

SUMMARY
Streptococcus pneumoniae (Spn) alone and during co-infection with influenza A virus (IAV) can result in severe pneumonia with mortality. Pneumococcal surface protein A (PspA) is an established virulence factor required for Spn evasion of lactoferricin and C-reactive protein-activated complement-mediated killing. Herein, we show that PspA functions as an adhesin to dying host cells. We demonstrate that PspA binds to host-derived glyceraldehyde-3-phosphate dehydrogenase (GAPDH) bound to outward-flipped phosphatidylserine residues on dying host cells. PspA-mediated adhesion was to apoptotic, pyroptotic, and necroptotic cells, but not healthy lung cells. Using isogenic mutants of Spn, we show that PspA-GAPDH-mediated binding to lung cells increases pneumococcal localization in the lower airway, and this is enhanced as a result of pneumolysin exposure or co-infection with IAV. PspA-mediated binding to GAPDH requires amino acids 230–281 in its α-helical domain with intratracheal inoculation of this PspA fragment alongside the bacteria reducing disease severity in an IAV/Spn pneumonia model.
In brief
Park et al. demonstrate that pneumococcal surface protein A (PspA) acts as a bacterial adhesin. They show that PspA mediates Streptococcus pneumoniae binding to dying host cells via interactions with surface-bound host GAPDH. Pneumococcal localization in the lower airway is enhanced following pneumolysin- or influenza A virus (IAV)-mediated damage in a PspA-dependent manner.
Graphical abstract

INTRODUCTION
Pneumonia as the result of influenza A virus (IAV) and Streptococcus pneumoniae (Spn, the pneumococcus) superinfection is characterized by its severity and high mortality rate (Didierlaurent et al., 2007; McCullers, 2006; Morens et al., 2008). For example, following retrospective examination of banked lung autopsy samples, the vast majority of individuals who died as result of the 1918 Spanish influenza pandemic had evidence of co-infection or secondary infection with Spn (Morens et al., 2008; Weinberger et al., 2012). Today, and despite access to antimicrobials, ~10% of IAV bacteria superinfections that require hospitalization result in death, with Spn being present in ~56% of these instances (Palacios et al., 2009). Notably, this high rate of morbidity and mortality persists despite widespread implementation of vaccines against both pathogens (Zhang et al., 2016).
Spn is a Gram-positive bacterium and the leading cause of community-acquired pneumonia (Feldman and Anderson, 2016); as such it has been extensively studied for over 130 years. In addition to its polysaccharide capsule, which protects against opsonophagocytosis, all invasive strains of Spn carry multiple other virulence determinants. Two critical virulence proteins carried by all Spn are pneumococcal surface protein A (PspA) and pneumolysin (Crain et al., 1990; Kanclerski and Möllby, 1987). PspA is a member of the choline-binding protein family (Talkington et al., 1991; Yother and White, 1994). Choline-binding proteins (CBPs) are surface exposed and have a wide variety of functions. They contain a typically C-terminal choline-binding domain that non-covalently attaches the protein to phosphorylcholine residues on cell wall-associated C-polysaccharide and teichoic acids. The N terminus of each CBP is distinct, providing each CBP with an independent function (Maestro and Sanz, 2016). PspA, at ~65–95 kDa, has a N-terminal α-helix region composed of the α-helix domain (αHD) and clade defining region (CDR). This is followed by a proline-rich domain (PRD) and then 6–10 repeats of the choline binding motif (Figure 1A) (Hollingshead et al., 2000). Previously we have shown that PspA binds to and neutralizes the cationic antimicrobial peptide lactoferricin (Håkansson et al., 2001; Senkovich et al., 2007; Shaper et al., 2004). As one of the most abundant CBPs, PspA also prevents recognition of otherwise unoccupied phosphorylcholine residues on the bacterial surface by host C-reactive protein (Mukerji et al., 2012). Pneumolysin is a highly conserved cholesterol-binding pore-forming toxin (Vögele et al., 2019). It induces airway epithelial and alveolar macrophage cell death by induction of apoptosis or necroptosis (González-Juarbe et al., 2015, 2017, 2018). Notably, we have recently demonstrated that IAV-induced oxidative stress profoundly enhanced the susceptibility of lung cells for pneumolysin-mediated necroptosis (Gonzalez-Juarbe et al., 2020). This is one of the many ways that IAV and Spn act synergistically to cause severe lung disease (LeMessurier et al., 2020; Rudd et al., 2016).
Figure 1. IAV-induced pneumococcal PspA contributes to IAV/Spn superinfection.
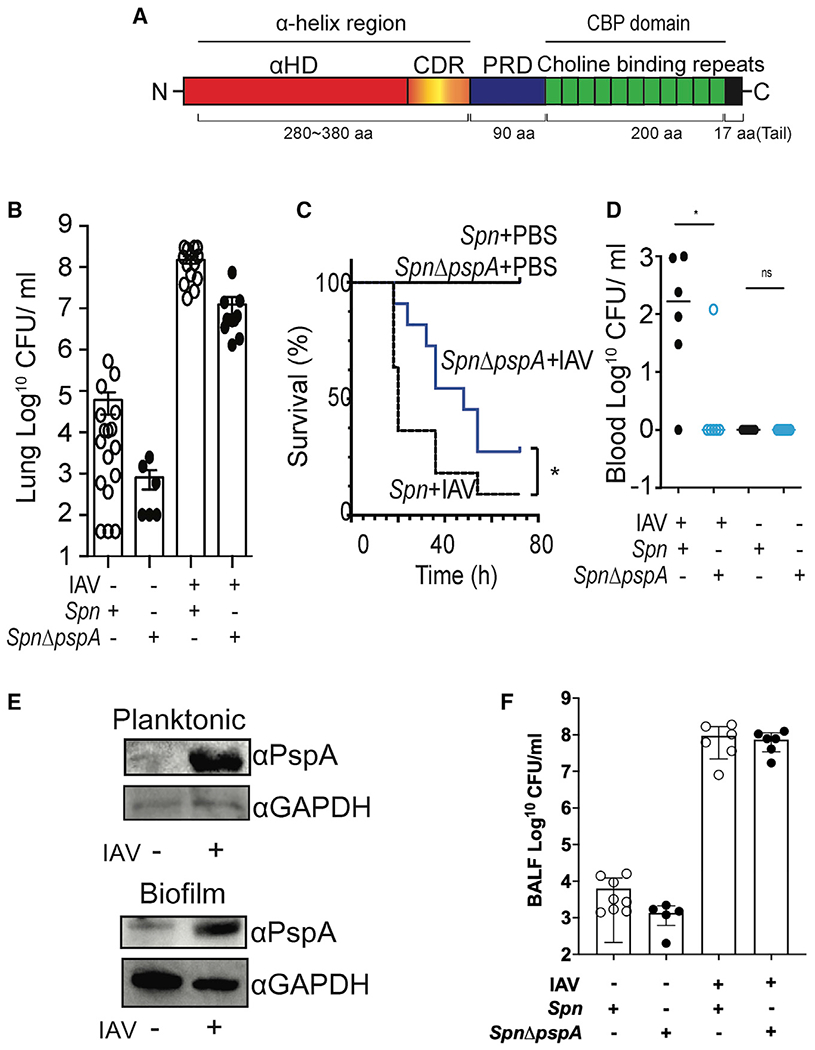
(A) Schematic diagram of PspA showing the N-terminal α-helix region, proline-rich domain (PRD), and C-terminal choline-binding domain. The α-helix region is composed of the α-helix domain (αHD) and clade defining region (CDR).
(B) Mice were infected intratracheally with 107 CFUs of Spn EF3030 or an isogenic pspA mutant (Spn ΔpspA) 7 days after intranasal challenge with 200 PFUs of IAV (PR8) or PBS. Mouse lungs were collected 1 day after pneumococcal challenge, and then total bacteria in whole lung homogenates was determined.
(C) Survival curves of IAV/Spn superinfected mice and PBS/Spn control mice. Survival rates were subsequently compared using a Gehan Breslow-Wilcoxon test (*p = 0.036; n = 11 per cohort).
(D) Bacterial titers in the blood of mice 32 h after IAV/Spn superinfection.
(E) Spn EF3030 (107 CFUs) was incubated in CDM media. Six hours (Planktonic) or 1 day (Biofilm) after pneumococcal culture, 109 IAVs were added and incubated for 2 h with the Spn. PspA expression in the Spn was examined by immunoblot.
(F) BALF was collected from Spn and IAV/Spn-infected mice 1 day after pneumococcal challenge, and then total bacterial titers in BALF were enumerated.
(A, D, and F) Circles represent individual mice. Statistical comparisons between infected cohorts were performed using a Kruskal-Wallis test with Dunn’s multiple-comparison post-test. *p < 0.05, ****p ≤ 0.0001.
Numerous investigators have demonstrated that PspA and pneumolysin are required for virulence, and that immunization with recombinant versions of these proteins, especially when mixed together, confers protective immunity (Bricker and Camilli, 1999; Briles et al., 2003; Coats et al., 2005; Håkansson et al., 2001; McDaniel et al., 1991; Ogunniyi et al., 2000; Shaper et al., 2004; Swiatlo et al., 2003; Talkington et al., 1991). Pertinent to this study, PspA has also been shown to be required during IAV/Spn superinfection (Greene et al., 2016; King et al., 2009; Roberts et al., 2019). Herein, we describe a new role for PspA that helps to explain why individuals with IAV have increased susceptibility to pneumococcal infection. Our results are broadly applicable to other conditions where lung epithelial cell death is occurring and has important implications on human susceptibility to Spn disease.
