZFN) directed at the CH2 domain of SHC1: frameshift leading to premature termination codon, p66Shc-KO (10); in-frame deletion of 9 amino acids surrounding Serine 36, p66Shc-Del; and knock-in substitution of Ser36Ala substitution, p66Shc-S36A (10). Pups negative for ZFN modification at the SHC1 locus (wildtype) were bred and designated p66Shc-WT. Breeders of SS transgenics were fed a normal salt diet (0.4% NaCl, Teklad 7034), and experimental animals were switched to a high salt diet (1% NaCl, Purina 5001) at three weeks of age. SS/BN2 rats (Dahl SS rats with second chromosome replaced by second chromosome of Brown Norway rats) were used in some experiments as an additional control.
Renal Smooth Muscle Cells Isolation and Shc Isoform Expression Analysis
Renal smooth muscle cells (SMC) were isolated and cultured as previously described (10). Briefly, the kidneys from 6- to 8-week-old male rats were harvested into dissociation buffer (3–6% BSA/ high-glucose DMEM with 10 mM HEPES pH 7.4 and antibiotic/antimycotic cocktail) on ice for each preparation. The medulla was removed and interlobar and arcuate arteries were dissected and put into fresh, ice-cold dissociation buffer. Arteries were digested by a serial treatment with 60U/mL DTT-activated papain, 240U/mL collagenase II, 15U/mL elastase and 0.1 mg/mL trypsin inhibitor (Worthington Biochemical Corporation, Lakewood NJ USA) at 37 °C until single cells began to be observed in solution, but for no longer than 30 minutes. Upon complete dispersion of arterial segments via gentle trituration with a pipette, the cells were washed with growth medium (dissociation buffer with 10% fetal bovine serum) to remove enzymes and debris. Single vascular SMC were resuspended in growth medium and cultured in 6-well plates at 37 °C, 5% CO2. Experiments were conducted on cultures beginning at passage 6 or 12–15 doublings. Cultured cells displayed the typical hill-and-valley growth pattern and stained positive for smooth muscle a actin.
Shc antibody (BD Transduction, USA) was used in Western blotting to detect total Shc isoform expression. To detect serine 36 phosphorylation of p66Shc, SMC from p66Shc-WT and p66Shc-Del were pre-incubated in 0.5% fetal bovine serum/DMEM for 2 days before treatment with 100 nM ET-1 for 5 min. 1 mg/mL whole cell lysate was subjected to immunoprecipitation using 4 µg/mL Shc antibody, separated by SDS-PAGE, transferred to PVDF membranes, and probed with site-specific antibody against p66Shc phosphoserine 36 (Calbiochem, USA).
Arterial Blood Pressure
Rats were placed on a 1% NaCl diet (Purina 5001) at 3 weeks of age for 17 weeks. Telemetry transmitters (Data Sciences International) were implanted into the carotid artery after approximately 15 weeks of 1% NaCl treatment and sealed subcutaneously. Animals were allowed to recover for 5–7 days, and blood pressure monitored for an additional 5 days, as previously described (10).
Urinary Albumin Excretion
After 17 weeks on either normal salt (0.4% NaCl) or high-salt (1% NaCl) diet, rats were housed in metabolic caging during a 16- to 24-hour overnight urine collection. Water and food were provided ad libitum. Urine output was determined by volume, taking care to account for debris, and samples were stored at −80°C until assayed. Urinary albumin was quantified with Albumin Blue 580 dye (Molecular Probes) using a fluorescent plate reader (FL600, Bio-Tek).
Glomerular Injury Scoring
Formalin-fixed, paraffin embedded kidney sections were stained with Periodic Acid Schiff reagent and counterstained with Harris hemotoxylin. Images were collected by a Nanozoomer whole slide scanner (Hamamatsu Photonics). The glomerular score of each animal was averaged between two investigators and derived as the mean of 50 glomeruli. The severity of glomerulosclerosis was expressed on a scale from grades 0 to 4. The glomerular score for individual glomeruli was: Grade 0 normal glomerulus; Grade 1 beginning of mesangial expansion and/or thickening of basement membrane; Grade 2 mild and moderate segmental sclerosis involving less than half of the glomerular tuft; Grade 3 diffuse glomerular sclerosis involving more than half of the tuft; Grade 4 diffuse glomerulosclerosis with total tuft obliteration and collapse.
Ex Vivo Measurement of Renal Interstitial H2O2 Levels
Renal levels of H2O2 was estimated by the reduction in the biosensors current in response to interstitial catalase infusion. Detailed application of the biosensor amperometry technique to assess renal H2O2 levels ex vivo was described previously (17,18). Briefly, animals were prepared using an anesthetic mixture of ketamine (20 mg/kg, intramuscularly) and inactin (50 mg/kg, i.p.) and placed on a temperature-controlled surgical table to maintain body temperature at 37°C throughout the experimental protocol. Supplementary anesthetic (inactin) was administered i.p., as required. A midline incision was made to expose and isolate the left kidney from surrounding fat before placing it in a stainless-steel kidney cup to reduce breathing artifacts (Figure 4B). The exposed kidneys were not denervated. Following surgery and a 20-min equilibrium period, control baseline measurements were recorded. H2O2-sensitive biosensors (sensitive tip - 0.5 mm, 50 µm diameter; # SBS-NUL-10–50 Sarissa Biomedical, UK) connected to dual channel potentiostat (DY2021, Digi-Ivy, Inc., USA) was inserted into the kidney cortex and medulla with a micromanipulators (MN-153, Narishige International USA, Inc). Interstitial infusion of catalase (dissolved in physiological saline 2 μg/ml; #C40, ≥10,000 units/mg protein, Sigma-Aldrich) was performed directly to the kidney via an implanted catheter connected to a peristaltic pump to scavenge and blocks the H2O2 signal detected by the biosensors. The obtained levels of the catalase sensitive current were recalculated as interstitial H2O2 concentrations corresponding to a linear calibration curve as previously described (17,18).