RESULTS
Pneumococcal PspA is required during Spn secondary infection following IAV
We first sought to confirm, under lab conditions, the increased disease severity observed in humans during IAV/Spn superinfection and the importance of PspA as a virulence determinant. Mice infected intranasally with IAV 7 days prior to intratracheal Spn challenge had a 5,600-fold increase in the number of Spn colony-forming units (CFUs) recovered from the lungs 24 h post-infection versus mice challenged with only Spn (Figure 1B). IAV/Spn-superinfected mice also had greater mortality versus control mice infected only with Spn (Figure 1C) and were far more likely to have developed bacteremia at 32 h post-infection (Figure 1D). Consistent with the known roles for PspA, pspA deficiency attenuated the pneumococcus across all in vivo conditions tested. We observed a 25- to 100-fold reduction in the number of CFUs in the lungs of challenged mice versus corresponding controls (Figure 1B), significantly reduced time until death (Figure 1C), and starkly reduced incidence of bacteremia in IAV-infected mice challenged with Spn ΔpspA (Figure 1D).
Greene et al. (2016) have previously reported that PspA levels were increased as a result of Spn exposure to IAV. This result was reproducible, and we observed via immunoblot that IAV-exposed Spn had markedly increased levels of PspA during planktonic and biofilm growth conditions (Figure 1E). To discern whether PspA specifically contributed to disease severity during IAV/Spn super-infection, we normalized Spn titers in the lungs of IAV super-infected mice to those observed in Spn-only-infected mice. The >10-fold fewer CFUs corresponding to Spn ΔpspA in comparison with wild type (WT) suggested this was indeed the case (Figure S1). Notably and forming the basis of our studies described hereafter, we observed that despite the greater number of pneumococci recovered from whole lungs of WT infected mice versus those challenged with Spn ΔpspA, there were no differences in the number of recovered Spn present in bronchoalveolar lavage fluid (BALF) samples (Figure 1F). This unexpected result was interpreted as meaning that WT Spn were more resistant to dislodgement than Spn ΔpspA and served as rationale for further experimentation. Finally, PspA deficiency had no effect on in vitro growth of Spn (Figure S2). Thus, the reduced titers seen for Spn ΔpspA in vivo were not the result of a general fitness defect.
PspA binds to host GAPDH
Numerous CBPs have been shown to serve as bacterial adhesins (Orihuela et al., 2009). Given that PspA is surface exposed, is abundant, and has conferred resistance to Spn dislodgement during bronchoalveolar lavage, we postulated that in addition to its known roles, PspA contributed to disease severity by functioning as an adhesin. To test this hypothesis, we performed pull-down assays with recombinant portions of PspA as bait using whole-cell lysates from human type II pneumocyte (A549) and human pharyngeal epithelial (D562) cell lines. These recombinant (r)PspAs corresponded to the αHD and PRD and were lacking the choline-binding domain and signal peptide. One strong band associated with rPspA and common to both cell lysates was detected following protein separation by SDS-PAGE (Figure 2A). Subsequently, using liquid chromatography tandem mass spectroscopy (LC-MS/MS), we identified the most abundant protein within this band as glyceraldehyde-3-phosphate dehydrogenase (GAPDH); a complete list of identified proteins is provided in Table S1. GAPDH is highly abundant and is therefore commonly detected in LC-MS/MS analyses of isolated host proteins. A specific interaction between PspA and human GAPDH (huGAPDH) was corroborated by immunoblot after Ni-NTA pull-down and detection with specific monoclonal antibodies against huGAPDH and PspA, respectively (Figure 2B). Likewise, using surface plasmon resonance (SPR), we observed that rPspA and huGAPDH interacted with high affinity (KD = 2.95 μM) (Figure 2C). Importantly, using fluorescein isothiocyanate (FITC)-conjugated huGAPDH, we observed by flow cytometry huGAPDH binding to both WU2 and EF3030, two unrelated strains of Spn. This was not observed when using their respective ΔpspA mutants (Figure 2D). Binding of huGAPDH to other bacteria (Streptococcus mutans, Staphylococcus aureus, or uropathogenic Escherichia coli) was not observed (Figure 2E). Thus, binding to huGAPDH was restricted to bacteria carrying PspA.
Figure 2.
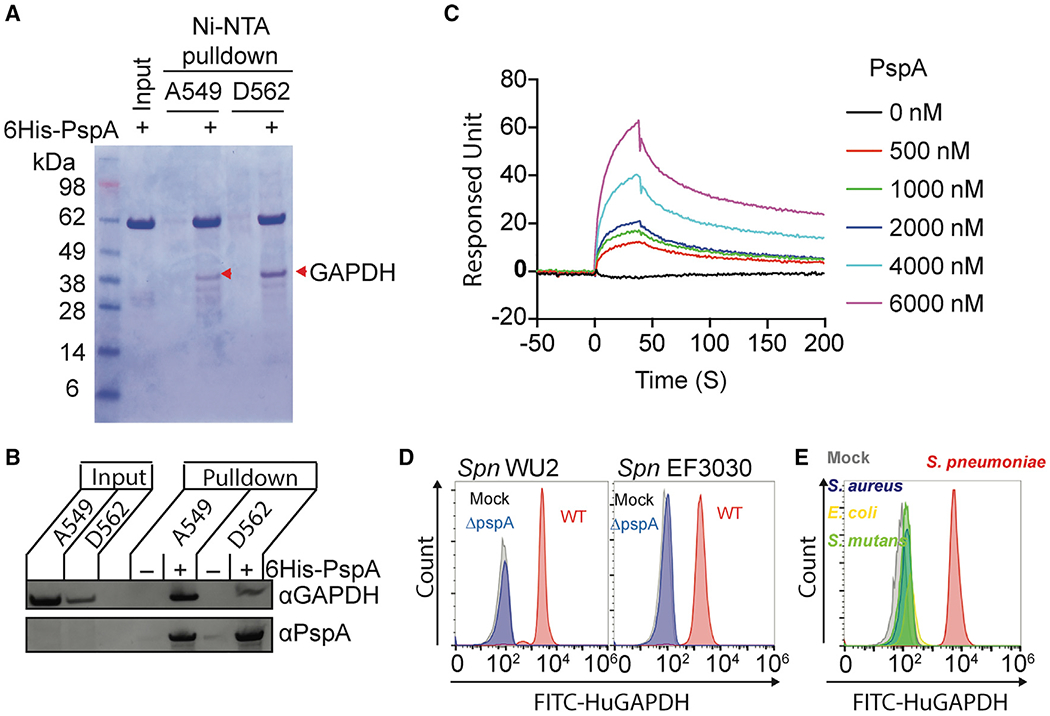
Pneumococcal PspA interacts with human GAPDH
(A) Recombinant His-tagged PspA protein composed of the αHD and PRD was used in pulldown experiments with A549 and D562 cell lysates. The PspA bound proteins were separated by SDS-PAGE and visualized by Coomassie blue dye. Interacted proteins were identified by liquid chromatography tandem mass spectrometry (Table S1). Red arrowheads indicate GAPDH.
(B) Interaction of PspA with host GAPDH was analyzed by immunoblot after A549 and D562 cell lysates were pulled down using PspA as bait.
(C) Interactions of PspA with huGAPDH were analyzed by surface plasmon resonance spectroscopy under different PspA concentrations. (D) Spn WU2, Spn EF3030, or its pspA isogenic mutants, and (E) S. aureus, E. coli, and S.mutans were incubated with FITC-conjugated huGAPDH. The huGAPDH binding to bacterium was measured by flow cytometry. Mock represents Spn bacteria only.
Data are representative of (A and B) two or (C–D) three independent experiments.
PspA binds to host-cell-derived GAPDH via PspA’s α-helical domain
To further confirm a specific interaction with GAPDH and identify the domain of PspA responsible, fragments of PspA corresponding to αHD, PRD, and sub-sections of αHD were expressed, purified, and used as bait in pull-down experiments with huGAPDH. Fragments (F)-1, F-2, F-6, and F-8, but not F-3 or F-7, were able to pull down huGAPDH. Even though F-7 did not pull down huGAPDH, only the PspA fragments that contained its corresponding amino acids (aa; 230–281) were able to pull down huGAPDH (Figure 3A; Figure S3). The simplest interpretation of these results is that aa 230–281 in the αHD contained the epitope of PspA binding huGAPDH, and that proximal regions, either N-terminal or C-terminal, were also required for conformation or stability of this interaction.
Figure 3.
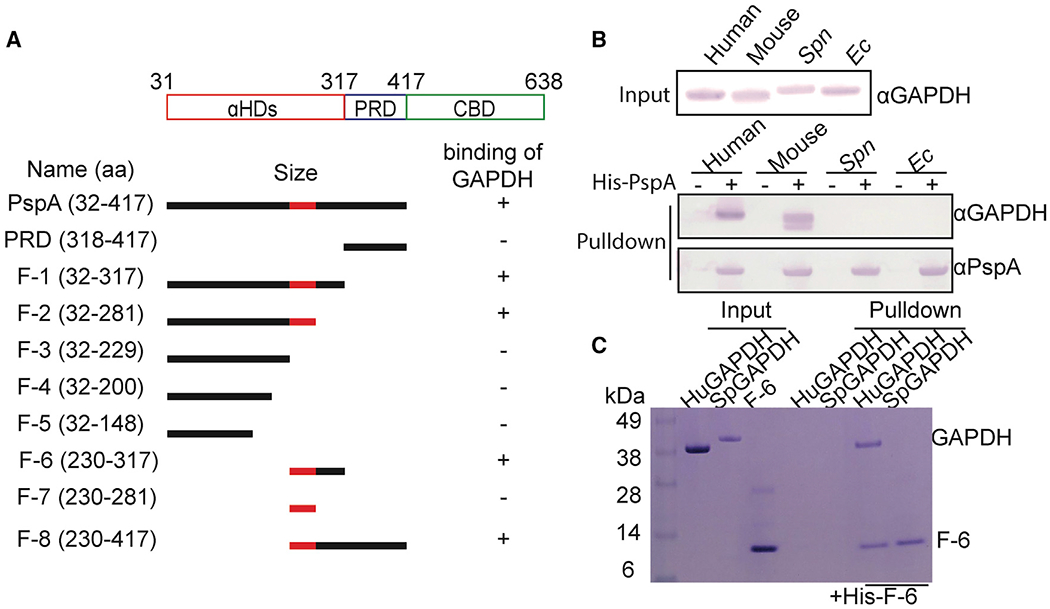
α-Helical domain (aa 230–281) of PspA is required for host GAPDH selectively
(A) Diagram of PspA and the corresponding rPspA fragments (F-) used in pull-down experiments. These truncated fragments were used to test PspA binding to GAPDH (see Figure S3). The region colored red within the αHD of PspA was identified as the subdomain that mediates GAPDH binding.