Figure 4. Renal production of H2O2 in genetically modified rats.
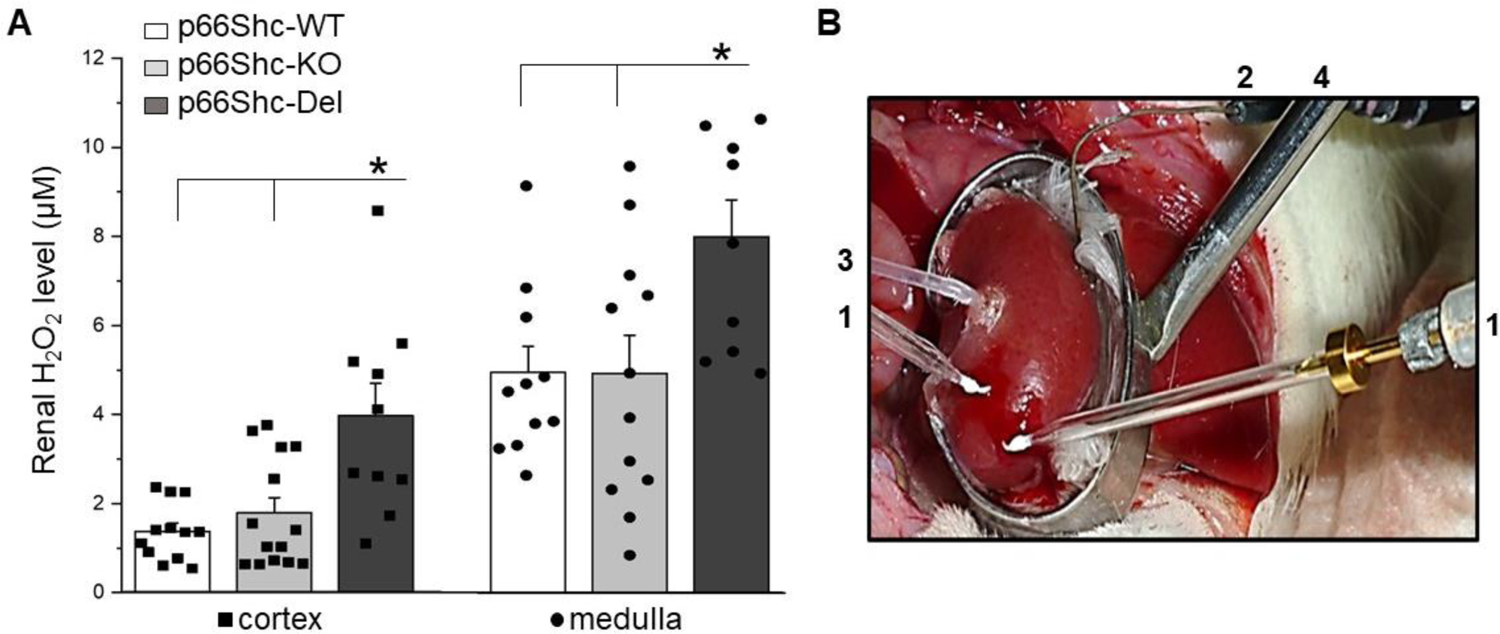
(a) Renal production of H2O2 was determined ex vivo using enzymatic microelectrode biosensors at the end of the 17 weeks of 1% NaCl treatment. H2O2 is significantly elevated in both renal cortex (*P<0.05) and medulla (#P<0.05) of p66Shc-Del compared to control WT and p66Shc-KO rats on 1% NaCl diet, suggesting increased oxidative stress. (b) An example of the preparation for the measurement of renal interstitial H2O2 levels ex vivo by the biosensors amperometry. The kidney was exposed and mounted in a kidney cup (#4). A pair of biosensors (#1) connected to a dual-channel potentiostat was inserted into the kidney cortical and medullar layer, correspondingly. The reference electrode (#2) was placed onto the kidney surface and attached to the potentiostat to ensure noise quality recordings. Interstitial perfusion (#4) of catalase was simultaneously performed.
Quantitation of Glomerular Number Via Acid Maceration
The right kidney of 3–6-week-old littermates of heterozygous p66Shc-Del rats were extracted, decapsulated, and cut into small pieces with scissors and razor blade. Tissue fragments were incubated in 5 mL of 6N HCl at 37˚C for 90 min. Every 30 min, the suspension was manually aspirated to dissociate glomeruli. The suspension was diluted to 50 mL with PBS and incubated overnight at 4˚C until quantification. 0.25 mL samples were placed into the wells of an inverted 12-well plate cover along with an equal volume of PBS. Glomeruli from 4 wells per kidney were counted with a stereoscope, averaged, and corrected for dilution.
Statistical Analysis
Data are presented as means ± SEM. Statistical analysis consisted of one-way ANOVA followed by Bonferroni or Tukey correction for multiple comparisons. (SigmaPlot 12.5 or OriginPro 9.0), with a p-value of <0.05 considered significant. Urinary albumin excretion and glomerular injury data was analyzed using Welch’s one-way ANOVA. For the analysis of redox-sensitive fluorescent data, the non-parametric Kruskal-Wallis test was performed.
Results
Generation of Genetically Modified Rats with a Deletion in a Regulatory Region of p66Shc
We have carried out Zinc Finger nucleases (ZFN)- and CRISPR/Cas9-mediated targeted editing of rat Shc1 gene on the genetic background of Dahl SS rats. Our genetically modified rats lack either p66Shc protein (p66Shc-KO) (10) or p52Shc protein (p52Shc-KO) (19), or express endogenous p66Shc with either Ser36Ala substitution (p66ShcS36A) (10), or deletion of 9 amino acids (AA) in the N-terminal region of the molecule, which also removes regulatory residue Ser36 (p66Shc-Del) (Figure 1A). Deletion of 9 AA in collagen homology domain 2 (CH2) of p66Shc results in the expression of the isoform with increased mobility of p66Shc in SDS-PAGE analysis (Figure 1B). Whether this shift in mobility results from diminished molecular mass or reflects reduced hydrophobicity caused by loss of four prolines in the deleted region of p66Shc molecule requires further investigation. Importantly, expression of other Shc isoforms p52Shc and p46Shc is not changed in p66Shc-Del rat renal tissues.