(B) Cell lysates from different sources were used in pull-down experiments using rPspA as bait. Versions of GAPDH that interacted with PspA were detected by immunoblot.
(C) F-6 was pulled down with purified human GAPDH and visualized by SDS-PAGE separation and staining with Coomassie blue dye. Results shown are representative with experiments performed three times with identical results.
The aa sequence of GAPDH is highly conserved across animals and plants. Yet host-cell-derived GAPDH has not previously been shown to be a target for bacterial adherence. This would be unrelated to the numerous reports that show bacterial GAPDH (bGAPDH) can serve as an adhesin in a moonlighting capacity (Andre et al., 2017; Wang and Lu, 2007). In ourstudies reported here, huGAPDH and mouse GAPDH, but not E. coli or Spn GAPDH, were pulled down with rPspA after their mixture with corresponding cell lysates (Figure 3B). Similarly, in separate pull-down experiments, F-6 bound to huGAPDH and not bGAPDH from Spn (Figure 3C). Thus, PspA interacted with host GAPDH specifically. It is of note that mixture of PspA at 1:1 ratio with huGAPDH did not negatively impact its enzymatic activity. Albeit at a 10:1 ratio, a ~35% reduction in GAPDH activity was observed (Figure S4). To further validate that PspA-GAPDH interactions were specific, we tested binding interaction with human serum albumin and found no interactions between GAPDH (Figure S5).
PspA mediates attachment of Spn to dying lung cells
Mammalian GAPDH is recognized to bind the surface of dying cells as result of its affinity for “flipped” phosphatidylserine residues newly presented on the outer cell membrane (Fadok et al., 1992; Martin et al., 1995). We detected endogenous huGAPDH on the surface of A549 cells 4 h after induction of cell death with H2O2 or pneumolysin and 4 days after IAV infection (Figure 4A). To further investigate potential interactions between PspA and dying cells, we induced A549 cells to undergo either apoptosis, pyroptosis, or necroptosis, and we tested the ability of rPspA to bind to them. In all instances, and as determined by immunoblots of whole-cell lysates (Figure 4B) and fluorescent microscopy with Annexin V (Figure 4C), rPspA bound to the dying cells. In contrast, recombinant PcpA, also a pneumococcal adhesin (Khan et al., 2012), bound to healthy and dying cells equally (Figure 4B). Similarly, using flow cytometry, rPspA was found to exclusively bind cells that stained positive with propidium iodide (Figure 4D), a marker of cell death. rPspA also bound to cells dying as a result of IAV infection (Figure 4E). Notably, PspA fragment F-6 bound to pneumolysin-killed cells, whereas F-3 did not (Figure 4F), reaffirming that aa 230–317 of PspA was required. Subsequently, validating the role of PspA-GAPDH interactions during infection with live bacteria, only WT and not PspA-deficient Spn adhered to pneumolysin-treated, Annexin V-positive, dying cells (Figure 4G). It is worth noting that we performed traditional in vitro adhesion assays using cell monolayers to test PspA’s contribution to Spn adhesion (Brissac and Orihuela, 2019). However, PspA’s requirement for host cell death made quantification unreliable. This was due to the detachment and subsequent loss of dying cells during washing steps to remove unadhered bacteria. Despite this limitation, the preponderance of evidence suggests that PspA acts as an adhesin to bind pneumococci to dying host cells.
Figure 4.
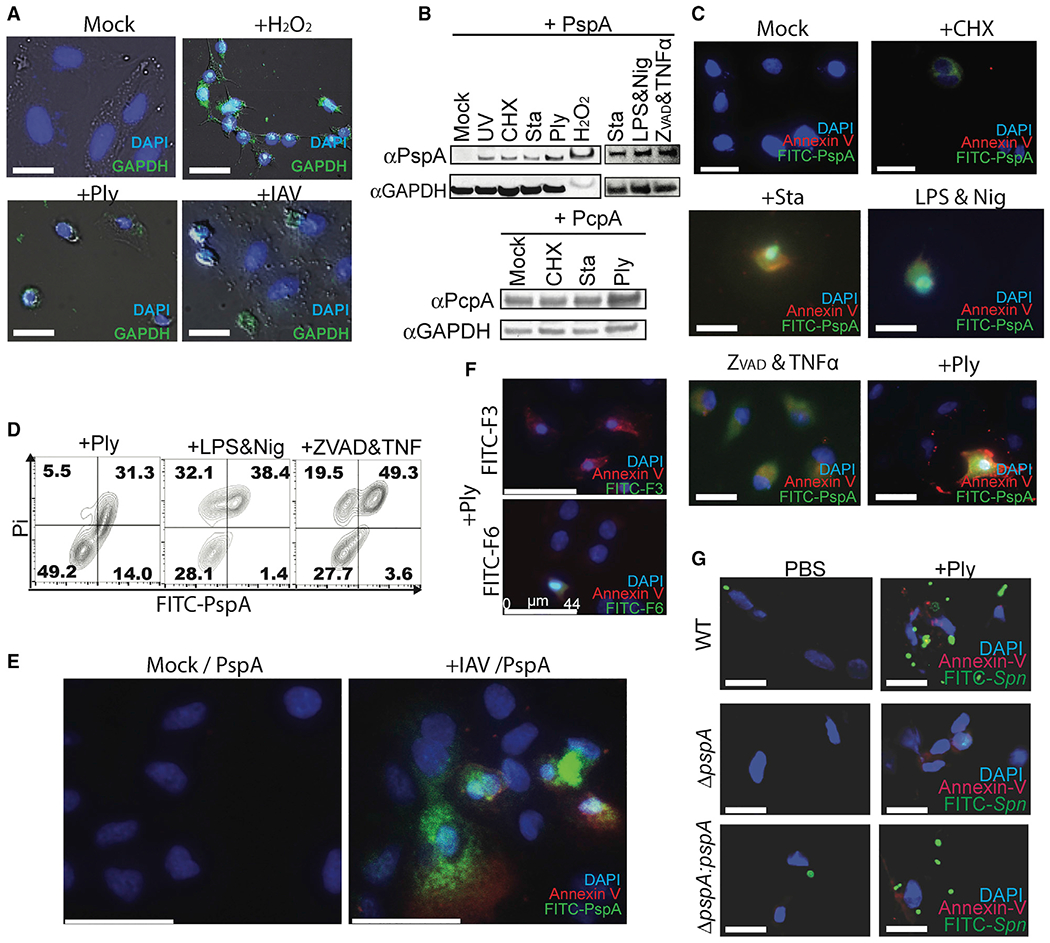
Spn binds to dying cells via PspA-GAPDH interaction
(A) A549 cells were incubated with H2O2, rPly, or influenza A virus (PR8, MOI 5). Surface-exposed GAPDH on unfixed cells was detected using mouse monoclonal anti-GAPDH antibody (green).
(B and C) Purified His6-PspA or PcpA proteins were incubated with healthy or dying cells (UV exposure; cycloheximide ; staurosporine; rPly; H2O2; lipopolysaccharide and nigericin; and zVAD-fmk, cycloheximide, and TNF-α). Bound PspA or PcpA were detected by (B) immunoblot and (C) fluorescent microscopy.
(D) Healthy or dying A549 cells were incubated with FITC-PspA. Adhered FITC-PspA to cells was analyzed by flow cytometry.
(E) A549 cells were infected with or without influenza A virus (PR8) and subsequently incubated with FITC-labeled PspA and Annexin V, then detected by fluorescent microscopy.
(F) Spn-infected A549 cells were incubated with F-3 or F-6, and Annexin V and PspA fragments bound to cells were detected by fluorescent microscopy.
(G) FITC-labeled Spn EF3030 (wild type [WT], pspA mutant [ΔpspA], or pspA revertant [ΔpspA:pspA]) were incubated with healthy or rPly-induced dying cells, and Spn adhesion was determined by fluorescent microscopy.
Data shown are representative of three independent experiments. Scale bars: 25 μm (A, C, and G); 44 μm (E and F).
PspA modulates IAV/Spn superinfection
We next examined the effects of potential PspA-GAPDH interactions within the airway. Adult C57BL/6 mice were challenged intratracheally with rPly. The next day, mice were treated with FITC-labeled derivatives of rPspA. We observed extensive cell death in pneumolysin-treated lungs and colocalization of FITC-rPspA with lung cells undergoing necroptosis, i.e., p-MLKL positive (Figure 5A). Using the same model, we observed that WT Spn and pspA revertant, but not an isogenic pspA mutant, were attached to necroptotic bronchial epithelial cells 1 h after intratracheal challenge (Figure 5B). Notably, PspA-dependent binding was also observed, albeit at starkly reduced rates, in control mice pretreated instead with PBS (Figure S6). In this instance, lung injury at the time of sample collection was far less than in rPly-treated animals. Thus, pneumolysin-mediated cell death within the airway enhanced PspA-mediated binding of Spn to host cells, and the extent to which this occurred seemed to be damage dependent.
Figure 5. Spn binds to dying cell on mouse lung.