Figure 1. Schematic diagram of deletion in CH2 protein-protein interaction domain of p66Shc and expression of Shc isoforms in genetically modified rats.
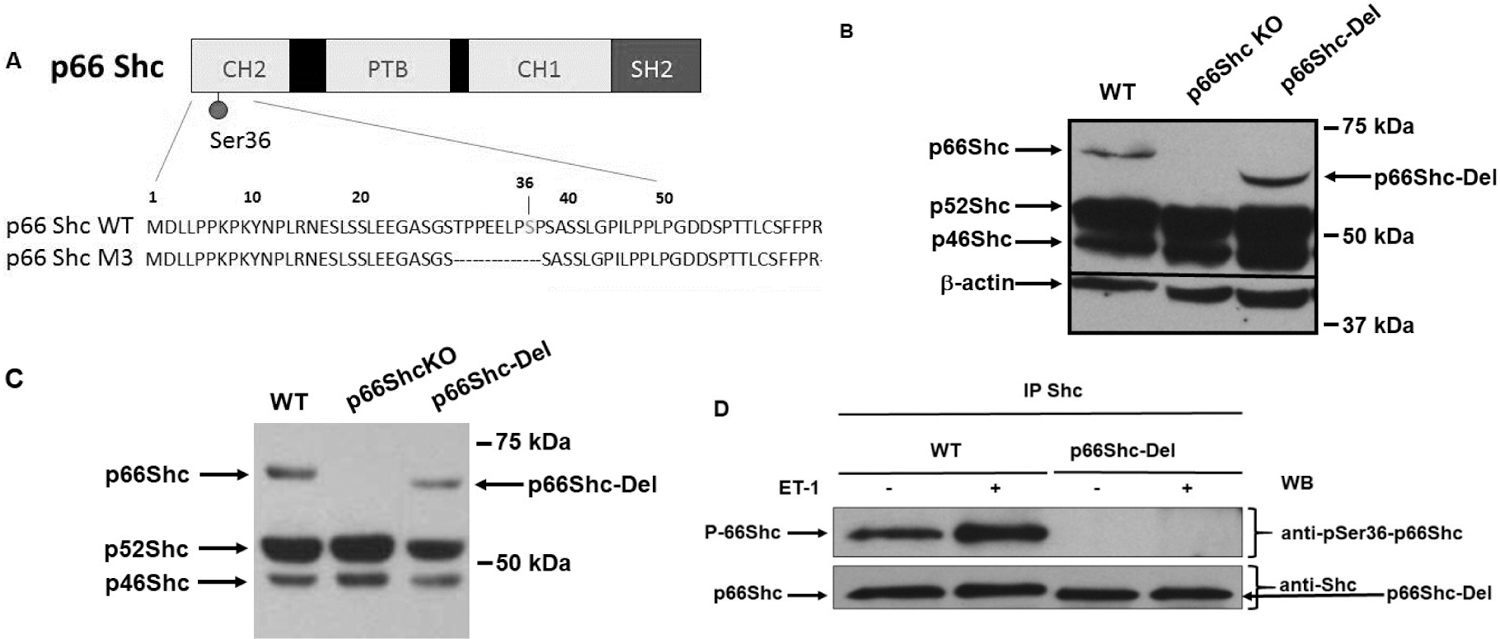
(a) Schematic of p66Shc-Del mutant rats. Protein-protein interaction domains Collagen Homology 1 and 2 (CH1 and CH2), Phosphotyrosine Binding (PTB), and Src Homology 2 (SH2) domains are shown. Amino acids sequence of the N-terminal portion of p66Shc is shown with an indication of missing amino acids in p66Shc-Del mutant. (b) Protein expression of Shc isoforms in wild type (WT) and genetically modified rats. Expression of three Shc isoforms in renal tissues of WT, p66Shc knockout (p66Shc KO), and p66Shc-Del genetically modified rats is shown by Western blot with anti-Shc antibodies following SDS-PAGE. Equal loading was verified by anti-b-actin antibodies. (c) Protein expression of Shc isoforms in smooth muscle cells (SMC) isolated from renal blood vessels of genetically modified rats. Increased mobility of p66Shc-Del mutant, caused by 9AA deletion, is shown by Western blot with anti-Shc antibodies following SDS-PAGE analysis of SMC cultures. (d) The absence of Ser-36 phosphorylation in SMC from p66Shc-Del is verified by Western blot analysis with phospho-Ser36 specific antibodies. Shc immunoprecipitates from either quiescent or Endothelin-1 (ET-1)-treated SMC cells subjected to SDS-PAGE analysis following immunoblotting with either anti-Shc or phospho-Ser36 antibodies. 9AA deletion results in the absence of Ser36 phosphorylation, despite the presence of p66Shc in immunoprecipitates.
Western blot analysis of primary smooth muscle cells (SMC), isolated from genetically modified rats, further confirmed the increased mobility of p66Shc isolated from p66Shc-Del rats, when compared with SMC isolated from wild type (WT) rats (Figure 1C). Phosphorylation of regulatory residue Ser-36 is triggered by activation of several signaling pathways, including those activated by biologically active peptide Endothelin-1 (ET-1) (20), which participates in homeostasis of glomerular structure and filtration function (21). Accordingly, the Ser36 phosphorylation was detected in ET-1 treated WT SMC by specific phospho-Ser36 antibodies but was not observed after treatment of p66Shc-Del SMC, even though the p66Shc-Del was present in immunoprecipitates (Figure 1D).