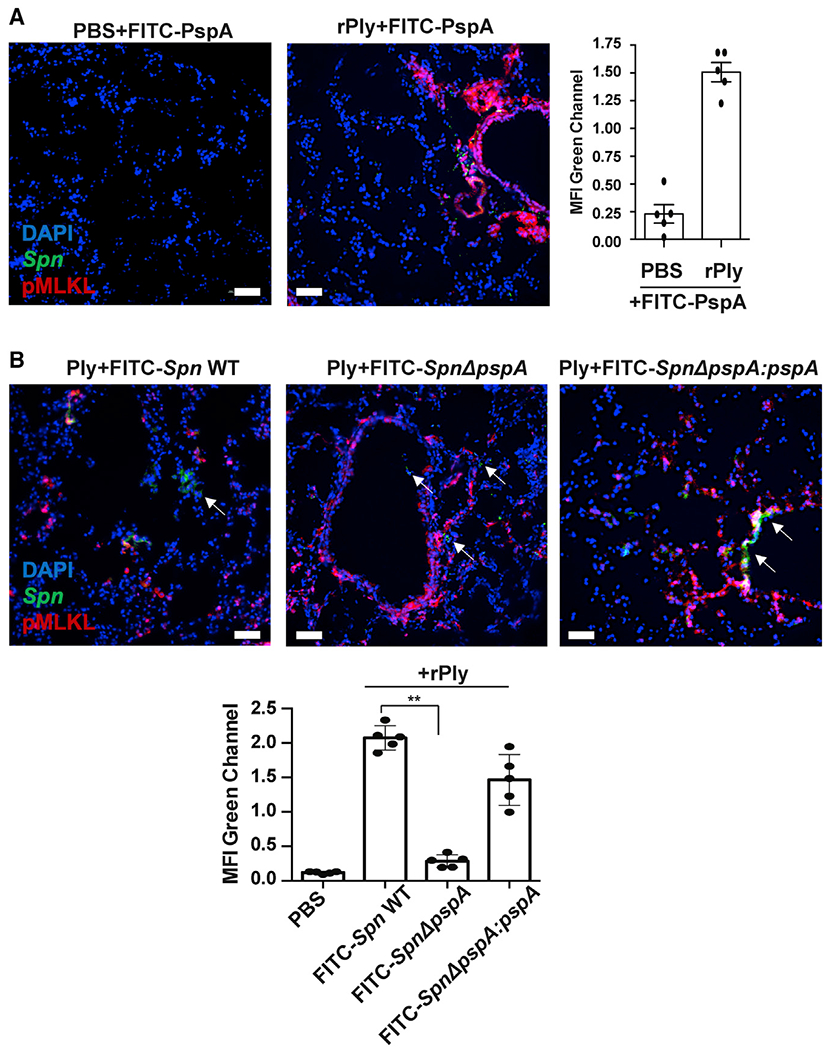
Mice were treated intratracheally with rPly (160 ng/mouse) or PBS. After 24 h, mice were challenged intratracheally with (A) FITC-rPspA or(B) 106 CFUs of FITC-labeled Spn EF3030 (WT, pspA mutant [ΔpspA], or pspA revertant [ΔpspA;pspA]) and then euthanized after 1 h (n = 5 per cohort). Lungs were collected and co-stained with anti-pMLKL (red) and DAPI (blue). White arrows indicate Spn. Mean fluorescence intensity (MFI) of the FITC-labeled rPspA (green) or Spn (green) was measured using ImageJ. Kruskal-Wallis test with Dunn’s multiple-comparison post-test. *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001. Scale bars: 50 μm for all microscopic images.
Immediately following aspiration in a healthy host, pneumococci do not normally first encounter dying lung epithelial cells. However, during a prior infection with IAV, any pneumococci entering the lung or upper airway encounter a high percentage of dying host cells. We observed that rPspA readily binds to necroptotic/IAV-infected cells in the mouse lungs (Figure 6A). In lung sections from IAV-infected mice that had also been treated with FITC-rPspA, FITC-F-3, or FITC-F-6, only FITC-rPspA and FITC-F-6 showed evidence of binding, and this was exclusive to influenza-induced necroptosis-positive cells (Figure 6A). We also observed that IAV enhanced binding of WT, but not PspA-mutant/deficient live pneumococci (Figure 6B). In vitro, addition of F-6 or GAPDH70, a GAPDH phosphatidylserine binding peptide, each reduced PspA-GAPDH interactions on the surface of UV-treated A549 cells by ~50%. If F-6 and GAPDH70 were given together, they reduced PspA binding to baseline levels (Figure 6C). In IAV-infected mice, suspension of bacteria in saline with F-6 or F-6 with GAPDH70 priorto its intratracheal instillation, but not GAPDH70 alone, resulted in a 20-fold reduction in the number of bacteria in the airway the next day. In this instance, an additive effect for F-6 and GAPDH70 was not observed (Figure 6D). Thus, F-6 alone, but not this fragment of GAPDH alone, had a therapeutic effect in mice.
Figure 6.
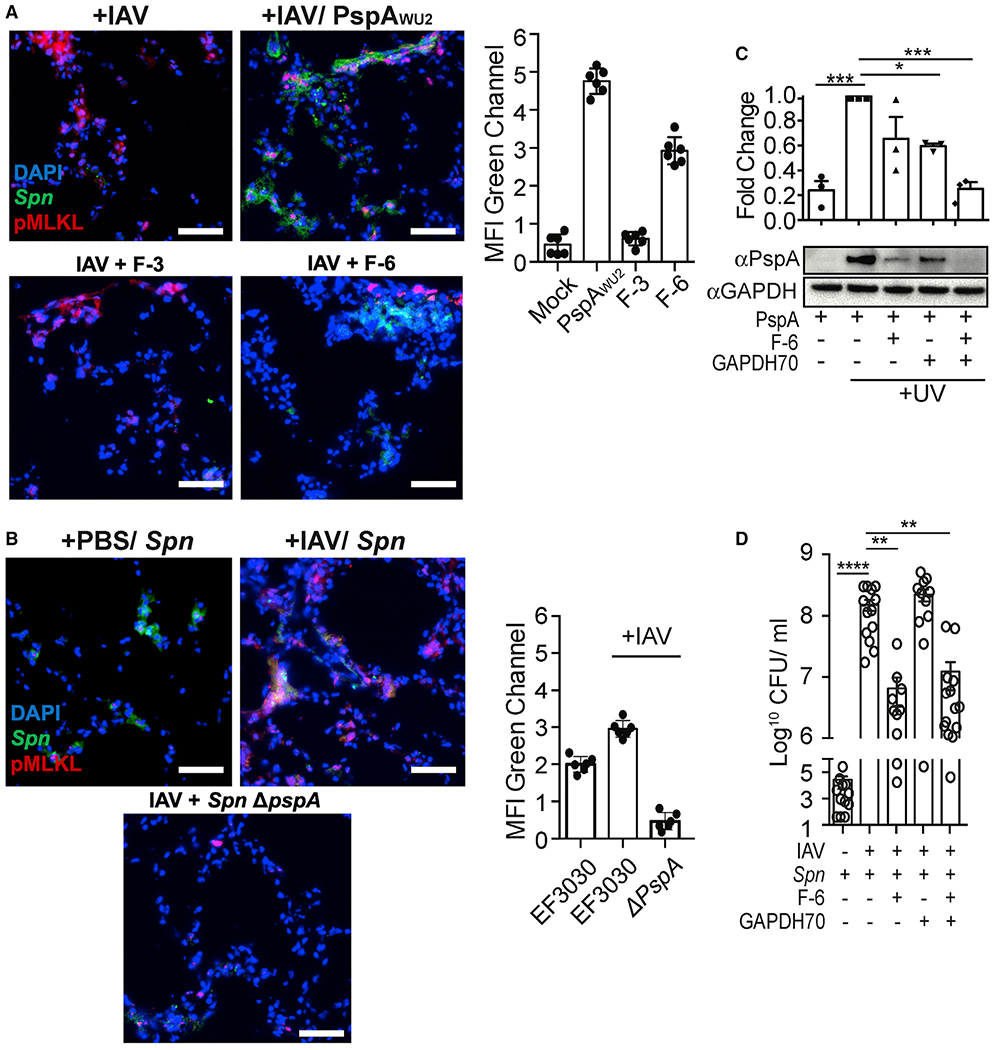
Spn binds influenza-mediated dying cells and induces pneumococcal superinfection
(A and B) Mice were challenged intratracheally with 200 PFUs of IAV (PR8) or saline (PBS) for 5 days and subsequently challenged intratracheally with (A) FITC-labeled whole rPspA, F3, and F6, or (B) infected with 106 CFUs of FITC-labeled Spn EF3030 (WT) or pspA isogenic mutant (Spn ΔpspA). One day after secondary challenge, lungs were collected andco-stained with anti-pMLKL (red) and DAPI (blue). MFI of the FITC-labeled (green) rPspA or adhered Spn was measured using ImageJ. Experiments were performed three times.
(C) UV-treated dying cells were incubated with GAPDH70 and subsequently with rPspA or F-6, and cell-bound PspA was assessed by immunoblot.
(D) Seven days following infection with IAV or control treated with PBS, mice were infected intratracheally with 107 CFUs of Spn EF3030 that had been mixed in suspension with F-6, GAPDH70, or F-6 with GAPDH70. The next day, Spn titers in whole-lung homogenates were determined. Open circles represent individual mice used for each experiment.
Kruskal-Wallis test with Dunn’s multiple-comparison post-test. *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.001. Scale bar: 50 μm for all microscopic images.
DISCUSSION
Although many different types of virus and bacteria can co-infect the airway, the morbidity and mortality associated with IAV/Spn superinfection far exceeds that of other well-studied combinations (McCullers, 2006). It is for this reason we expected that the virulence determinants particular to these pathogens might synergistically complement one other. Along such lines, we have recently discovered that IAV profoundly enhances the susceptibility of lung epithelial cells for pneumolysin-mediated necroptosis (Gonzalez-Juarbe et al., 2020). Building upon this, herein we have demonstrated that PspA mediates bacterial binding to dying lung epithelial cells, and this contributes to the severity of IAV/Spn superinfection. Importantly, and despite >30 years since its discovery, PspA has not previously been shown to function as an adhesin. Thus, our finding of PspA’s role in adherence substantially advances our knowledge on the interactions of Spn with its host.
IAV primes the airway for Spn infection by various means. Free sialic acid, generated by the IAV neuraminidase, is a nutrient for Spn and an environmental cue that enhances its virulence and propensity to invade tissues (Hatcher et al., 2016; Siegel et al., 2014). The removal of sialic acid from the host cell surface exposes otherwise cryptic ligands that Spn uses for attachment (Tong et al., 2002). IAV infection has been shown to inhibit ion transport in bronchial lung epithelial cells. This causes a change in the pH and viscosity of mucus resulting in reduced antimicrobial peptide efficacy and ineffective ciliary clearance (Brand et al., 2018). IAV infection also has other profound immunological consequences that worsen Spn disease. For example, IAV-induced interferon-gamma results in downregulation of MARCO, a macrophage scavenger receptor that is critical in defense of the airways against encapsulated bacteria (Sun and Metzger, 2008). These and other sensitizing events all occur simultaneously and help to explain why pneumonia as result of superinfection can be so severe. They also explain why PspA deficiency alone does not completely abrogate the exacerbated disease observed in our co-infection model.
PspA is found in all clinical isolates of Spn and is expressed across a variety of anatomical sites within the host (Shenoy et al., 2017). We have previously shown that PspA inhibits binding of C-reactive protein on the surface of the bacterium (Mukerji et al., 2012) and PspA protects Spn from killing by the antimicrobial peptides of lactoferrin (Shaper et al., 2004), and we now recognize that PspA also functions as an adhesin. These properties, along with the fact that immunization of mice with recombinant PspA elicits antibodies that are protective against normally lethal Spn challenge (Greene et al., 2016; Roberts et al., 2019), explain why and demonstrate that PspA is vital to the bacterium. IAV and Spn are each capable of inducing programmed cell death by various means. In such instances, and as demonstrated, host GAPDH binds to phosphatidylserine residues that have been flipped to the outer cell membrane, and these now become targets for PspA-mediated adhesion. In vivo, GAPDH would presumably be released by host cells that have already died. Importantly, our results showed that endogenous GAPDH was present on the surface of cells that were dying as a result of pharmacologically induced apoptosis, pyroptosis, or necroptosis. GAPDH was also detectable on the surface of cells infected by IAV and those challenged with pneumolysin. These findings, along with the recently recognized fact that IAV sensitizes cells for pneumolysin-mediated necroptosis (Gonzalez-Juarbe et al., 2020), suggest that IAV and pneumolysin independently, but especially together, set the stage for PspA-mediated adhesion via induction of cell death. Alternatively, there are complementary roles for pneumolysin and PspA, which are amplified by IAV. These relationships help to explain why immunity to the two proteins has synergetic protective effects against intraperitoneal (i.p.) and lung challenge with Spn (Briles et al., 2003; Ogunniyi et al., 2000).
PspA binds to both huGAPDH and mouse GAPDH; these have 94% aa sequence identity (Zheng et al., 2014). GAPDH consists of two highly conserved domains: an NAD+ binding domain (aa 1–150) and the catalytic glyceraldehyde-3-phosphate domain (aa 151–335). The NAD+ binding domain contains the phosphatidylserine binding motif (aa 70–94), glutathione binding motif (aa 67–77), and an RNA binding domain (respective aa responsible are unknown). The catalytic domain has a nuclear export domain (aa 258–327). We would predict that PspA binds the catalytic glyceraldehyde-3-phosphate domain of GAPDH, because the NAD+ binding domain contains the phosphatidylserine binding motif and therefore would be facing inward to the cell and unexposed. Importantly, GAPDH also normally forms a tetramer. Future studies are warranted to identify the domain of GAPDH bound by PspA, and whether PspA binds to individual GAPDH molecules or the GAPDH tetramer.
It is notable that GAPDH binds to lactoferrin; the latter has been demonstrated to be a mechanism by which host cells acquire iron (Chauhan et al., 2015). Lactoferrin can be cleaved by a protease into bactericidal peptides that are amphipathic and cationic (Gifford et al., 2005). Because PspA both binds GAPDH and protects against killing by lactoferricin, it now seems highly unlikely that these roles for PspA are not linked. PspA’s protection against lactoferricin was observed to occur in the absence of host GAPDH. So, this property is independent of GAPDH, albeit the responsible domains on PspA are in proximity within the αHD. It may be that Spn uses GAPDH to capture lactoferrin and therefore iron. Alternatively, because GAPDH is negatively charged, it may serve as a sink for lactoferricin that is initially captured by PspA. These activities may or may not be independent of PspA’s simultaneous ability to mediate bacterial attachment.
The ability of PspA to mediate bacterial attachment to dying cells is perhaps the most straightforward explanation for how it contributes to enhanced pneumonia severity. We speculate that in addition to helping the bacteria avoid mucociliary clearance, the close physical interaction of Spn with dying lung cells also serves to enhance the bacterium’s accessibility to any released nutrients or host proteins that could be co-opted by Spn (Park et al., 2021; Sender et al., 2020). These would otherwise be diluted within the mucosal secretions. This adhesive property of PspA is also most likely an explanation for the increased susceptibility of individuals with pre-existing lung damage, not IAV-induced, for pneumococcal pneumonia. Most obvious are cigarette smokers, who routinely experience bronchial epithelial damage and are recognized to be at elevated risk for pneumococcal infection. The specificity of PspA for dying cells also suggests that this property confers benefit only once a certain level of airway damage has been incurred. We propose this is why PspA’s role as an adhesin has not been previously described. Most investigators, including ourselves, typically use healthy cell monolayers for adhesion experiments.
Instillation of influenza-infected mice with a recombinant PspA fragment containing the GAPDH binding motif, i.e., F-6, reduced disease severity. This presumably occurred as a result of F-6 competitive inhibition of Spn binding to cells via PspA/GAPDH. Whether antibodies against the same domain confer protection is an important question. Antibodies against pneumolysin have been shown to reduce disease severity, albeit not prevent overall disease (García-Suárez et al., 2004; Musher et al., 2001). Prior work has shown that injection of pneumolysin toxoid covalently linked at the N terminus with sections of PspC (also known as CbpA) that mediate bacterial translocation across vascular endothelial cells conferred protection against bacterial invasion of the central nervous system and heart during bacteremia (Brown et al., 2014; Chen et al., 2015). We propose that an expanded version containing the 230–281 aa GAPDH binding motif of PspA on the carboxy terminus may be a means to add to the protective benefits of this immunizing peptide.
In conclusion, we identified a new molecular mechanism behind PspA’s promotion of Spn virulence in hosts with pulmonary damage and during secondary bacterial infections to influenza. Our findings support the targeting of regions of PspA for therapeutic and vaccine development against IAV/Spn superinfections.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to Dr. Carlos J. Orihuela (corihuel@uab.edu).
Materials availability
Bacterial strains and new recombinant DNA constructs are available upon request.
Data and code availability
This study did not generate/analyze datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacteria culture
All Spn strains used in this study are listed in the Key resources table. Spn was cultured on blood agar plates at 37°C for 24 h in candle jar for anaerobic. Colonies were inoculated into Todd-Hewitt-broth with 0.5% yeast extract and grown until exponential phase (OD621 = 0.3 ~1 X 108 CFU/ ml). Isogenic mutants were cultivated using appropriate antibiotics. E. coli strains were cultured in Luria-Bertani (Aki et al., 1997) broth supplemented with ampicillin.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-PspA (1b2.21) | (Genschmer et al., 2013) | N/A |
| Anti-PcpA | (Glover et al., 2008) | N/A |
| Annexin V, APC conjugate | Invitrogen™ | Cat#A35110 |
| Anti-pMLKL | Abcam | ab196436; RRID:AB_2687465 |
| Mouse Anti-GAPDH | Abcam | Cat#Ab8245; RRID:AB_2107448 |
| Rabbit Anti-GAPDH | Abcam | Cat#Ab181602; RRID:AB_2630358 |
| Bacterial and virus strains | ||
| S. pneumoniae strain WU2 and EF3030 | David E Briles’s lab, University of Alabama at Birmingham | N/A |
| Staphylococcus aureus | Orihuela Lab, University of Alabama at Birmingham | PMC4676650 |
| Uropathogenic E. coli | Orihuela Lab, University of Alabama at Birmingham | PMC4676650 |
| Streptococcus mutants UA159 | Dr. Hui Wu Lab, University of Alabama at Birmingham | PMC6763767 |
| NEB®Express Iq Competent E. coli | NEB | Cat#C3037I |
| E. coli strain BL21(DE3) | Novagen | Cat#69450-4 |
| E. coli strain NovaBlue | Novagen | Cat#70181-3 |
| Influenza A virus (A/PR/8/34, PR8) | ATCC | ATCC® VR-95 |
| Chemicals, peptides, and recombinant proteins | ||
| FluoroTag FITC Conjugation Kit | Sigma-Aldrich | Cat#FITC1 |
| InSolution Staurosporine, Streptomyces sp. | Sigma-Aldrich | Cat#569396 |
| Lipopolysaccharides from Escherichia coli O111:B4 | Sigma-Aldrich | Cat#L2630 |
| Nigericin sodium salt Ready Made Solution | Sigma-Aldrich | Cat#SML1779 |
| Caspase inhibitor I - CAS 187389-52-2-Calbiochem | Sigma-Aldrich | Cat#627610 |
| TNF-a | Sigma-Aldrich | Cat#H8916 |
| RIPA buffer | Sigma-Aldrich | Cat#R0277 |
| Cycloheximide | Sigma-Aldrich | Cat#01810 |
| Propidium iodide | ThermoFisher | P1304MP |
| GAPDH70: N’- KPITIFQERDPVKIKWG DAGAEYVC - C’ | (Kaneda et al., 1997) | N/A |
| CSP-1:competence stimulating peptide −1 | AnaSpec Inc. | Cat#AS-63779 |
| Recombinant human GAPDH | Abcam | Cat#ab82633 |
| Experimental models: cell lines | ||
| A549 | ATCC | ATCC® CCL-185 |
| D562 | ATCC | ATCC® CCL-138 |
| Experimental models: organisms/strains | ||
| C57BL/6 mice, adult, female | Jackson labs | C57BL/6 |
| Oligonucleotides | ||
| Primer for PspA and PspA fragments, see Table S2 | This study | N/A |
| Primer for S. pneumoniae mutation, see Table S2 | This study | N/A |
| Primer for S. pneumoniae GAPDH expression, see Table S2 | This study | N/A |
| Recombinant DNA | ||
| pQE30 | QIAGEN | Cat#32915 |
| pET32b | MilliporeSigma | Cat#690163 |
| pDL276 | (Dunny et al., 1991) | N/A |
| pCR2.1:ermBR | (Embry et al., 2007) | N/A |
| pCR2.1:kanR | This study | N/A |
| pET30-2-GAPDH | (Pacold et al., 2016) | Addgene (Cat#83910) |
| pET32b-spGAPDH | This study | N/A |
| pQE30-SS01 | This study | N/A |
| pQE30-SS02 | This study | N/A |
| pQE30-SS04 | This study | N/A |
| pQE30-SS10 | This study | N/A |
| pQE30-SS11 | This study | N/A |
| pQE30-SS12 | This study | N/A |
| pQE30-SS13 | This study | N/A |
| pQE30-SS14 | This study | N/A |
| pQE30-SS16 | This study | N/A |
| pQE30-SS17 | This study | N/A |
| Software and algorithms | ||
| Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | Becton, Dickinson & Company | https://www.flowjo.com |
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
Cell lines
Low passage, A549 human type II alveolar epithelial cells (Cat #CCL-185), and MDCK canine kidney cells (#Cat CCL-34) were purchased from ATCC (Manassas, VA). For passage and maintenance, cells were cultured in F12 (GIBCO) or EMEM (ATCC) media), respectively, supplemented with 10% FBS (R&D systems) and Antibiotic-Antimycotic (GIBCO). Cells were not used beyond passage 30.