Increased Renal H2O2 Production in p66Shc-Del Rats
One of the compelling reasons for generating p66Shc mutants in the SS rat has been the noteworthy similarities of hypertension-induced nephropathy with phenotypic traits seen in the African American form of hypertension (22). We have previously reported that p66Shc knockout prevented onset of vascular dysfunction in SS rats and mitigated the development of progressive glomerular disease (10). Even though p66Shc-KO mitigated kidney damage, it did not prevent the development of hypertension in SS rats maintained on a high salt diet (1% NaCl) (10). In our experiments, p66Shc-Del rats, likewise, displayed increased mean arterial blood pressure (MAP) following chronic exposure to a high salt diet (Figure 2A). Increased MAP was caused by the elevation of both systolic and diastolic pressure (Figure 2B). Rat Shc gene is located on chromosome 2. Thus, the consomic rat strain SS/BN2, which did not display renal damage when maintained on a high salt diet, despite increased mean arterial pressure, was included in the analysis as an additional control. Notably, p66Shc-KO rats are protected from renal injury development as determined by measurement of urinary albumin excretion, whereas p66Shc-Del and WT rats are not (Figure 3). Thus, the deletion of the region of p66Shc N-terminal CH2 domain, containing regulatory Ser36 residue, is not analogous to the complete deletion of p66Shc.
Figure 2. Measurement of Mean Arterial Pressure in genetically modified rats maintained on 1% NaCl diet.
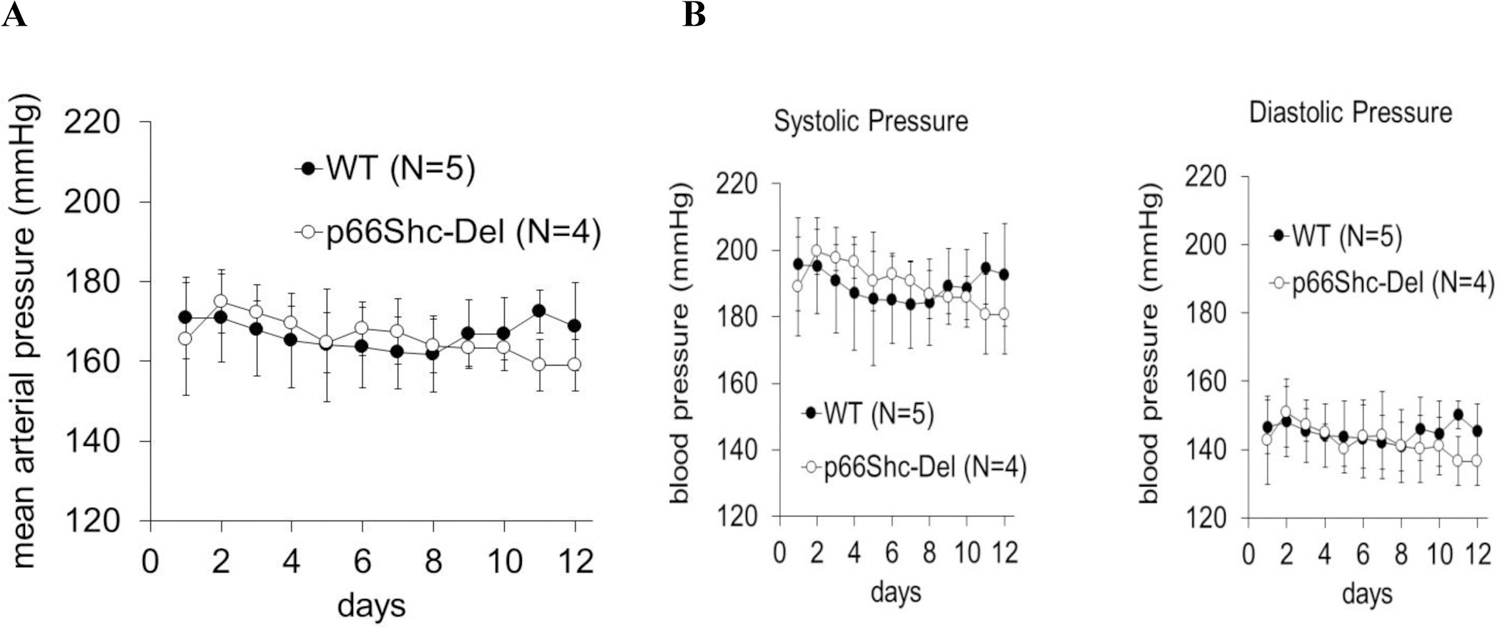
(a) Mean Arterial Pressure was evaluated in male WT and p66Shc-Del mutant rats maintained on a 1% NaCl diet for 20 weeks. (b) Measurement of Systolic and Diastolic Arterial Pressure in male WT and p66Shc-Del rats maintained on 1% salt diet. Shown are the days of continuous blood measurement.
Figure 3. Albuminuria, extracellular matrix accumulation and glomerular injury in wild type and genetically modified hypertensive rats maintained on a 1% NaCl salt diet for 17 weeks.