Ethics statement
Animal experiments were performed in compliance with the federal regulations set forth in the Animal Welfare Act, the recommendations in the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the University of Alabama at Birmingham Institutional Animal Use and Care Committee (IACUC). All protocols used in this study were approved by the IACUC at the University of Alabama at Birmingham (protocols #20090 and #22157).
Animals
Female 8-12-week-old C57BL/6 mice were obtained from The Jackson Laboratories (Maine, US) and allowed to acclimate for one week before experimental challenge. At all times mice received food and water ad libitum.
METHOD DETAILS
Bacteria cloning and plasmids
All plasmid and primer information are listed in Key resources table and Table S2. Isogenic Spn mutants deficient in pspA were generated by insertion of an erythromycin or kanamycin resistance cassette using homologous recombination method. For each PspA locus mutation, DNA flanking regions of pspA were amplified by PCR (primers listed in Table S2), restriction enzyme digested, and cloned upstream and downstream, respectively, of either ermB or kanR in pCR2.1. Purified plasmid isolated from transformed Escherichia coli was subsequently used to amplify the mutagenic PCR constructs and transform Spn made competent with competence stimulating peptide (CSP-1 or CSP-2) (Bricker and Camilli, 1999). Mutagenic Spn were selected on blood agar plates containing the corresponding antibiotic with isolated mutants confirmed by PCR amplification and sequencing of pspA locus. The PspA and PspA fragments were amplified (primers in Table S2) and inserted into pQE30 or pET32b vectors for purification of proteins.
In vitro influenza virus infection
Influenza A H1N1 A/Puerto Rico/8/1934 (PR8) stocks were propagated in MDCK cells and subsequently titered by plaque assay in MDCK cells to determine plaque forming units (PFUs). A549 cells were infected with PR8 at a MOI of 5 for 4 days then treated FITC-PspA for 30 min and then staining dying cell by Annexin-V, or treated anti-GAPDH and then added FITC-conjugated secondary antibody for fluorescent microscopy.
Induction of cell death and assessment of cell viability
A549 cells were cultured in DMEM with 10% FBS and challenged with apoptosis insults H2O2 (5 mM) for 2 h or UV (1,000 mJ/cm2) for 2 h. For pyroptosis cells were treated with lipopolysaccharide (10 ng/mL) for 4 h then nigericin (10 μM) for 6 h. For induction of necroptosis cells were administered rPly (0.1 μg/mL) for 4 h or zVAD-fmk (20 μM) and cycloheximide (1 μg/ml) for 1 hour before addition of TNF (100 ng/ml) (Coats et al., 2005). Host cell cytotoxicity of recombinant PspA and Ply were evaluated by WST-1 solution according to the manufacturer’s recommendations (Roche, Germany). In other instances, cell viability was measured using trypan blue staining method according to the manufacturer’s instructions (ThermoFisher, USA).
Adhesion assay
Pneumococcus adhesion to A549 cells was assessed using standard methods. Briefly, heathy or dying A549 cells pellet were washed in PBS and then incubated with FITC-labeled Spn (MOI of 10:1) for 30 min after which they were washed once again and analyzed using flow cytometry (BD, AccuriTHC6 Flow Cytometer).
Ni-NTA pull-down assay
Cultured cells (A549 and D562) and bacteria (E. coli or Spn WU2) were lysed by sonication in RIPA buffer containing 1X protease inhibitors (Sigma). Cell lysates (20 μg), bacterial lysates (20 μg) or 2 μg of recombinant huGAPDH were mixed with 2 μg of 6His-PspA or 6His-PspA fragments on buffer A (PBS, 0.1% Triton X-100, 0.5 mM DTT and 10 mM imidazole) and then incubated for 1hourat 4°C on rotator (6 rpm). Ni-NTA resin (20 μl) was equilibrated in buffer A. Subsequently, Ni-NTA resin and mixtures of proteins were mixed together and incubated for 1 hour at 4°C on a rotator (6 rpm). Incubated Ni-NTA resins were washed four times with 1 mL of buffer A containing 20 mM imidazole at 2,000 X g. 2X SDS loading buffer was added to the Ni-NTA resins and then incubated at 90°C for 5 min. Proteins were separated on 4%–12% gradient SDS-PAGE (Invitrogen) and visualized by Coomassie blue staining or immunoblots. For protein identification, the Coomassie blue stained bands were cut out of the gel and digested with trypsin overnight. Elution of the peptides were identified using LC-MS at the Cancer Center at University of Alabama at Birmingham, USA.
Protein purification
Recombinant proteins PspA, PspA fragments, Ply, human GAPDH (huGAPDH) and Spn GAPDH (SpGAPDH) were expressed in E. coli BL21 and purified by cobalt resin. The his- and trx-tags on SpGAPDH were removed by enterokinase and cobalt resin. Purified proteins were loaded on the Superdex S200 or S75 columns. Collected protein fractions were checked on SDS-PAGE and concentrated by centrifugal concentrator tube. Protein concentration was measured by UV 280nm with aliquots stored in Dulbecco’s Phosphate buffered Saline (PBS) with 20% glycerol at −70°C.
Fluorescein isothiocyanate conjugation
Purified proteins or Spn were conjugated by FluoroTag FITC Conjugation Kit (Sigma) following the manufacturer’s recommendation. Briefly, proteins or Spn were incubated FITC for 120 min or 30 min, respectively. Conjugated proteins were dialyzed with 20% glycerol in PBS and stored on −70°C. Spn were washed using PBS and centrifugation twice. Bacteria were resuspended in PBS with 10% glycerol and stored on −70°C.
Surface plasmon resonance analysis (SPR)
SPR experiments were run on Biacore 2000 (GE healthcare) at room temperature with PBS as running buffer. huGAPDH was diluted to 50 μg/ml in 10 mM NaOAc pH 6.0 and immobilized on CM5 chips. Analytes were injected at 5 μl/min and surface regenerated using 1M NaCl with 10mM EDTA. Data for injections of PspA over huGAPDH were fitted to a Langmuir 1:1 interaction.
Peptide synthesis
The synthetic peptide GAPDH70 (Aki et al., 1997) (KPITIFQERDPVKIKWGDAGAEYVC) was manufactured and purified to a purity > 95% by LifeTein (USA).
Flow cytometry
Spn was cultured OD600 0.3 (~1.0 X 108 CFU/ml) and harvested using centrifugation (3,500 g, 10 min). Spn was washed and resuspended in FACS buffer (PBS, 3% FBS and 0.1% sodium azide). Supernatant of Spn cultures were diluted in FACS buffer to approximately 5.0 X 106 CFU/ml. Then 50 μL of cell solution was incubated on ice for 30 min with 0.5 μg of FITC-labeled huGAPDH, washed and analyzed using flow cytometry (BD, AccuriTHC6 Flow Cytometer).
Dying A549 cells were harvested by cell dissociation solution (Sigma, USA). The Cell pellets were washed three times with PBS and incubated at room temperature for 30 min with 50 μg / ml of FITC-PspA and necrotic or late apoptosis cell death was stained by propidium iodide (PI) (ImmunoChemistry Technologies, USA) according to manufacturer’s instructions. Incubated cells were analyzed using flow cytometry (BD, AccuriTHC6 Flow Cytometer).
Fluorescent microscopy
The healthy or dying cells were incubated with FITC-PspA, PspA fragments or FITC-Spn for 30 min. Dying cells were stained using annexin-V staining according to manufacturer’s instructions (Thermo Fisher). Cells were fixed with 4% paraformaldehyde (PFA) in PBS for 10 min. Surface exposed GAPDH was stained using mouse anti-GAPDH antibody (Abcam) and then washed three times with PBS between the primary- and secondary-antibody incubation. As a negative control, FITC-labeled human serum albumin (Abcam, #8030) was used to evaluate non-specific binding of a host protein to healthy and dying cells in vitro and in the lungs. Cell nucleus was stained using 4,6-diamidino-2-phenylindole (DAPI; Thermo Fisher). Images were acquired using a Leica LMD6.