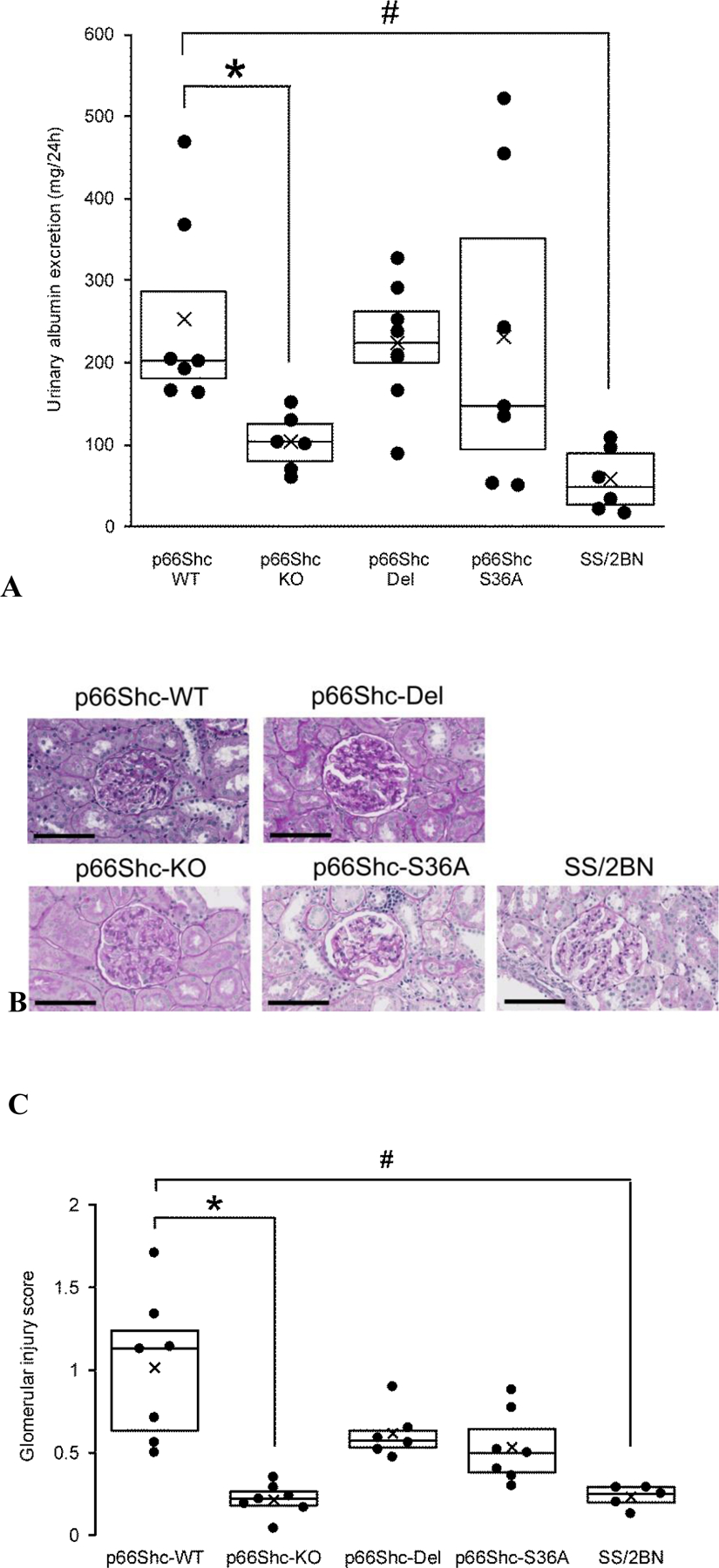
(a) Urinary albumin excretion is significantly greater in WT compared p66Shc KO (*P<0.05) and SS/BN2 (#P<0.01). p66Shc-Del rats are not protected from hypertension-induced renal damage. Boxplots show 25–75% percentiles with dividing line representing median and mean indicated by the black cross. A scatterplot presents individual data. (b) Extracellular matrix accumulation and mesangial expansion in glomeruli of rats fed a high salt, as detected by PAS staining (red purple). Scale bar is 100µm. (c) Summary of glomerular injury scoring indicates glomerular damage is significantly greater in WT compared to p66Shc-KO (*P=.016) and SS/2BN (#P=.018) but not p66Shc-Del (P=.27) and p66Shc-S36A (P=.16). Boxplots show 25–75% percentiles with dividing line representing median and mean indicated by the black cross. A scatterplot presents individual data.
The striking difference between p66Shc-Del rats and both WT and p66Shc-KO rats was revealed when we measured H2O2 renal production in rats maintained on a high salt diet (Figure 4A). We observed increased H2O2 renal production in p66Shc-Del rats. p66Shc-Del rats display increased H2O2 renal production, as measured in vivo by enzymatic microelectrode biosensors and catalase perfusion control (Figure 4B), when compared with parental WT rats and p66Shc-KO rats. This statistically significant difference in renal H2O2 production was observed in both cortex and medulla of p66Shc-Del. p66Shc is known to promote deviation of a fraction of mitochondrial electron flow to the production of H2O2, but also is capable to activate the plasma membrane bound NADPH oxidase to generate ROS through Grb2 mediated activation of Rac1 (7). Regardless of the source of ROS production, p66Shc-Del rats are different from wild type p66Shc rats with regard to renal ROS production, which causes the renal oxidative stress.
p66Shc-Del Mutant Causes Reduced Number of Glomeruli
The process of nephrogenesis requires a combination of apoptosis and proliferation events during renal development. Fine regulation of ROS production is imperative for normal ureteric bud branching and nephrogenesis (13). Since we observed abnormal renal production of H2O2 by p66Shc-Del rats, we tested the possibility that it might affect kidney development, resulting in impaired renal function. To this end, we calculated nephron numbers in WT, p66Shc-Del, and p66Shc-KO rats (Figure 5). Glomeruli were extracted from littermate neonatal kidneys using the acid maceration technique. p66Shc-Del rats demonstrated a significant loss of nephron number when compared with WT and p66Shc-KO rats. Heterozygote p66Shc-Del/WT rats showed the tendency to fewer glomeruli, albeit the difference did not reach statistical significance.
Figure 5. Nephron numbers in genetically modified rats.