Immunoblotting
For assessment of PspA production following exposure to IAV, Spn EF3030 was grown to early exponential phase in chemically defined media (planktonic) for 6h and as a 24h (biofilm) in a polystyrene 6-well plate. IAV was added to a concentration of 109 PFU/mL and bacteria incubated for an additional 2h at 37 ° C with 5% CO2. Spn were collected and centrifuged at 7,000 g for 10 min. Cells were resuspend and lysed by RIPA buffer (Sigma) with 1X protease inhibitors. Total protein was separated from cell debris by centrifugation at 15,000 g for 30 min. Protein concentrations were measured by BCA assay (Bio-Rad). Lysates were boiled for 5 min at 95°C with NuPAGE LDS sample buffer and electrophoresed in 4%–12% Bis-Tris gel (Invitrogen). Subsequently, the proteins were transferred to phenylmethylsulfonyl fluoride membrane and blocked with 3% bovine serum albumin (BSA) in PBS with 0.1% Tween 20 (sigma). Expressed PspA were detected by using mouse anti-PspA (Walker et al., 2016) and anti-mouse IgG conjugated HRP used as secondary antibodies. Images were obtained by using a ChemiDoc (Bio-Rad). For experiments examining PspA adhesion to heathy or dying host cells, A549 cells were incubated with 40 μg/ ml of PspA or PcpA for 30 min in DMEM with 10% FBS and washed by PBS for 3 times. To measure PspA-binding inhibition of F-6 and GAPDH 70, GAPDH 70 (1.25 mg / ml) were pre-incubation after UV exposer and F-6 (100 μg/ ml) incubated with PspA. Cells were lysed by RIPA buffer (Sigma) containing 1X protease inhibitors. Total protein was separated from cell debris by centrifugation at 15,000 g for 30 min. Protein concentrations were measured by BCA assay (Bio-Rad). Lysates were boiled for 5 min at 95°C with NuPAGE LDS sample buffer and electrophoresed in 4%–12% Bis-Tris gel (Invitrogen). Subsequently, the proteins were transferred to phenylmethylsulfonyl fluoride membrane and blocked with 3% bovine serum albumin (BSA) in PBS with 0.1% Tween 20 (Sigma). Target proteins were detected by using each antibody: mouse anti-PspA (Walker et al., 2016), rabbit- anti-GAPDH (Abcam) and mouse anti-PcpA (Novicket al., 2017). Anti-rabbit IgG or anti-mouse IgG conjugated to alkaline phosphatase or HRP were used as secondary antibodies. Images were obtained by using a ChemiDoc (Bio-Rad).
Pulmonary injury and pneumonia induction
To induce lung cell damage, mice were administered 160 ng of recombinant Ply in 100 μl saline via intratracheal instillation. After 24 hours, mice were subsequently challenged with 3 μg of FITC-PspA, FITC-F-3, FITC-F-6 or FITC-conjugated Spn (1.0 X 106 CFU) via intratracheal route for 1 hour before euthanasia. Subsequently, nasal washes or lung tissue were collected for flow cytometry or fluorescent microscopy, respectively. For the influenza mice model, C57BL/6 mice were infected via intranasal route with 200 PFU of PR8 in 50 μL PBS or 50 μL normal PBS for control. On day 5 post-influenza or PBS inoculation, mice were challenged with 3 μg of FITC-PspA, FITC-F-3, FITC-F-6 or FITC-conjugated Spn (1.0 X 106 CFU) via forced inhalation. An hour later mice were sacrificed and tissue was collected for flow cytometry or fluorescent microscopy, respectively. For experiments involving secondary bacterial infection to influenza, C57BL/6 mice were infected via intranasal route with 200 PFU of PR8 in 50 μL PBS or 50 μL normal PBS for control. On day 7 post-influenza or PBS inoculation, mice were infected via intratracheal route with 1.0 X 107 CFU of Spn EF3030 or EF3030 ΔpspA with 40 μg of F-3 or F-6, 20 μg GAPDH70 with or without of 40 μg F-6 in 40 μL PBS. In some instances, whole lungs or BALF were collected from IAV/Spn infected mice as described (Gonzalez-Juarbe et al., 2020). CFU in these samples were enumerated by serial dilution and plating of lung homogenates or BALF, respectively, and extrapolation from colony counts. For experiments examining duration of survival, mice were monitored at 6h intervals and euthanized when deemed moribund.
QUANTIFICATION AND STATISTICAL ANALYSIS
For multiple group analyses, we used a nonparametric Kruskal-Wallis H test with Dunn’s post hoc analysis; grouped analyses were performed using a Two-Way ANOVA with Sidak’s post hoc analysis. For data with a single independent factor of two groups, we used a Mann-Whitney U test. For survival curve analysis, a Gehan-Breslow-Wilcoxon test was used. The number of biological and technical replicates for each experiment, as appropriate, are provided within the figure legend. Error bars on all main and supplemental figures indicate standard deviation. Statistical analyses were performed using Prism 8 (GraphPad Software: La Jolla, CA), with p values noted within the figure panels and legends.
Supplementary Material
Highlights.
Pneumococcal surface protein A (PspA) functions as an adhesin
PspA binds host GAPDH, present on the surface of dying host cells
PspA influences localization of Streptococcus pneumoniae in the airway
PspA effects are exacerbated by damage caused by influenza A virus infection
ACKNOWLEDGMENTS
N.G.-J. and A.N.R. were supported by National Institutes of Health (NIH) Immunologic Diseases and Basic Immunology grant 5T32AI007051-38. N.G-J. received support from J. Craig Venter Institute Start-up funds. D.E.B. received support from NIH grant AI118805. C.J.O. receives support from NIH grants AI156898, AI146149, AI148368, and AI114800 and also from AHA grant 16GRNT30230007.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109267.
DECLARATION OF INTERESTS
S.-S.P., N.G.-J., D.E.B., and C.J.O. hold a provisional patent on PspA.
REFERENCES
- Aki T, Yanagisawa S, and Akanuma H (1997). Identification and characterization of positive regulatory elements in the human glyceraldehyde 3-phosphate dehydrogenase gene promoter. J. Biochem. 122, 271–278. [DOI] [PubMed] [Google Scholar]
- Andre GO, Converso TR, Politano WR, Ferraz LF, Ribeiro ML, Leite LC, and Darrieux M (2017). Role of Streptococcus pneumoniae Proteins in Evasion of Complement-Mediated Immunity. Front. Microbiol. 8, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand JD, Lazrak A, Trombley JE, Shei RJ, Adewale AT, Tipper JL, Yu Z, Ashtekar AR, Rowe SM, Matalon S, and Harrod KS (2018). Influenza-mediated reduction of lung epithelial ion channel activity leads to dysregulated pulmonary fluid homeostasis. JCI Insight 3, 123467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker AL, and Camilli A (1999). Transformation of a type 4 encapsulated strain of Streptococcus pneumoniae. FEMS Microbiol. Lett. 172, 131–135. [DOI] [PubMed] [Google Scholar]
- Briles DE, Hollingshead SK, Paton JC, Ades EW, Novak L, van Ginkel FW, and Benjamin WH Jr. (2003). Immunizations with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J. Infect. Dis. 188, 339–348. [DOI] [PubMed] [Google Scholar]
- Brissac T, and Orihuela CJ (2019). In Vitro Adhesion, Invasion, and Transcytosis of Streptococcus pneumoniae with Host Cells. Methods Mol. Biol. 1968, 137–146. [DOI] [PubMed] [Google Scholar]
- Brown AO, Mann B, Gao G, Hankins JS, Humann J, Giardina J, Faverio P, Restrepo MI, Halade GV, Mortensen EM, et al. (2014). Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan AS, Rawat P, Malhotra H, Sheokand N, Kumar M, Patidar A, Chaudhary S, Jakhar P, Raje CI, and Raje M (2015). Secreted multifunctional Glyceraldehyde-3-phosphate dehydrogenase sequesters lactoferrin and iron into cells via a non-canonical pathway. Sci. Rep. 5, 18465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Mann B, Gao G, Heath R, King J, Maissoneuve J, Alderson M, Tate A, Hollingshead SK, Tweten RK, et al. (2015). Multivalent Pneumococcal Protein Vaccines Comprising Pneumolysoid with Epitopes/Fragments of CbpA and/or PspA Elicit Strong and Broad Protection. Clin. Vaccine Immunol. 22, 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coats MT, Benjamin WH, Hollingshead SK, and Briles DE (2005). Antibodies to the pneumococcal surface protein A, PspA, can be produced in splenectomized and can protect splenectomized mice from infection with Streptococcus pneumoniae. Vaccine 23, 4257–4262. [DOI] [PubMed] [Google Scholar]
- Crain MJ, Waltman WD 2nd, Turner JS, Yother J, Talkington DF, McDaniel LS, Gray BM, and Briles DE (1990). Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect. Immun. 58, 3293–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didierlaurent A, Goulding J, and Hussell T (2007).The impact of successive infections on the lung microenvironment. Immunology 122, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunny GM, Lee LN, and LeBlanc DJ (1991). Improved electroporation and cloning vector system for gram-positive bacteria. Appl. Environ. Microbiol. 57, 1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embry A, Hinojosa E, and Orihuela CJ (2007). Regions of Diversity 8, 9 and 13 contribute to Streptococcus pneumoniae virulence. BMC Microbiol. 7, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, and Henson PM (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216. [PubMed] [Google Scholar]
- Feldman C, and Anderson R (2016). The Role of Streptococcus pneumoniae in Community-Acquired Pneumonia. Semin. Respir. Crit. Care Med. 37, 806–818. [DOI] [PubMed] [Google Scholar]