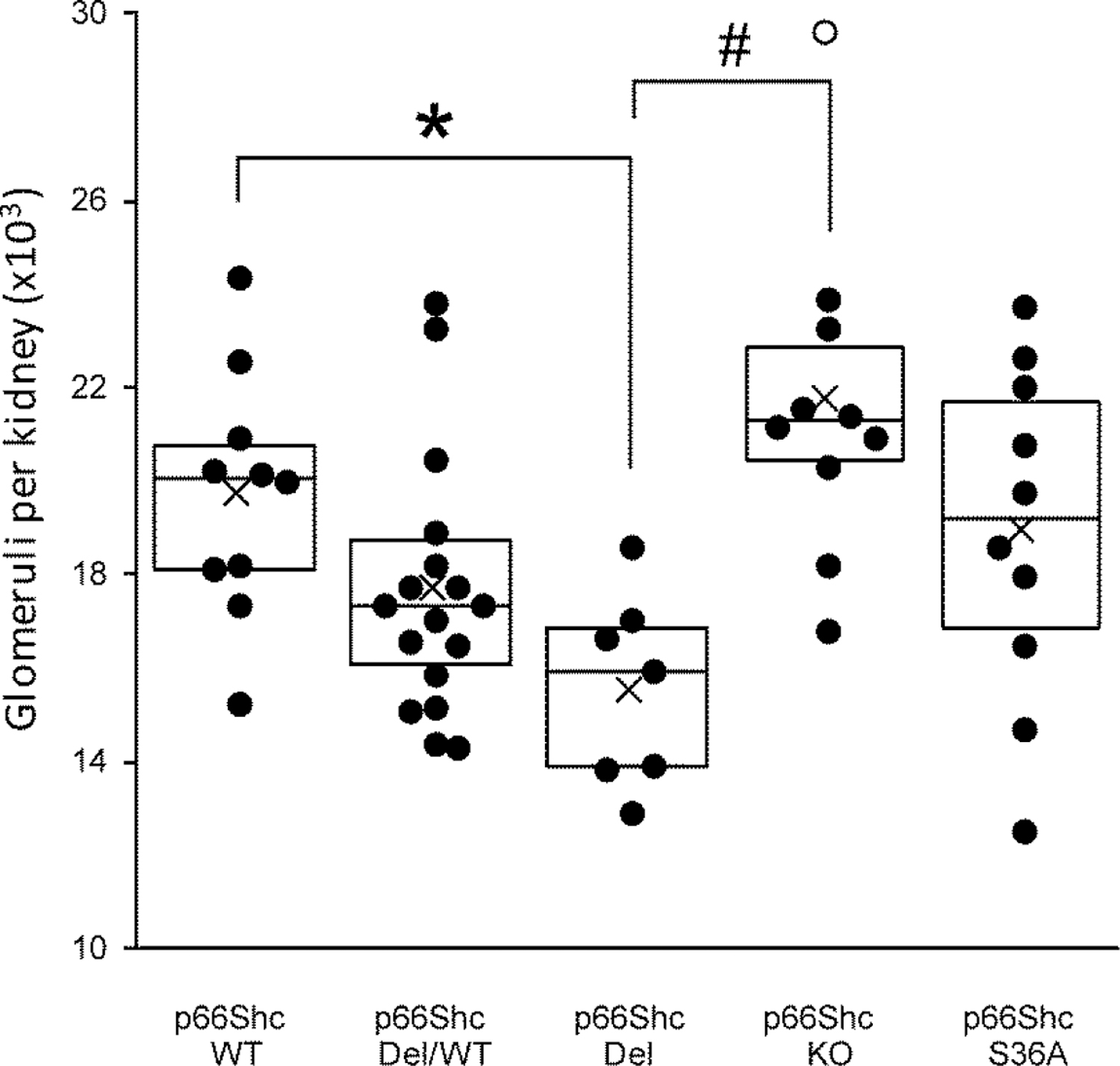
Nephron numbers in neonatal WT, heterozygotes of WT/p66Shc-Del, p66Shc-Del, and p66Shc KO rats counted using the acid maceration technique. The glomerular count of p66Shc-Del is significantly reduced compared to WT (*P<0.01) and p66Shc-KO (#P<0.0001). Boxplots show 25–75% percentiles with dividing line representing median and mean indicated by the black cross. Individual data is presented as a scatterplot with outliers indicated by open circles.
Discussion
It is generally accepted that adaptor protein p66Shc plays an essential role in the sensitivity of cells and organs to oxidative stress (3). Contrary to two other Shc isoforms, namely p46Shc and p52Shc, p66Shc isoform, as a rule, is not constitutively expressed, but is upregulated in injured tissues (7). However, p66Shc is expressed and controls the oxidative stress response in early mammalian embryos (23). p66Shc also regulates oxidative stress during the preimplantation development of sheep embryos (24). Developmental regulation of p66Shc also takes place during lung development (4). Our finding suggests that p66Shc-Del rats display increased H2O2 renal production, as measured by enzymatic microelectrode biosensors when compared with parental SS rats. Notably, the number of glomeruli was significantly reduced in adults p66Shc-Del rats when compared to SS rats.
It has been established that oxidative stress is an important pathogenetic link to developmental origins of renal diseases (9). Our data obtained with p66Shc-Del mutant indicates the role of p66Shc in nephron number regulation and nephrogenesis. It is of note, that reduced nephron number is an important factor in influencing susceptibility to developing hypertension and diabetes (25,26), whereas p66Shc was shown to contribute to pathophysiology of both hypertension-induced nephropathy and diabetes (7). We have previously reported that p66Shc plays an important role in microvascular reactivity of renal blood vessels (10,11), and is involved in the control of renal vessel vasomotion (27). Our observation that the reduction in nephron number accompanies expression of p66Shc-Del mutant suggests that Shc1 gene could operate in organogenesis and particularly in nephrogenesis. Low nephron number is a risk factor for hypertension and chronic kidney disease, as well as for the progression of these diseases (28).
The induction of ureteric bud into the metanephric mesenchyme is controlled by complex interactions among neurotrophic factors, such as glial-derived neurotrophic factor (Gdnf) and signaling molecules, such as tyrosine kinase receptor c-Ret (25). The expression of Gdnf is regulated by multiple transcription and growth factors (29). ROS produced by p66Shc-Del might either prevent normal regulation of expression of molecules required for normal kidney development or interfere with signaling by molecules responsible for nephrogenesis. Whereas p66Shc-mediated mitochondrial ROS formation is widely appreciated (30), the involvement of cytosolic p66Shc in Rac1 activation and ROS production via NADPH oxidase should be also taken into account (31). Deletion of 9 amino acids in the N-terminal portion of p66Shc molecule in p66Shc-Del mutant removes Ser36, phosphorylation of which is required for prolyl isomerase 1 (Pin1)-mediated change of p66Shc conformation resulting in translocation into mitochondria (32). We have previously shown that Ser36 phosphorylation is triggered by activation of MEK-ERK pathway (20). Other laboratories demonstrated that phosphorylation of Ser36 by protein kinase C-b makes p66Shc accessible for Pin-1 interaction (33,34). Accordingly, from one point of view, p66Shc-Del mutant should be retained in cytoplasm and act via NADPH oxidase to produce ROS. Mitochondria-independent action of p66Shc is well documented and includes formation of multiunit complexes with such signling molecules as bPix and FOXO3a (35,36). However, it is also sensible to suggest that 9 amino acid deletion is sufficient to change p66Shc conformation, independently of Ser36 phosphorylation and interaction with Pin1, resulting in p66Shc-Del constitutive translocation into mitochondria. The origin of increased H2O2 production in p66Shc-Del rats should be the subject of future studies. Exact causes of oxidative stress in humans with hypertension remain unclear (37).
The relationship between reduced nephron number and hypertension in humans and animals is well established. Multiple human cohorts have shown that kidneys from subjects with hypertension has lower number of nephrons. It was showed that subjects with hypertension have significantly lower nephron number than normotensive subjects (38). Human syndromic disorders with reduced nephron number like renal coloboma syndrome, Wolf-Hirschhorn syndrome, and Townes-Brock syndrome all have hypertension. This data has been shown multiple times in animal models of reduced nephron number despite that variation in the causative mutation and events that led to reduce nephron number. The limitation of our study is that similar p66Shc deletions has not been identified in humans. Nevertheless, the resulting reduction in nephron number indicate that function variation of Shc1 gene is highly relevant to hypertension in humans.
Conclusions
In short, our data suggest that H2O2 renal production is associated with signaling by p66Shc and that p66Shc-mediated abnormal ROS production negatively impacts kidney development and renal function, increasing susceptibility to diabetic nephropathy and hypertension-induced nephropathy.
Acknowledgments
Authors thank Perrin Schupbach, B.S. (Department of Medicine, Medical College of Wisconsin, Milwaukee, WI) for help in editing and drafting the manuscript.
Funding
This work was supported by the National Institutes of Health NHLBI grants R01 HL147976 (to Sorokin) and R35 HL135749 (to Staruschenko), and Department of Veteran Affairs I01 BX004024 (to Staruschenko). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- AA
amino acids
- CH1
collagen homology domain 1
- CH2
collagen homology domain 2
- Gdnf
glial-derived neurotrophic factor
- ET-1
endothelin 1
- MEK-ERK pathway
MAP Kinase Kinase-MAP kinase pathway
- p66Shc-Del
rat which express endogenous p66Shc with 9 amino acid deletion
- p66Shc-KO
rat with p66Shc knockout
- p66Shc-S36A
rats which express endogenous p66Shc with Ser36Ala substitution
- p52Shc-KO
rat with p52Shc knockout
- PBS
phosphate buffer solution
- PTB
phosphotyrosine binding domain
- ROS
reactive oxygen species
- SH2
Src homology 2
- Ser
serine
- SS
salt-sensitive
- SMC
smooth muscle cells
- SS/BN2
Dahl SS rats with second chromosome replaced by second chromosome of Brown Norway rats
- WT
wild type
- ZFN
zinc finger nucleases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Francia P, Cosentino F, Schiavoni M, Huang Y, Perna E, Camici GG, Luscher TF, and Volpe M (2009) p66(Shc) protein, oxidative stress, and cardiovascular complications of diabetes: the missing link. J Mol Med (Berl) 87, 885–891 [DOI] [PubMed] [Google Scholar]
- 2.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, and Pelicci PG (1999) The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402, 309–313 [DOI] [PubMed] [Google Scholar]
- 3.Purdom S, and Chen QM (2003) p66(Shc): at the crossroad of oxidative stress and the genetics of aging. Trends Mol Med 9, 206–210 [DOI] [PubMed] [Google Scholar]
- 4.Lee MK, Pryhuber GS, Schwarz MA, Smith SM, Pavlova Z, and Sunday ME (2005) Developmental regulation of p66Shc is altered by bronchopulmonary dysplasia in baboons and humans. Am J Respir Crit Care Med 171, 1384–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MK, Zhao J, Smith SM, Tefft JD, Bringas P, Hwang C, and Warburton D (1998) The Shc 66 and 46 kD isoforms are differentially downregulated at parturition in the fetal mouse lung. Pediatr Res 44, 850–859 [DOI] [PubMed] [Google Scholar]
- 6.Edwards NA, Watson AJ, and Betts DH (2018) Knockdown of p66Shc Alters Lineage-Associated Transcription Factor Expression in Mouse Blastocysts. Stem Cells Dev 27, 1479–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright KD, Staruschenko A, and Sorokin A (2018) Role of adaptor protein p66Shc in renal pathologies. Am J Physiol Renal Physiol 314, F143–F153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, and Pelicci PG (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122, 221–233 [DOI] [PubMed] [Google Scholar]