- García-Suárez, Mdel M, Cima-Cabal MD, Flórez N, García P, Cernuda-Cernuda R, Astudillo A, Vázquez F, De los Toyos JR, and Mèndez FJ (2004). Protection against pneumococcal pneumonia in mice by monoclonal antibodies to pneumolysin. Infect. Immun. 72, 4534–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genschmer KR, Accavitti-Loper MA, and Briles DE (2013). A modified surface killing assay (MSKA) as a functional invitro assay for identifying protective antibodies against pneumococcal surface protein A (PspA). Vaccine 32, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford JL, Hunter HN, and Vogel HJ (2005). Lactoferricin: a lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell. Mol. Life Sci. 62, 2588–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover DT, Hollingshead SK, and Briles DE (2008). Streptococcus pneumoniae surface protein PcpA elicits protection against lung infection and fatal sepsis. Infect. Immun. 76, 2767–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzáilez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, and Orihuela CJ (2015). Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 11, e1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzáilez-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, Restrepo MI, Dube PH, Bergman MA, and Orihuela CJ (2017). Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 24, 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzáilez-Juarbe N, Bradley KM, Riegler AN, Reyes LF, Brissac T, Park SS, Restrepo MI, and Orihuela CJ (2018). Bacterial Pore-Forming Toxins Promote the Activation of Caspases in Parallel to Necroptosis to Enhance Alarmin Release and Inflammation During Pneumonia. Sci. Rep. 8, 5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Juarbe N, Riegler AN, Jureka AS, Gilley RP, Brand JD, Trombley JE, Scott NR, Platt MP, Dube PH, Petit CM, et al. (2020). Influenza-Induced Oxidative Stress Sensitizes Lung Cells to Bacterial-Toxin-Mediated Necroptosis. Cell Rep. 32, 108062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene CJ, Marks LR, Hu JC, Reddinger R, Mandell L, Roche-Hakansson H, King-Lyons ND, Connell TD, and Hakansson AP (2016). Novel Strategy To Protect against Influenza Virus-Induced Pneumococcal Disease without Interfering with Commensal Colonization. Infect. Immun. 84, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håkansson A, Roche H, Mirza S, McDaniel LS, Brooks-Walter A, and Briles DE (2001). Characterization of binding of human lactoferrin to pneumococcal surface protein A. Infect. Immun. 69, 3372–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatcher BL, Hale JY, and Briles DE (2016). Free Sialic Acid Acts as a Signal That Promotes Streptococcus pneumoniae Invasion of Nasal Tissue and Nonhematogenous Invasion of the Central Nervous System. Infect. Immun. 84, 2607–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingshead SK, Becker R, and Briles DE (2000). Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect. Immun. 68, 5889–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanclerski K, and Möllby R (1987). Production and purification of Streptococcus pneumoniae hemolysin (pneumolysin). J. Clin. Microbiol. 25, 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Takeuchi K, Inoue K, and Umeda M (1997). Localization ofthe phosphatidylserine-binding site of glyceraldehyde-3-phosphate dehydrogenase responsible for membrane fusion. J. Biochem. 122, 1233–1240. [DOI] [PubMed] [Google Scholar]
- Khan MN, Sharma SK, Filkins LM, and Pichichero ME (2012). PcpA of Streptococcus pneumoniae mediates adherence to nasopharyngeal and lung epithelial cells and elicits functional antibodies in humans. Microbes Infect. 14, 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King QO, Lei B, and Harmsen AG (2009). Pneumococcal surface protein A contributes to secondary Streptococcus pneumoniae infection after influenza virus infection. J. Infect. Dis. 200, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMessurier KS, Tiwary M, Morin NP, and Samarasinghe AE (2020). Respiratory Barrier as a Safeguard and Regulator of Defense Against Influenza A Virus and Streptococcus pneumoniae. Front. Immunol 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro B, and Sanz JM (2016). Choline Binding Proteins from Streptococcus pneumoniae: A Dual Role as Enzybiotics and Targets for the Design of New Antimicrobials. Antibiotics (Basel) 5, E21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, and Green DR (1995). Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 182, 1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA (2006). Insights intothe interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 19, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel LS, Sheffield JS, Delucchi P, and Briles DE (1991). PspA, a surface protein of Streptococcus pneumoniae, is capable of eliciting protection against pneumococci of more than one capsular type. Infect. Immun. 59, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens DM, Taubenberger JK, and Fauci AS (2008). Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198, 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukerji R, Mirza S, Roche AM, Widener RW, Croney CM, Rhee DK, Weiser JN, Szalai AJ, and Briles DE (2012). Pneumococcal surface protein A inhibits complement deposition on the pneumococcal surface by competing with the binding of C-reactive protein to cell-surface phosphocholine. J. Immunol. 189, 5327–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musher DM, Phan HM, and Baughn RE (2001). Protection against bacteremic pneumococcal infection by antibody to pneumolysin. J. Infect. Dis. 183, 827–830. [DOI] [PubMed] [Google Scholar]
- Novick S, Shagan M, Blau K, Lifshitz S, Givon-Lavi N, Grossman N, Bodner L, Dagan R, and Mizrachi Nebenzahl Y (2017). Adhesion and invasion of Streptococcus pneumoniae to primary and secondary respiratory epithelial cells. Mol. Med. Rep. 15, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunniyi AD, Folland RL, Briles DE, Hollingshead SK, and Paton JC (2000). Immunization of mice with combinations of pneumococcal virulence proteins elicits enhanced protection against challenge with Streptococcus pneumoniae. Infect. Immun. 68, 3028–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orihuela CJ, Mahdavi J, Thornton J, Mann B, Wooldridge KG, Abouseada N, Oldfield NJ, Self T, Ala’Aldeen DA, and Tuomanen EI (2009). Laminin receptor initiates bacterial contact with the blood brain barrier in experimental meningitis models. J. Clin. Invest. 119, 1638–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ, Possemato R, Chen WW, Sullivan LB, Fiske BP, et al. (2016). A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 12, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios G, Hornig M, Cisterna D, Savji N, Bussetti AV, Kapoor V, Hui J, Tokarz R, Briese T, Baumeister E, and Lipkin WI (2009). Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS ONE 4, e8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-S, Gonzalez-Juarbe N, Martínez E, Hale JY, Lin Y-H, Huffines JT, Kruckow KL, Briles DE, and Orihuela CJ (2021). Streptococcus pneumoniae binds to host lactate dehydrogenase via PspA and PspC to enhance virulence. mBio 12, e00673–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S, Williams CM, Salmon SL, Bonin JL, Metzger DW, and Furuya Y (2019). Evaluation of Pneumococcal Surface Protein A as a Vaccine Antigen against Secondary Streptococcus pneumoniae Challenge during Influenza A Infection. Vaccines (Basel) 7, E146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd JM, Ashar HK, Chow VT, and Teluguakula N (2016). Lethal Synergism between Influenza and Streptococcus pneumoniae. J. Infect. Pulm. Dis. 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender V, Hentrich K, Pathak A, Tan Qian Ler A, Embaie BT, Lundström SL, Gaetani M, Bergstrand J, Nakamoto R, Sham LT, et al. (2020). Capillary leakage provides nutrients and antioxidants for rapid pneumococcal proliferation in influenza-infected lower airways. Proc. Natl. Acad. Sci. USA 117,31386–31397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkovich O, Cook WJ, Mirza S, Hollingshead SK, Protasevich II, Briles DE, and Chattopadhyay D (2007). Structure of a complex of human lactoferrin N-lobe with pneumococcal surface protein a provides insight into microbial defense mechanism. J. Mol. Biol. 370, 701–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaper M, Hollingshead SK, Benjamin WH Jr., and Briles DE (2004). PspA protects Streptococcus pneumoniae from killing by apolactoferrin, and antibody to PspA enhances killing of pneumococci by apolactoferrin [corrected]. Infect. Immun. 72, 5031–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy AT, Brissac T, Gilley RP, Kumar N, Wang Y, Gonzalez-Juarbe N, Hinkle WS, Daugherty SC, Shetty AC, Ott S, et al. (2017). Streptococcus pneumoniae in the heart subvert the host response through biofilmmediated resident macrophage killing. PLoS Pathog. 13, e1006582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SJ, Roche AM, and Weiser JN (2014). Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe 16, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, and Metzger DW (2008). Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 14, 558–564. [DOI] [PubMed] [Google Scholar]
- Swiatlo E, King J, Nabors GS, Mathews B, and Briles DE (2003). Pneumococcal surface protein A is expressed in vivo, and antibodies to PspA are effective for therapy in a murine model of pneumococcal sepsis. Infect. Immun. 71, 7149–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkington DF, Crimmins DL, Voellinger DC, Yother J, and Briles DE (1991).A 43-kilodalton pneumococcal surface protein, PspA: isolation, protective abilities, and structural analysis of the amino-terminal sequence. Infect. Immun. 59, 1285–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong HH, Liu X, Chen Y, James M, and Demaria T (2002). Effect of neuraminidase on receptor-mediated adherence of Streptococcus pneumoniae to chinchilla tracheal epithelium. Acta Otolaryngol. 122, 413–419. [DOI] [PubMed] [Google Scholar]
- Vögele M, Bhaskara RM, Mulvihill E, van Pee K, Yildiz Ö, Kühlbrandt W, Müller DJ, and Hummer G (2019). Membrane perforation by the pore-forming toxin pneumolysin. Proc. Natl. Acad. Sci. USA 116, 13352–13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MM, Novak L, Widener R, Grubbs JA, King J, Hale JY, Ochs MM, Myers LE, Briles DE, and Deshane J (2016). PcpA Promotes Higher Levels of Infection and Modulates Recruitment of Myeloid-Derived Suppressor Cells during Pneumococcal Pneumonia. J. Immunol. 196, 2239–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, and Lu C (2007). Adhesion activity ofglyceraldehyde-3-phosphate dehydrogenase in a Chinese Streptococcus suis type 2 strain. Berl. Munch. Tierarztl. Wochenschr. 120, 207–209. [PubMed] [Google Scholar]
- Weinberger DM, Simonsen L, Jordan R, Steiner C, Miller M, and Viboud C (2012). Impact of the 2009 influenza pandemic on pneumococcal pneumonia hospitalizations in the United States. J. Infect. Dis. 205, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yother J, and White JM (1994). Novel surface attachment mechanism of the Streptococcus pneumoniae protein PspA. J. Bacteriol. 176, 2976–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YY, Tang XF, Du CH, Wang BB, Bi ZW, and Dong BR (2016). Comparison of dual influenza and pneumococcal polysaccharide vaccination with influenza vaccination alone for preventing pneumonia and reducing mortality among the elderly: A meta-analysis. Hum. Vaccin. Immunother. 12,3056–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Wang Q, Yun C, Wang Y, Smith WW, and Leng J (2014). Identification of glyceraldehyde 3-phosphate dehydrogenase sequence and expression profiles in tree shrew (Tupaia belangeri). PLoS ONE 9, e98552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.