- 9.Hsu CN, and Tain YL (2020) Developmental Origins of Kidney Disease: Why Oxidative Stress Matters? Antioxidants (Basel) 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller B, Palygin O, Rufanova VA, Chong A, Lazar J, Jacob HJ, Mattson D, Roman RJ, Williams JM, Cowley AW Jr., Geurts AM, Staruschenko A, Imig JD, and Sorokin A (2016) p66Shc regulates renal vascular tone in hypertension-induced nephropathy. J Clin Invest 126, 2533–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller BS, Blumenthal SR, Shalygin A, Wright KD, Staruschenko A, Imig JD, and Sorokin A (2018) Inactivation of p66Shc Decreases Afferent Arteriolar KATP Channel Activity and Decreases Renal Damage in Diabetic Dahl SS Rats. Diabetes 67, 2206–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee MK, Smith SM, Banerjee MM, Li C, Minoo P, Volpe MV, and Nielsen HC (2014) The p66Shc adapter protein regulates the morphogenesis and epithelial maturation of fetal mouse lungs. Am J Physiol Lung Cell Mol Physiol 306, L316–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kondo S, Matsuura S, Ariunbold J, Kinoshita Y, Urushihara M, Suga K, Ozaki N, Nagai T, Fujioka K, and Kagami S (2019) Expression of NADPH oxidase and production of reactive oxygen species contribute to ureteric bud branching and nephrogenesis. J Med Invest 66, 93–98 [DOI] [PubMed] [Google Scholar]
- 14.Chen YW, Chenier I, Chang SY, Tran S, Ingelfinger JR, and Zhang SL (2011) High glucose promotes nascent nephron apoptosis via NF-kappaB and p53 pathways. Am J Physiol Renal Physiol 300, F147–156 [DOI] [PubMed] [Google Scholar]
- 15.Coats LE, Davis GK, Newsome AD, Ojeda NB, and Alexander BT (2019) Low Birth Weight, Blood Pressure and Renal Susceptibility. Curr Hypertens Rep 21, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schreuder MF (2012) Safety in glomerular numbers. Pediatr Nephrol 27, 1881–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palygin O, Evans LC, Cowley AW Jr., and Staruschenko A (2017) Acute In Vivo Analysis of ATP Release in Rat Kidneys in Response to Changes of Renal Perfusion Pressure. J Am Heart Assoc 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palygin O, Levchenko V, Evans LC, Blass G, Cowley AW Jr., and Staruschenko A (2015) Use of Enzymatic Biosensors to Quantify Endogenous ATP or H2O2 in the Kidney. J Vis Exp [DOI] [PMC free article] [PubMed]
- 19.Wright KD, Miller BS, El-Meanawy S, Tsaih SW, Banerjee A, Geurts AM, Sheinin Y, Sun Y, Kalyanaraman B, Rui H, Flister MJ, and Sorokin A (2019) The p52 isoform of SHC1 is a key driver of breast cancer initiation. Breast Cancer Res 21, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foschi M, Franchi F, Han J, La Villa G, and Sorokin A (2001) Endothelin-1 induces serine phosphorylation of the adaptor protein p66Shc and its association with 14–3–3 protein in glomerular mesangial cells. J Biol Chem 276, 26640–26647 [DOI] [PubMed] [Google Scholar]
- 21.Barton M, and Sorokin A (2015) Endothelin and the glomerulus in chronic kidney disease. Semin Nephrol 35, 156–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cowley AW Jr. (2006) The genetic dissection of essential hypertension. Nat Rev Genet 7, 829–840 [DOI] [PubMed] [Google Scholar]
- 23.Betts DH, Bain NT, and Madan P (2014) The p66(Shc) adaptor protein controls oxidative stress response in early bovine embryos. PLoS One 9, e86978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang T, Zhao X, Hai R, Li R, Zhang W, and Zhang J (2019) p66Shc is associated with hydrogen peroxide-induced oxidative stress in preimplantation sheep embryos. Mol Reprod Dev 86, 342–350 [DOI] [PubMed] [Google Scholar]
- 25.Wang X, and Garrett MR (2017) Nephron number, hypertension, and CKD: physiological and genetic insight from humans and animal models. Physiol Genomics 49, 180–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gross ML, Amann K, and Ritz E (2005) Nephron number and renal risk in hypertension and diabetes. J Am Soc Nephrol 16 Suppl 1, S27–29 [DOI] [PubMed] [Google Scholar]
- 27.Palygin O, Miller BS, Nishijima Y, Zhang DX, Staruschenko A, and Sorokin A (2019) Endothelin receptor A and p66Shc regulate spontaneous Ca(2+) oscillations in smooth muscle cells controlling renal arterial spontaneous motion. FASEB J 33, 2636–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanzaki G, Tsuboi N, Shimizu A, and Yokoo T (2020) Human nephron number, hypertension, and renal pathology. Anat Rec (Hoboken) 303, 2537–2543 [DOI] [PubMed] [Google Scholar]
- 29.Brodbeck S, and Englert C (2004) Genetic determination of nephrogenesis: the Pax/Eya/Six gene network. Pediatr Nephrol 19, 249–255 [DOI] [PubMed] [Google Scholar]
- 30.Di Lisa F, Kaludercic N, Carpi A, Menabo R, and Giorgio M (2009) Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res Cardiol 104, 131–139 [DOI] [PubMed] [Google Scholar]
- 31.Oshikawa J, Kim SJ, Furuta E, Caliceti C, Chen GF, McKinney RD, Kuhr F, Levitan I, Fukai T, and Ushio-Fukai M (2012) Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells. Am J Physiol Heart Circ Physiol 302, H724–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinton P, and Rizzuto R (2008) p66Shc, oxidative stress and aging: importing a lifespan determinant into mitochondria. Cell Cycle 7, 304–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehta KD (2014) Emerging role of protein kinase C in energy homeostasis: A brief overview. World J Diabetes 5, 385–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowski MR, Del Sal G, Pelicci PG, and Rizzuto R (2007) Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 315, 659–663 [DOI] [PubMed] [Google Scholar]
- 35.Chahdi A, and Sorokin A (2008) Endothelin-1 couples betaPix to p66Shc: role of betaPix in cell proliferation through FOXO3a phosphorylation and p27kip1 down-regulation independently of Akt. Mol Biol Cell 19, 2609–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Staruschenko A, and Sorokin A (2012) Role of betaPix in the Kidney. Front Physiol 3, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Touyz RM, Rios FJ, Alves-Lopes R, Neves KB, Camargo LL, and Montezano AC (2020) Oxidative Stress: A Unifying Paradigm in Hypertension. Can J Cardiol 36, 659–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keller G, Zimmer G, Mall G, Ritz E, and Amann K (2003) Nephron number in patients with primary hypertension. N Engl J Med 348, 101–108 [DOI] [PubMed] [Google Scholar]